
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibacterial properties of main-chain cationic polymers prepared through amine–epoxy ‘Click’ polymerization†
Junki Oh,
Seung-Jin Kim,
Min-Kyu Oh and
Anzar Khan*
and
Anzar Khan*
Department of Chemical and Biological Engineering, Korea University, Seoul, 02841, South Korea. E-mail: anzar@korea.ac.kr
First published on 17th July 2020
Abstract
Poly(β-hydroxyl amine)s are prepared through an amine–epoxy ‘click’ polymerization process in water under ambient conditions. These materials could be subjected to a post-polymerization protonation/alkylation reaction at the nitrogen atom to yield quaternary ammonium salts in the polymer backbone. The antimicrobial activities indicated that polymers carrying butyl chains at the nitrogen atom are effective towards Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus), as only 10–20 μg mL−1 polymer concentrations are required to inhibit the bacterial growth by >90%. One of the candidates was also found to be effective towards Mycobacterium smegmatis (M. smegmatis) – a model organism to develop drugs against rapidly spreading tuberculosis (TB) infections. The hemolysis assay indicated that a majority of antimicrobial agents did not disrupt red blood cell membranes. The mechanistic studies suggested that cell wall disruption by the cationic polymers was the likely cause of bacterial death.
The concept of ‘click’ chemistry, as introduced by Sharpless and coworkers, refers to high efficiency, regioselective, modular, and atom-economic chemical reactions with a high degree of orthogonality and simple purification protocols.1 Besides the preparation of small molecules, such reactions can also be applied as a polymerization process. To describe this variation, Tang and coworkers coined the term ‘click’ polymerization.2–5 In this context, the reaction between the amine and epoxide functionalities is known to proceed under ambient and aqueous conditions to afford poly(β-hydroxyl amine)s.6–9 The amine–epoxy reaction is catalyst-free and generates no by-product.10,11 Therefore, a post-polymerization purification protocol is not necessary and the polymers can be produced in a quantitative yield. The polymers also have the advantage of carrying a tertiary amine site in the backbone. Paulusse and coworkers have shown that such amines have a large (up to 64%) buffering capacity in the pH range of 7.4–5.1. They harnessed this property to form complexes of the cationic polymers with anionic plasmid DNA useful in gene delivery applications.12 Inspired by this study, we hypothesized that such main-chain amine groups could be employed in interacting with the anionic bacterial cell membrane to design antimicrobial polymers.13–18 The avenue for alkylation at the nitrogen atoms also offers an opportunity for preparing quaternary ammonium analogues and comparing their properties with the tertiary amine polymers.19 To investigate this, we prepared a new family of poly(β-hydroxyl amine)s differing in their chemical composition and studied their antibacterial and hemolytic properties.
The polymer synthesis follows a simple sequence (Scheme 1). Initially, commercially available primary amine and di-epoxide monomers are stirred in water for 48 hours under ambient conditions. This procedure leads to the formation of poly(β-hydroxyl amine)s polymer chains, which can be isolated through a simple freeze-drying procedure (Fig. 1 and S1–S27†). Typically, the molecular weights of these polymers range from 1–6 kDa with dispersity (Mw/Mn) in the range of 1.2–1.8. A post-polymerization treatment with an aqueous hydrochloric acid solution leads to protonation of the backbone amines. Alternatively, alkyl iodide under silver tetrafluoroborate catalysis can yield alkylation at the nitrogen atom. The quaternization of the nitrogen atoms can be observed with the help of 1H-NMR spectroscopy (Fig. S1–S19†). The methylene groups adjacent to the nitrogen atoms in the precursor polymers can be seen at 2.6 ppm. Upon quaternization, these signals shift downfield by 1 ppm due to electron-deficient nature of the positively charged nitrogen atom. In all cases, these signals moved completely and a good area integration ratio could be observed between the side-chain protons and main-chain protons indicating a quantitative transformation of the tertiary amine atoms to quaternary amines in the polymer backbone.
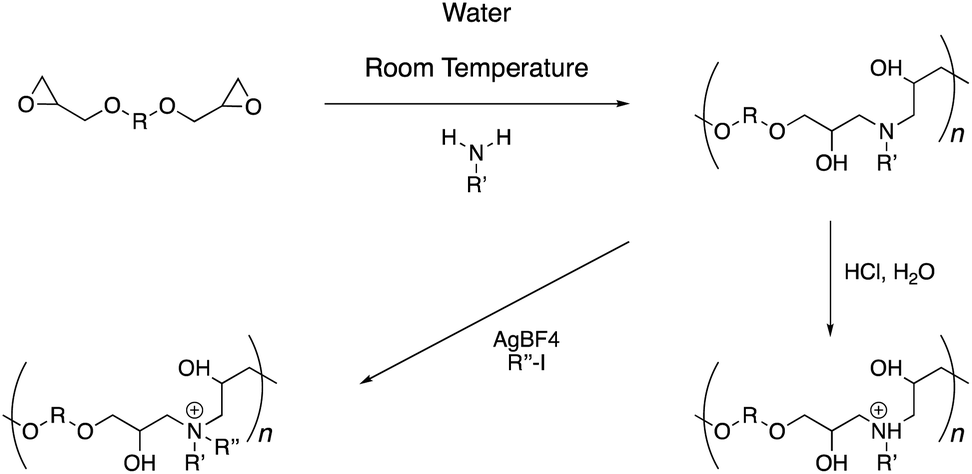 | ||
| Scheme 1 Synthesis and post-polymerization modification of poly(β-hydroxyl amine)s. | ||
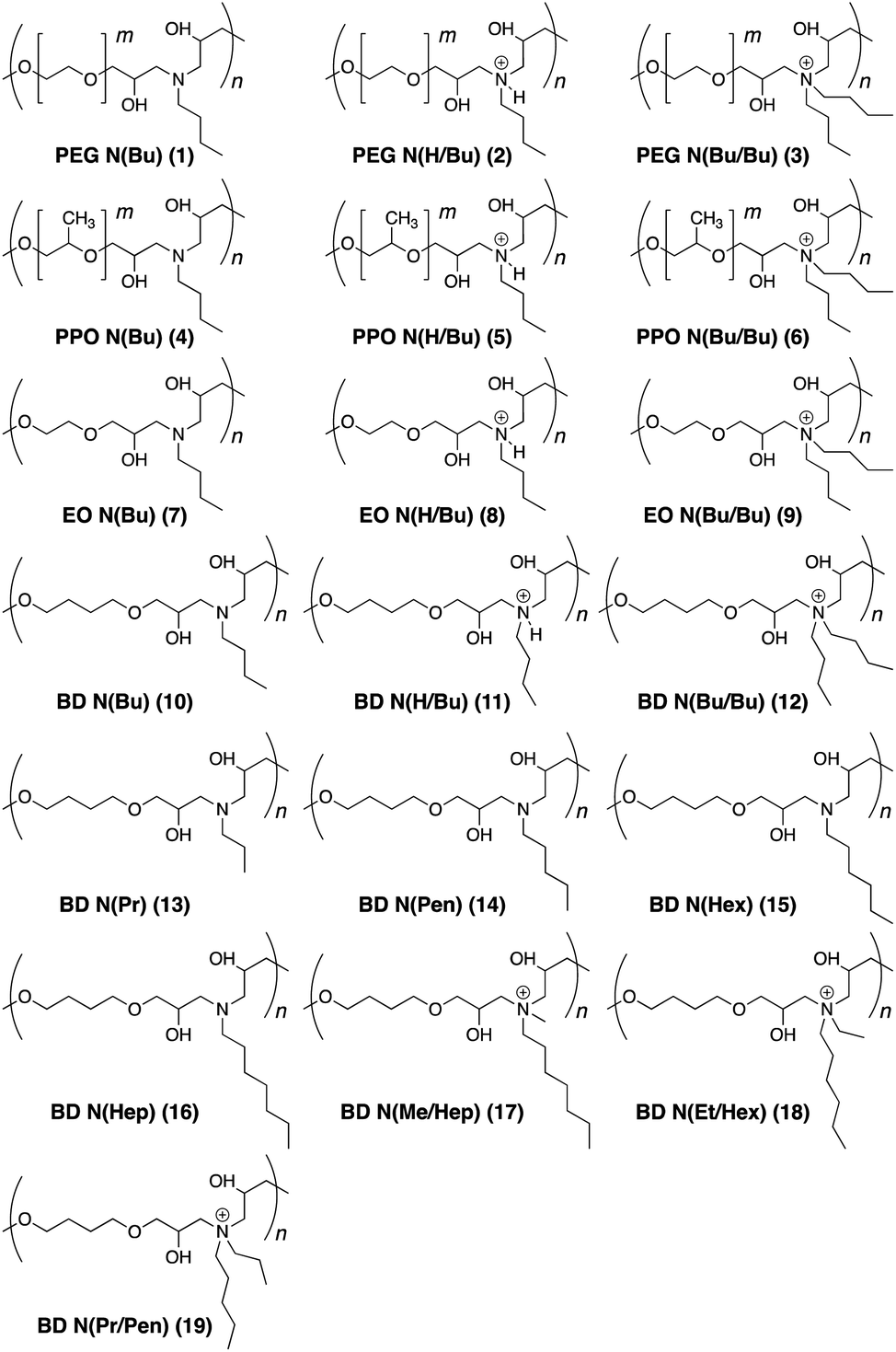 | ||
| Fig. 1 Chemical structure of the polymers. The polymer names indicate the linker (R) in the diglycidyl ether monomers and the substituent(s) (R′/R′′) on the nitrogen atom. R = PEG (polyethylene glycol, m = 8–9), PPO (polypropylene oxide, m = 4–5), EO (ethylene oxide), BD (butanediol). R′ = Pr (C3H7), Bu (C4H9), Pen (C5H11), Hex (C6H13), Hep (C7H15). R′′ = Me (CH3), Et (C2H5), Pr (C3H7), Bu (C4H9). | ||
The antimicrobial activity of the synthesized polymers was investigated by the broth microdilution method to determine the minimum inhibitory concentration (MIC>90) against E. coli, S. aureus, and M. smegmatis bacterial species (Table 1). MIC>90 values were noted as the lowest polymer concentration to inhibit the visible bacterial growth after overnight incubation. The polyethylene glycol (PEG)-based polymers (1–3) did not display any antimicrobial activities against any bacteria irrespective of their tertiary or quaternary amine structures. Polypropylene oxide (PPO)-based polymers (4–6) were effective against E. coli and S. aureus in the concentration range of 10–50 μg mL−1. The quaternary ammonium salt (6) was found to be better than the protonated and neutral polymer forms (4 and 5, respectively). In general, PPO polymers were more effective towards E. coli than S. aureus. The ethylene oxide polymers (7–9) were effective only in their quaternary ammonium salt form (9) as protonated (8) and neutral (7) forms required high concentrations of 100–250 μg mL−1 to kill 90% of the bacteria. The butanediol (10–12) polymers displayed good activity against E. coli with MIC>90 values of 10–20 μg mL−1. However, the neutral (10) and protonated (11) polymers remained completely ineffective towards S. aureus. The cationic form (12), however, was as potent towards S. aureus as it was against E. coli. Interestingly, this polymer could also inhibit the growth of M. smegmatis at a low concentration of 20 μg mL−1.21
| Polymer | MIC>90, μg mL−1 | HC50, μg mL−1 | Selectivity HC50/MIC>90 | |||||
|---|---|---|---|---|---|---|---|---|
| E. coli | S. aureus | M. smegmatis | E. coli | S. aureus | M. smegmatis | |||
| 1 | PEG N(Bu) | — | — | — | 5000 | — | — | — |
| 2 | PEG N(H/Bu) | — | — | — | 5000 | — | — | — |
| 3 | PEG N(Bu/Bu) | — | — | — | 5000 | — | — | — |
| 4 | PPO N(Bu) | 20 | 50 | 250 | 10 | 0.5 | 0.2 | 0.04 |
| 5 | PPO N(H/Bu) | 20 | 50 | 250 | 10 | 0.5 | 0.2 | 0.04 |
| 6 | PPO N(Bu/Bu) | 10 | 20 | 100 | 5 | 0.5 | 0.25 | 0.05 |
| 7 | EO N(Bu) | 100 | 250 | 100 | 2000 | 20 | 8 | 8 |
| 8 | EO N(H/Bu) | 100 | 250 | 100 | 2000 | 20 | 8 | 8 |
| 9 | EO N(Bu/Bu) | 20 | 10 | 50 | 2000 | 100 | 200 | 40 |
| 10 | BD N(Bu) | 20 | — | 250 | 2000 | 100 | — | 8 |
| 11 | BD N(H/Bu) | 20 | — | 250 | 2000 | 100 | — | 8 |
| 12 | BD N(Bu/Bu) | 10 | 20 | 20 | 2000 | 200 | 100 | 100 |
| 13 | BD N(Pr) | — | — | — | 2000 | — | — | — |
| 14 | BD N(Pen) | 10 | 20 | 50 | 100 | 10 | 5 | 2 |
| 15 | BD N(Hex) | 100 | 100 | — | 2000 | 20 | 20 | — |
| 16 | Bd N(Hep) | — | — | — | 2000 | — | — | — |
| 17 | BD N(Pr/Pen) | 10 | 10 | 20 | 50 | 5 | 5 | 2.5 |
| 18 | BD N(Et/Hex) | 10 | 5 | 5 | 50 | 5 | 10 | 10 |
| 19 | BD N(Me/Hep) | 5 | 5 | 5 | 20 | 4 | 4 | 4 |
| 20 | Kanamycin | 20 | 10 | 5 | — | — | — | — |
| 21 | Carbenicillin | 5 | 10 | 50 | — | — | — | — |
To further examine the success of the butanediol-based polymers, the length of the alkyl chains in the neutral form was varied from three carbon atoms to seven carbon atoms. Polymer 13 carrying propyl chains was ineffective towards all bacterial strains. Polymer 14 with pentyl chains was active against all bacterial strains. Although, not as potent against M. smegmatis as that of 12. Polymers 15 and 16 carrying hexyl and heptyl carbon chains were largely ineffective.
Finally, isomeric versions of the most successful polymer 12 were studied. Polymers 17–19 carried a total number of 8 carbon atoms at the ammonium cation. However, the carbon chain length varied. These polymers were relatively more potent than polymer 12 in killing all three strains of bacteria.
As a measure of biocompatibility, a hemolysis assay was performed to assess the lytic activity of the polymers using sheep red blood cells (RBCs). The polymer concentrations that resulted in 50% hemolysis of RBCs are indicated as HC50 values in Table 1. The PEG polymers (1–3) exhibited minimal mammalian cell toxicity. In contrast, PPO polymers (4–6) displayed significantly high hemolytic activity. The ethylene oxide and butanediol polymers (7–13, 15–16) were compatible with mammalian cells with HC50 value of 2000 μg mL−1. Interestingly, the neutral polymer with pentyl chain (13) was significantly toxic. In addition, the isomeric versions of 12 (polymers 17–19) led to high hemolysis at low concentrations of 20–50 μg mL−1.
Having studied the antimicrobial and hemolytic aspects, certain general conclusions can be drawn. The polymers display properties that are related to the chemical nature of their backbone and the side-chains. The PEG carrying backbone (1–3) is the most hydrophilic and displays no antibacterial and hemolytic properties. The most hydrophobic backbone containing PPO leads to highly toxic materials (4–6), especially towards mammalian cells. A reduction in the PEG segment to just one repeating unit, as seen in polymers 7–9 (having one ethylene oxide unit in between the amines), leads to good compatibility with RBCs and moderate antibacterial capacity. A good balance is offered by an increase in the backbone hydrophobicity by the incorporation of two additional carbon atoms (butanediol segments) in the backbone (10–12) as compared to polymers 7–9. One of these polymers, 12, exhibited the best performance as the mammalian cells tolerated a high concentration (2000 μg mL−1) while the bacteria were killed by a very low concentration (10–20 μg mL−1) of this polymer.
In comparing neutral and protonated polymers (4, 7, and 10 to 5, 8, and 11, respectively), the properties appear similar most likely due to protonation of the neutral polymers under the experimental conditions (PBS buffer, pH = 7.4). Therefore, it is reasonable to assume that in such polymers, carrying tertiary amines in the backbone, a separate protonation step is not required as the backbone amines can get protonated in situ. However, it is likely that as the alkyl chain attains a certain length, the steric hindrance may be unhelpful towards such protonation. This can be seen in the inactivity of polymer 16 towards any bacterial strain.
The carbon length of the polymer side-chain is found to be important as well. For instance, while a 3-carbon atom side-chain is ineffective towards any bacteria (polymer 13), the 5-carbon atom long side-chain is toxic to mammalian cells (polymer 14). A balance is offered by the 4-atom long side-chain (polymer 10), which is compatible with RBCs and highly effective against E. coli.
In comparing protonated to permanently charged ammonium cations (5, 8, and 11 to 6, 9, and 12, respectively), the latter are much more effective not only as a relatively lower amount is required to kill >90% of the bacteria but that they are potent against a more difficult Gram positive strains which are protected by a thicker cell wall.14–18
Finally, within the group of permanently charged ammonium polymers with a butanediol backbone, longer alkyl chains, such as in polymers 17, 18, and 19 as compared to 12, leads to higher hemolytic activity. The butyl chains can therefore be considered optimum in designing such main-chain ammonium polymers for antimicrobial purposes. The performance of the butyl chain-containing polymer 12 was found to be in a similar range as bactericidal antibiotics such as kanamycin and carbenicillin (Table 1, entries 20–21). Interestingly, an antibiotic producing bacteria, Streptomyces venezuelae,22 could also be targeted by polymer 12 and MIC>90 could be achieved at a low concentration of 5 μg mL−1 (Fig. S28†).
Next, to understand the antimicrobial mechanism, live/dead bacterial viability assays against E. coli were performed using polymers 7 and 10 (Fig. 2).20 In this assay, bacterial cells were stained using SYTO and propidium iodide to distinguish live (i.e. green fluorescence) from dead (i.e. red fluorescence) cells by confocal laser scanning microscopy (CLSM) observations. This study indicated that 4 hours of incubation with E. coli in the presence of 7 and 10 resulted in significant levels of cell death. After 24 hours of incubation, primarily red signals were recorded from both polymers, indicating the death of nearly the whole bacterial colonies. Scanning electron microscopy (SEM) imaging was then utilized to compare differences in cell morphology before and after treatment with polymer 12 (Fig. 3). The significantly damaged bacterial membranes were observed from E. coli after 24 hours of incubation, while the bacterial cells from the control group remained intact and rod-shaped. For S. aureus, the control group indicated an intact spherical morphology. However, the polymer treated samples lost their shape and smoothness and aggregated into larger lumps. These results indicated that the polymers caused cell death through cell membrane disruption mechanism.20
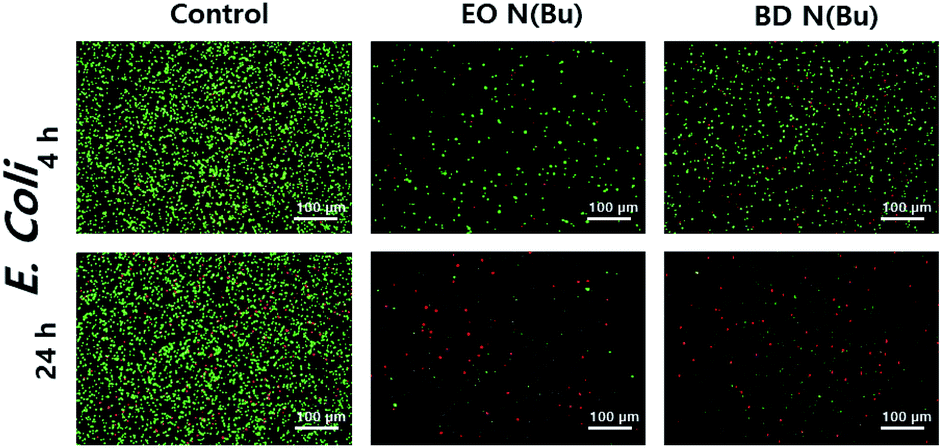 | ||
| Fig. 2 Confocal laser scanning microscopy (CLSM) merged images of the green (SYTO, indicates live cells) and red (propidium iodide, indicates dead cells) channels. The bacteria suspension without polymers was used as control. | ||
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of E. coli and S. aureus cells after treatment with the cationic polymer. | ||
A kinetic study was then undertaken. This study indicated that two hours of exposure time to polymer 12 was sufficient for killing >90% of the bacteria (Fig. S29†).
Finally, outer- and inner-membrane permeabilization assays were performed (Fig. S30†). For the outer membrane, 1-N-phenyl-naphthylamine (NPN) was used as a fluorescent probe. This dye emits strongly when part of the hydrophobic environment such as those created by the lipid membrane. In presence of polymer 12, a rapid rise in the emission could be observed from NPN indicating outer-membrane permealization. Propidium iodide, another dye molecule earlier used in the live/dead cell assay can enter once the outer-membrane is compromised and allows for studying the inner-membrane permealization. In presence of polymer 12, a slow but steady increase in the emission from propidium iodide is observed indicating a successful inner-membrane permealization albeit with a slower kinetics than the outer-membrane permealization.
To conclude, a new polymer family is synthesized through the amine–epoxy ‘click’ polymerization in water under ambient conditions. A post-synthesis modification of the nitrogen atoms leads to the formation of main-chain cationic polymers. A properties study indicated that out of 19 polymers, butanediol polymer 12, carrying two butyl chains on the quaternary nitrogen, offers a good balance between antimicrobial activity and biocompatibility. The mammalian cells tolerated a high concentration (2000 μg mL−1) while the bacteria were killed by a very low concentration (5–20 μg mL−1) of this polymer. The fundamental polymer design developed here is now being examined in epoxy coatings to create large area antibacterial surfaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by National Research Foundation of Korea grant funded by the Korean government (MSIP) (NRF18R1D1A1B07048527).Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- A. J. Qin, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2010, 43, 8693–8702 CrossRef CAS.
- Y. Shi, J. Z. Sun and A. J. Qin, J. Polym. Sci. Pol. Chem., 2017, 55, 616–621 CrossRef CAS.
- D. Huang, Y. Liu, A. Qin and B. Tang, Polym. Chem., 2018, 9, 2853–2867 RSC.
- B. X. Li, D. Huang, A. J. Qin and B. Z. Tang, Macromol. Rapid Commun., 2018, 39, 1800098–18000112 CrossRef PubMed.
- J. Hong and A. Khan, Polymers, 2019, 11, 1941–1953 CrossRef CAS PubMed.
- J. Hong, J. K. Oh and A. Khan, J. Macromol. Sci., Part A: Pure Appl.Chem., 2020, 57, 472–478 CrossRef CAS.
- J. Oh, J. Hong and A. Khan, J. Macromol. Sci., Part A: Pure Appl.Chem., 2020, 57, 685–690 CrossRef.
- J. Oh, K. I. Jung, H. W. Jung and A. Khan, Polymers, 2019, 11, 1491–1493 CrossRef CAS PubMed.
- Z. L. Su and X. S. Jiang, Polymer, 2016, 93, 221–239 CrossRef CAS.
- A. Saha, S. De, M. C. Stuparu and A. Khan, J. Am. Chem. Soc., 2012, 134, 17291–17297 CrossRef CAS PubMed.
- G. Y. Si, M. R. Elzes, J. F. J. Engbersen and J. M. J. Paulusse, Polymers, 2018, 10, 687–700 CrossRef PubMed.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46–71 CrossRef CAS.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- G. J. Gabriel, A. Som, A. E. Madkour, T. Eren and G. N. Tew, Mater. Sci. Eng. R Rep., 2007, 57, 28–64 CrossRef PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- A. C. Engler, N. Wiradharma, Z. Y. Ong, D. J. Coady, J. L. Hedrick and Y. Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS.
- M. S. Ganewatta and C. B. Tang, Polymer, 2015, 63, A1–A29 CrossRef CAS.
- Y. Jiao, L. N. Niu, S. Ma, J. Li, F. R. Tay and J. H. Chen, Prog. Polym. Sci., 2017, 71, 53–90 CrossRef CAS PubMed.
- G. N. Tew, D. H. Liu, B. Chen, R. J. Doerksen, J. Kaplan, P. J. Carroll, M. L. Klein and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5110–5114 CrossRef CAS PubMed.
- D. J. Phillips, J. Harrison, S. J. Richards, D. E. Mitchell, E. Tichauer, A. T. M. Hubbard, C. Guy, I. Hands-Portman, E. Fullam and M. I. Gibson, Biomacromolecules, 2017, 18, 1592–1599 CrossRef CAS PubMed.
- J. Y. Song, Y. J. Yoo, S.-K. LimS, H. Cha, J.-E. Kim, J.-H. Roe, J. F. Kim and Y. J. Yoon, J. Biotech, 2016, 219, 57–58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04625f |
| This journal is © The Royal Society of Chemistry 2020 |