
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-Ni cathode material improved with Zr for stable cycling of Li-ion rechargeable batteries†
Kwangjin Park‡a,
Dong Jin Ham‡
b,
Seong Yong Parkc,
Jihyun Jangb,
Dong-Hee Yeonb,
San Moon *b and
Sung Jin Ahn*b
*b and
Sung Jin Ahn*b
aDepartment of Mechanical Engineering, Gachon University, 1342 Sungnamdaero, Sujeong-Gu, Sungnam Si, Gyeonggi-do 13120, Republic of Korea. E-mail: ydmj79@gachon.ac.kr; Tel: +82-31-750-5708
bEnergy Laboratory, Samsung Advanced Institute of Technology, Samsung Electronics Co. Ltd., 130, Samsung-ro, Yeongtong-gu, Suwon-Si, Gyeonggi-do 16678, Republic of Korea
cAnalytical Engineering Group, Samsung Advanced Institute of Technology, 130 Samsung-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do 443-803, Republic of Korea
First published on 17th July 2020
Abstract
The Zr solvent solution method, which allows primary and secondary particles of LiNi0.90Co0.05Mn0.05O2 (NCM) to be uniformly doped with Zr and simultaneously to be coated with an Li2ZrO3 layer, is introduced in this paper. For Zr doped NCM, which is formed using the Zr solvent solution method (L-NCM), most of the pinholes inside the precursor disappear owing to the diffusion of the Zr dopant solution compared with Zr-doped NCM, which is formed using the dry solid mixing method from the (Ni0.90Co0.05Mn0.05)(OH)2 precursor and the Zr source (S-NCM), and Li2ZrO3 is formed at the pinhole sites. The mechanical strength of the powder is enhanced by the removal of the pinholes by the formation of Li2ZrO3 resulting from diffusion of the solvent during the mixing process, which provides protection from cracking. The coating layer functions as a protective layer during the washing process for removing the residual Li. The electrochemical performance is improved by the synergetic effects of suitable coatings and the enhanced structural stability. The capacity-retentions for 2032 coin cells are 86.08%, 92.12%, and 96.85% at the 50th cycle for pristine NCM, S-NCM, and L-NCM, respectively. The superiority of the liquid mixing method is demonstrated for 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 full cells. In the 300th cycle in the voltage range of 2.8–4.35 V, the capacity-retentions for S-NCM and L-NCM are 77.72% and 81.95%, respectively.
650 full cells. In the 300th cycle in the voltage range of 2.8–4.35 V, the capacity-retentions for S-NCM and L-NCM are 77.72% and 81.95%, respectively.
1. Introduction
For high-power applications such as electric vehicles (EVs) and hybrid EVs (HEVs), Li-ion batteries (LIBs) with a high power density have attracted attention.1–5 However, the energy density and cycle retention of LIBs are far from the required levels for practical EVs, limiting the use of LIBs. In particular, existing cathode materials such as LiCoO2 (LCO) and LiMnO2 (LMO) have a low capacity that is less than two-thirds of the specific capacity of conventional carbon-based anodes.1,3,5–7 The widely commercialized cathode materials cannot satisfy the high energy density of LIBs required for EVs and HEVs. Therefore, studies have been actively performed to improve the capacity of the cathode.Ni-rich layered oxides with a Ni content of ≥80 wt% can satisfy the high-energy demands as cathode materials. In contrast to the classical layered oxides, Ni-rich materials exhibit capacities that exceed conventional capacity values.4,6,8,9 However, the control of the cationic stoichiometry in the Li layer is difficult. Thus, the addition of excessive Li is essential for reducing the cation mixing in the Li layer. Ni-rich cathode materials with excess Li retain their well-ordered layer structure with a low degree of cation mixing; thus, the materials can deliver a high capacity upon cycling. Because an excessive amount of Li is needed to synthesize a highly ordered structure for Ni-rich layered materials, residual Li in an oxide form, i.e., Li2O, can remain on the surface of the cathode material.4,9–11
For practical batteries, excessive Li residues in Ni-rich materials may cause more serious problems.9,11–13
(1) Ni-rich compounds usually exhibit a highly alkaline state (pH > 11) owing to the remaining Li salts. This initiates defluorination of the polyvinylidene fluoride (PVDF) binder, which leads to particle agglomeration and slurry gelation during cathode fabrication and poor mechanical properties of the cathode.
(2) The carbonate species in the Li residues easily decompose under the attack of HF in the electrolyte, which may cause gas evolution and cell swelling, hampering the safe operation of the battery.
(3) Through the decomposition of the electrolyte solvent, the produced CO and CO2 tend to bond with the hydrated Li2O layer (LiOH), forming an Li2CO3 layer in the outermost surface, which is accompanied by the formation of water molecules. This accelerates the decomposition of the electrolytic salt, LiPF6, leading to more formation of insulating LiF in the outermost surface, which impedes the diffusion of Li ions owing to its insulating properties and thus deteriorates the electrochemical performance of the active materials.
(4) At high voltages, the oxidative decomposition of LiOH and Li2CO3 causes an irreversible capacity.
Therefore, the amount of residual Li on the surface of active materials should be reduced for preventing these side reactions from occurring at the interface between the cathode materials and the electrolyte during cycling. Considerable effort has been directed toward enhancing the electrochemical performance of the high-Ni cathode material for improved cells. Although surface coating has been conventionally applied to overcome residual-Li issues in Ni-rich cathode materials, this method has the effect of reducing the reaction between the surface materials and the electrolyte and is not a fundamental solution. Industrially, the most effective method is to wash away the residual Li.11,14 However, this significantly degrades the capacity and cycle retention by damaging the structure of the materials. Assuming that the washing process is essential for the Ni-rich material production process, studies were conducted to minimize the damage to the cathode material in the washing process.15–17 The most effective method is to increase the structural stability using a dopant. Among the many dopants that have been employed, Zr exhibits good electrochemical properties owing to its structural stability.6,8,9,18,19 A. Yu et al. improved the performance by simultaneously doping and coating LiNi0.6Co0.2Mn0.2O2 with Zr.8 However, in the conventional simple mixing method, it was difficult to uniformly dope the primary particles.
Herein, we introduce a solvent solution method for uniform Zr doping of an LiNi0.90Co0.05Mn0.05O2 (NCM) cathode material. The Zr solvent solution method involves wet processing using deionized (DI) water as a solvent during mixing of the (Ni0.90Co0.05Mn0.05)(OH)2 precursor and the Zr source. For Zr doped NCM, which is formed using Zr solvent solution method (L-NCM), uniform Zr doping was achieved, and the grain boundary and surface of the cathode material were well coated with Li2ZrO3 compared with Zr-doped NCM, which is formed using the dry solid mixing method of the (Ni0.90Co0.05Mn0.05)(OH)2 precursor and the Zr source (S-NCM). The coating layer functioned as a protective layer during the washing process. Thus, the electrochemical performance was improved by the synergetic effects of the suitable coating and residual-Li reduction compared to comparative sample witch was fabricated by for Zr doping solid mixing method (S-NCM). The coating quality was examined using transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The phases of the coating material were characterized via X-ray diffraction (XRD) measurements and density functional theory calculations, and the electrochemical performance of the double coating was systematically evaluated via electrochemical measurements using 2032 coin cells and 18680 full cells.
2. Experiments and computations
2.1. Sample preparation and coating process
Ni0.90Co0.05Mn0.05(OH)2 (Reshine New Material Co., Ltd., China) powder was prepared as a precursor. For the Zr solid mixing method, zirconium hydroxide (Zr(OH)4) and (NiMnCo)(OH)2 were mixed using a planetary mill for 5 min. For fabrication of L-NCM, zirconium(IV) chloride (ZrCl4) dissolved in DI water was used. The (Ni0.90Co0.05Mn0.05)(OH)2 precursor powder was added to the ZrO(NO3)2 solution for coprecipitation of Zr compounds on the (Ni0.90Co0.05Mn0.05)(OH)2 precursor via the increase in the pH. The solution was then stirred and filtered. After the filtering, the precursor was dried at 120 °C until the solvent completely evaporated. The molar ratio of the precursor to the zirconium oxynitrate in the second aqueous solution was approximately 1:0.003. For fabrication of S-NCM, the ZrCl4 powder and the (Ni0.90Co0.05Mn0.05)(OH)2 precursor in the same proportion as L-NCM were mixed for 5 min using planetary mixer. Finally, the Zr-mixed (Ni0.90Co0.05Mn0.05)(OH)2 precursors using two different methods were ground with a stoichiometric amount of LiOH and calcined at 750 °C for 24 h under flowing O2 in order to obtain L-NCM.
The sintered NCM active material was washed using DI water for removing Li residuals on the surface. The NCM cathode active material and DI water were mixed and stirred for approximately 20 min and then filtered to isolate a precipitate. The filtered precipitate was dried in an oven at approximately 150 °C for approximately 3 h to prepare a dried product. The dried powder was thermally treated at approximately 720 °C for ∼5 h under flowing O2.
Composite positive electrodes with 92 wt% active material, 4 wt% Denka black, and 4 wt% PVDF were fabricated and placed on an Al foil current collector. The electrodes were dried at 120 °C in vacuo and then pressed. Metallic Li was used as the counter electrode. The electrolyte solution comprised 1.0 M LiPF6 dissolved in a solution of fluoroethylene carbonate and dimethylene carbonate. CR2032-type coin cells were assembled in a dry room, and the cells were discharged and charged galvanostatically. The loading level for the active ingredient was set as 10 mg cm−2.
Cylindrical 18650 cells were assembled to evaluate the long-term cyclability of the NCM/graphite full cells. Galvanostatic charge–discharge cycling of the cells was performed between 2.8 V and a cutoff voltage of 4.3 V at 200 mA h g−1 in a temperature-controlled chamber at 25 °C.
2.2. Instrumental method and characterization
XRD patterns of samples were acquired using a Bruker AXS D8 Advance X-ray diffractometer (Cu Kα radiation, λ = 1.5406 Å) to identify the different phases present. The morphologies of the products were observed via SEM (Hitachi, S-5500) and high-resolution TEM (FEI, TITAN-80-300). The electrochemical activity of the cathodes was analyzed via electrochemical impedance spectroscopy (EIS) using a Solartron 1260 frequency-response analyzer (Solartron Analytical, Farnborough, U.K.). Impedance measurements were conducted over an applied-frequency range of 10 mHz to 1 MHz.2.3. Simulation
First-principles calculations were performed using the Vienna Ab initio Simulation Package via the projector augmented wave method. The exchange–correlation interactions were included with the generalized gradient approximation Perdew–Burke–Ernzerhof functional, and the plane wave cutoff energy was set as 500 eV. The structural relaxations were conducted with the criteria of 10−4 eV for the total energy and 0.02 eV Å−1 for the forces on each atom. The effective onsite Hubbard Ueff corrections were 6.885 and 2.00 eV on the 3d electrons for Ni and Zr atoms, respectively.20–223. Results and discussion
Fig. 1(a) shows the XRD patterns of pristine, S-NCM, and L-NCM samples. After Zr mixing and heat treatment, the NCM particles retained their initial crystal structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), suggesting that the Zr mixing did not induce significant structural changes or lead to the formation of additional foreign products. All the peaks were indexed to the hexagonal α-NaFeO2 structure with the space group Rm. The lattice parameters and the peak intensity ratio of (003)/(104) of the Zr-doped samples calculated using the XRD measurements are presented in Table S1 (ESI).† A comparison of the lattice parameters of the Zr-added and pristine samples revealed that the lattice parameters increased owing to the Zr. Thus, the NCM structure was successfully doped with Zr, because the ionic radius of Zr4+ (0.79 Å) is larger than those of transition metals, such as Ni2+ (0.69 Å) and Co3+ (0.545 Å), which is consistent with previous reports.23 The cation mixing, which is the Li+/Ni2+ exchange is also deeply related to the ratio of c/a. The general trend of the changes of this ratio c/a is also similar to that of the peak intensity ratio of (003)/(104). The peak intensity ratio of (003)/(104) decide the ordering of Li and transition metals. The intensity ratio will decide the positioning of transition metal ion and lithium in the host matrix.24 The Zr doped samples represented the higher peak intensity ratio, which indicated that cation mixing has decreased due to Zr doping. The cation mixing of S-NCM and L-NCM was similar. Additionally, there were no changes in the crystal structure of NCM upon doping, primarily owing to the low content (2 mol%) of the doping materials.
m), suggesting that the Zr mixing did not induce significant structural changes or lead to the formation of additional foreign products. All the peaks were indexed to the hexagonal α-NaFeO2 structure with the space group Rm. The lattice parameters and the peak intensity ratio of (003)/(104) of the Zr-doped samples calculated using the XRD measurements are presented in Table S1 (ESI).† A comparison of the lattice parameters of the Zr-added and pristine samples revealed that the lattice parameters increased owing to the Zr. Thus, the NCM structure was successfully doped with Zr, because the ionic radius of Zr4+ (0.79 Å) is larger than those of transition metals, such as Ni2+ (0.69 Å) and Co3+ (0.545 Å), which is consistent with previous reports.23 The cation mixing, which is the Li+/Ni2+ exchange is also deeply related to the ratio of c/a. The general trend of the changes of this ratio c/a is also similar to that of the peak intensity ratio of (003)/(104). The peak intensity ratio of (003)/(104) decide the ordering of Li and transition metals. The intensity ratio will decide the positioning of transition metal ion and lithium in the host matrix.24 The Zr doped samples represented the higher peak intensity ratio, which indicated that cation mixing has decreased due to Zr doping. The cation mixing of S-NCM and L-NCM was similar. Additionally, there were no changes in the crystal structure of NCM upon doping, primarily owing to the low content (2 mol%) of the doping materials.
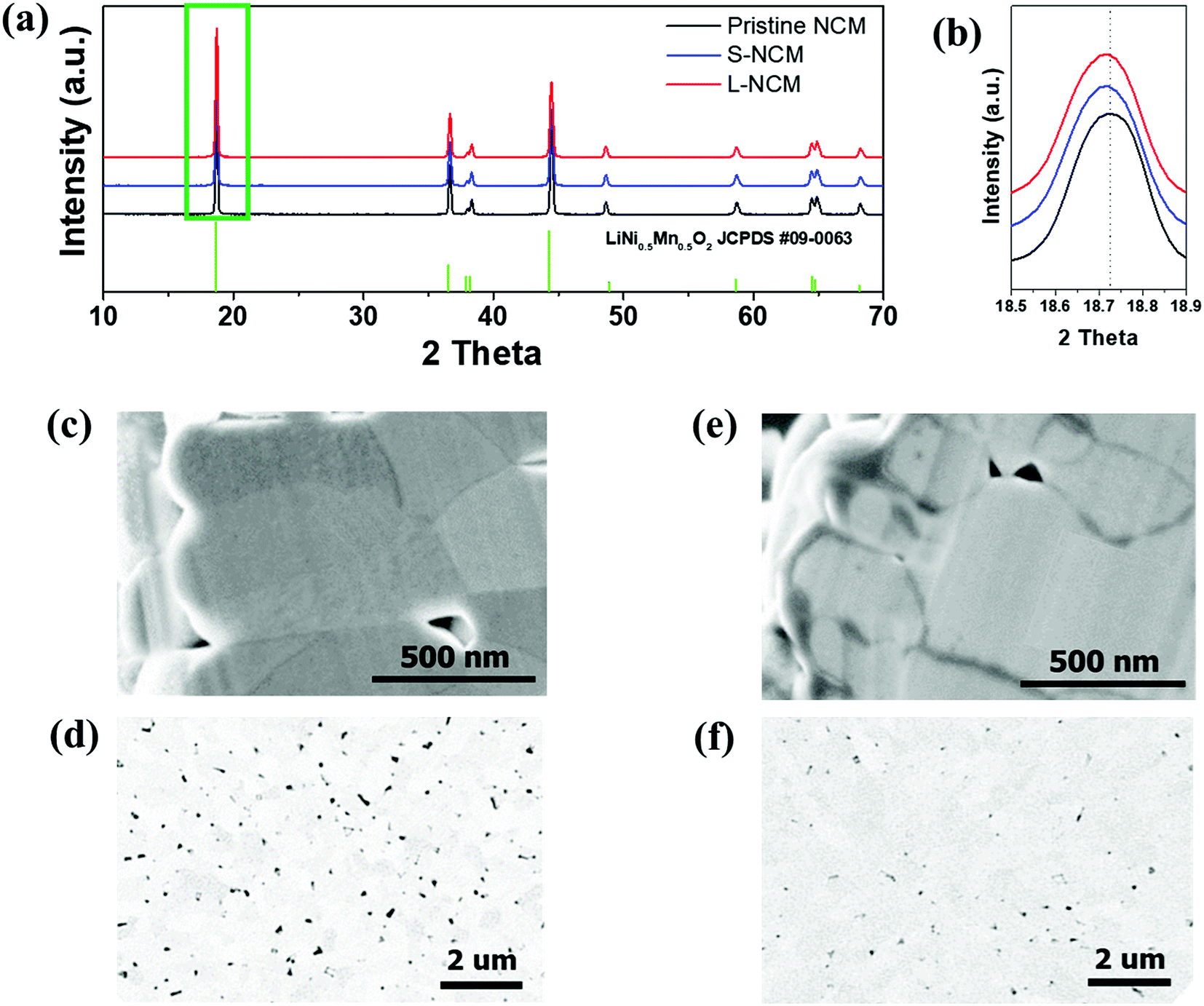 | ||
| Fig. 1 (a) XRD patterns of the pristine NCM, S-NCM, and L-NCM. (b) Enlarged XRD pattern of the green box in (a). Cross-sectional SEM and BSE images of (c and d) S-NCM and (e and f) L-NCM. | ||
Fig. 1(c)–(f) show SEM secondary and backscattered electron (BSE) images of S-NCM and L-NCM. Pinholes were observed in both particles. The ratio of the pinhole area to the powder cross-sectional area in the BSE images was 5.44% and 0.76% for the S-NCM and L-NCM samples, respectively. The pinholes were larger and more numerous for S-NCM than for L-NCM. When the Zr source material was added to the precursor by using the solution, the solution containing the material was diffused between the pinholes existing on the precursor to block the pinholes. After the sintering process, the internal pinholes disappeared owing to the formation of Li2ZrO3.
For first-principles calculations, bulk LiNiO2 supercell structures and the surface LiNiO2 were considered. The supercells selected for the bulk LiNiO2 structures contained 36 unit cells of LiNiO2, in which 36 Li sites, 36 Ni sites, and 72 O sites were considered. The Zr doping was implemented by substituting Ni ions with Zr atoms in the supercell. To consider the Zr dopant on the NCM surfaces, the surfaces of LiNiO2 were modeled using LiNiO2 slabs, and the first-principles calculations were performed. The LiNiO2 slabs contained 6 Li layers, 6 Ni layers, and 12 O layers, and the O layers were located between the Li and Ni layers. The slabs had a (003) surface of a (Rm) LiNiO2 structure. Each layer had four atoms; thus, the LiNiO2 slabs were composed of 24 Li atoms, 24 Ni atoms, and 48 O atoms. The positions of the atoms in the lower three Li layers, three Ni layers, and six O layers were fixed to reflect the bulk properties, and the other atoms in the upper parts were relaxed. For blocking the interaction between two surfaces generated by the periodic boundary conditions. To investigate the possibility of 2nd-phase formation, first-principles calculations for Li–Zr–O, e.g., Li2ZrO3 and Li6Zr2O7, were performed.
To elucidate the behavior of the Zr dopants, the heat of formation was calculated for both bulk-doped and surface-doped Zr atoms. The heat of formation for the bulk-doped Zr atoms was calculated using the following reaction.
| 35LiNiO2 + Li + ZrO2 ↔ Li36Ni35ZrO72 | (1) |
The heat of formation energy ΔHf is given as follows:
| ΔHf = E[Li36Ni35ZrO72] − {35 × E[LiNiO2] + E[Li] + E[ZrO2]}, | (2) |
| (Li24Ni24O48)slabs + Li + ZrO2 ↔ (Li24Ni23ZrO48)slabs + LiNiO2, | (3) |
| ΔHf = {E[(Li24Ni23ZrO48)slabs] + E[LiNiO2]} − {E[(Li24Ni23ZrO48)slabs] + E[Li] + E[ZrO2]} | (4) |
The heat of formation for bulk doping was −2.054 eV. When Zr atoms were doped at the Ni sites of the 1st and 2nd transition-metal layers, the heat of formation was −2.584 and −2.724 eV, respectively, and Zr atoms tended to be doped at the surface rather than inside the electrode particles.
To investigate the possibility of 2nd-phase formation instead of bulk doping or surface doping, two Li–Zr–O compounds—Li2ZrO3 and Li6Zr2O7—were considered, according to the following reactions.
| Li36Ni36−xZrxO72 + x(Li + 1/2O2) ↔ (36 − x)LiNiO2 + xLi2ZrO3 | (5) |
| Li36Ni36−xZrxO72 + x(2Li + 3/4O2) ↔ (36 − x)LiNiO2 + x/2Li6Zr2O7 | (6) |
These reactions indicate whether the bulk-doped LiNiO2 was stable rather than 2nd-phase formation. The reaction enthalpy is presented in Table 1. The first-principles calculations indicated that Zr preferred the 2nd-phase formation of Li2ZrO3 rather than being doped into the electrode. The formation of Li6Zr2O7 was unstable in this system. Similar calculations were performed for the surface-doped system, as follows.
| (Li24Ni23ZrO48)slab + LiNiO2 + Li + 1/2O2 ↔ (Li24Ni24O48)slab + Li2ZrO3 | (7) |
| (Li24Ni23ZrO48)slab + LiNiO2 + 2Li + 3/4O2 ↔ (Li24Ni24O48)slab + 1/2Li6Zr2O7 | (8) |
| Sample | 1st layer | 2nd layer | Bulk 1 | Bulk 2 | Bulk 3 | Bulk 6 |
|---|---|---|---|---|---|---|
| ΔH1 | −4.273 | −4.131 | −4.802 | −4.634 | −4.581 | −4.319 |
| ΔH2 | 9.044 | 9.186 | 8.515 | 17.365 | 26.207 | 53.989 |
The calculation results were identical for the bulk-doped system, although the surface provided more stable sites for Zr atoms. According to the first-principles calculations, Zr was doped at the surface initially, and after the Zr concentration exceeded a certain value for the nucleation of Li2ZrO3, the 2nd-phase Li2ZrO3 was formed at the surfaces. This not only increased the density of the electrode, resulting in a higher capacity in the same area, but also reduced the penetration of the electrolyte into the powder during operation, making it easier to maintain the secondary particles without breaking the powder.
To confirm the distribution and composition of the dopant in the NCM particles, energy-dispersive X-ray spectroscopy (EDS) was performed, and the results are shown in Fig. 2(a) and (b). The elemental-mapping images indicate that all the Zr was distributed uniformly in the particles. However, in L-NCM, the density of Zr was very high at the positions where the primary particles met. This is supported by the aforementioned SEM results, which indicated that the pinholes were filled by the diffusion of the solution during doping with the dopant solution. Furthermore, Zr exhibited high concentrations as if the surface had been coated. Although it is solution type for doping, not only doping but also a Zr surface coating effect was observed.
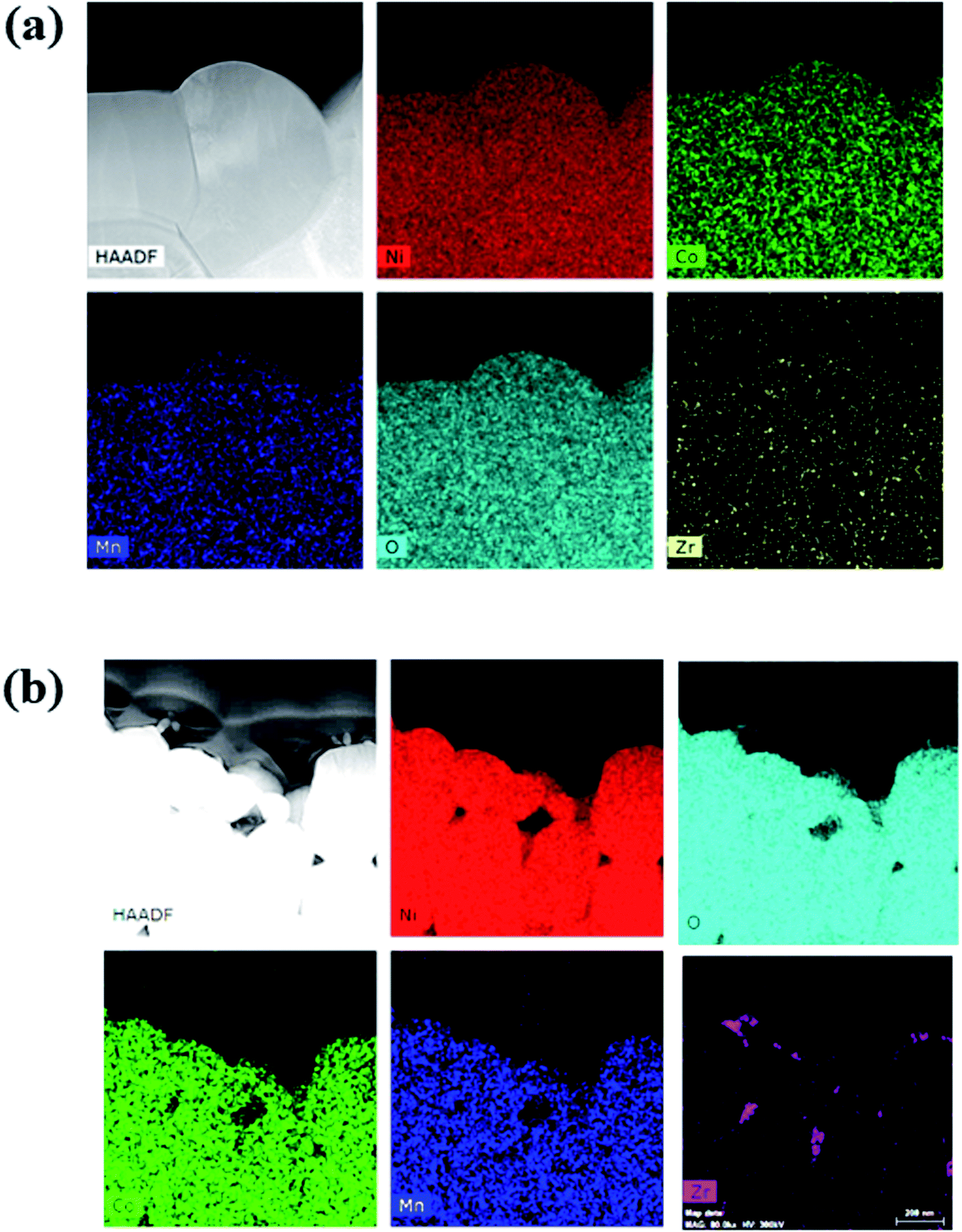 | ||
| Fig. 2 EDS mapping of (a) S-NCM and (b) L-NCM NCM. | ||
Electrochemical tests using a CR2032-type coin cell were conducted to investigate the effects of the doping. The capacities for various applied currents are presented in Table 2. At a cutoff voltage of 2.8–4.35 V, the cells containing pristine NCM, S-NCM, and L-NCM delivered reversible capacities of 225.6, 223.7, and 215.1 mA h g−1 at 0.1C, respectively. The Zr-doped NCMs exhibited a slightly lower capacity than the pristine NCM, possxibly owing to the Zr coating.12,25,26 However, the capacity retention of the Zr-doped samples was high at high applied currents. This is because the slab size was increased by the Zr doping, which facilitated the diffusion of Li and by the removing the Li residuals as shown in Table 3.
| Sample | 0.1C (mA h g−1) | 0.2C (mA h g−1) | 0.33C (mA h g−1) | 0.5C (mA h g−1) | 1C (mA h g−1) | 2C (mA h g−1) |
|---|---|---|---|---|---|---|
| Pristine NCM | 225.6 | 220.2 | 215.4 | 205.8 | 200.0 | 196.7 |
| S-NCM | 223.7 | 219.7 | 215.9 | 206.4 | 201.2 | 198.1 |
| L-NCM | 215.1 | 212.8 | 209.9 | 201.3 | 196.4 | 193.6 |
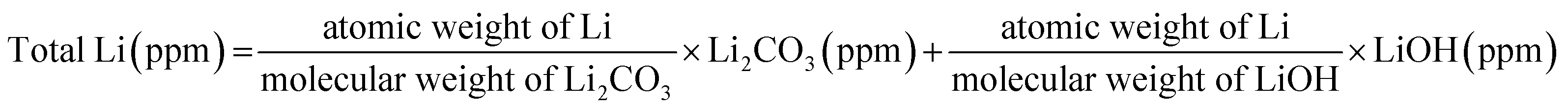
Fig. 3(a) compares the cycling performance of the cells at 25 °C and at a charging/discharging rate of 1C. Here, a 2032 coin half-cell was used. The capacity-retention values were 86.08%, 92.12%, and 96.85% at the 50th cycle for the pristine NCM, S-NCM, and L-NCM, respectively. This retention value for L-NCM was much higher than the previous work.27,28 All the Zr-doped samples exhibited higher capacity retention than the pristine NCM owing to the enhancement of the structural stability caused by the doping.23 In particular, the capacity retention of the L-NCM sample was remarkably high, as the reaction between the electrolyte and the cathode was blocked by the uniform Zr coating layer, as reported previously.12
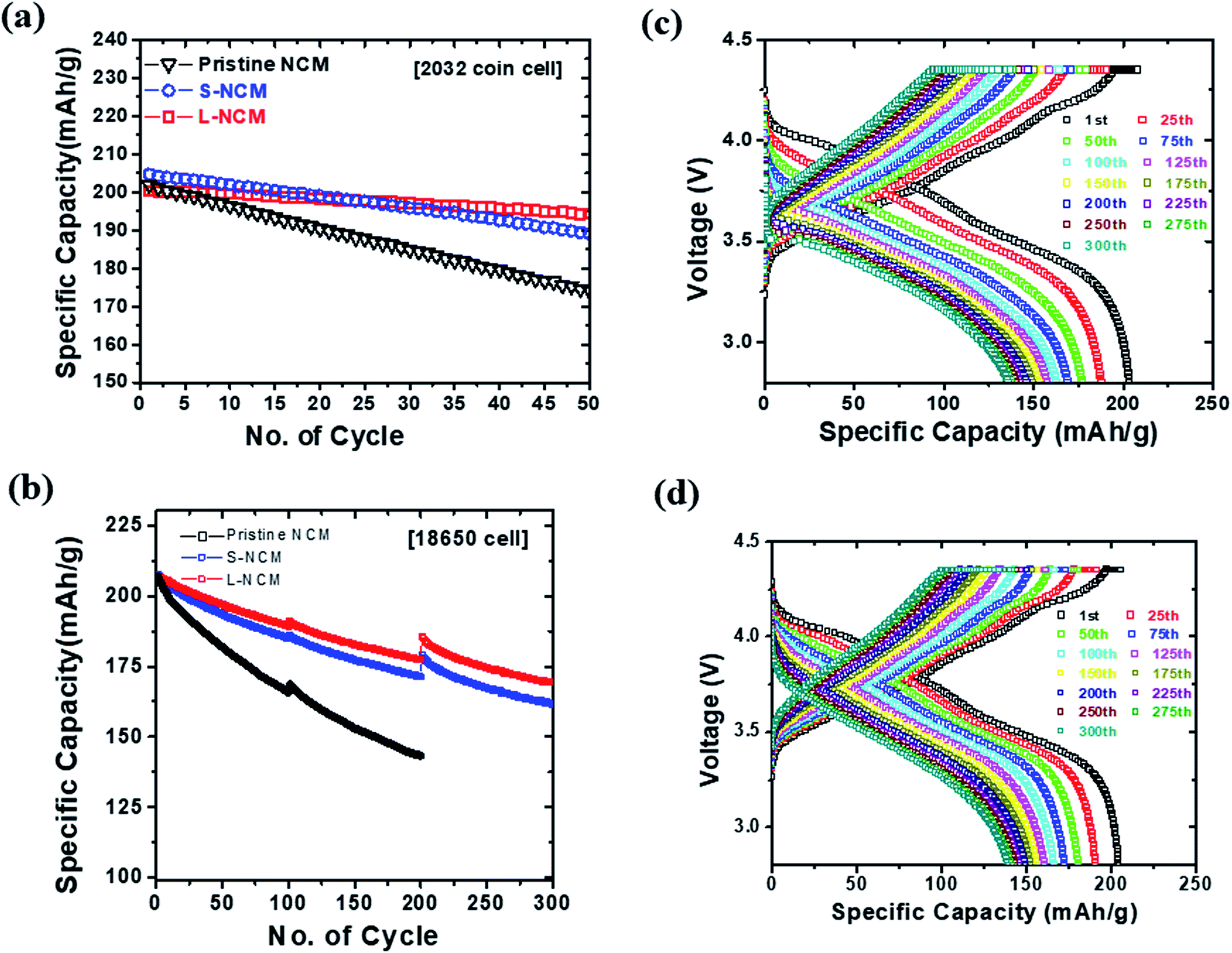 | ||
| Fig. 3 Comparisons of (a) the cycle retention of the 2032 coin half-cell for pristine NCM, S-NCM, and L-NCM and (b) the cycling performance of the 18650 full cell for pristine NCM, S-NCM, and L-NCM. Charge–discharge characteristics at intervals of 25 cycles for (c) S-NCM and (d) L-NCM. | ||
To examine the effect of Zr in a full cell, the electrochemical performance of cylindrical 18650 cells was evaluated. The cyclability of pristine NCM, S-NCM and L-NCM cells at 1C and 45 °C is shown in Fig. 3(b). The discontinuous points every 100 cycles in the cycling data shown in Fig. 3(b) coincide with the capacity check at 0.1C. In the 300th cycle in the voltage range of 2.8–4.35 V, the capacity-retention values for S-NCM and L-NCM were 77.72% and 81.95% after 300 cycles, respectively. The capacity ratio of pristine was 69.2% after 200 cycles. The evaluation criteria of 18650 full cell are less than 70% of the initial capacity. The evaluation was stopped because the pristine NCM was below the evaluation criteria after 200 cycles. The capacities of S-NCM and L-NCM in the first cycle were 207.57 and 206.58 mA h g−1 at 1C, respectively. Similar to the coin-cell results, the L-NCM sample exhibited a slightly lower initial capacity but far better capacity retention after cycling. A comparison of the charge–discharge behaviors and plots of dQ/dV with respect to the voltage of the cells in the first cycle and every 25 cycles until the 300th cycle are presented in Fig. 3(c), (d), S2(a) and (b).† In the 1st cycle, the anodic and cathodic peaks for the two samples appeared at similar voltages, as shown in Fig. S2(a) and (b).† However, after 25 cycles, the peak of S-NCM around 4.2 V had disappeared. Moreover, most of the peak positions for S-NCM shifted toward higher resistance values compared with those for L-NCM, as shown in Fig. S2(a) and (b).† However, the L-NCM sample exhibited lower redox voltages owing to the suppression of the side reaction between the cathode and the electrolyte due to the filling of pinholes between primary particles and the formation of the Li2ZrO3 layer on the surface.
To investigate the high resistance of L-NCM after cycling, EIS was performed, as shown in Fig. 4(a)–(d). The impedance spectra of the two samples in the initial state were measured before the formation process of the 18650 cell. It is known that direct-current (DC) resistance arises mainly from the resistance of the electrolyte; however, the DC resistances of the two samples before and after the experiment were similar.29,30 The impedances in the high- and low-frequency regions were closely related to the impedances from the electrochemical reactions at the anode and cathode, respectively. When the impedance spectra were divided into the solution resistance (Rs), negative electrode resistance (Rn), and positive electrode resistance (Rp), Rp was the most critical component of the three. Both the high- and low-frequency regions were larger for S-NCM than for L-NCM. The increase in the lower frequency range was more pronounced. The intensity in the low-frequency region, which was related to the electrochemical reaction at the cathode, was increased. The impedance may have been affected by the percolation of the electrolyte into the cathode and the resistive layer caused by the filling of pinholes in the particles and the Li2ZrO3 layer on the surface. This led to a large impedance for the L-NCM sample. These findings support the aforementioned dQ/dV results and provide sufficient evidence that the largest proportion of the resistance came from the diffusion resistance at the cathode.
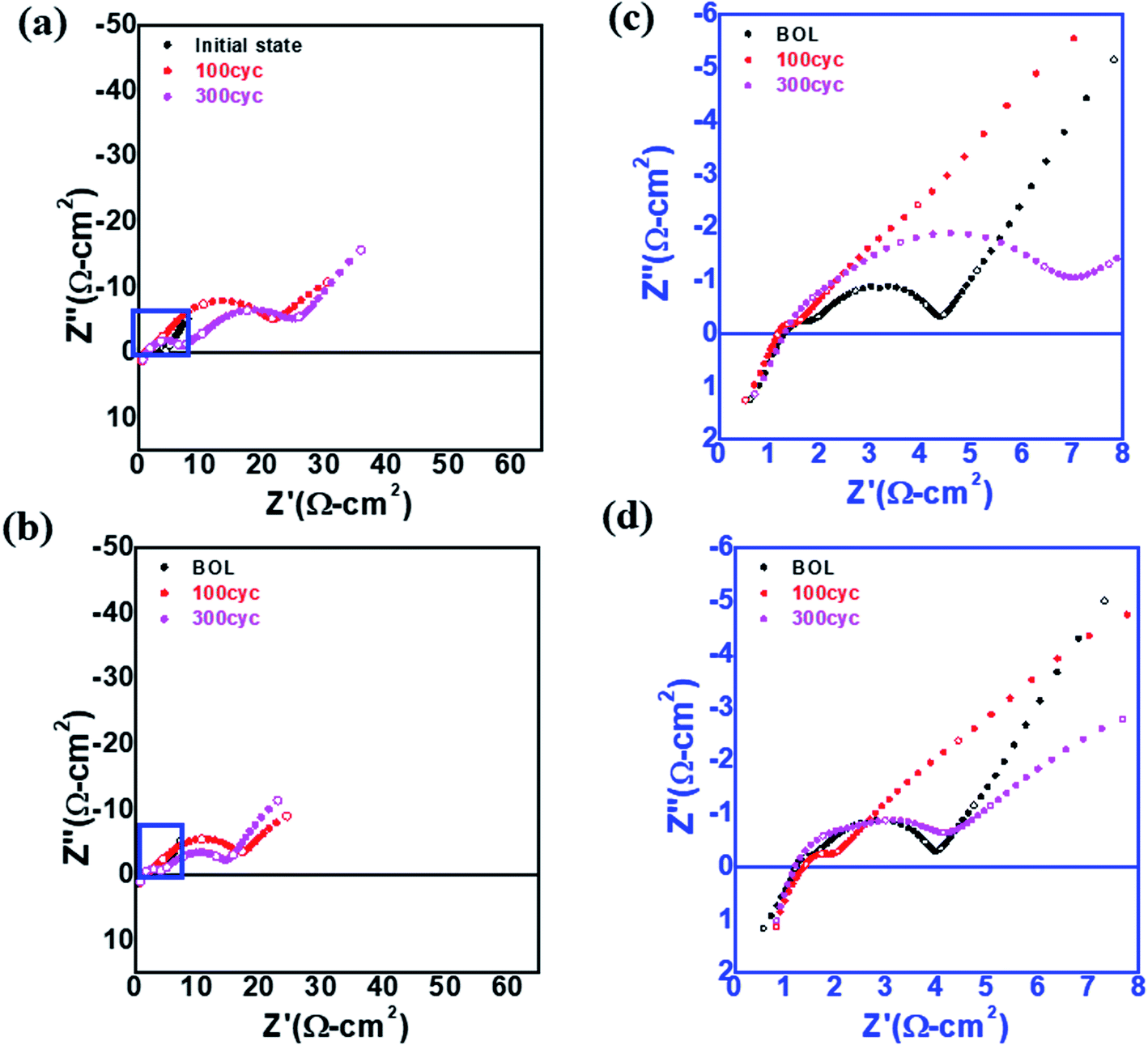 | ||
| Fig. 4 EIS spectra of (a) S-NCM and (b) L-NCM. Enlarged EIS spectrum of the blue box in (a) for (c) S-NCM and (d) L-NCM. | ||
Although the L-NCM sample exhibited better electrochemical properties, it was important to determine whether this approach prevented the percolation of the electrolyte through the filling of pinholes in the NCM particles. Hence, the crack generation inside the secondary particles of NCM (between the primary particles) was traced by obtaining SEM images of the S-NCM and L-NCM particles after 300 cycles, as shown in Fig. 5(a) and (b). There was no SEM image of pristine NCM because the pristine NCM was stopped at 200 cycles. For comparison, the cross-sectional SEM images of samples the first cycle were shown in Fig. S3.† The cracks of pristine NCM and S-NCM were similar. The intergranular cracking behavior of the cycled S-NCM was clearly evident and dominant. However, for the L-NCM sample, although slight separation between the primary particles was observed after 300 cycles, the secondary particles were well-formed, and the initial shape was well-maintained. In general, the Ni rich layered oxide material generates a more severe phase transition near the end of the charge, which increases the anisotropic volume change, and generates a newly exposed surface area.31,32 However, a severe anisotropic volume change may be suppressed by Zr coating layer, which acts as adhesive. The crack of the samples provides more channels for electrolyte penetration. The fresh cathode surface, which had not reacted with electrolyte and revealed by cracking, may form the cathode electrolyte inter-phase (CEI) caused by reaction with electrolyte and cathode material. The CEI layer led to inhibition of Li ion diffusion and that appeared as an increase in impedance. Thus, it was considered that the Zr solvent solution method suppressed the permeation of the electrolyte and/or the formation of the Zr layer on the surfaces of the secondary particles, enhancing the structural stability during the charge−discharge cycling.
 | ||
| Fig. 5 SEM images of (a) S-NCM and (b) L-NCM, obtained after 300 cycles. | ||
4. Conclusion
We introduced a solvent solution mixing method for uniform Zr doping and coating of an LiNi0.90Co0.05Mn0.05O2 (NCM) cathode material. The method involves wet processing using DI water as a solvent during the mixing of the (Ni0.90Co0.05Mn0.05)(OH)2 precursor and the Zr source. Zr compounds were formed on the NCM precursor powder via coprecipitation. To confirm the superiority of Zr doped NCM, which is formed using Zr solvent solution method (L-NCM), it was compared with Zr-doped NCM, which is formed using the dry solid mixing method of the (Ni0.90Co0.05Mn0.05)(OH)2 precursor and the Zr source (S-NCM). The amount of pinholes inside the L-NCM particles was significantly reduced by the formation of Li2ZrO3. The coating layer functioned as a protective layer during the washing process for removing the residual Li. Moreover, the liquid mixing method significantly reduced the crack generation, because the Zr compounds removed the pinholes inside the NCM powder of the secondary particles and functioned as a buffer layer against the volume change during cycling. Therefore, the electrochemical performance was improved by the synergetic effects of the suitable coating and enhanced structural stability. The capacity-retention values were 86.08%, 92.12%, and 96.85% at the 50th cycle for pristine NCM, S-NCM, and L-NCM, respectively.Conflicts of interest
There is no conflicts to declare.Acknowledgements
This work was supported by the Gachon University research fund of 2019 (GCU-2019-0356) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07049165).References
- J. Tollefson, Nature, 2008, 456, 436–440 CrossRef CAS PubMed
.
- M. Armand and J. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
- J. Yang, M. Hou, S. Haller, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2016, 189, 101–110 CrossRef CAS
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS
- J.-G. Han, J. Bin Lee, A. Cha, T. K. Lee, W. Cho, S. Chae, S. J. Kang, S. K. Kwak, J. Cho, S. Y. Hong and N.-S. Choi, Energy Environ. Sci., 2018, 11, 1552–1562 RSC
- C. S. Yoon, U. H. Kim, G. T. Park, S. J. Kim, K. H. Kim, J. Kim and Y. K. Sun, ACS Energy Lett., 2018, 3, 1634–1639 CrossRef CAS
- X. Xiong, Z. Wang, P. Yue, H. Guo, F. Wu, J. Wang and X. Li, J. Power Sources, 2013, 222, 318–325 CrossRef CAS
- S. Liu, Z. Dang, D. Liu, C. Zhang, T. Huang and A. Yu, J. Power Sources, 2018, 396, 288–296 CrossRef CAS
- S. Gao, X. Zhan and Y. T. Cheng, J. Power Sources, 2019, 410–411, 45–52 CrossRef CAS
- H.-H. Sun and A. Manthiram, Chem. Mater., 2017, 29, 8486–8493 CrossRef CAS
- J. Kim, Y. Hong, K. S. Ryu, M. G. Kim and J. Cho, Electrochem. Solid-State Lett., 2006, 9, A19–A23 CrossRef CAS
- K. Park, J. H. Park, S. G. Hong, B. Choi, S. W. Seo, J. H. Park and K. Min, Phys. Chem. Chem. Phys., 2016, 18, 29076–29085 RSC
- M.-J. Lee, M. Noh, M.-H. Park, M. Jo, H. Kim, H. Nam and J. Cho, J. Mater. Chem. A, 2015, 3, 13453–13460 RSC
- J. Hyeon, C. Jo, H. Yashiro, S. Kim and S. Myung, J. Power Sources, 2016, 313, 1–8 CrossRef
- G. Cao, J. Zhu and Y. Li, RSC Adv., 2020, 10, 9917–9923 RSC
- W. Wang, H. Yanjie, W. Haijiang, Y. Zhong, G. Wang, B. Zhong, B. Fang, X. Guo and S. Liao, Chin. J. Chem., 2015, 33, 261–267 CrossRef
- W. B. Hua, X. D. Guo, Z. Zheng, Y. J. Wang, B. H. Zhong, B. Fang, J. Z. Wang, S. L. Chou and H. Liu, J. Power Sources, 2015, 275, 200–206 CrossRef CAS
- F. Schipper, H. Bouzaglo, M. Dixit, E. M. Erickson, T. Weigel, M. Talianker, J. Grinblat, L. Burstein, M. Schmidt, J. Lampert, C. Erk, B. Markovsky, D. T. Major and D. Aurbach, Adv. Energy Mater., 2018, 8, 1701682 CrossRef
- F. Ulu Okudur, J. D'Haen, T. Vranken, D. De Sloovere, M. Verheijen, O. M. Karakulina, A. M. Abakumov, J. Hadermann, M. K. Van Bael and A. Hardy, RSC Adv., 2018, 8, 7287–7300 RSC
- P. W. Tasker, J. Phys. C Solid State Phys., 1979, 12, 4977–4984 CrossRef CAS
- G. Kressea and J. Furthmülle, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 558–561 CrossRef CAS PubMed
- S. Liu, Z. Dang, D. Liu, C. Zhang, T. Huang and A. Yu, J. Power Sources, 2018, 396, 288–296 CrossRef CAS
- J. Cho, z. G. Kim and H. S. Lim, J. Electrochem. Soc., 1999, 146, 3571–3576 CrossRef CAS
- X. Zhang, Y. Yin, Y. Hu, Q. Wu and Y. Bai, Electrochim. Acta, 2016, 193, 96–103 CrossRef CAS
- S. M. Lee, S. H. Oh, J. P. Ahn, W. Il Cho and H. Jang, J. Power Sources, 2006, 159, 1334–1339 CrossRef CAS
- K. Park, J. H. Park, S. G. Hong, B. Choi, S. Heo, S. W. Seo, K. Min and J. H. Park, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed
- K. Min, K. Park, S. Y. Park, S.-W. Seo, B. Choi and E. Cho, J. Electrochem. Soc., 2018, 165, A79–A85 CrossRef CAS
- K. Park, D. Han, J. Shon, S. G. Doo and S. Lee, RSC Adv., 2015, 5, 6340–6344 RSC
- K. Park, D. Han, J. Shon, S. G. Doo and S. Lee, RSC Adv., 2015, 5, 6340–6344 RSC
- K. Min and E. Cho, Phys. Chem. Chem. Phys., 2018, 20, 9045–9052 RSC
- A. Manthiram, J. C. Knight, S. Myung and S. Oh, Adv. Energy Mater., 2016, 6, 1501010 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01543a |
| ‡ K. Park and D. Ham equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |