
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeeking potent anti-tubercular agents: design and synthesis of substituted-N-(6-(4-(pyrazine-2-carbonyl)piperazine/homopiperazine-1-yl)pyridin-3-yl)benzamide derivatives as anti-tubercular agents†
Singireddi Srinivasaraoa,
Adinarayana Nandikollaa,
Amaroju Suresha,
Kevin Van Calsterb,
Linda De Voogtb,
Davie Cappoenb,
Balaram Ghoshc,
Himanshu Aggarwala,
Sankaranarayanan Murugesand and
Kondapalli Venkata Gowri Chandra Sekhar *a
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Hyderabad-500078, Telangana, India. E-mail: kvgc@hyderabad.bits-pilani.ac.in; kvgcs.bits@gmail.com; Tel: +91 40 66303527
bDepartment of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, B-9000, Ghent, Belgium
cDepartment of Pharmacy, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Hyderabad-500078, Telangana, India
dMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science, Pilani, 333031, India
First published on 25th March 2020
Abstract
Pyrazinamide is an important first-line drug used in shortening TB therapy. In our current work, a series of novel substituted-N-(6-(4-(pyrazine-2-carbonyl)piperazine/homopiperazine-1-yl)pyridin-3-yl)benzamide derivatives were designed, synthesized, and evaluated for their anti-tubercular activity against Mycobacterium tuberculosis H37Ra. Among the tested compounds, five compounds (6a, 6e, 6h, 6j and 6k) from Series-I and one compound (7e) from Series-II exhibited significant activity against Mycobacterium tuberculosis H37Ra with 50% inhibitory concentrations (IC50) ranging from 1.35 to 2.18 μM. To evaluate the efficacy of these compounds, we examined their IC90 values. Five of the most active compounds were found to be more active with IC90s ranging from 3.73 to 4.00 μM and one compound (6e) showed an IC90 of 40.32 μM. Moreover, single crystals were developed for 6d, 6f and 6n. In addition, most active compounds were evaluated for their cytotoxicity on HEK-293 (human embryonic kidney) cells. Our results indicate that the compounds are nontoxic to human cells. The molecular interactions of the derivatised conjugates in docking studies reveal their suitability for further development.
1. Introduction
Tuberculosis (TB) is an infectious disease, caused by the Gram positive aerobic acid fast bacillus Mycobacterium tuberculosis (MTB) and it is one of the major cause of deaths. MDR-TB patients still develop resistance when treated with two of the most effective first-line drugs (rifampicin and isoniazid) along with second-line drugs such as the fluoroquinolones and injectable aminoglycoside or polypeptide drugs leading to extensively drug resistant TB. Hence there is an urgent need for new and effective anti-TB drugs.1,2 Pyrazinamide (PZA) is well-known front line prodrug and is used in the treatment of active TB. New WHO guidelines recommend PZA for isoniazid-resistant and rifampicin susceptible tuberculosis for six months.1 Reports reveal that the analogues of pyrazine and pyrazinamide can exhibit higher anti-TB activity against MTB.3–16 Substituted N-phenylpyrazine-2-carboxamides synthesized by Dolezal et al. were evaluated in vitro for anti-mycobacterial activity. It was shown that compound N-(4-(trifluoromethyl)phenyl)pyrazine-2-carboxamide (A), N-(2-bromo-3-methylphenyl)pyrazine-2-carboxamide (B) and N-(3-iodo-4-methylphenyl)pyrazine-2-carboxamide (C) exhibited four times higher activity (MIC ≤ 2 μg L−1) than the standard drug PZA (=8 μg L−1).17 5-(tert-Butyl)-6-chloro-N-(3-iodo-4-methylphenyl)pyrazine-2-carboxamide (D), another active compound also showed high anti-mycobacterial activity with an IC50 of 0.728 μg mL−1; IC90 = 0.819 μg mL−1 (PZA IC90 > 20 μg mL−1).17 Zhou et al. also reported PZA analogues of which N-(2-(piperazin-1-yl)ethyl)pyrazine-2-carboxamide (E) and N-(2-morpholinoethyl)pyrazine-2-carboxamide (F) were shown to exhibit MICs of 12.2 μg mL−1 and 8.0 μg mL−1 respectively against MTB H37Rv (Fig. 1).18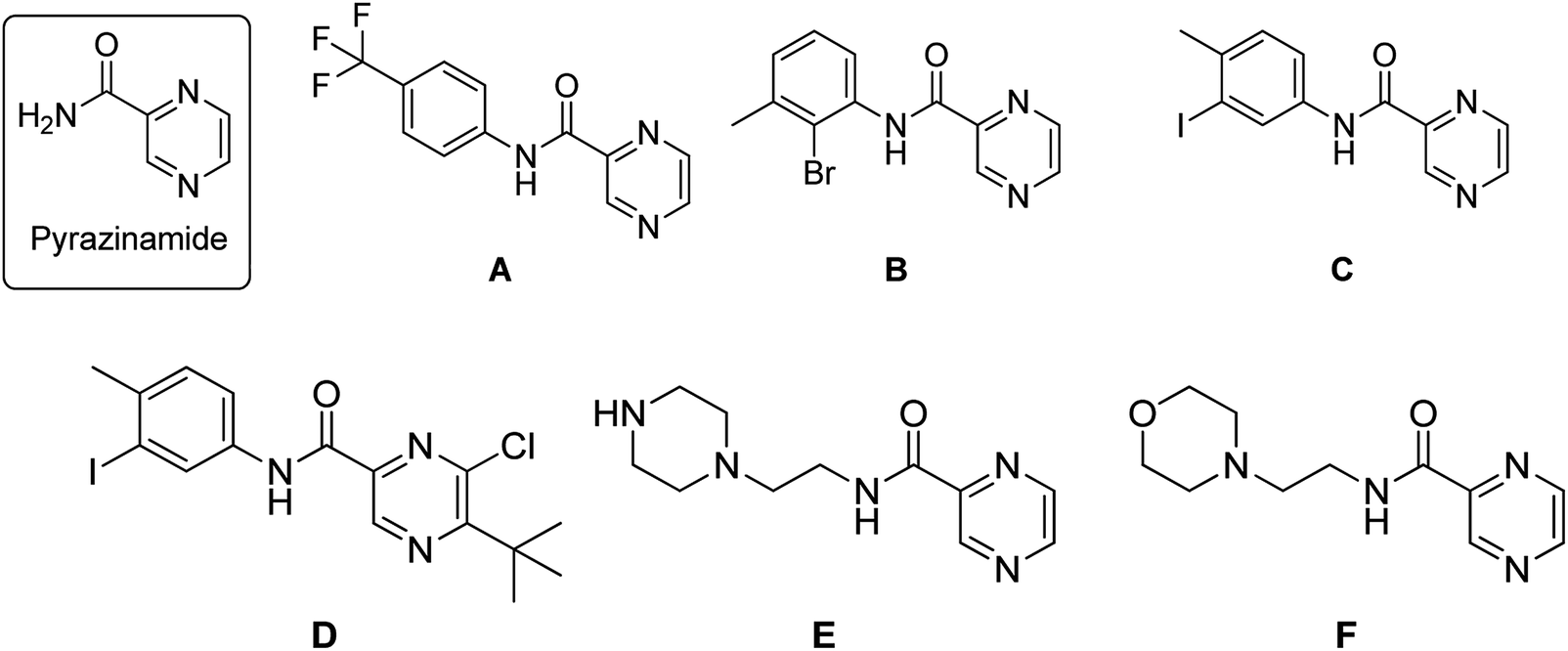 | ||
| Fig. 1 Pyrazinamide based anti-tubercular agents. | ||
Piperazine rings are a central core for several antibiotics which include ciprofloxacin, sparfloxacin, levofloxacin. PBTZ169 is a piperazine based clinical candidate which inhibits DprE1 and has synergic effect with either bedaquiline, clofazimine, delamanid or sutezolid.19 AX-35 is another piperazine based potent anti-TB agent with MIC 0.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μM against M. bovis BCG and MTB H37Rv targeting QcrB.20 Bobesh et. al., and Chandran et. al., reported several piperazine based active anti-tubercular agents of which 4-(2-(7-methoxy-2-oxo-1,5-naphthyridin-1-(2H)-yl)ethyl)-N-(4-nitrophenyl)piperazine-1-carboxamide21 and N-(4-chlorophenyl)-4-(6-nitro-4-oxo-4H-benzo[e][1,3]thiazin-2-yl)piperazine-1-carbothioamide18 were the most active compounds with IC50 0.29 μM and 0.51 μM in the DNA supercoiling assay and MTB MIC 3.45 μM and 4.41 μM respectively.21,22 In our previous work, we linked a piperazine moiety to the pyridine nucleus of phenanthridine and these compounds were shown to exhibit good to excellent anti-TB activity with MICs ranging from 1.56 μg mL−1 to 50 μg mL−1. Compounds 6-(4-((1-(phenylsulfonyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)phenanthridine (G) and 6-(4-(pyridin-2-yl)piperazin-1-yl)phenanthridine (H) were the most active compounds with MTB MIC 1.56 μg mL−1 against H37Rv.23,24 Compounds I and J are homo-piperazine based anti-TB agents with MIC 1.56 and 24.03 μM respectively25,26 (Fig. 2). Few nicotine and nicotinamide based anti-TB agents are also reported in literature.27–30 Recently Reddyrajula et. al., reported several 1,2,3-triazole based pyrazine, pyrazinamide and nicotinamide analogues as good anti-TB agents and most of the compounds exhibited promising activity with MIC 1.56 μg mL−1 to 25.0 μg mL−1.30 Compound K is one of the most active nicotinamide analogue with MTB MIC 1.56 μg mL−1 (Fig. 3).
μM against M. bovis BCG and MTB H37Rv targeting QcrB.20 Bobesh et. al., and Chandran et. al., reported several piperazine based active anti-tubercular agents of which 4-(2-(7-methoxy-2-oxo-1,5-naphthyridin-1-(2H)-yl)ethyl)-N-(4-nitrophenyl)piperazine-1-carboxamide21 and N-(4-chlorophenyl)-4-(6-nitro-4-oxo-4H-benzo[e][1,3]thiazin-2-yl)piperazine-1-carbothioamide18 were the most active compounds with IC50 0.29 μM and 0.51 μM in the DNA supercoiling assay and MTB MIC 3.45 μM and 4.41 μM respectively.21,22 In our previous work, we linked a piperazine moiety to the pyridine nucleus of phenanthridine and these compounds were shown to exhibit good to excellent anti-TB activity with MICs ranging from 1.56 μg mL−1 to 50 μg mL−1. Compounds 6-(4-((1-(phenylsulfonyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)phenanthridine (G) and 6-(4-(pyridin-2-yl)piperazin-1-yl)phenanthridine (H) were the most active compounds with MTB MIC 1.56 μg mL−1 against H37Rv.23,24 Compounds I and J are homo-piperazine based anti-TB agents with MIC 1.56 and 24.03 μM respectively25,26 (Fig. 2). Few nicotine and nicotinamide based anti-TB agents are also reported in literature.27–30 Recently Reddyrajula et. al., reported several 1,2,3-triazole based pyrazine, pyrazinamide and nicotinamide analogues as good anti-TB agents and most of the compounds exhibited promising activity with MIC 1.56 μg mL−1 to 25.0 μg mL−1.30 Compound K is one of the most active nicotinamide analogue with MTB MIC 1.56 μg mL−1 (Fig. 3).
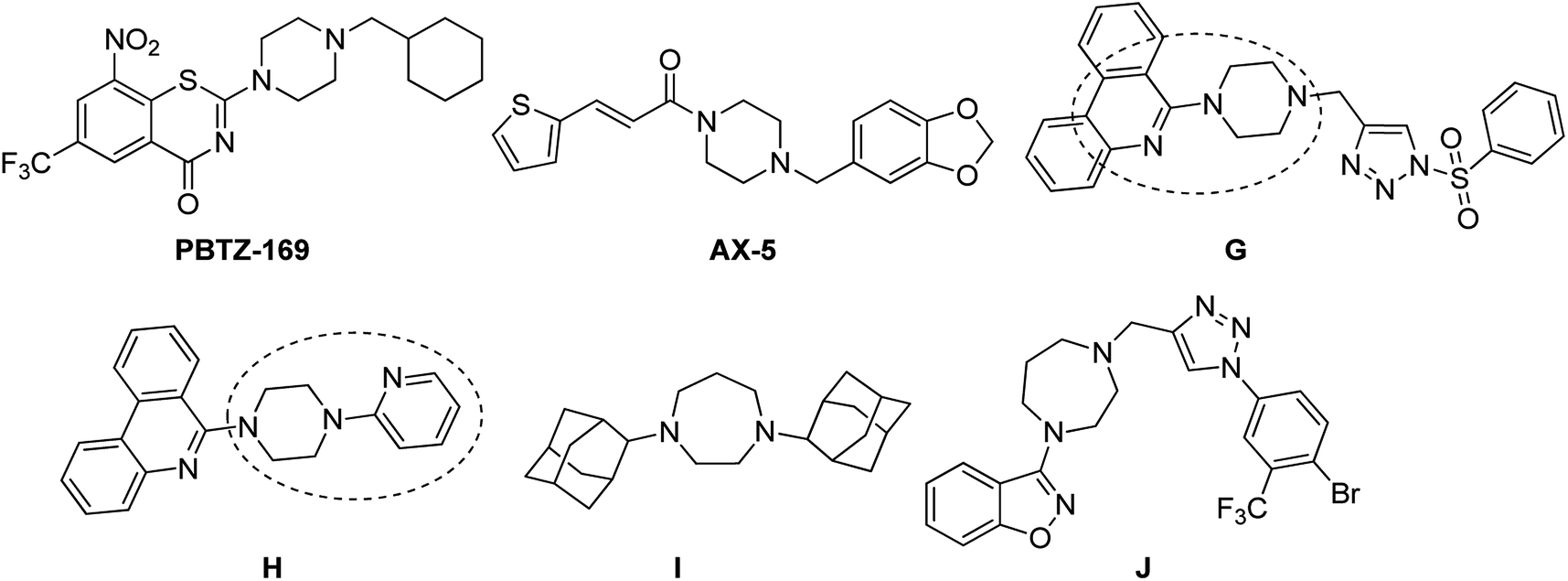 | ||
| Fig. 2 Piperazine/homopiperazine based anti-tubercular agents. | ||
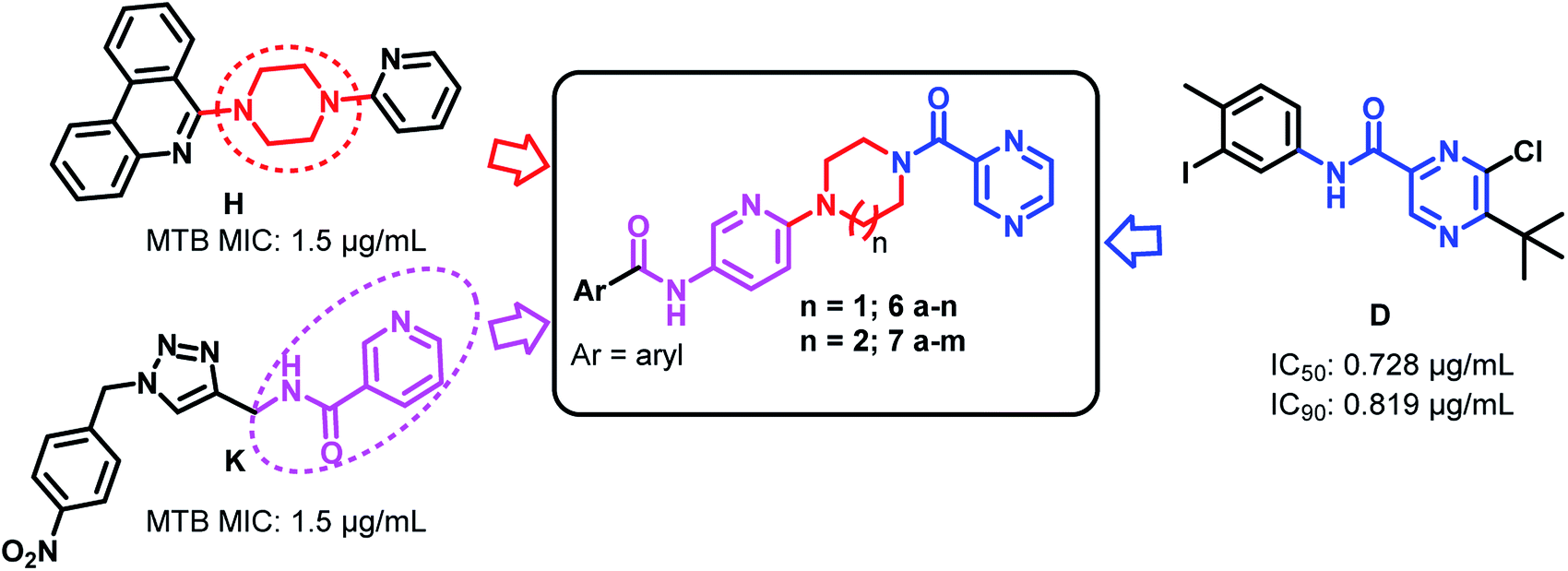 | ||
| Fig. 3 Design of the titled compounds 6a–n and 7a–m. | ||
In our present work, we focused our efforts towards a synthetic strategy involving the incorporation of the active pharmacophore units of pyrazinamide, piperazine/homopiperazine and pyridine (Fig. 3) into one single molecule.
2. Results and discussion
Synthesis
As shown in Scheme 1, treatment of 2-chloro-5-nitropyridine (1) with 1-Boc-piperazine/1-Boc-homopiperazine at 110 °C yielded compounds 2a and 2b.29 Intermediate compounds 2a and 2b were dissolved in CH2Cl2 and then treated with 4 N HCl in dioxane to get BOC protected compounds 3a and 3b. The amine compounds 3a and 3b were coupled with pyrazinoic acid using EDC·HCl, HOBt, N,N-diisopropylethylamine in DMF to yield compounds 4a and 4b respectively. Next, we prepared pre-final compounds 5a and 5b by reducing the nitro group of 4a and 4b with hydrogen gas in the presence of Pd catalyst. Finally, we performed acid–amine coupling by treating amines compounds 5a and 5b with different substituted benzoic acid, EDC·HCl, HOBt and N,N-diisopropylethylamine in DMF to yield the title compounds 6a–n and 7a–m respectively.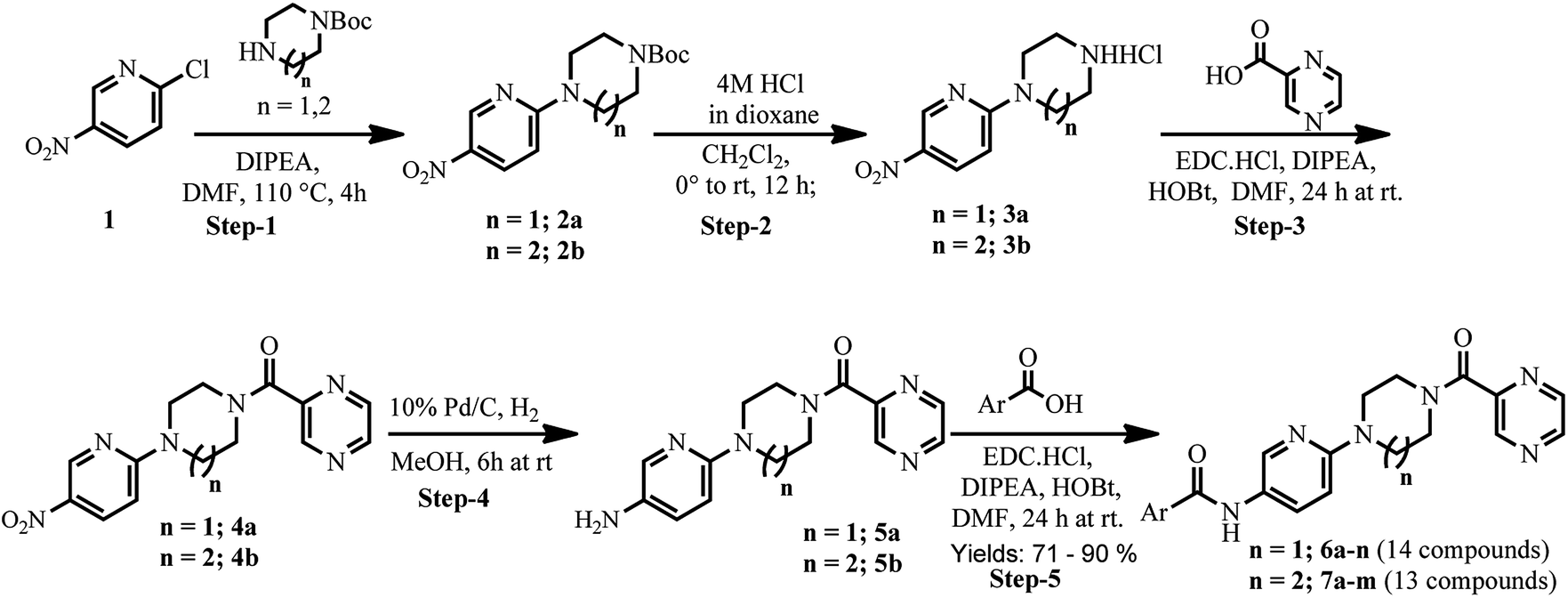 | ||
| Scheme 1 Synthesis of titled compounds 6a–n and 7a–m. | ||
The purity of synthesized final compounds 6a–n and 7a–m was checked by LC-MS and elemental analyses. Structures of the compounds were confirmed by spectral data. In 1H NMR and 13C NMR, the signals of the corresponding protons and carbon atoms were verified on the basis of their chemical shifts, multiplicities, and coupling constants. The results of elemental analysis were within ±0.05 of the theoretical values.
1H NMR of compounds from Scheme 1 showed singlets in the range 9.5 to 11 ppm due to amide proton and another singlet of triazole nucleus was observed between 8.5 to 9 ppm. Further, we observed a multiplet of eight protons in the aliphatic region (3 to 4.5 ppm) for 6a–n and ten protons for compounds 7a–m, confirming the presence of piperazine and homopiperazine respectively. All the compounds showed two peaks between 160 to 170 ppm in 13C NMR indicating the presence of two amide carbons.
Biological activity
The pyrazinamide derivatives 6a–n and 7a–m were screened against the Gram-negative Escherichia coli, the Gram-positive Staphylococcus aureus and the acid-fast Mtb H37Ra. Results showed that the novel compounds maintained their specific antimicrobial activity against Mtb H37Ra as E. coli and S. aureus showed low susceptibility towards the final compounds, in contrast to the susceptibility of Mtb (Table 1). All compounds (6a–n and 7a–m) were screened for their in vitro anti-TB activity towards Mtb H37Ra.31 H37Ra is an attenuated tubercle bacillus strain closely related to the virulent type strain M. tuberculosis H37Rv. Several compounds significantly inhibited the growth at micro molar to submicromolar levels. We evaluated the IC50 and IC90 values for the final compounds. Among the twenty-seven compounds tested, eight compounds (6a, 6e, 6h, 6j, 6k and 7e) exhibited excellent anti-mycobacterial activity against Mtb H37Ra with IC50 ranging from 1.35 μM to 2.18 μM and IC90 ranging from 3.73 μM to 40.32 μM as shown in Table 1.| Entry | Ar | n | S. aureus IC50 (μM) | E. coli IC50 (μM) | M. tuberculosis H37Ra-lux | |
|---|---|---|---|---|---|---|
| IC50 (μM) | IC90 (μM) | |||||
| Series-I | ||||||
| 6a | 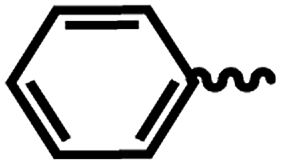 |
1 | >64 | >64 | 1.46 | 3.73 |
| 6b | 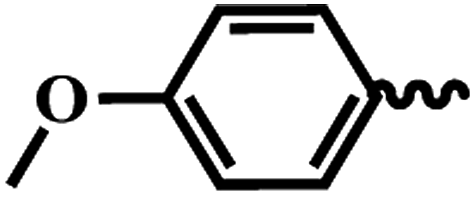 |
1 | >64 | >64 | >64 | >64 |
| 6c | 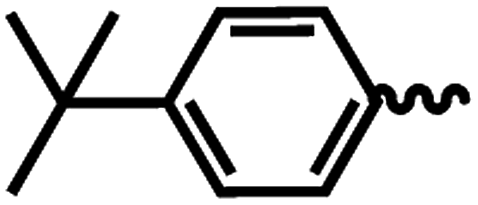 |
1 | >64 | >64 | >64 | >64 |
| 6d | 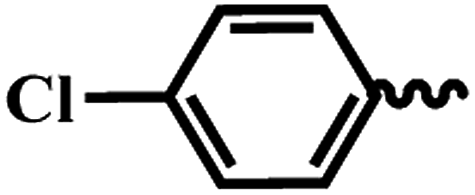 |
1 | >64 | >64 | >64 | >64 |
| 6e | 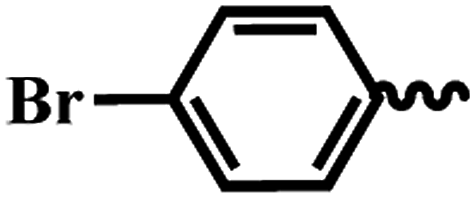 |
1 | >64 | >64 | 2.18 | 40.32 |
| 6f | 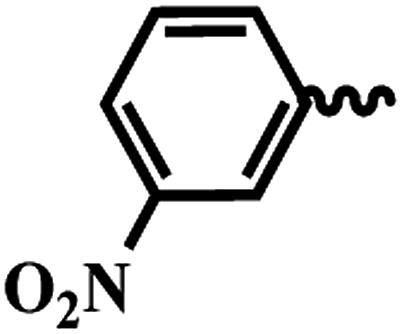 |
1 | >64 | >64 | >64 | >64 |
| 6g | 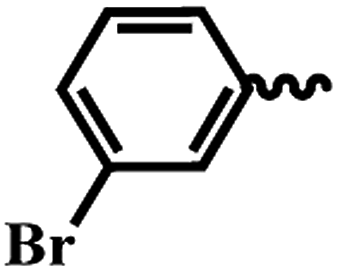 |
1 | >64 | >64 | >64 | >64 |
| 6h | 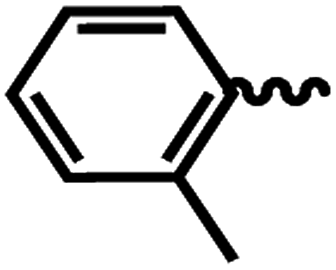 |
1 | >64 | >64 | 1.82 | 3.85 |
| 6i | 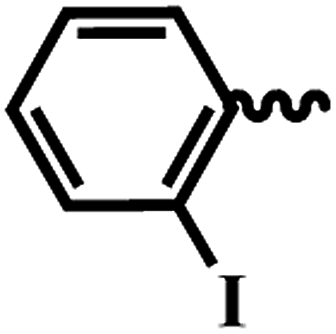 |
1 | >64 | >64 | >64 | >64 |
| 6j | 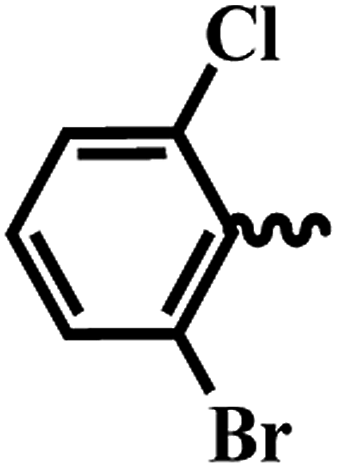 |
1 | >64 | >64 | 2.14 | 3.94 |
| 6k | 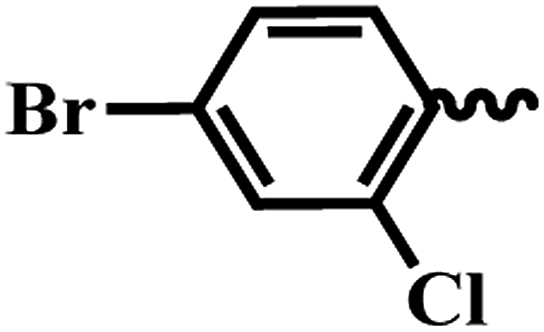 |
1 | >64 | >64 | 1.35 | 4.00 |
| 6l | 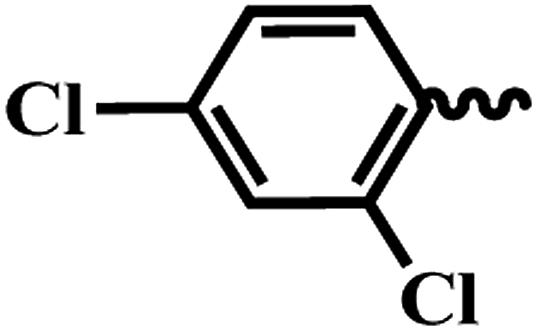 |
1 | >64 | >64 | >64 | >64 |
| 6m |  |
1 | >64 | >64 | >64 | >64 |
| 6n |  |
1 | >64 | >64 | >64 | >64 |
|
||||||
| Series-II | ||||||
| 7a |  |
2 | >64 | >64 | >64 | >64 |
| 7b |  |
2 | >64 | >64 | >64 | >64 |
| 7c | 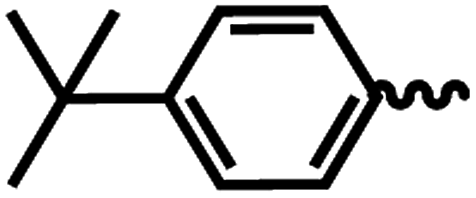 |
2 | >64 | >64 | >64 | >64 |
| 7d | 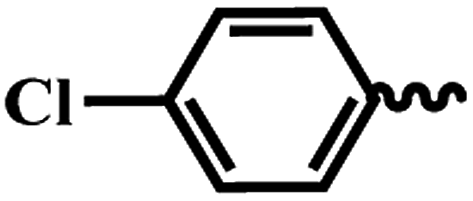 |
2 | >64 | >64 | >64 | >64 |
| 7e | 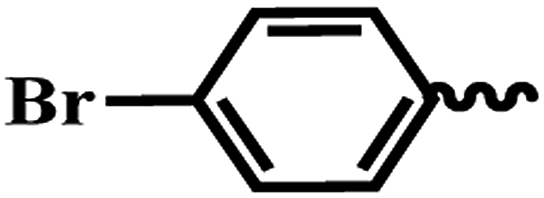 |
2 | >64 | >64 | 2.12 | 3.88 |
| 7f | 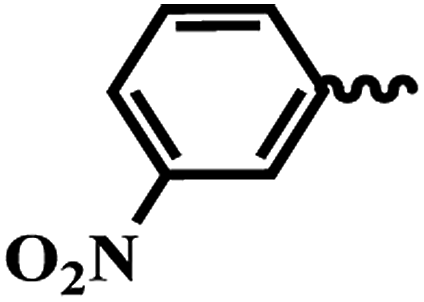 |
2 | >64 | >64 | >64 | >64 |
| 7g |  |
2 | >64 | >64 | >64 | >64 |
| 7h |  |
2 | >64 | >64 | >64 | >64 |
| 7i |  |
2 | >64 | >64 | >64 | >64 |
| 7j |  |
2 | >64 | >64 | >64 | >64 |
| 7k | 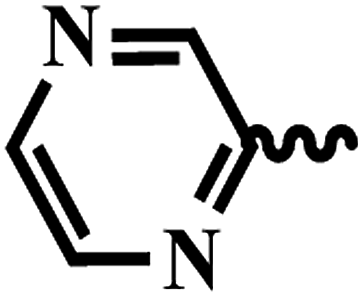 |
2 | >64 | >64 | >64 | >64 |
| 7l | 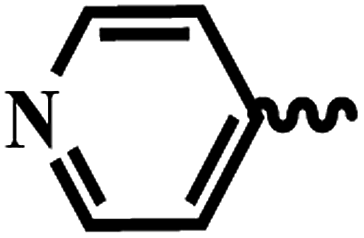 |
2 | >64 | >64 | >64 | >64 |
| 7m | 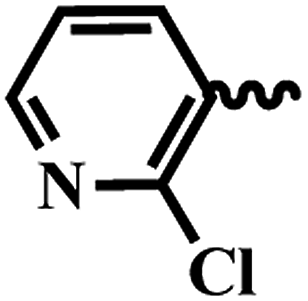 |
2 | >64 | >64 | >64 | >64 |
| Doxycyclin | — | — | 0.09 | — | — | — |
| Norfloxacin | — | — | — | 0.38 | — | — |
| Isoniazid | — | — | — | — | 0.013 | — |
SAR of substituted-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide/isonicotinamide/pyrazine-2-carboxamide (6a–n). Here, we evaluated fourteen compounds against M. tuberculosis H37Ra and SAR is explained on the basis of 6a. Compound 6a with no substituent on phenyl ring exhibited promising anti-TB activity with IC50 and IC90 1.46 μM and 3.73 μM respectively. We introduced a strong electron donating group (–OMe), alkyl group (t-butyl) and halogen (Cl & Br) at the para position (6b–e) and noticed that the bromo substituted compound 6e exhibited good anti-TB activity with an IC50 of 2.18 μM and a moderate IC90 of 40.32 μM. Meta-substituted nitro compound 6f and bromo-compound 6g were inactive. Ortho substituted compound 6h (methyl) is another potent anti-TB agent with IC50 and IC90 values of 1.82 μM and 3.82 μM respectively. Further, when we screened di-halogen substituted compounds 6j, 6k and 6l, two of the derivatives with different halogens (Cl and Br) (6j and 6k) exhibited good activity with MICs irrespective of their position while di-chloro compound (6l) was found to be inactive. With Cl and Br at ortho positions (6j), the IC50 and IC90 were 1.82 μM and 3.82 μM, whereas, bromo at para position and chloro at ortho position (6k) the IC50 and IC90 were 1.35 μM and 4.00 μM. When the phenyl group is replaced with a heterocyclic moiety (6m and 6n) it resulted in loss of activity. In conclusion, from this series (6a–n), we noticed that unsubstituted phenyl (6a), p-bromo (6e), and o-methyl (6h) are well tolerated and compounds with three groups exhibited best activity. Di-halo substituted phenyl derivatives (6j and 6k) were also found to be active against M. tuberculosis H37Ra.
SAR of substituted-N-(6-(4-(homopyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide/isonicotinamide/pyrazine-2-carboxamide (7a–m). Like piperazine analogues, various substituted homopiperazine analogues were also prepared. Ortho-, para- and meta-substituted derivatives, di-substituted halogen compounds and heterocyclic compounds were screened. Unfortunately, except the bromo-substituted compound (7e), all the other compounds were inactive (IC50 > 64 μM). In this series, para substituted bromo compound (7e) exhibited most potent activity with IC50 and IC90 values of 2.12 μM and 3.88 μM respectively.
Cytotoxicity studies
The most potent five compounds (6a, 6e, 6h, 6j and 6k) were subjected to cytotoxicity studies against normal human cell lines, human embryonic kidney (HEK) cells. Cell viability was measured by in vitro MTT assay.32 Cells were exposed to compound treatment at of the following concentrations; 2000 μM, 1000 μM, 500 μM, 250 μM, 125 μM, 62.5 μM, 31.25 μM, 15.62 μM and 7.81 μM to determine their IC50 values from the dose–response curve. Data represent mean values of measurements ± SD (Fig. 4).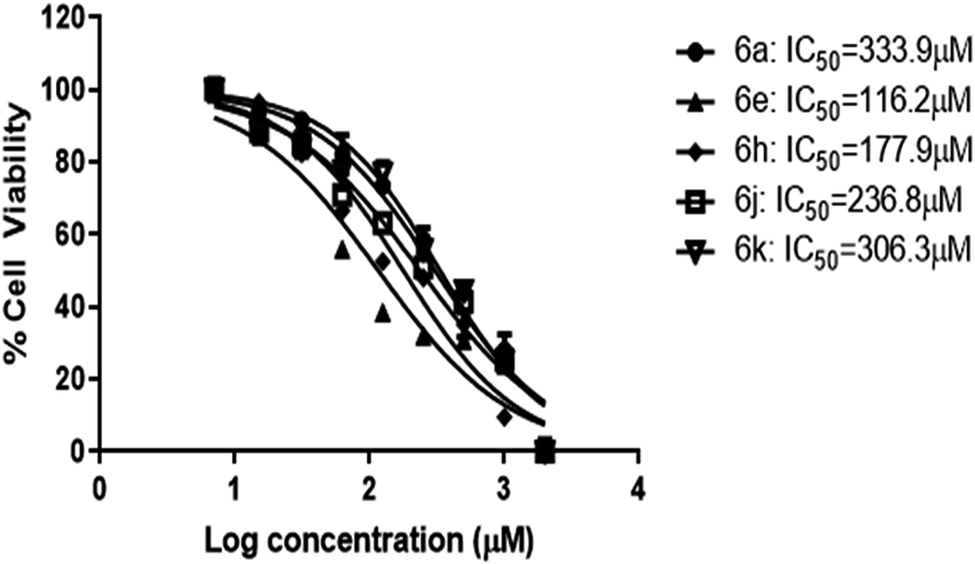 | ||
| Fig. 4 IC50 results of the compounds by MTT assay. HEK-293 cells were treated with the compounds at concentrations ranging from 7–2000 μM (n = 2) for 24 h. Data represents mean ± SD. | ||
Docking studies
Docking studies were carried out using Schrödinger software33 and the docking of significantly active molecules was performed using the Glide module34 in Schrödinger. All docking calculations were performed using Extra Precision (XP) mode. A scaling factor of 0.8 and a partial atomic charge of less than 0.15 was applied to the atoms of the protein. The Glide docking score was used to determine the best-docked structure from the output. The interactions of these docked complexes were investigated further using XP visualizer.In silico prediction of physico-chemical parameters
Physicochemical parameters of the designed compounds were in silico predicted using the QikProp module of Schrödinger. The different parameters predicted were; molecular weight (MW), total solvent accessible volume (TSAV), number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA), van der Waals polar surface area (PSA) of nitrogen and oxygen atoms, octanol/water partition coefficient (logP), aqueous solubility (logS), predicted apparent Caco-2 cell permeability in nm s−1 (PCaco), apparent Madin Darby Canine Kidney (MDCK) permeability and percentage of human oral absorption.
| S. no. | Code | MWa | Volumeb | Donor HBc | Accpt. HBd | PSAe |
|---|---|---|---|---|---|---|
| a Molecular weight, in Da (range for 95% of drugs: 130–725 Da).b Total solvent-accessible volume in cubic angstroms using a probe with a 1.4 Å radius (500–2000).c No. of hydrogen bonds donated by the molecule (range for 95% of drugs: 0–6).d No. of hydrogen bonds accepted by the molecule (range for 95% of drugs: 2–20).e van der Waals surface area of polar nitrogen and oxygen atoms (7–200). | ||||||
| 1 | 6a | 388.43 | 1237.83 | 1 | 9.5 | 100.903 |
| 2 | 6b | 418.45 | 1315.53 | 1 | 10.25 | 109.088 |
| 3 | 6c | 444.54 | 1449.71 | 1 | 9.5 | 103.189 |
| 4 | 6d | 422.87 | 1282.72 | 1 | 9.5 | 100.928 |
| 5 | 6e | 467.32 | 1291.59 | 1 | 9.5 | 100.922 |
| 6 | 6f | 433.43 | 1316.55 | 1 | 10.5 | 146.004 |
| 7 | 6g | 467.32 | 1293.06 | 1 | 9.5 | 101.176 |
| 8 | 6h | 402.46 | 1294.67 | 1 | 9.5 | 98.368 |
| 9 | 6i | 514.33 | 1300.64 | 1 | 9.5 | 99.719 |
| 10 | 6j | 501.77 | 1331.50 | 1 | 9.5 | 98.263 |
| 11 | 6k | 501.77 | 1333.22 | 1 | 9.5 | 99.503 |
| 12 | 6l | 457.32 | 1324.40 | 1 | 9.5 | 99.494 |
| 13 | 6m | 389.42 | 1225.41 | 1 | 11 | 113.971 |
| 14 | 6n | 390.40 | 1219.58 | 1 | 12 | 126.298 |
| 15 | 7a | 402.46 | 1254.20 | 1 | 9.5 | 101.174 |
| 16 | 7b | 446.51 | 1374.94 | 1 | 10.25 | 103.684 |
| 17 | 7c | 458.56 | 1452.16 | 1 | 9.5 | 98.396 |
| 18 | 7d | 436.90 | 1298.40 | 1 | 9.5 | 101.24 |
| 19 | 7e | 481.35 | 1307.04 | 1 | 9.5 | 101.25 |
| 20 | 7f | 447.45 | 1301.85 | 1 | 10.5 | 141.108 |
| 21 | 7g | 481.35 | 1279.93 | 1 | 9.5 | 96.051 |
| 22 | 7h | 528.35 | 1312.31 | 1 | 9.5 | 100.504 |
| 23 | 7i | 515.80 | 1348.48 | 1 | 9.5 | 100.169 |
| 24 | 7j | 471.35 | 1339.35 | 1 | 9.5 | 100.18 |
| 25 | 7k | 404.43 | 1209.67 | 1 | 12 | 122.659 |
| 26 | 7l | 403.44 | 1241.43 | 1 | 11 | 114.283 |
| 27 | 7m | 454.89 | 1310.31 | 1 | 9.5 | 99.284 |
| S. no. | Code | logPo/wa |
logSb |
PCacoc | PMDCKd | Percent human oral absorptione |
|---|---|---|---|---|---|---|
| a Predicted octanol/water partition co-efficient logP (acceptable range: −2.0 to 6.5).b Predicted aqueous solubility; S in mol L−1 (acceptable range: −6.5 to 0.5).c Apparent Caco-2 permeability (nm s−1) (<25 poor, >500 great).d Apparent MDCK permeability (nm s−1) (<25 poor, >500 great).e Percentage of human oral absorption (<25% poor and >80% is high). |
||||||
| 1 | 6a | 2.617 | −5.101 | 518.855 | 243.416 | 90.861 |
| 2 | 6b | 2.716 | −5.324 | 528.557 | 248.339 | 91.584 |
| 3 | 6c | 3.71 | −6.648 | 384.776 | 176.201 | 94.936 |
| 4 | 6d | 3.106 | −5.833 | 516.592 | 597.193 | 93.69 |
| 5 | 6e | 3.181 | −5.944 | 516.69 | 642.193 | 94.131 |
| 6 | 6f | 1.939 | −5.296 | 63.379 | 25.084 | 57.594 |
| 7 | 6g | 3.19 | −5.952 | 515.4 | 640.157 | 94.163 |
| 8 | 6h | 3.036 | −5.633 | 725.254 | 349.588 | 95.917 |
| 9 | 6i | 3.271 | −6 | 593.317 | 669.692 | 82.777 |
| 10 | 6j | 3.661 | −6.454 | 664.701 | 1490.213 | 85.946 |
| 11 | 6k | 3.706 | −6.608 | 647.779 | 1756.658 | 86.006 |
| 12 | 6l | 3.631 | −6.497 | 647.197 | 1632.012 | 100 |
| 13 | 6m | 1.668 | −4.415 | 278.843 | 124.409 | 80.479 |
| 14 | 6n | 1.052 | −4.03 | 184.188 | 79.466 | 73.651 |
| 15 | 7a | 2.717 | −4.828 | 525.517 | 246.796 | 91.549 |
| 16 | 7b | 3.212 | −5.149 | 906.183 | 444.739 | 100 |
| 17 | 7c | 3.972 | −6.094 | 859.818 | 420.196 | 100 |
| 18 | 7d | 3.202 | −5.552 | 525.206 | 607.987 | 94.385 |
| 19 | 7e | 3.275 | −5.66 | 523.982 | 652.031 | 94.791 |
| 20 | 7f | 1.984 | −4.505 | 105.505 | 43.514 | 61.814 |
| 21 | 7g | 3.249 | −5.191 | 873.968 | 1133.125 | 100 |
| 22 | 7h | 3.294 | −5.708 | 559.505 | 597.557 | 82.453 |
| 23 | 7i | 3.758 | −6.416 | 602.205 | 1614.986 | 85.746 |
| 24 | 7j | 3.682 | −6.301 | 601.965 | 1501.502 | 100 |
| 25 | 7k | 1.146 | −3.284 | 301.161 | 135.205 | 78.021 |
| 26 | 7l | 1.767 | −4.143 | 281.02 | 125.458 | 81.119 |
| 27 | 7m | 3.422 | −5.748 | 637.589 | 1033.633 | 100 |
 | ||
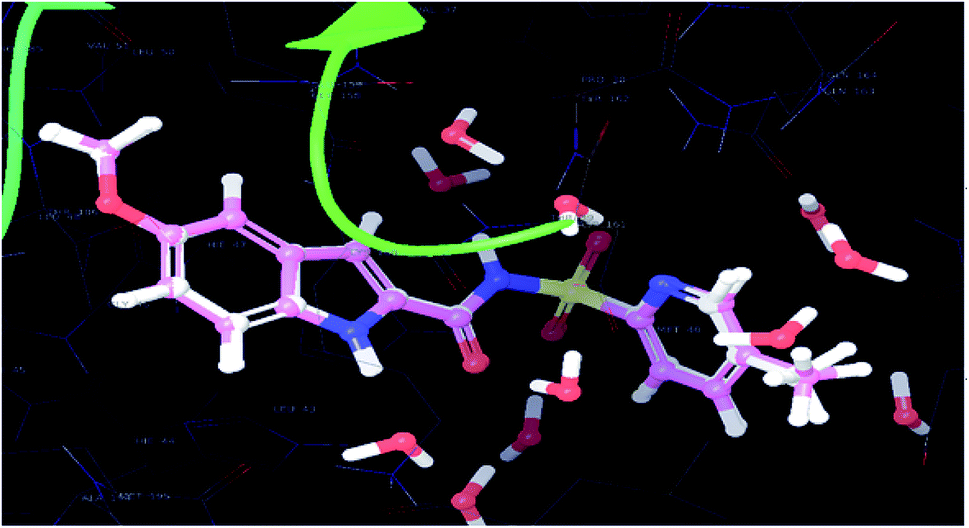
| Fig. 5 Superimposed view of the native pose of ligand (X-ray crystallized pose) and docked pose of the same ligand in the active site of the protein (3IUB) (root mean square deviation 0.22 Å) (color interpretation – pink color – binding pose after docking, white color – X-ray native pose of ligand). | ||
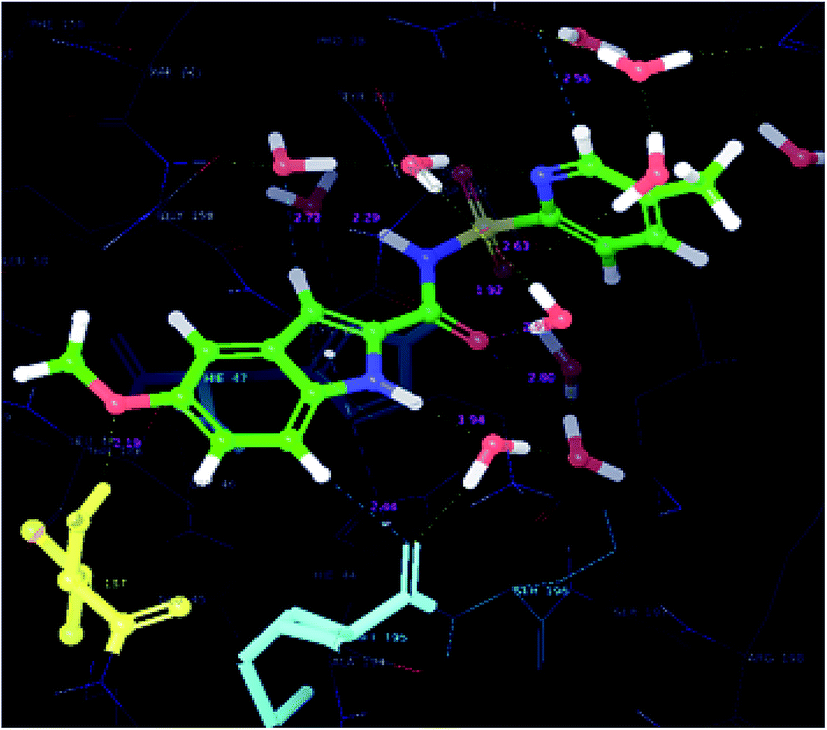
Results and discussion of the molecular docking studies
In order to find the putative binding mode of the significantly active molecule, a docking study was carried out and the docked poses were analyzed using XP visualizer. The docked values along with their interactions are discussed in the Table 4. By scrutinizing the 3D and 2D (Fig. 6 and 7) poses of the co-crystal ligand, water molecules played a vital role in the hydrogen bond formation and formed six bonds. Amino acid residues MET-40, HIS-47 and VAL-187, contributed their part in the hydrogen bond formation with the co-crystal ligand and showed a docking score and energy of −9.6 and −70.6 kcal mol−1, respectively. Apart from the hydrogen bond interactions, the co-crystal ligand also exhibited three aromatic bonds with the HIS-47, MET-195 and GLN-164 amino acid residues. HIS-44 amino acid residue revealed π–π stacking interaction with the co-crystal ligand.| S. no. | Code | Hydrogen-bond | Aromatic bond | π–π interactions | Glide score (kcal mol−1) | Glide energy (kcal mol−1) |
|---|---|---|---|---|---|---|
| 1 | Co-crystal ligand (FG-2), (PDB 3IUB) | MET-40, HIS-47, VAL-187, H2O bonds (6) | HIS-47, GLN-164, MET-195 | HIS-44 (π–π stacking) | −9.6 | −70.6 |
| 2 | 6k | H2O | THR-276 | ARG-198 | −2.3 | −32.10 |
 | ||
| Fig. 6 Docked pose of co-crystallized ligand and its interactions in the active site of the protein-3IUB (color interpretation yellow – hydrogen bond, blue – aromatic bond). | ||
 | ||
| Fig. 7 2D representation of the docked pose of the co-crystallized ligand (color interpretation magenta – hydrogen bond, green – π–π stacking). | ||
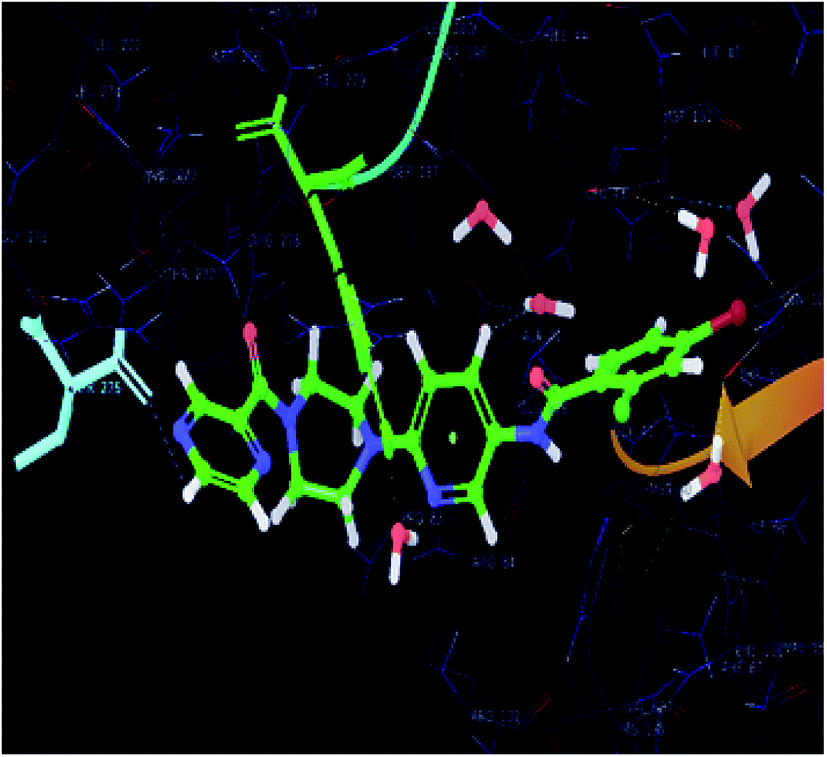
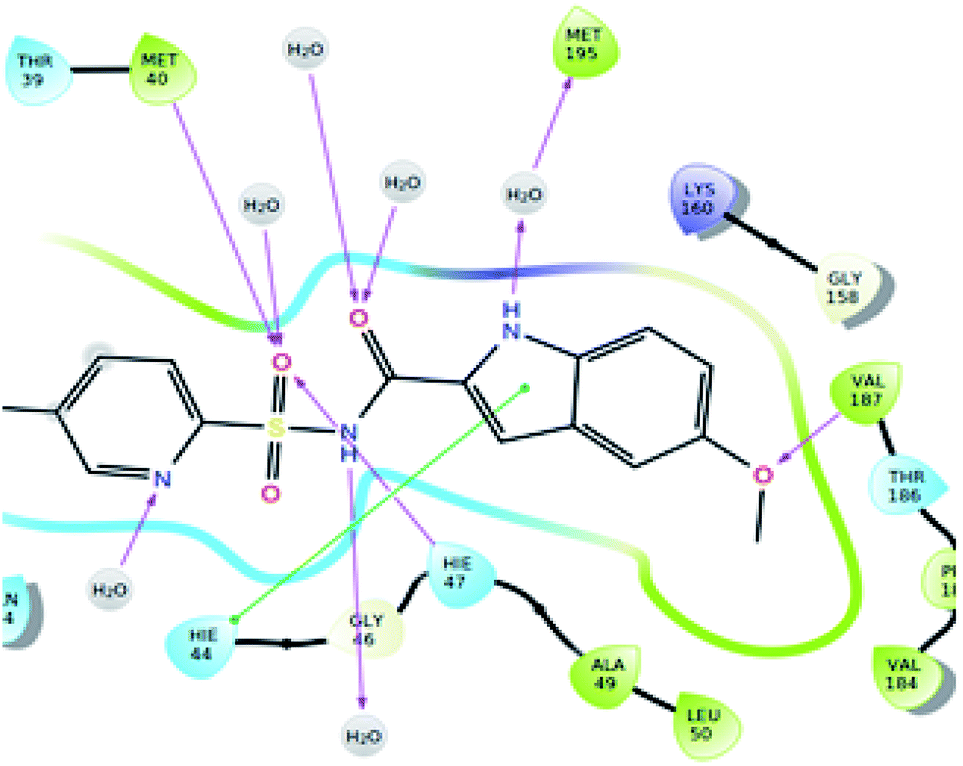
The significantly active compound 6k demonstrated a lower docking score when compared to the co-crystal ligand showing a docking score of −2.3 kcal mol−1 and docking energy of −32.10 kcal mol−1. 3D picture of the compound 6k (Fig. 8) showed the aromatic interaction with the amino acid residue THR-276. The pyridine nitrogen played an essential role in the hydrogen bond formation with the water molecule and in 2D representation (Fig. 9), chlorine atom which is present in the bromo benzene moiety actively participated in the halogen bond formation with a water molecule. In vitro studies of titled molecules revealed a correlation between the docking studies and most of the compounds were found to be less active.
 | ||
| Fig. 8 Collaboration of the significantly active compound 6k exhibiting various interactions in the active site of the protein (3IUB) (color interpretation yellow – hydrogen bond, blue – aromatic bond). | ||
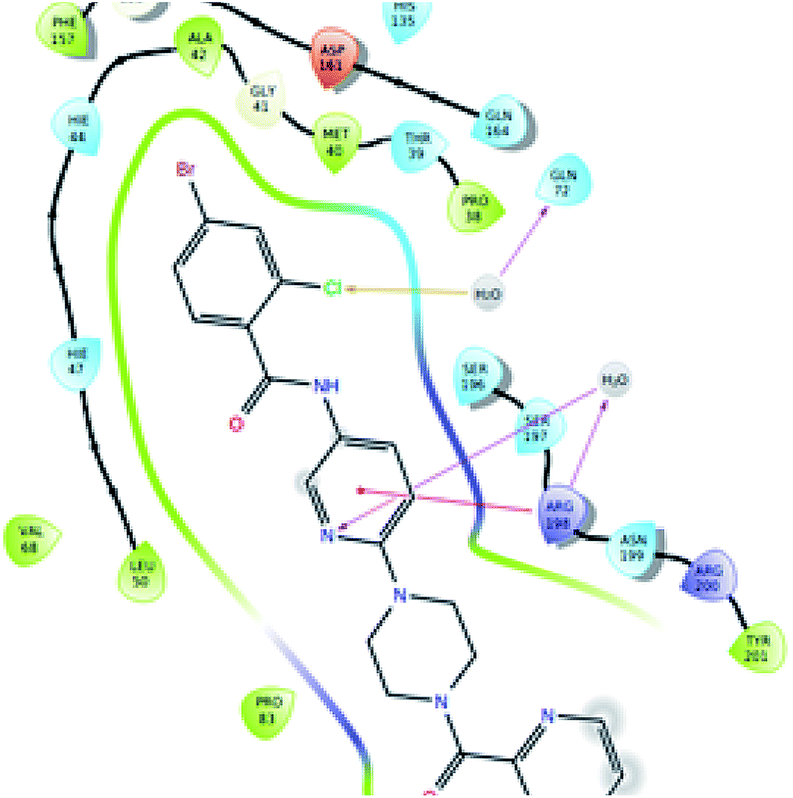 | ||
| Fig. 9 2D representation of the docked pose of the significantly active compound 6k (color interpretation magenta – hydrogen bond, red – π-cation and yellow – halogen bond). | ||
X-ray crystallographic studies
| Identification code | 6d | 6f | 6n |
| Empirical formula | C21H19ClN6O2 | C21H19N7O4 | C19H18N8O2 |
| Formula weight | 422.88 | 433.43 | 390.41 |
| Temperature/K | 100 | 100 | 100 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 5.69780(10) | 8.2628(2) | 30.7919(5) |
| b/Å | 21.2347(3) | 9.9451(2) | 5.80932(8) |
| c/Å | 16.1672(2) | 12.8729(3) | 9.90381(15) |
| α/° | 90 | 74.307(2) | 90 |
| β/° | 98.5610(10) | 74.059(2) | 96.4675(15) |
| γ/° | 90 | 79.348(2) | 90 |
| Volume/Å3 | 1934.29(5) | 972.22(4) | 1760.32(5) |
| Z | 4 | 2 | 4 |
| ρcalc g cm−3 | 1.4520 | 1.481 | 1.473 |
| μ/mm−1 | 2.024 | 0.890 | 0.843 |
| F(000) | 884.1 | 452.0 | 816.0 |
| Crystal size/mm3 | 0.2 × 0.1 × 0.05 | 0.13 × 0.1 × 0.05 | 0.2 × 0.1 × 0.05 |
| Radiation | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| 2Θ range for data collection/° | 8.32 to 160.48 | 9.304 to 158.86 | 8.67 to 159.206 |
| Index ranges | −6 ≤ h ≤ 5 | −6 ≤ h ≤ 10 | −38 ≤ h ≤ 38 |
| −26 ≤ k ≤ 24 | −12 ≤ k ≤ 12 | −5 ≤ k ≤ 7 | |
| −20 ≤ l ≤ 20 | −11 ≤ l ≤ 16 | −12 ≤ l ≤ 12 | |
| Reflections collected | 10531 |
10168 |
9609 |
| Independent reflections | 4009 [Rint = 0.0330, Rsigma = 0.0373] | 4089 [Rint = 0.0215, Rsigma = 0.0248] | 3735 [Rint = 0.0264, Rsigma = 0.0334] |
| Data/restraints/parameters | 4009/0/271 | 4089/0/289 | 3735/0/316 |
| Goodness-of-fit on F2 | 1.028 | 1.061 | 1.028 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0419, wR2 = 0.1168 | R1 = 0.0365, wR2 = 0.0980 | R1 = 0.0430, wR2 = 0.1147 |
| Final R indexes [all data] | R1 = 0.0448, wR2 = 0.1189 | R1 = 0.0382, wR2 = 0.0996 | R1 = 0.0461, wR2 = 0.1173 |
The structure of 6n crystallizes in monoclinic P21/c space group with C19H18N8O2 empirical formula. The terminal pyrazine ring in the structure is disordered over 2 positions with 0.60 and 0.40 occupancies. Moreover, the carbonyl oxygen adjacent to this pyrazine ring is also disordered over 2 positions with 0.34 and 0.66 occupancies (Fig. 10). However, such disorder was not observed in the case of compounds 6d and 6f with the empirical formulas C22H20N2O2Cl and C21H19N7O4 and, respectively.
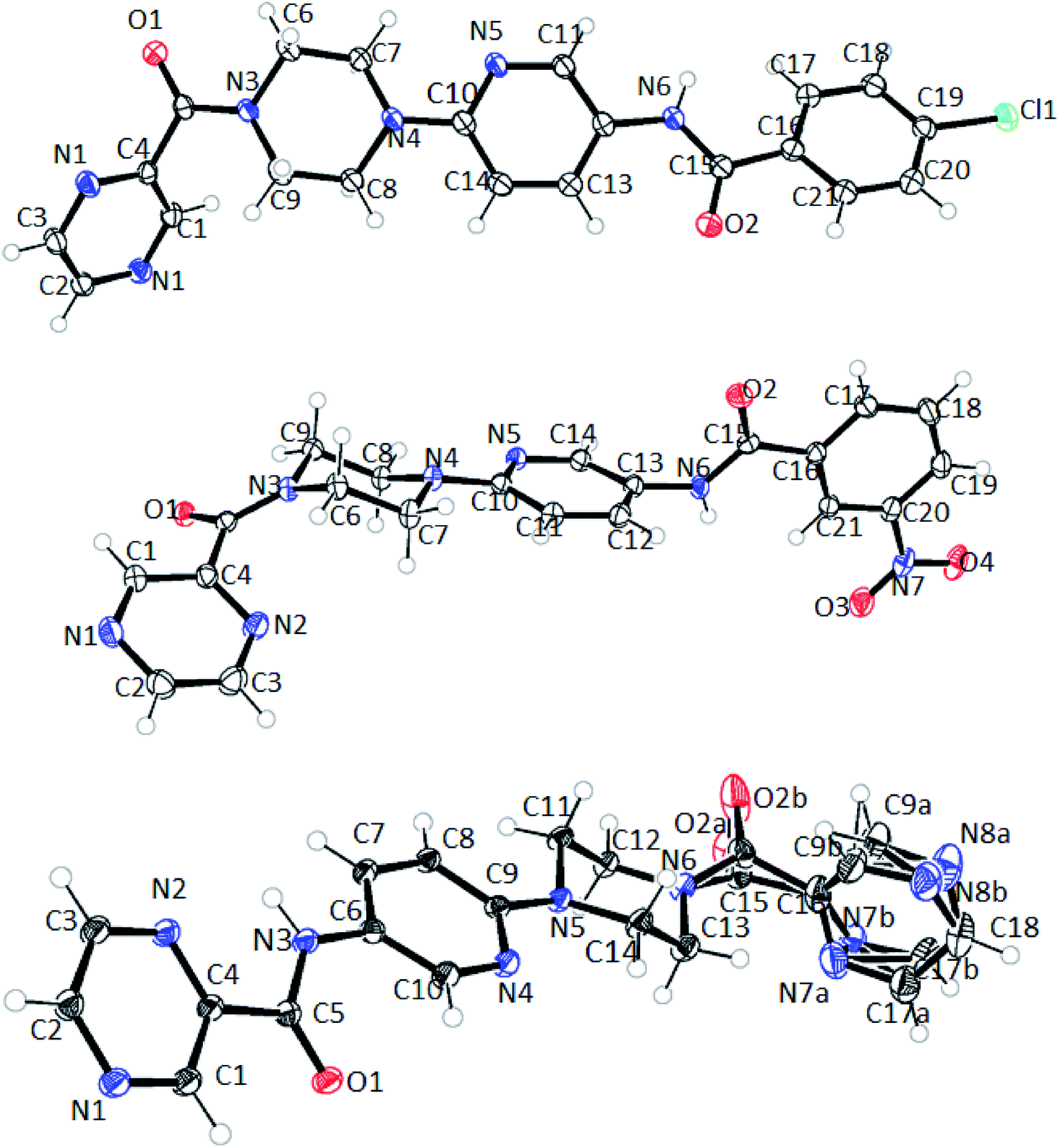 | ||
| Fig. 10 ORTEP diagram of compound 6d (top), 6f (middle) and 6n (bottom). | ||
Interestingly, compounds 6d and 6f show intermolecular H-bonding through amidic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups. Compound P9 6d displays intermolecular H-bonding. However, in this case each molecule is H-bonded to two more molecules through amidic oxygen and nitrogen atoms whereby amidic oxygen is H-bonded to one molecule whereas the amidic N–H is H-bonded to a second molecule. This results in each molecule being H-bonded to two other molecules as shown in Fig. 11. The H-bonding distance in this case is about 2.99 Å.
O and N–H groups. Compound P9 6d displays intermolecular H-bonding. However, in this case each molecule is H-bonded to two more molecules through amidic oxygen and nitrogen atoms whereby amidic oxygen is H-bonded to one molecule whereas the amidic N–H is H-bonded to a second molecule. This results in each molecule being H-bonded to two other molecules as shown in Fig. 11. The H-bonding distance in this case is about 2.99 Å.
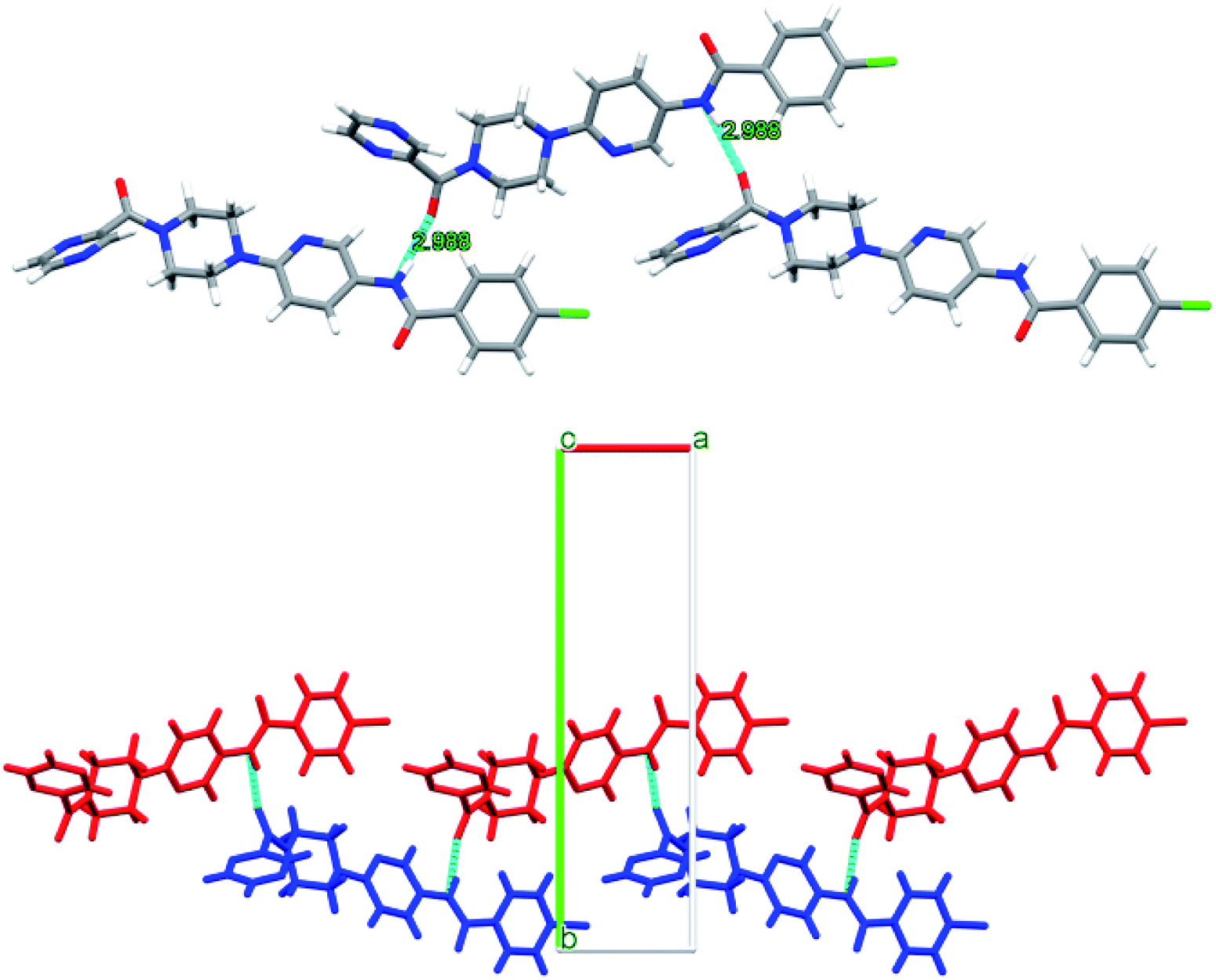 | ||
| Fig. 11 Intermolecular H-bonding arrangement in compound 6d (top) where C (grey), N (blue), O (red) and H (white), and packing arrangement in 6d (bottom) along c-axis. | ||
Similarly, in the case of 6n, each ligand molecule is H-bonded to another ligand molecule through 2 hydrogen bonds. The H-bonding distance between N6 of one molecule and O1 of the other molecule is about 2.8 Å. This gives rise to AB stacking of the molecules when viewed along a-axis where pyrazine rings of the molecules A and B point in opposite directions, thus resulting in the two molecules being non-superimposable upon each other (Fig. 12).
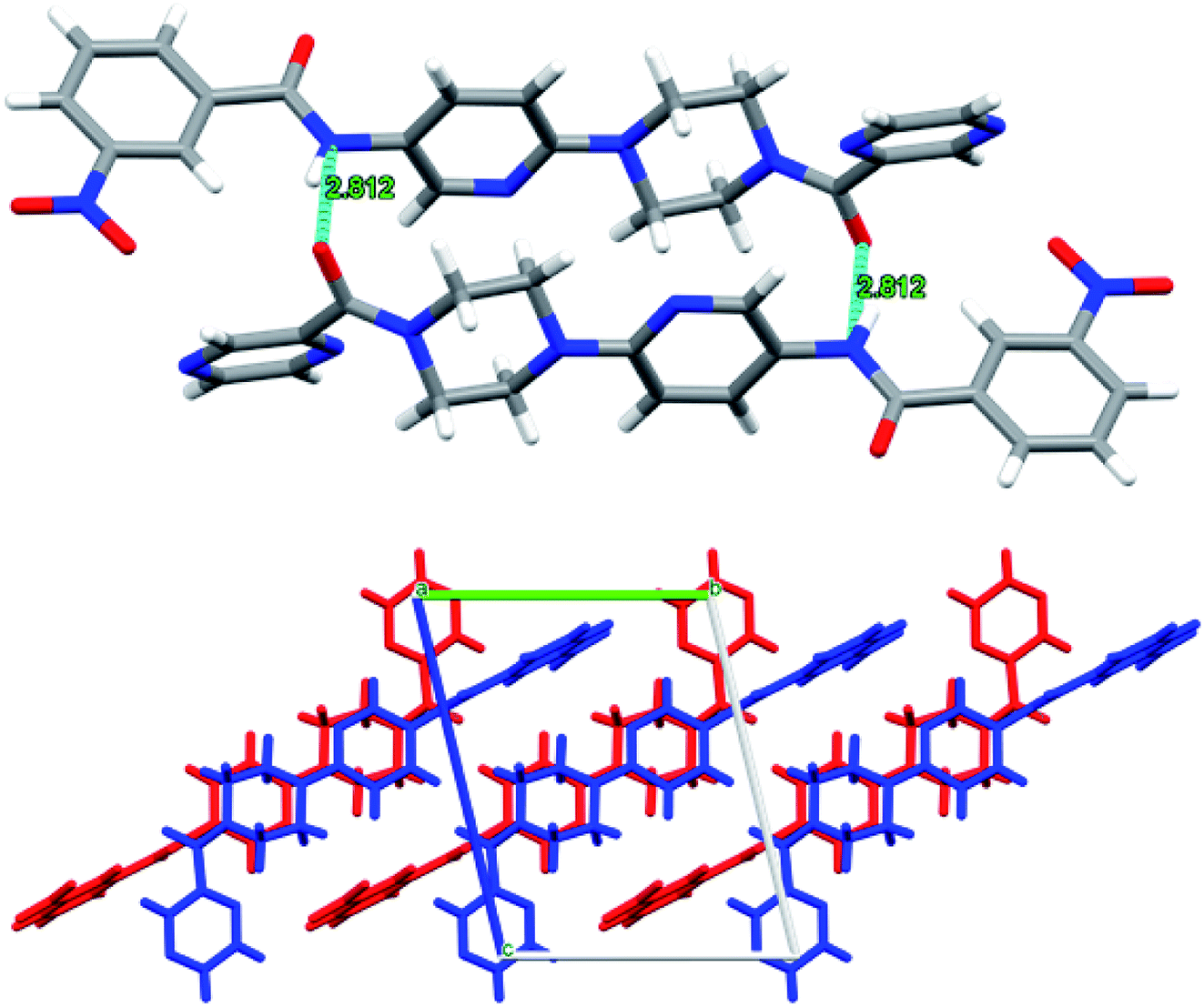 | ||
| Fig. 12 Intermolecular H-bonding arrangement in 6f (top) where C (grey), N (blue), O (red) and H (white), and view of H-bonded molecules along a-axis (bottom). The molecules A and B have been shown in red and blue colours for the ease of visualization. | ||
3. Conclusion
In conclusion, we identified five potential hit compounds from Series-I and one compound from Series-II. Compounds 6a, 6e, 6h, 6j, 6k and 7k exhibited greatest activity with IC50 values of 1.46, 2.18, 1.82, 2.14, 1.35 and 2.12 μM respectively and IC90 of 3.73, 40.32, 3.85, 3.94, 4.00 and 3.88 respectively against M. tuberculosis H37Ra. Further, docking studies were carried out for all the compounds using Schrödinger software and the docking of significantly active molecules was performed using the Glide module in Schrödinger. The most active compounds were found to be less toxic against HEK 298 cells. We developed single crystals for 6d, 6f and 6n.4. Experimental section
Materials and methods
Compound 2a. Yellow solid (9.0 g, yield: 92%); melting point of 2a: 168–170 °C; ESI-MS showed 309.16 (M + H)+. 1H NMR (400 MHz, DMSO) δ 8.97 (s, 1H), 8.25 (dd, J = 9.6, 2.9 Hz, 1H), 6.94 (d, J = 9.6 Hz, 1H), 3.83–3.72 (m, 4H), 3.51–3.41 (m, 4H), 1.43 (s, 9H).
Compound 2b. Yellow solid (8.9 g, yield: 89%); melting point of 2b: 157–159 °C; ESI-MS showed 323.20 (M + H)+. 1H NMR (400 MHz, DMSO) δ 8.97 (s, 1H), 8.21 (dd, J = 9.2, 2.6 Hz, 1H), 6.86 (d, J = 9.6 Hz, 1H), 4.05–3.65 (m, 4H), 3.63–3.43 (m, 2H), 3.33–3.30 (m, 2H), 1.77 (q, 2H), 1.25 (s, 9H).
Compound 3a. Pale yellow solid (7.1 g, yield: 98%); melting point of 3a: 84–86 °C (salt free compound); ESI-MS showed 209.20 (M + H)+. 1H NMR (400 MHz, DMSO) δ 9.69 (bs, 2H), 8.99 (s, 1H), 8.39–8.22 (m, 1H), 7.04 (d, J = 9.6 Hz, 1H), 4.45–4.38 (m, 4H), 4.11–3.95 (m, 4H).
Compound 3b. Pale yellow solid (7.20 g, yield: 100%); melting point of 3b: 79–81 °C (salt free compound); ESI-MS showed 223.20 (M + H)+. 1H NMR (400 MHz, DMSO) δ 9.47 (bs, 2H), 8.99 (s, 1H), 8.27 (dd, J = 9.6, 2.8 Hz, 1H), 6.90 (d, J = 9.6 Hz, 1H), 3.93–3.96 (m, 4H), 3.34–3.07 (m, 4H), 2.12 (q, 2H).
Compound 4a. Yellow gummy solid; yield: 83%; ESI-MS showed 315.16 (M + H)+. 1H NMR (400 MHz, DMSO) δ 8.91 (s, 1H), 8.61–8.52 (m, 3H), 8.11 (dd, J = 9.6, 2.9 Hz, 1H), 6.79 (d, J = 9.6 Hz, 1H), 3.88–3.63 (m, 4H), 3.50–3.08 (m, 4H).
Compound 4b. Yellow gummy solid; yield: 73%; ESI-MS showed 329.20 (M + H)+. 1H NMR (400 MHz, DMSO) δ 8.92 (s, 1H), 8.67–8.56 (m, 3H), 8.17 (dd, J = 9.6, 2.9 Hz, 1H), 6.85 (d, J = 9.6 Hz, 1H), 3.83–3.71 (m, 4H), 3.60–3.03 (m, 4H), 1.78 (q, 2H).
Compound 5a. Dark black and gummy solid; yield: 82%; ESI-MS showed 285.16 (M + H)+.
Compound 5b. Dark brown and gummy solid; yield: 70%; ESI-MS showed 399.20 (M + H)+.
Antitubercular activity. In vitro anti-mycobacterial activity of the compounds was evaluated by a luminometric assay based on a M. tuberculosis H37Ra laboratory strain transformed with a pSMT1 luciferase reporter plasmid (H37Ra-lux). A twofold serial dilution of each compound was made in complete 7H9 broth with final concentrations ranging from 128 to 0.125 μm. Volumes of 100 μL of the serial dilutions were added in triplicate to black, flat-bottomed 96-well plates. As a positive control, isoniazid, a first-line anti-mycobacterial drug, was included. The mycobacterial suspension was made by thawing a frozen glycerol stock of H37Ra-lux and, subsequently, diluting it in complete H9 broth to obtain a suspension with 10
000 relative light units (RLU) per mL. A volume of 100 μL of bacteria was added to each well. All of the outer-perimeter wells were filled with 200 μL of sterile deionized water to minimize evaporation of the medium in the test wells during incubation. After 7 days, the bacterial replication was analyzed by luminometry. To evoke a luminescent signal, 25 μL of 1% n-decanal in ethanol was added to each well, where after light emission was measured.
Materials and methods of docking. Docking studies were carried out using Schrödinger software (Version 2019-1, Schrödinger) installed on Intel Xenon W 3565 processor and Ubuntu enterprise version 18.04 as the operating system. Targeted ligands were drawn in ChemDraw 18.0. The result of the docking results was analyzed with the help of XP Visualiser (Version 2019-1, Schrödinger).
Ligand preparation. The ligands used as inputs for docking were sketched using ChemDraw software and cleaned up the structure for the bond alignment; ligands were incorporated into the workstation, the energy was minimized using OPLS3e force field in Ligprep (Version 2019-1, Schrödinger). This minimization helps to assign bond orders, the addition of the hydrogens to the ligands and conversion of 2D to 3D structure for the docking studies. The generated output file (best conformations of the ligands) was used for docking studies.
Receptor grid generation. A receptor grid was generated around the protein by picking the inhibitory ligand (X-ray pose of the ligand in the protein). The centroid of the ligand is selected to create a grid box around it, and van der Waals radius of receptor atoms was scaled to 1.00 Å with a partial atomic charge of 0.25.
Protein preparation. Protein was prepared using the protein preparation wizard (Version 2019-1, Schrödinger). Hydrogen atom was added to the proteins and charges were assigned. Generated het states using Epik at pH 7.0 ± 2.0. Pre-process the protein and refine, modify the protein by analyzing the workspace, water molecules and other heteroatoms were examined and the non-significant atoms were excluded from the crystal structure of the protein. Finally, the protein was minimized using OPLS3e force filed. A grid was created by considering co-crystal ligand, which is included in the active site of the selected target pantothenate synthetase (PDB 3IUB) from Mycobacterium tuberculosis.
Chemistry. Chemicals and solvents were procured from commercial source. The solvents and reagents were of LR grade and if necessary purified before use. Thin-layer chromatography (TLC) was carried out on aluminum-supported silica gel plates (Merck 60 F254) with visualization of components by UV light (254 nm). Column chromatography was carried out on silica gel (Merck 100–200 mesh). 1H NMR and 13C NMR spectra were recorded at 400 MHz and 101 MHz respectively using a Bruker AV 400 spectrometer (Bruker Co., Switzerland) in CDCl3 and DMSO-d6 solution with tetramethylsilane as the internal standard and chemical shift values (δ) were given in ppm. Melting points were determined on an electro thermal melting point apparatus (Stuart-SMP30) in open capillary tubes and are uncorrected. Elemental analyses were performed by Elementar Analysensysteme GmbH vario MICRO cube CHN Analyzer.
Spectral data
N-(6-(4-(Pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6a). Off white solid (88%); mp 137–139 °C; IR (KBr) νmax/cm−1 3573, 3032, 2923, 1675, 1445, 1052. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.5, 1.5 Hz, 1H), 8.49 (d, J = 2.5 Hz, 1H), 8.03–7.90 (m, 3H), 7.64–7.49 (m, 3H), 6.91 (d, J = 9.1 Hz, 1H), 3.84–3.77 (m, 2H), 3.65–3.48 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 165.3, 156.0, 149.7, 146.0, 145.0, 143.7, 140.8, 135.0, 132.0, 131.7, 128.9, 128.0, 127.5, 107.5, 46.6, 45.9, 45.3, 41.90. EI-MS m/z 389.17 (M + H)+; anal. calcd for C21H20N6O2: (%) C, 64.94; H, 5.20; N, 21.64; found: C, 64.95; H, 5.21; N, 21.65.
4-Methoxy-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6b). White solid (87%); mp 188–190 °C; (KBr) νmax/cm−1 3579, 3029, 2853, 1687, 1408, 1344, 500. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.79 (d, J = 2.6 Hz, 1H), 8.72 (dd, J = 2.6, 1.5 Hz, 1H), 8.49 (d, J = 2.6 Hz, 1H), 7.97–7.91 (m, 2H), 7.90 (s, 1H), 7.78–7.73 (m, 2H), 6.92 (d, J = 9.1 Hz, 1H), 3.83 (s, 3H), 3.81–3.76 (m, 2H), 3.63–3.48 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 165.1, 162.3, 155.9, 149.7, 146.0, 145.0, 143.7, 140.8, 131.7, 129.9, 127.7, 127.1, 114.0, 107.5, 55.9, 46.6, 46.0, 45.3, 41.9. EI-MS m/z 418.18 (M + H)+; anal. calcd for C22H22N6O2: (%) C, 63.15; H, 5.31; N, 20.08; found: C, 63.17; H, 5.32; N, 20.09.
4-(tert-Butyl)-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6c). White solid (91%); mp 156–158 °C; IR (KBr) νmax/cm−1 3576, 3031, 2925, 1685, 1421, 1330, 1050. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.5, 1.5 Hz, 1H), 8.48 (d, J = 2.6 Hz, 1H), 7.98–7.85 (m, 3H), 7.58–7.50 (m, 2H), 6.90 (d, J = 9.1 Hz, 1H), 3.81 (dd, J = 13.1, 8.4 Hz, 2H), 3.67–3.46 (m, 6H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 163.7, 152.1, 149.0, 146.2, 145.1, 143.7, 140.8, 134.1, 131.9, 131.7, 130.1, 127.3, 125.1, 107.5, 46.6, 45.9, 45.2, 41.9, 35.9, 30.8. EI-MS m/z 445.27 (M + H)+; anal. calcd for C25H28N6O2: (%) C, 67.56; H, 6.35; N, 18.92; found: C, 67.57; H, 6.37; N, 18.93.
4-Chloro-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6d). Light yellow solid (81%); mp 144–146 °C; IR (KBr) νmax/cm−1 3585, 3031, 2922, 1675, 1532, 1372, 1027, 575. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.90 (d, J = 1.3 Hz, 1H), 8.78 (d, J = 2.5 Hz, 1H), 8.71 (dd, J = 2.4, 1.5 Hz, 1H), 8.47 (d, J = 2.5 Hz, 1H), 8.04–7.91 (m, 3H), 7.61 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 9.1 Hz, 1H), 3.83–3.75 (m, 2H), 3.57 (ddd, J = 22.2, 7.7, 4.1 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 164.6, 156.1, 149.7, 146.0, 145.0, 143.7, 140.8, 136.8, 133.7, 131.7, 130.1, 128.9, 127.3, 107.5, 46.6, 46.27–45.97 (m), 45.6, 41.9. EI-MS m/z 423.87 (M + H)+; anal. calcd for C21H19ClN6O2: (%) C, 59.65; H, 4.54; N, 19.87; found: C, 59.66; H, 4.54; N, 19.88.
4-Bromo-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6e). White solid (90%); mp 119–121 °C; IR (KBr) νmax/cm−1 3590, 3021, 2843, 1687, 1410, 1340, 570. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.72 (dd, J = 2.6, 1.5 Hz, 1H), 8.47 (d, J = 2.6 Hz, 1H), 7.97–7.91 (m, 2H), 7.90 (s, 1H), 7.78–7.72 (m, 2H), 6.92 (d, J = 9.1 Hz, 1H), 3.81–3.76 (m, 2H), 3.64–3.48 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 164.7, 156.1, 149.7, 146.0, 145.0, 143.7, 140.8, 134.1, 131.9, 131.7, 130.1, 127.3, 125.8, 107.5, 46.6, 45.9, 45.3, 41.9. EI-MS m/z 468.08 (M + H)+; 469.07 (M + H)2+; anal. calcd for C21H19BrN6O2: (%) C, 53.97; H, 4.10; N, 17.98; found: C, 53.98; H, 4.11; N, 17.99.
3-Nitro-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6f). Yellow solid (82%); mp 202–203 °C; IR (KBr) νmax/cm−1 3590, 3021, 2833, 1515, 1679, 1410, 1340, 1120, 1061. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.90 (d, J = 1.2 Hz, 1H), 8.83–8.76 (m, 2H), 8.74–8.69 (m, 1H), 8.50 (d, J = 2.4 Hz, 1H), 8.47–8.39 (m, 2H), 7.96 (dd, J = 9.1, 2.5 Hz, 1H), 7.85 (t, J = 8.0 Hz, 1H), 6.94 (d, J = 9.1 Hz, 1H), 3.83–3.77 (m, 2H), 3.66–3.51 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 163.6, 156.2, 149.6, 148.2, 146.0, 144.9, 143.7, 140.9, 136.3, 134.5, 131.9, 130.7, 127.6, 126.7, 122.7, 107.5, 46.6, 45.8, 45.2, 41.9. EI-MS m/z 434.15 (M + H)+; anal. calcd for C21H19N7O4: (%) C, 58.19; H, 4.42; N, 22.63; found: C, 58.20; H, 4.43; N, 22.64.
3-Bromo-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6g). White solid (76%); mp 246–248 °C; IR (KBr) νmax/cm−1 3595, 3059, 2933, 1680, 1412, 1357, 1279, 1045, 612. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.5, 1.5 Hz, 1H), 8.48 (d, J = 2.6 Hz, 1H), 8.14 (t, J = 1.7 Hz, 1H), 8.00–7.90 (m, 2H), 7.80 (ddd, J = 8.0, 1.9, 0.9 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.84–3.76 (m, 2H), 3.65–3.50 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 164.1, 156.1, 149.7, 146.0, 145.0, 143.7, 140.8, 137.1, 134.7, 131.7, 131.1, 130.6, 127.2, 122.2, 107.5, 46.6, 45.9, 45.2, 41.9. EI-MS m/z 467.09 (M + H)+; 469.08 (M + H)2+; anal. calcd for C21H19BrN6O2: (%) C, 53.98; H, 4.10; N, 17.98; found: C, 53.99; H, 4.11; N, 17.99.
2-Methyl-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6h). Brown solid (80%); mp 165–167 °C; IR (KBr) νmax/cm−1 3542, 3027, 2832, 1675, 1424, 1365, 1034, 1020. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.90 (s, 1H), 8.78 (d, J = 2.4 Hz, 1H), 8.71 (s, 1H), 8.47 (d, J = 2.3 Hz, 1H), 7.95 (dd, J = 9.0, 2.4 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.39 (t, J = 7.3 Hz, 1H), 7.34–7.25 (m, 2H), 6.90 (d, J = 9.1 Hz, 1H), 3.79 (d, J = 5.0 Hz, 2H), 3.63–3.55 (m, 4H), 3.50 (d, J = 2.8 Hz, 2H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 165.3, 155.9, 149.7, 146.0, 145.0, 143.7, 140.0, 137.4, 135.8, 131.0, 130.8, 130.1, 127.8, 127.7, 126.1, 107.7, 46.6, 46.1, 45.4, 41.9, 19.8. EI-MS m/z 403.20 (M + H)+; anal. calcd for C22H22N6O2: (%) C, 65.66; H, 5.52; N, 20.89; found: C, 65.67; H, 5.53; N, 20.90.
2-Iodo-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6i). White solid (89%); mp 165–167 °C; (KBr) νmax/cm−1 3599, 3027, 2834, 1677, 1412, 1343, 1043, 615. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.90 (d, J = 1.3 Hz, 1H), 8.78 (d, J = 2.5 Hz, 1H), 8.74–8.69 (m, 1H), 8.44 (d, J = 2.4 Hz, 1H), 7.97–7.90 (m, 2H), 7.50 (q, J = 7.2 Hz, 2H), 7.26–7.20 (m, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.83–3.77 (m, 2H), 3.63–3.55 (m, 4H), 3.53–3.48 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 165.3, 155.9, 146.0, 145.0, 143.7, 140.0, 137.4, 135.8, 131.0, 130.8, 130.0, 127.8, 127.3, 122.3, 107.7, 93.8, 46.6, 46.1, 45.4, 41.9. EI-MS m/z 515.08 (M + H)+; anal. calcd for C21H19IN6O2: (%) C, 49.05; H, 3.73; N, 16.34; found: C, 49.07; H, 3.75; N, 16.35.
2-Bromo-6-chloro-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6j). P-7 pale yellow solid (87%); mp 150–152 °C; IR (KBr) νmax/cm−1 3582, 3022, 2973, 1671, 1422, 1332, 1055, 680. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.90 (d, J = 1.3 Hz, 1H), 8.78 (d, J = 2.5 Hz, 1H), 8.71 (dd, J = 2.5, 1.5 Hz, 1H), 8.42 (d, J = 2.6 Hz, 1H), 7.91 (dd, J = 9.1, 2.7 Hz, 1H), 7.84 (d, J = 2.4 Hz, 1H), 7.71 (dd, J = 8.6, 2.4 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.82–3.78 (m, 2H), 3.64–3.55 (m, 4H), 3.53–3.49 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 163.5, 156.2, 149.7, 146.0, 145.0, 143.7, 139.9, 138.9, 134.3, 132.2, 131.9, 130.8, 129.9, 127.2, 120.4, 107.7, 46.6, 45.9, 45.3, 41.9. EI-MS m/z 501.04 (M + H)+; 503.06 (M + H)2+; anal. calcd for C21H18BrClN6O2: (%) C, 50.27; H, 3.64; N, 16.75; found: C, 50.28; H, 3.65; N, 16.76.
4-Bromo-2-chloro-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6k). Light brown solid (80%); mp 147–149 °C; IR (KBr) νmax/cm−1 3575, 30
311, 2925, 1673, 1402, 1043, 560. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.90 (s, 1H), 8.78 (d, J = 2.3 Hz, 1H), 8.71 (s, 1H), 8.42 (d, J = 2.2 Hz, 1H), 7.91 (dd, J = 9.0, 2.3 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.71 (dd, J = 8.5, 2.1 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.80 (s, 2H), 3.66–3.54 (m, 5H), 3.51 (d, J = 4.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 161.8, 156.2, 148.7, 148.2, 147.0, 145.4, 145.0, 144.7, 143.9, 143.8, 141.0, 131.7, 126.6, 107.4, 46.6, 45.7, 45.2, 41.8. EI-MS m/z 501.04 (M + H)+; 503.06 (M + H)2+; anal. calcd for C21H18BrClN6O2: (%) C, 50.27; H, 3.64; N, 16.75; found: C, 50.28; H, 3.65; N, 16.76.
2,4-Dichloro-N-(6-(4-(pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)benzamide (6l). Off solid (80%); mp 197–199 °C; IR (KBr) νmax/cm−1 3595, 3029, 2945, 1683, 1422, 1053, 560. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.95 (d, J = 1.5 Hz, 1H), 8.88 (d, J = 2.5 Hz, 1H), 8.73 (dd, J = 2.6, 1.5 Hz, 1H), 8.46 (d, J = 2.6 Hz, 1H), 7.89 (dd, J = 9.1, 2.7 Hz, 1H), 7.80 (d, J = 2.4 Hz, 1H), 7.69 (dd, J = 8.6, 2.4 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.78 (t, J = 5.2 Hz, 2H), 3.60–3.49 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 161.8, 156.2, 148.7, 148.2, 147.0, 145.4, 145.0, 144.7, 143.9, 143.8, 141.0, 131.7, 126.6, 107.4, 46.6, 45.7, 45.2, 41.8. EI-MS m/z 558.10 (M + H)+; anal. calcd for C21H18Cl2N6O2: (%) C, 55.16; H, 3.97; N, 18.38; found: C, 55.18; H, 3.98; N, 18.39.
N-(6-(4-(Pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)isonicotinamide (6m). Off white solid (83%); mp 212–213 °C; IR (KBr) νmax/cm−1 3510, 3021, 2865, 1670, 1410, 1340, 1060. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.93 (d, J = 2.5 Hz, 2H), 8.91 (d, J = 1.6 Hz, 2H), 8.80 (dd, J = 2.5, 1.7 Hz, 2H), 8.78 (d, J = 2.6 Hz, 1H), 8.70 (dd, J = 2.6, 1.5 Hz, 1H), 8.66 (d, J = 2.7 Hz, 1H), 8.07 (dd, J = 9.1, 2.7 Hz, 1H), 6.90 (d, J = 9.1 Hz, 1H), 3.79 (dd, J = 6.6, 4.2 Hz, 2H), 3.63 (dd, J = 6.6, 4.2 Hz, 2H), 3.60–3.53 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 162.9, 156.1, 149.7, 146.1, 145.3, 145.1, 143.7, 143.6, 140.9, 131.2, 126.2, 121.2, 107.5, 46.7, 45.8, 45.1, 41.9. EI-MS m/z 389.19 (M + H)+; anal. calcd for C21H20N6O2: (%) C, 64.94; H, 5.19; N, 21.64; found: C, 64.95; H, 5.21; N, 21.65.
N-(6-(4-(Pyrazine-2-carbonyl)piperazin-1-yl)pyridin-3-yl)pyrazine-2-carboxamide (6n). Off white solid (87%); mp 205–206 °C; (KBr) νmax/cm−1 3525, 3025, 2867, 16
775, 1412, 1363, 1022. 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 10.73 (s, 1H), 9.28 (d, J = 1.5 Hz, 1H), 8.93 (d, J = 2.5 Hz, 1H), 8.90 (d, J = 1.5 Hz, 1H), 8.80 (dd, J = 2.5, 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.5, 1.5 Hz, 1H), 8.63 (d, J = 2.6 Hz, 1H), 8.32 (d, J = 1.5 Hz, 1H), 8.07 (dd, J = 9.1, 2.7 Hz, 1H), 6.92 (d, J = 9.1 Hz, 1H), 3.82–3.77 (m, 2H), 3.67–3.50 (m, 7H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 161.9, 156.1, 149.7, 148.1, 146.0, 145.4, 145.0, 144.4, 143.7, 143.7, 141.0, 131.6, 126.6, 107.4, 46.6, 45.8, 45.2, 41.9. EI-MS m/z 391.16 (M + H)+; anal. calcd for C19H18N8O2: (%) C, 58.45; H, 4.65; N, 28.70; found: C, 58.46; H, 4.67; N, 28.71.
N-(6-(4-(Pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7a). White solid (86%); mp 147–149 °C; IR (KBr) νmax/cm−1 3610, 3019, 2917, 1668, 1484, 1326, 1032. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.91 (d, J = 1.5 Hz, 1H), 8.76 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.6, 1.5 Hz, 1H), 8.47 (d, J = 2.8 Hz, 1H), 7.97–7.91 (m, 3H), 7.90 (s, 1H), 7.78–7.72 (m, 2H), 6.92 (d, J = 9.2 Hz, 1H), 3.71–3.66 (m, 8H), 2.44–2.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.8, 163.7, 152.2, 148.9, 147.9, 145.2, 143.6, 140.7, 134.5, 131.9, 131.7, 130.2, 127.8, 125.8, 107.7, 67.3, 56.4, 52.1, 49.1, 29.7. EI-MS m/z 403.19 (M + H)+; anal. calcd for C22H22N6O2: (%) C, 65.66; H, 5.51; N, 20.88; found: C, 65.67; H, 5.52; N, 20.89.
4-Ethoxy-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7b). Brown solid (88%); mp 207–209 °C; IR (KBr) νmax/cm−1 3560, 3021, 2835, 1684, 1411, 1365, 1024. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.98 (d, J = 1.6 Hz, 1H), 8.73 (d, J = 2.5 Hz, 2H), 8.57 (d, J = 2.6 Hz, 1H), 7.90 (dd, J = 9.2, 2.3 Hz, 3H), 7.10–7.01 (m, 2H), 6.90 (d, J = 9.0 Hz, 1H), 4.15–4.07 (m, 2H), 3.79–3.75 (m, 4H), 3.60–3.54 (m, 4H), 3.50–3.45 (m, 2H), 1.31 (t, J = 7.0, 1.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 165.2, 161.6, 155.6, 149.7, 146.1, 145.0, 143.7, 140.2, 132.0, 129.9, 127.7, 126.9, 114.5, 107.8, 63.9, 58.8, 55.4, 46.6, 46.0, 45.4, 41.3, 28.1, 15.0. EI-MS m/z 446.21 (M + H)+; anal. calcd for C24H26N6O3: (%) C, 64.57; H, 5.87; N, 18.82; found: C, 64.58; H, 5.88; N, 18.83.
4-(tert-Butyl)-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7c). Off white solid (83%); mp 228–230 °C; IR (KBr) νmax/cm−1 3573, 3029, 2912, 1672, 1420, 1335, 1066. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.93 (d, J = 1.5 Hz, 1H), 8.78 (d, J = 2.6 Hz, 1H), 8.71 (dd, J = 2.6, 1.5 Hz, 1H), 8.57 (d, J = 2.6 Hz, 1H), 7.87–7.91 (m, 2H), 7.91 (s, 1H), 7.76–7.70 (m, 2H), 6.91 (d, J = 9.1 Hz, 1H), 3.68–3.62 (m, 8H), 2.54–2.38 (m, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 164.6, 153.1, 148.9, 145.8, 145.0, 143.9, 141.1, 134.2, 131.7, 131.2, 130.2, 127.7, 125.4, 107.7, 67.3, 55.4, 53.8, 46.9, 38.7, 35.1, 30.2, 25.6. EI-MS m/z 458.25 (M + H)+; anal. calcd for C26H30N6O2: (%) C, 68.11; H, 6.59; N, 18.33; found: C, 68.13; H, 6.60; N, 18.34.
4-Chloro-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7d). White solid (77%); mp 240–242 °C; IR (KBr) νmax/cm−1 3575, 3039, 2940, 1684, 1412, 13
495, 1188, 1021, 782. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.76–8.65 (m, 2H), 8.64 (dd, J = 9.1, 2.8 Hz, 1H), 8.00–7.58 (m, 3H), 7.55–7.51 (m, 2H), 6.72 (dd, J = 9.2, 2.4 Hz, 1H), 3.84–3.50 (m, 8H), 1.93 (q, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.5, 164.8, 156.2, 149.8, 146.4, 145.5, 143.1, 140.8, 136.8, 133.7, 131.8, 129.2, 128.4, 127.7, 107.5, 55.9, 53.4, 50.7, 47.8, 25.8. EI-MS m/z 436.14 (M + H)+; anal. calcd for C22H21ClN6O2: (%) C, 60.48; H, 4.84; N, 19.25; found: C, 60.49; H, 4.85; N, 19.50.
4-Bromo-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7e). White solid (90%); mp 119–121 °C; IR (KBr) νmax/cm−1 3590, 3021, 2843, 1687, 1410, 1340, 570. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.79–8.68 (m, 2H), 8.65 (dd, J = 9.1, 2.8 Hz, 1H), 8.42–8.34 (m, 1H), 7.98–7.90 (m, 2H), 7.89 (m, 1H), 7.79–7.74 (m, 2H), 6.73 (dd, J = 9.2, 2.4 Hz, 1H), 3.85–3.52 (m, 8H), 1.96 (q, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.7, 166.6, 164.6, 154.5, 150.4, 150.1, 145.9, 144.3, 143.5, 141.4, 134.2, 132.1, 131.9, 130.1, 125.8, 125.6, 105.7, 48.4, 47.7, 46.2, 45.2, 29.5. EI-MS m/z 481.19 (M + H)+; 483.21 (M + H)2+; anal. calcd for C22H21BrN6O2: (%) C, 54.91; H, 4.40; N, 17.46; found: C, 54.93; H, 4.42; N, 17.47.
3-Nitro-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7f). Yellow solid (78%); mp 212–213 °C; IR (KBr) νmax/cm−1 3591, 3041, 2893, 1689, 1411, 1351, 1120, 1061. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.90–8.76 (m, 2H), 8.63 (s, 1H) 8.51–8.32 (m, 4H) 7.97–7.90 (m, 2H), 6.73 (d, J = 9.3 Hz, 1H), 3.85–3.32 (m, 8H), 1.97 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.3, 168.1, 159.4, 154.9, 153.0, 150.6, 149.0, 148.2, 146.1, 141.3, 139.2, 136.8, 135.4, 131.3, 127.5, 110.55, 53.32, 52.8, 59.0, 50.0, 29.3. EI-MS m/z 447.18 (M + H)+; anal. calcd for C22H21N7O4: (%) C, 59.05; H, 4.73; N, 21.91; found: C, 58.20; H, 4.43; N, 22.64.
3-Bromo-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7g). White solid (71%); mp 246–248 °C; IR (KBr) νmax/cm−1 3590, 3050, 2931, 1682, 1410, 1333, 1276, 1035, 615. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.87 (d, J = 1.6 Hz, 1H), 8.76 (d, J = 2.6 Hz, 1H), 8.70 (dd, J = 2.6, 1.6 Hz, 1H), 8.49 (d, J = 2.6 Hz, 1H), 8.24 (t, J = 1.8 Hz, 1H), 7.99–7.90 (m, 2H), 7.83 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.54 (t, J = 7.9 Hz, 1H), 6.93 (d, J = 9.1 Hz, 1H), 3.57 3–3.50 (m, 4H), 3.45–3.37 (m, 6H), 1.85 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.5, 168.3, 159.3, 155.1, 154.9, 152.9, 150.5, 149.1, 148.1, 141.2, 139.1, 136.9, 135.2, 131.3, 130.1, 127.6, 110.4, 53.1, 52.6, 51.8, 50.1, 30.0. EI-MS m/z 480.09 (M + H)+; 482.08 (M + H)2+; anal. calcd for C22H21BrN6O2: (%) C, 54.91; H, 4.40; N, 17.46; found: C, 54.92; H, 4.41; N, 17.48.
2-Iodo-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7h). Off white solid (80%); mp 226–228 °C; IR (KBr) νmax/cm−1 3581, 3032, 2916, 1689, 1417, 1325, 1130, 1060, 634. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.92 (d, J = 1.5 Hz, 1H), 8.88 (d, J = 2.6 Hz, 1H), 8.80–8.71 (m, 2H), 7.95 (dd, J = 9.0, 2.7 Hz, 2H), 7.49 (dd, J = 8.5, 6.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 2H), 6.91 (d, J = 9.1 Hz, 1H), 3.82–3.78 (m, 2H), 3.59 (t, 4H), 3.50–3.41 (m, 2H), 1.77 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 165.3, 155.9, 146.0, 145.0, 143.7, 139.9, 137.4, 135.8, 131.0, 130.8, 130.1, 127.8, 127.3, 122.3, 107.7, 93.8, 57.0, 53.2, 50.9, 47.2, 25.9. EI-MS m/z 528.17 (M + H)+; anal. calcd for C22H21IN6O2: (%) C, 50.01; H, 4.02; N, 15.91; found: C, 50.03 H, 4.04; N, 15.92.
4-Bromo-2-chloro-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7i). White solid (77%); mp 221–223 °C; IR (KBr) νmax/cm−1 3560, 3029, 2864, 1686, 1415, 1323, 1109, 1031, 623. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.87–8.76 (dd, J = 2.6, 1.5 Hz, 2H), 8.52 (s, 1H), 7.90 (dd, J = 9.1, 2.7 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.69 (dd, J = 8.6, 2.4 Hz, 1H), 7.57 (d, J = 8.6 Hz, 1H), 6.73 (d, J = 9.1 Hz, 1H), 3.80–3.41 (m, 8H), 1.73 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 161.8, 156.2, 148.7, 148.2, 147.0, 145.4, 145.0, 144.7, 143.9, 143.8, 140.9, 131.7, 126.6, 107.4, 57.0, 53.2, 50.9, 47.2, 29.8. EI-MS m/z 514.06 (M + H)+; 516.08 (M + H)2+; anal. calcd for C22H20BrClN6O2: (%) C, 51.23; H, 3.92; N, 16.29; found: C, 51.24; H, 3.93; N, 16.30.
2,4-Dichloro-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7j). Off white solid (80%); mp 197–199 °C; IR (KBr) νmax/cm−1 3597, 3028, 2947, 1687, 1427, 1023, 569. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.89 (m. 2H), 8.78 (d, J = 2.5 Hz, 1H), 8.73 (dd, J = 2.6, 1.5 Hz, 1H), 87.89–7.5 (m, 3H), 7.42 (m, 1H), 7.78 (d, J = 2.4 Hz, 1H), 7.51 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 9.0 Hz, 1H), 3.89–3.36 (m, 8H), 1.71 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.4, 161.8, 156.2, 148.7, 148.2, 147.0, 145.4, 146.0, 145.7, 143.9, 143.8, 141.0, 132.9, 126.7, 109.4, 58.9, 54.2, 51.9, 48.2, 32.8. EI-MS m/z 470.12 (M + H)+; anal. calcd for C22H20Cl2N6O2: (%) C, 56.06; H, 4.28; N, 17.83; found: C, 56.08; H, 4.29; N, 17.84.
N-(6-(4-(Pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)pyrazine-2-carboxamide (7k). Brown solid (79%); mp 135–136 °C; IR (KBr) νmax/cm−1 3572, 3021, 2915, 1677, 1454, 1350, 1090. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.30 (d, J = 1.6 Hz, 1H), 8.94 (d, J = 2.7 Hz, 1H), 8.89 (d, J = 1.5 Hz, 1H), 8.81 (dd, J = 2.5, 1.7 Hz, 1H), 8.75 (d, J = 2.6 Hz, 1H), 8.70 (dd, J = 2.6, 1.7 Hz, 1H), 8.64 (d, J = 2.6 Hz, 1H), 8.17 (dd, J = 9.1, 2.7 Hz, 1H), 6.90 (d, J = 9.1 Hz, 1H), 3.66–3.60 (m, 8H), 2.47–2.28 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.5, 162.0, 157.0, 149.8, 148.2, 146.2, 145.3, 145.2, 144.7, 143.3, 143.0, 140.6, 130.1, 126.6, 109.1, 67.3, 56.4, 52.8, 49.9, 28.7. EI-MS m/z 405.17 (M + H)+; anal. calcd for C20H20N8O2: (%) C, 59.40; H, 4.98; N, 27.71; found: C, 59.42; H, 4.90; N, 27.72.
N-(6-(4-(Pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)isonicotinamide (7l). White solid (82%); mp 139–141 °C; IR (KBr) νmax/cm−1 3585, 3029, 2840, 1684, 1415, 1342, 1070. 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.93 (d, J = 2.5 Hz, 2H), 8.91 (d, J = 1.6 Hz, 2H), 8.81 (dd, J = 2.5, 1.7 Hz, 2H), 8.78 (d, J = 2.5 Hz, 1H), 8.70 (dd, J = 2.6, 1.6 Hz, 1H), 8.66 (d, J = 2.7 Hz, 1H), 8.07 (dd, J = 9.1, 2.5 Hz, 1H), 6.89 (d, J = 9.2 Hz, 1H), 3.69–3.51 (m, 2H), 3.51 (t, 2H), 3.40–3.33 (m, 6H), 1.88 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 162.9, 156.0, 149.7, 146.3, 145.3, 145.1, 143.7, 143.6, 140.9, 131.2, 126.2, 121.2, 107.5, 67.3, 55.4, 53.8, 46.9, 25.6. EI-MS m/z 404.18 (M + H)+; anal. calcd for C21H21N7O2: (%) C, 62.53; H, 5.25; N, 24.31; found: C, 62.54; H, 5.26; N, 24.33.
2-Chloro-6-fluoro-N-(6-(4-(pyrazine-2-carbonyl)-1,4-diazepan-1-yl)pyridin-3-yl)benzamide (7m). Off white solid (81%); mp 198–199 °C; IR (KBr) νmax/cm−1 3594, 3042, 2913, 1687, 1412, 1036, 654. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.90 (d, J = 1.6 Hz, 1H), 8.80 (d, J = 2.5 Hz, 1H), 8.74 (dd, J = 2.6, 1.6 Hz, 1H), 8.50 (d, J = 2.6 Hz, 1H), 7.61–7.44 (m, 4H), 6.92 (d, J = 9.1 Hz, 1H), 3.67–3.52 (m, 4H), 3.55–3.47 (m, 6H), 1.87 (p, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 164.6, 159.3, 156.2, 149.6, 146.3, 145.1, 143.4, 141.8, 137.8, 136.7, 135.7, 131.2, 129.2, 128.1, 127.2, 113.2, 58.2, 56.2, 52.9, 46.2, 30.8. EI-MS m/z 454.14 (M + H)+; anal. calcd for C22H20ClFN6O2: (%) C, 58.09; H, 4.43; N, 18.49; found: C, 58.10; H, 4.44; N, 18.50.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
KVGCS and SM thank DBT, New Delhi [BT/IN/Spain/39/SMl2017-18] for providing financial support. The financial assistance provided by DST FIST grant (SR/FST/CSI-240/2012), New Delhi is gratefully acknowledged. SS thanks CSIR for providing SRF fellowship. Central analytical lab facilities of BITS Pilani Hyderabad campus are gratefully acknowledged.References
- https://apps.who.int/iris/bitstream/handle/10665/311389/9789241550529-eng.pdf.
- https://www.atsjournals.org/doi/full/10.1164/rccm.167.4.603#readcube-epdf.
- W. J. Chung, A. Kornilov, B. H. Brodsky, M. Higgins, T. Sanchez, L. B. Heifets, M. H. Cynamon and J. Welch, Tuberculosis, 2008, 88, 410 CrossRef CAS PubMed.
- V. Judge, B. Narasimhan and M. Ahuja, Hygeia, 2012, 4, 1 CAS.
- M. Dolezal, P. Cmedlova, L. Palek, J. Vinsova, J. Kunes, V. Buchta, J. Jampilek and K. Kralova, Eur. J. Med. Chem., 2008, 43, 1105 CrossRef CAS PubMed.
- F. M. Vergara, C. H. Lima, M. D. Henriques, A. L. Candéa, M. C. Lourenço, L. M. Ferreira, C. R. Kaiser and M. V. de Souza, Eur. J. Med. Chem., 2009, 44, 4954 CrossRef CAS PubMed.
- D. Sriram, P. Yogeeswari and S. P. Reddy, Bioorg. Med. Chem. Lett., 2006, 16, 2113 CrossRef CAS PubMed.
- A. Imramovský, S. Polanc, J. Vinsová, M. Kocevar, J. Jampílek, Z. Recková and J. Kaustová, Bioorg. Med. Chem., 2007, 15, 2551 CrossRef PubMed.
- M. Dolezal, L. Palek, J. Vinsova, V. Buchta, J. Jampilek and K. Kralova, Molecules, 2006, 11, 242 CrossRef CAS PubMed.
- L. Semelková, O. Janďourek, K. Konečná, P. Paterová, L. Navrátilová, F. Trejtnar, V. Kubíček, J. Kuneš, M. Doležal and J. Zitko, Molecules, 2017, 22, E495 CrossRef PubMed.
- B. Servusová, J. Vobicková, P. Paterová, V. Kubíček, J. Kuneš, M. Doležal and J. Zitko, Bioorg. Med. Chem. Lett., 2013, 23, 3589 CrossRef PubMed.
- N. R. Gangarapu, A. Ranganatham, E. K. Reddy, H. D. Surendra, A. M. Sajith, S. Yellappa and K. B. Chandrasekhar, J. Heterocycl. Chem., 2019, 56, 1117 CrossRef CAS.
- B. Servusova-Vanaskova, O. Jandourek, P. Paterova, J. Kordulakova, P. Magdalena, K. Vladimir Kubicek, R. Kucera, V. Garaj, L. Naesens, J. Kunes, M. Dolezala and J. Zitko, RSC Med. Chem., 2015, 6, 1311 RSC.
- T. Stringer, R. Seldon, N. Liu, D. F. Warner, C. Tam, W. Cheng, L. M. Land, P. J. Smith, K. Chibale and G. S. Smith, Dalton Trans., 2017, 46, 9875 RSC.
- J. Zitko, O. Jand'ourek, P. Paterová, L. Navrátilová, J. Kuneš, J. Vinšová and M. Doležal, RSC Med. Chem., 2018, 9, 685 RSC.
- B. Servusová, P. Paterová, J. Mandíková, V. Kubíček, R. Kučera, J. Kuneš, M. Doležal and J. Zitko, Bioorg. Med. Chem. Lett., 2014, 24, 450 CrossRef PubMed.
- M. Dolezal, J. Zitko, D. Kesetovicová, J. Kunes and M. Svobodová, Molecules, 2009, 14, 4180 CrossRef CAS PubMed.
- S. Zhou, S. Yang and G. Huang, J. Enzyme Inhib. Med. Chem., 2017, 32, 1183 CrossRef CAS PubMed.
- A. Lupien, A. Vocat, C. S. Foo, E. Blattes, J. Y. Gillon, V. Makarov and S. T. Cole, Antimicrob. Agents Chemother., 2018, 62, e00840 CrossRef PubMed.
- C. S. Foo, A. Lupien, M. Kienle, A. Vocat, A. Benjak, R. Sommer, D. A. Lamprecht, A. J. C. Steyn, K. Pethe, J. Piton, K. H. Altmann and S. T. Cole, mBio, 2018, 9, e01276 CrossRef CAS PubMed.
- K. A. Bobesh, J. Renuka, R. R. Srilakshmi, S. Yellanki, P. Kulkarni, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 42 CrossRef CAS PubMed.
- M. Chandran, J. Renuka, J. P. Sridevi, G. S. Pedgaonkar, V. Asmitha, P. Yogeeswari and D. Sriram, Int. J. Mycobact., 2015, 4, 104 CrossRef PubMed.
- H. N. Nagesh, K. M. Naidu, D. H. Rao, J. P. Sridevi, D. Sriram, P. Yogeeswari and K. V. G. Chandra Sekhar, Bioorg. Med. Chem. Lett., 2013, 23, 6805 CrossRef CAS PubMed.
- H. N. Nagesh, N. Suresh, K. M. Naidu, B. Arun, J. P. Sridevi, D. Sriram, P. Yogeeswari and K. V. G. Chandra Sekhar, Eur. J. Med. Chem., 2014, 74, 333 CrossRef CAS PubMed.
- E. Bogatcheva, C. Hanrahan, B. Nikonenko, R. Samala, P. Chen, J. Gearhart, F. Barbosa, L. Einck, C. A. Nacy and M. Protopopova, J. Med. Chem., 2006, 49, 3045 CrossRef CAS PubMed.
- K. M. Naidu, S. Srinivasarao, N. Agnieszka, A. K. Ewa, M. M. Kumar and K. V. G. Chandra Sekhar, Bioorg. Med. Chem. Lett., 2016, 26, 2245 CrossRef CAS PubMed.
- P. T. Gandhi, N. T. Athmaram and G. R. Arunkumar, Bioorg. Med. Chem., 2016, 24, 1637 CrossRef CAS PubMed.
- M. M. Patel and M. T. Chhabriya, Am. J. Infect. Dis., 2011, 7, 61 CrossRef CAS.
- P. C. Ting, J. F. Lee, N. Zorn, H. M. Kim, R. G. Aslanian, M. Lin, M. Smith, S. S. Walker, J. Cook, M. Van Heek and J. Lachowicz, Bioorg. Med. Chem. Lett., 2013, 23, 985 CrossRef CAS PubMed.
- R. Reddyrajula and U. Dalimba, Bioorg. Med. Chem. Lett., 2020, 30, 126846 CrossRef PubMed.
- E. Torfs, J. Vajs, M. B. de Macedo, F. Cools, B. Vanhoutte, Y. Gorbanev, A. Bogaerts, L. Verschaeve, G. Caljon, L. Maes, P. Delputte, P. Cos, J. Košmrlj and D. Cappoen, Chem. Biol. Drug Des., 2018, 91, 631 CrossRef CAS PubMed.
- T. Prakruti, N. Adhikari, S. A. Amin, T. Jha and B. Ghosh, Eur. J. Pharm. Sci., 2018, 124, 165 CrossRef PubMed.
- Schrödinger Release 2019-1, Maestro, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, J. Med. Chem., 2004, 47, 1739 CrossRef CAS PubMed.
- A. w. Hung, H. L. Silvestre, S. Wen, A. Ciulli, T. L. Blundell and C. Abell, Angew. Chem., Int. Ed., 2009, 48, 8452 CrossRef CAS PubMed.
- S. K. Burley, H. M. Berman, C. Bhikadiya, C. Bi, L. Chen, L. Di Costanzo, C. Christie, K. Dalenberg, J. M. Duarte and S. Dutta, Nucleic Acids Res., 2019, 47, 464 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1979943–1979945. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01348j |
| This journal is © The Royal Society of Chemistry 2020 |