
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling protein-hosted organocatalytic transformations
Alexander R. Nödling
 ,
Nicolò Santi
,
Thomas L. Williams
,
Yu-Hsuan Tsai
and
Louis Y. P. Luk
*
,
Nicolò Santi
,
Thomas L. Williams
,
Yu-Hsuan Tsai
and
Louis Y. P. Luk
*
School of Chemistry, Cardiff University, Main Building, Cardiff, CF10 3AT, UK. E-mail: lukly@cardiff.ac.uk
First published on 23rd April 2020
Abstract
In this review, the development of organocatalytic artificial enzymes will be discussed. This area of protein engineering research has underlying importance, as it enhances the biocompatibility of organocatalysis for applications in chemical and synthetic biology research whilst expanding the catalytic repertoire of enzymes. The approaches towards the preparation of organocatalytic artificial enzymes, techniques used to improve their performance (selectivity and reactivity) as well as examples of their applications are presented. Challenges and opportunities are also discussed.
 Alexander R. Nödling | Alexander R. Nödling is a Postdoctoral Research Associate in the groups of Louis Luk and Yu-Hsuan Tsai. He was born in Frankenthal, Germany, in 1986. He obtained his Diploma in Chemistry at the Philipps-Universität Marburg (2011), and his Dr rer. nat. (2016) under the guidance of Gerhard Hilt with a focus on QSAR of Lewis acid catalysis. His rising interest in tackling biological questions led to his current position at Cardiff University. His current research focuses on artificial enzyme design and biomolecule delivery. |
 Nicolò Santi | Nicolò Santi was born in Vignola, Italy, in 1992. He received his MSc degree in Medicinal Chemistry from Università degli studi di Modena e Reggio Emilia (UNIMORE, 2016). During this time, he spent 7 months at Cardiff University under the supervision of Fabrizio Pertusati working on the preparation of phosphonamidates (ProTides) as potential antiviral compounds. In 2017, he began his postgraduate studies at Cardiff University under the supervision of Louis Luk. His research includes the design and optimisation of organocatalytic artificial enzymes. |
 Thomas L. Williams | Thomas L. Williams graduated from the University of South Wales with a BSc in Forensic Science (2012) followed by a MSc in Forensic and Analytical Science (2014). He joined Cardiff University as an analytical technician specialising in mass spectrometry of proteins and peptides (2014). Thomas then joined the research groups of Louis Luk and Yu-Hsuan Tsai as a PhD student in Chemical Biology (2015). His research focuses on designing and characterising artificial enzymes using advanced synthetic biology techniques. |
 Yu-Hsuan Tsai | Yu-Hsuan Tsai is a Lecturer in the Cardiff School of Chemistry. He obtained his BSc at the National Taiwan University (2006), MSc at the ETH Zurich (2008), and Dr rer. nat. at the Freie Universität Berlin (2012). He was a graduate student under the guidance of Peter H. Seeberger at the ETH Zurich and Max Planck Institute of Colloids and Interfaces, and a post-doctoral researcher in the group of Jason W. Chin at the MRC Laboratory of Molecular Biology. Since 2015 he has become an independent researcher in Cardiff. His research focuses on developing tools to study and control protein functions. |
 Louis Y. P. Luk | Louis YP Luk is a Lecturer at the School of University of Cardiff University. He obtained his BSc in Chemistry and Microbiology & Immunology at the University of British Columbia. He obtained his PhD in the same university under the supervisor of Martin E. Tanner, followed by post-doctoral studies in the laboratories of Stephen BH Kent at the University of Chicago and Rudolf K. Allemann at Cardiff University. Louis became a University Research Fellow in 2015 and was recently tenured at Cardiff University. Combining his training in enzymology and peptide chemistry, Louis' current research focuses on artificial enzyme design and protein bioconjugation chemistry. |
Introduction
Biocompatible organocatalysis
Serving as a major tool for asymmetric chemical transformations,1–3 organocatalysis has now matured to a point where its bio-orthogonality can be exploited for important chemical and synthetic biology applications. Catalysts such as imidazolidinone,4–6 proline,7 thiourea,8 and anion–π9 derivatives have been used to mediate reactions that have no parallel in nature. Provided its bio-orthogonality, organocatalysis can be used in biological contexts for valuable chemical and biological applications.10–17 For instance, organocatalysts can serve to mediate labelling of biomolecules,11,12 analogous to existing approaches that use metals for reactions.18–20 Also, it is worth considering to merge organocatalysis and biocatalysis for the production of chiral synthons in a one-pot and atom economic fashion.13–17 However, there are only a few examples where organocatalysts function along with biomolecules or under biological conditions. In fact, taking into account the aqueous reaction medium, physiological pH (near 7.4) and temperature (near 37 °C),20 many of the reported organocatalysts do not function under biocompatible conditions.8,10,17,21–31 These boundaries vastly narrow the number of organocatalytic reactions applicable and, in response, efforts have been made to overcome limitations related to biocompatibility.17,32,33The use of proteins to host organocatalysts
To enhance the biocompatibility of organocatalysis, biomolecules including DNA, RNA and proteins can be used to host the reactions.34–44 Among them, proteins are particularly suitable. Most proteins can be made recombinantly, correctly folded in an aqueous environment under physiological conditions and are thus inherently biocompatible. Yet, the outer surfaces and interior of most proteins possess relatively low dielectric constants, which are similar to those found in many organic solvents.45 Consequently, proteins can provide a microenvironment that can stabilise the transition state during chemical transformation.34,46,47 Furthermore, superior to most organic solvents, proteins are inherently chiral – the scaffold where the catalytic motif is located can be modelled and/or genetically modified for improved selectivity.48 Most importantly, the protein host can be further refined by laboratory evolution, which has become increasingly facile as molecular cloning and screening techniques have become user-friendly.49,50 In contrast, it is relatively difficult to incorporate such “evolvability” in traditional catalyst design.34,36,48,49,51–54 To this end, the creation of genetically encoded protein scaffolds is a promising avenue to develop biocompatible stereoselective organocatalytic reactions.36,55Artificial organocatalytic enzymes
The term “artificial enzyme” has been widely used for any macromolecular complex designed to catalyse chemical reactions.54 Herein, we describe artificial enzymes as protein-based systems that have been genetically or chemically altered, repurposed or designed de novo to catalyse a reaction. While contemporary artificial enzyme design is mainly focused on metallo-enzymes or redesign of reaction-promiscuous natural enzymes,49,56 we set the scope of this review to the development of artificial organocatalytic enzymes based on recombinant proteins. These enzymes were categorised based on their design (Table 1). We will describe relevant examples of each strategy and the success in their approach. Engineering of natural cofactors in their native enzymes will be briefly discussed.17,32| Systems | Features | Advantages | Potential challenges | Reactions tested |
|---|---|---|---|---|
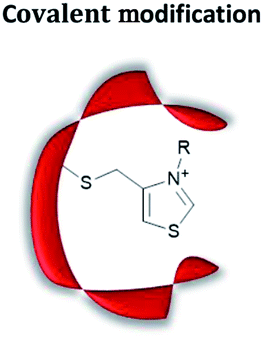 |
Site-selective modification with catalytically active motifs | Relative ease of preparation; quick screening of different catalytic moieties possible | Site-specific labelling can be challenging | Reduction;57,58 cyclisation;59 reductive amination;60 C–C bond formation61 |
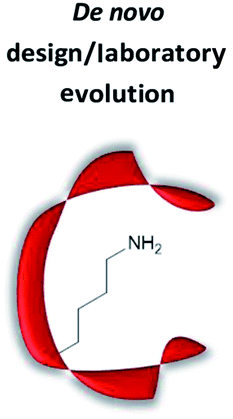 |
Computational design of active site, creating “theozyme” | High probability to create a novel active site as a consequence of precise design, and hence screening time is minimised | Mechanistic and structural knowledge needed; knowledge in computational chemistry needed | (Retro-)aldol reaction;62–68 Henry reaction;41 Knoevenagel condensation;24,43 conjugate addition40,42,44 |
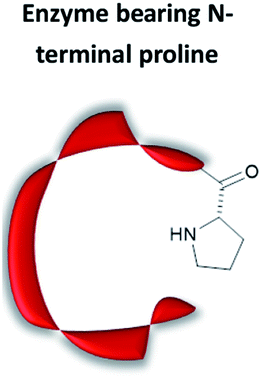 |
Use of substrate promiscuous N-terminal proline | No need of chemical modification or computer modelling | Limited to secondary amine organocatalysis at the N-terminal position; is it very easy to generate protein with N-terminal Pro recombinantly | Conjugate addition;38,69–71 intramolecular and intermolecular aldol condensation37,72,73 |
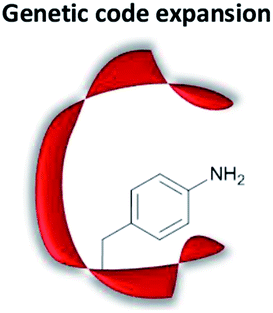 |
Genetic incorporation of an unnatural amino acid which bears (part of the) catalytically active motif | Wide selection of catalytically active amino acids; no chemical protein modification needed | Recombinant expression might be low yielding | Ester hydrolysis;74 oxime/hydrazone conjugation50,75 |
 |
Binding of a ligand bearing catalytically active motifs | Many catalytically active moieties can be attached; enable quick screening of different catalytic moieties | Limited to proteins with high affinity ligand(s) | Conjugate addition;39 decarboxylative Michael addition;36,55 domino aldol–Michael reaction76 |
Chemical modification
Site-selective chemical modification of proteins
Prior to the onset of modern molecular biology technologies, proteins were often chemically modified to purposefully alter their activity.77–79 Initially, modified enzymes were made by single atom replacement. Serine protease subtilisin was converted to its cysteine equivalent by a three-step chemical protocol (tosylation, followed by replacement with thioacetate and hydrolysis).80 The resulting “thiol-subtilisin” could hydrolyse activated aryl substituted ester bonds. However, this cysteine variant lost its protease activity and was found to be 100-fold less active than the parental enzyme towards activated esters, despite the higher nucleophilicity of the free thiol.81,82 In another study, selenosubtilisin was created by converting the active site serine residue into selenocysteine.57,58,83 The selenium-containing protein was shown to be a reductase; alkyl peroxides could be converted to their alcohol equivalents under the action of this modified enzyme using thiophenol as a source of oxidant (Fig. 1a).57 The artificial enzyme exhibits reaction rates comparable to those of natural enzymes. While mechanistic insights are not available, the selenosubtilisin displays an inverted enantioselectivity in the kinetic resolution of racemic peroxides when compared to the native enzymes (Fig. 1b).58 | ||
| Fig. 1 Selenosubtilisin catalyses the reduction of (a) tert-butyl hydroperoxide and (b) secondary alkyl hydroperoxide. MES = 2-(N-morpholino)ethanesulfonic acid, EDTA = ethylenediaminetetraacetic acid. | ||
Cysteine, due to its nucleophilic nature, is most frequently modified with cofactors for the creation of new organocatalytic artificial enzymes. An artificial oxidoreductase was created by linking the catalytically active cysteine residue of the protease papain to flavins. Using oxygen for oxidation, the resulting “flavopapain” was able to oxidise NADH and its derivatives at a rate 50-fold higher than that by flavin alone (Fig. 2a).84 Similarly, the natural cofactor thiamine was introduced to papain. The resulting “thiazolopapain” was one of the early artificial enzymes that can mediate C–C bond formation.59 Nevertheless, activity was suboptimal, as the model cyclisation reaction of 6-oxo heptanal required six days to reach completion with a significant portion of substrate transformed in dimerisation byproduct (Fig. 2b). A third cofactor, pyridoxamine, was used to label the adipocyte lipid binding protein.60 The pyridoxamine protein complex could successfully mediate the production of a wide range of amino acids with modest to excellent enantioselectivity with enantiomeric excess (ee) up to 94% (Fig. 2c).
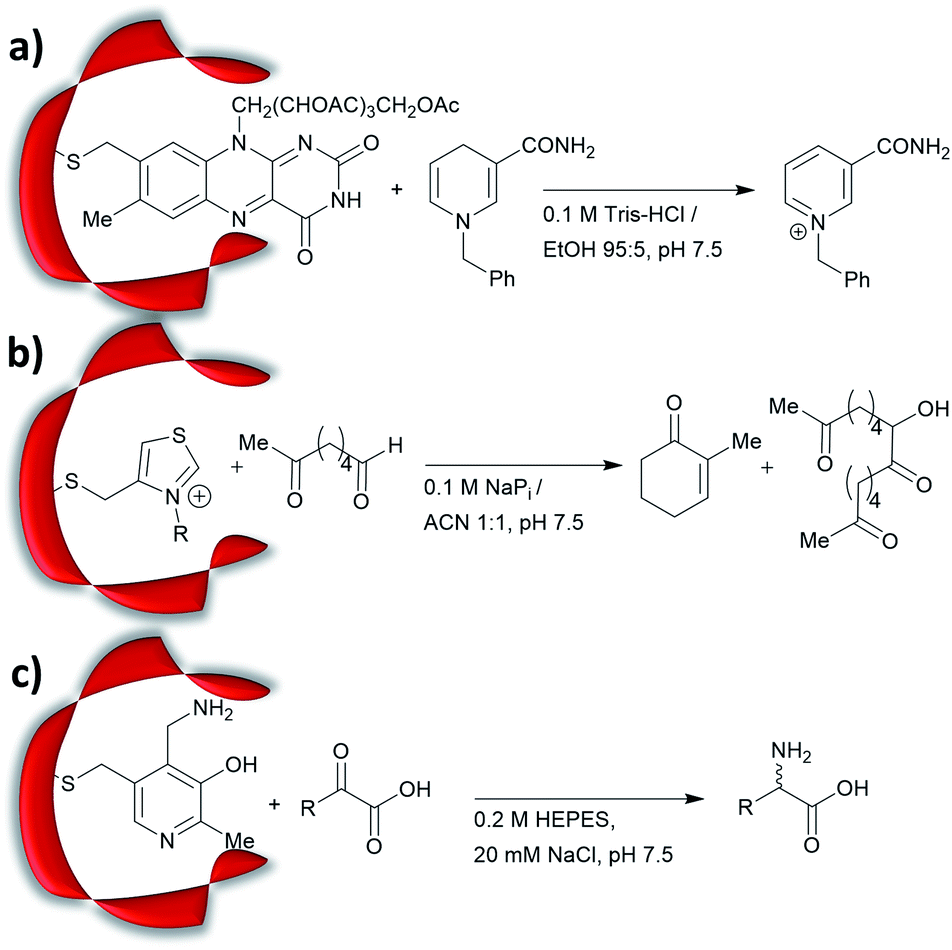 | ||
| Fig. 2 Artificial enzymes created by covalent modification of cofactors: (a) artificial flavopapain used for the oxidation of BNAH; (b) artificial thiazolopapains for C–C bond formation; and (c) artificial ALBP–pyridoxamine for enantioselective reductive amination. Tris = tris(hydroxymethyl)aminomethane, HEPES = (4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid), ALBP = adipocyte lipid binding protein. | ||
Recently, an alternative labelling strategy based on the metabolism of carbapenems by penicillin binding proteins was developed.61 In this work, secondary amine containing penicillin derivatives were anchored to beta-lactamase and the covalently modified protein was employed in a conjugate addition of nitromethane to cinnamaldehyde, giving moderate yields and low enantioselectivities (20–27%, e.r. ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45).
:45).
These studies lay the foundations for the future development of protein-hosted organocatalysis.57,84,85 Chemical methodologies for protein labelling have vastly diversified and improved in recent years, showing fine-tuned reactivity and biocompatibility with labelling achieved within live cells.86–94 One can anticipate that efficient artificial enzymes can be made by adapting these novel technologies.
De novo design/laboratory evolution
Development of de novo enzymes
The increase in computational power and applicable software, including Rosetta and ORBIT,95 has accelerated the development of de novo enzyme design.96,97 The first stage of de novo enzyme design is the in silico generation of a “theozyme,” a theoretical arrangement of side chain residues and bioavailable molecules (water and ions) that can stabilise the rate-limiting transition state(s) of a chosen reaction.98 This assembly of theozyme is subsequently transformed into an experimentally tangible protein structure through evaluations based on calculated parameters (e.g. geometry and energy) by screening of available protein structures available in repositories.49,95 Eventually, the best options are recombinantly produced for characterisations. The initial de novo enzymes are typically inefficient and are not selective. Thus, laboratory evolution is used to enhance both catalytic activity and reaction profile. This pathway led to the formation of a highly competent and promiscuous de novo Kemp eliminase,99,100 retro-aldolases (RA)40,43,44,63,65–68 and Diels–Alderases.52,101 Here, we will focus on retro-aldolases which bear a catalytically active lysine for iminium and enamine catalysis.Retro-aldolases are a class of de novo designed enzymes capable of catalysing retro-aldol reactions via formation of an iminium intermediate.68 Retro-aldolases have been created from a theozyme that is able to mediate cleavage of the fluorogenic compound methodol (1, Fig. 3).68 The reaction was selected to allow for facile screening as the retro-aldol product naphtaldehyde (2) is fluorescent.
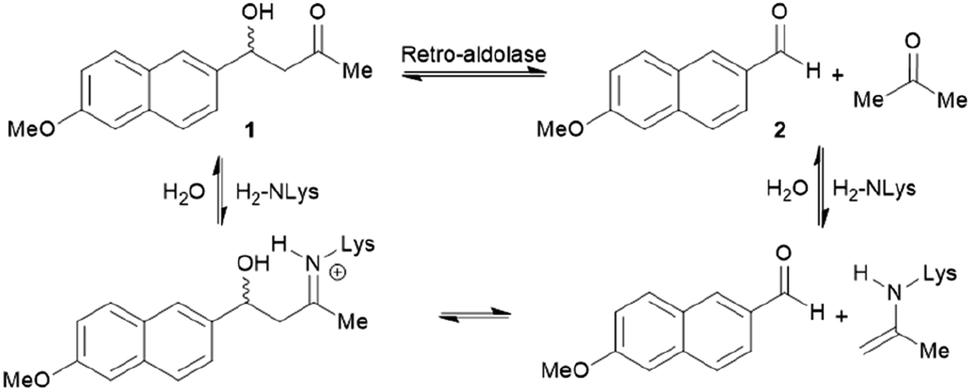 | ||
| Fig. 3 Net reaction of methodol (1) cleavage catalysed by the de novo designed retro-aldolases and important intermediates. | ||
The most effective theozyme in terms of rate enhancement in the recombinantly produced protein contains a catalytically active lysine residue within a hydrophobic binding pocket and a strategically positioned water molecule that helps mediating formation of the Schiff base intermediate.68 Interestingly, this artificially designed network was found to be catalytically more active than those made based on naturally found proton shuffle networks. Computational tools, such as RosettaMatch,96 were recruited to dock the theozyme into a protein scaffold, creating a suitable host for the artificial active site.
Indole-3-glycerol phosphate synthase,102 a TIM-barrel protein fold, was identified for hosting the theozyme. Further adjustment of the residues surrounding the transition state was made using RosettaDesign, which among other purposes enables optimisation of residue interactions around the active site.103 Among these active models, the variant RA95.0 with the catalytically active lysine at position 210 (apparent pKa = 8.1, Fig. 4) was identified as the most promising candidate. Experimentally, RA95.0 is able to mediate cleavage of methodol (1) with catalytic efficiency (kcat/KM) of ∼0.19 M−1 s−1 and selectivity for S over R (2.3:1).66,67 To create an enzyme with improved performance, regions at and around the active site of RA95.0 were subjected to iterative cassette mutagenesis, a form of saturation mutagenesis where pre-synthesised and mutated DNA strands are inserted into the gene by restriction enzyme digest and ligation.104 By combining mutations of the most active single variants, a highly improved variant RA95.5, which has six mutations in total, showed 73-fold increase in catalytic efficiency when compared to RA95.0 (kcat/KM = 14 M−1 s−1 with 3:1 R-to-S selectivity). Crystallographic studies revealed that the T83K mutation in RA95.5 created a second reaction centre, in addition to Lys210, both capable of forming Schiff base intermediates. This finding indicated that the active site underwent restructuring, and further refinement was needed (see below).66,67 In particular, the replacement of T83K mutation shifted the pKa of Lys210 to 7.6, to which the authors attributed the improved performance.
 | ||
| Fig. 4 Overlay of crystal structures of the first retro-aldolase RA95.0 (gold & orange, PDB: 4A29), in which Lys210 is responsible for forming the Schiff base intermediate with a 1,3-diketone inhibitor and the evolved variant RA95.5-8 (light & dark blue, PDB: 5AN7) that carries a novel catalytically active Lys83 (in complex with a 1,3-diketone inhibitor). | ||
Additional laboratory evolution studies of the entire gene (error-prone PCR and DNA shuffling) created the variant RA95.5-5 that has an additional six mutations (compared to RA95.5) and demonstrated significantly improved activity (>20-fold, kcat/KM = 320 M−1 s−1, and selectivity 5:1 R over S). Crystallographic studies illustrated that Lys83 transformed into the only reaction centre for the methodol (1) cleavage, indicating that there is a switch in location of the residue responsible for catalysis.66 Restructuring of the active site was likely unpredictable during the initial design, highlighting that randomness is a key element during the evolution of an efficient enzyme. Finally, a last three rounds of laboratory evolution yielded the variant RA95.5-8 (Fig. 4), which contains substitutions at both the active site and distal positions, and its catalytic efficiency (kcat/KM) was measured to be 850 M−1 s−1.66
Aldolase evolution
Showcasing the power of ultra-high throughput screening methods, microfluidic fluorescence-activated droplet sorters (FADS) were used to further improve the performance of the retro-aldolase. The resulting variant RA95.5-8F displayed 13 mutations and a 30-fold higher activity (kcat/KM = 34000 M−1 s−1 for (R)-methodol (R-1) with 480:1 R over S selectivity).64 Such impressive improvement was attributed to the genesis of a catalytic Lys-Tyr-Asn-Tyr tetrad for proton shuffling. The tetrad forms a hydrogen bonding network which transfers proton to and from the reaction centre, stabilising formation of reaction transition states. RA95.5-8F was the first RA to be able to mediate aldol reactions between acetone and various aldehydes (i.e. an aldolase). It should be noted that previous RA's were inhibited by the formation of Schiff base with these aldehydes, whereas RA95.5-8F selectively forms enamines with acetone.
Expanding the reaction profile of the RA95 family
A series of studies were conducted to expand the versatility of the RA95 family to catalyse different reactions (Fig. 5). Iminium catalysis mediated by RA95.5-8 was used as a means to mediate carbon–carbon bond formation, including conjugate additions (Fig. 5a and b),40,42,44 Knoevenagel (Fig. 5c)43 and Henry condensations (Fig. 5d).41 Enamine catalysis was also explored in the nitro-Michael addition of acetone to nitrostyrenes (Fig. 5e).42 In some cases, formation of reactive iminium species was verified by reduction of the intermediates followed by mass spectrometric analysis.43,44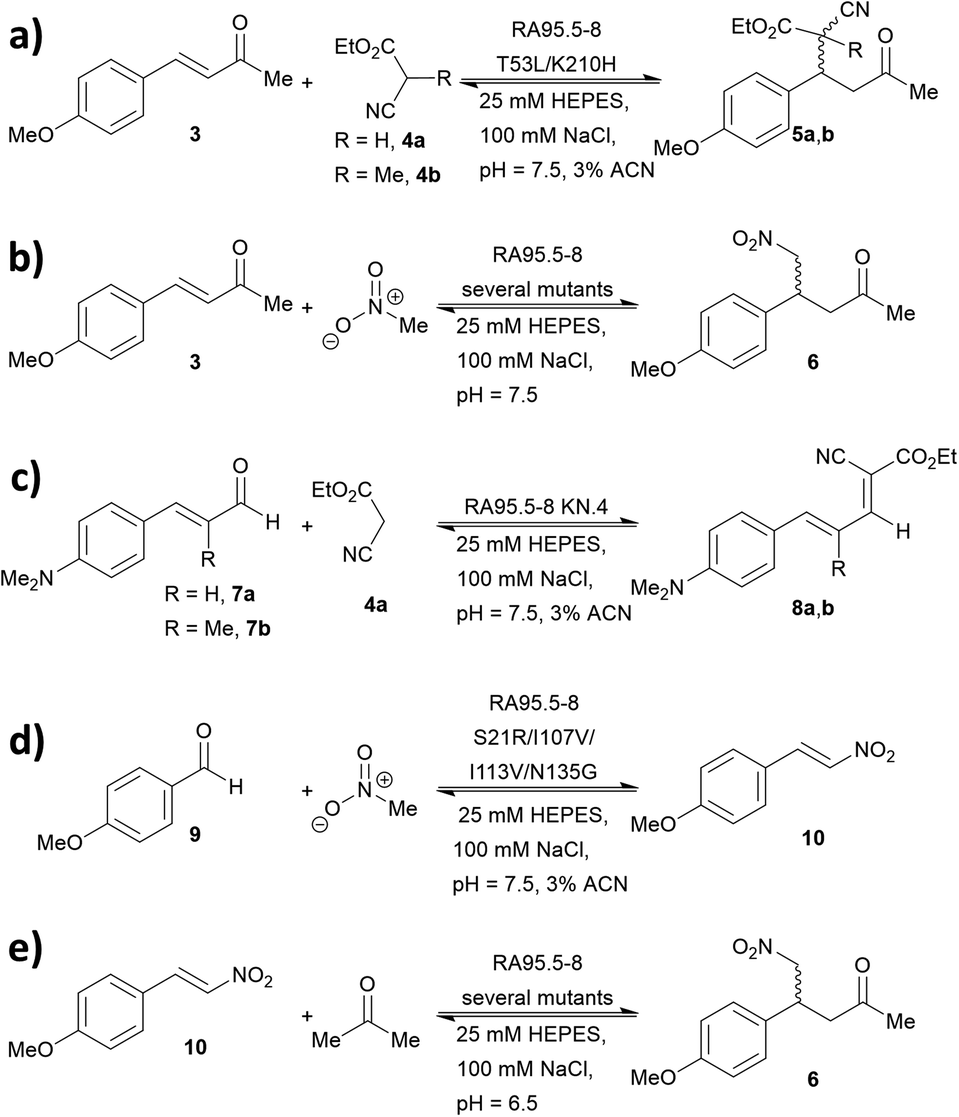 | ||
| Fig. 5 Promiscuity of RA95.5-8 and variants for carbon–carbon bond forming reactions. Iminium catalysis includes: (a) conjugate addition of carbon nucleophiles; (b) conjugate addition of nitromethane; (c) Knoevenagel condensations of carbon nucleophiles with α,β-unsaturated aromatic aldehydes and (d) Henry addition of nitromethane to aromatic aldehydes. Enamine catalysis includes: (e) conjugate addition of acetone to nitrostyrene. | ||
During the course of optimising RA95.5 to mediate different transformation (Fig. 5), several notes have been learned. Firstly, there is a positive correlation between stereoselectivity and catalytic efficiency.63–68 Nevertheless, it should be noted that, during the optimisation process, stereoselectivity may be weak because the active site undergoes reconstruction (e.g. re-locating the catalytic residue);40,41,44 eventually, stereoselectivity resumes and variants with kinetic parameters and selectivity similar to those of natural enzymes can be achieved. Furthermore, refined artificial enzymes often possess properties similar to those of natural enzymes. For instance, loop flexibility and residues that are distant from the active site (secondary shell and protein surface) could greatly affect the performance of the catalysis.41,42,44,105,106 In another instance, it was indicated that catalysis is partially driven by a negative activation heat capacity, which is considered as a result of tight binding to the transition state forming an ordered complex.62 Finally, the computationally designed 248-residued RA can be modified at approximately 30 positions. This signifies the genetic “plasticity”107,108 of RA and echoes the fact that TIM-barrel fold is found in at least 15 families of enzymes.109–111 The coupling of computational design (rational) and laboratory evolution with high-throughput screening (randomness) has proven to be an effective approach to create de novo enzyme. In recent years, this technology has been combined with others, including genetic code expansion (see below). We anticipate that the family of de novo enzymes will soon be vastly expanded.
N-Terminal proline
4-Oxalocrotonate tautomerase
When located at the N-terminus of a protein, proline offers a secondary amine that can be used for iminium- and enamine-based organocatalysis. One such example is 4-oxalocrotonate tautomerase (4-OT) from M. putida, which is composed of six homologous monomers carrying a catalytic N-terminal proline (Fig. 6a).112 Naturally, this residue acts as a general base, catalysing the tautomerisation of a dienol into an unsaturated ketone (Fig. 6b).38,113,114 Interestingly, this proline residue forms iminium intermediates with various carbonyl substrates.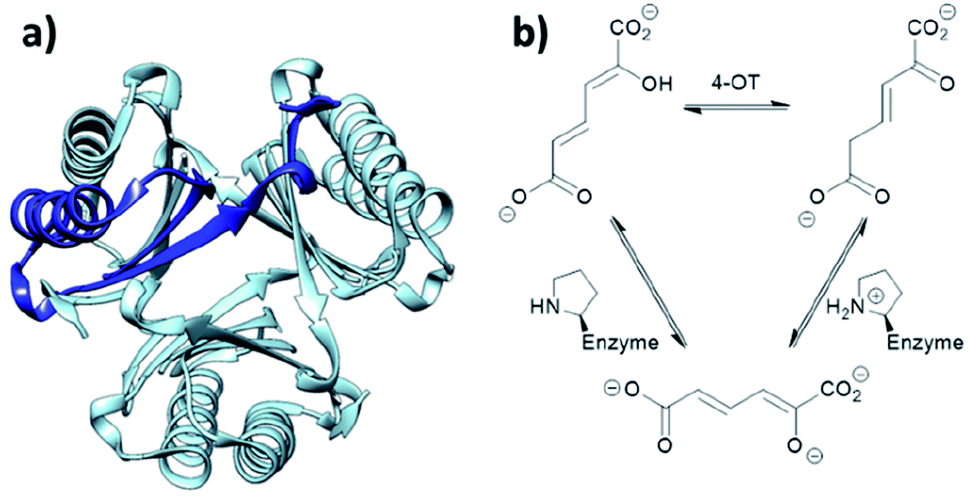 | ||
| Fig. 6 (a) Hexameric crystal structure of 4-OT (PDB: 4X19), monomer subunit highlighted in dark blue. (b) Native reaction of 4-OT, showing the net reaction in the upper part and the function of the N-terminal proline as general base below. | ||
Because of its significant substrate promiscuity, 4-OT has been used as an organocatalyst for chemical transformations. It has been demonstrated that 4-OT is able to catalyse enamine-based aldol reactions (Fig. 7a and b)37,70 and conjugate additions (Fig. 7c).71 Additionally, 4-OT has been exploited for iminium catalysis, including the conjugate addition of nitromethane (Fig. 7d).115 Reduction of the intermediate iminium ion by sodium cyanoborohydride and subsequent mass spectrometry analysis provide evidence that supports the formation of the iminium intermediate.73
 | ||
| Fig. 7 Reactions catalysed by 4-OT include (a) intermolecular aldol; (b) intramolecular aldol; (c) nitro-Michael addition; and (d) enamine catalysed conjugate addition. 4-OT = 4-oxalocrotonate tautomerase. | ||
Mutagenesis via a combined computational and experimental approach has led to the identification of enhanced variants. Three residues in proximity were found to be crucial for catalysis, including Phe50, Met45 and Ala33. Mutability landscapes were used to determine ‘residue hotspots.’ The experiment consisted of singly mutating all amino acids with the exception of the catalytic Pro1.
Protein solubility of single point mutations was first assessed, followed by an activity screen of the tautomerization reaction and subsequently the Michael addition. An F50A mutation resulted in an increase of catalytic efficiency by a factor of 600 for cross-coupling aldol reactions.72 In contrast, when both Phe50 and Met45 were replaced with valine and tyrosine respectively, the resulting variant was more effective at self-condensation reactions. The F50V/M45Y double mutant resulted predominantly in the R product, whereas a third mutant A33D selectively yielded the S enantiomer in the conjugate additions of acetaldehyde to β-nitrostyrenes.69 Crystal structures of the two mutants have been obtained, but the N-terminal region was not resolved likely due to its inherent flexibility. Hence, the actual assembly in the active site remains unclear.
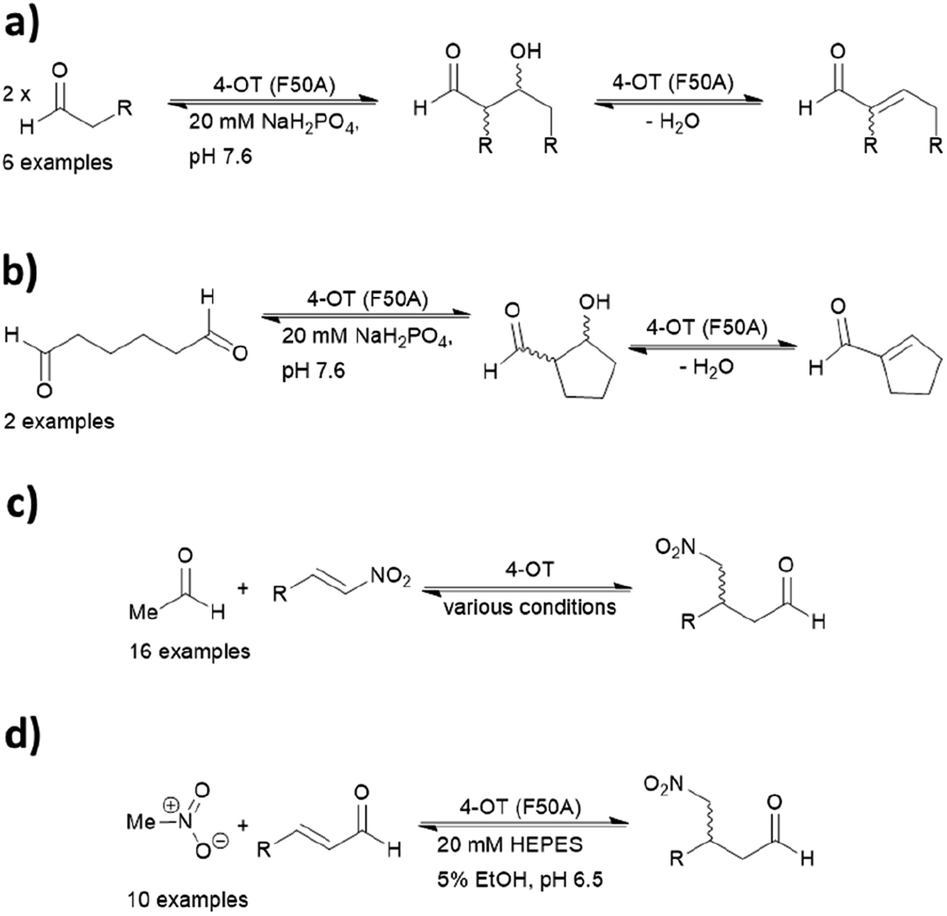
4-OT and its variants have been used for a range of applications including enzymatic115 and chemoenzymatic cascades,116 alongside whole cell catalytic systems.117–119 The anti-anxiety drug pregabalin and three of its analogues were synthesised by coupling the 4-OT reaction with catalysis by aldehyde dehydrogenase (ALDH, Fig. 8).
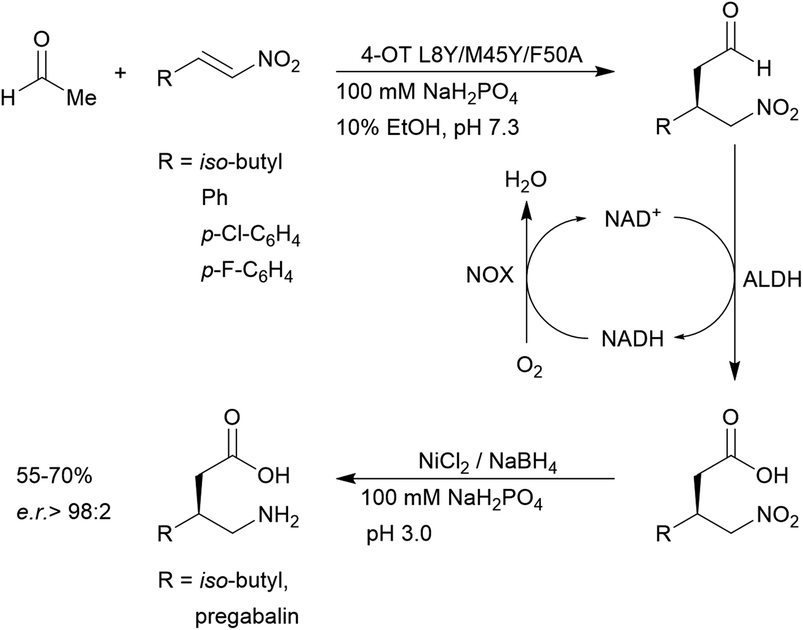 | ||
| Fig. 8 Chemoenzymatic synthesis of pregabalin and three of its analogues using 4-OT. NAD = nicotinamide adenine dinucleotide, ALDH = aldehyde dehydrogenase, NOX = NADH oxidase. | ||
Acetaldehyde was added stereoselectively to α,β-unsaturated nitro substrates under the action of a 4-OT variant, followed by oxidation by ALDH to yield the corresponding carboxylic acid (Fig. 8).116 To recycle NADH, a cofactor recycling system operated by NAD oxidase (NOX) was included. Lastly, the nitro group was reduced to the amine using sodium borohydride in the presence of nickel chloride. These applications present evidence that protein-based organocatalysis can be used in combined synthesis which may not be readily achievable using traditional organocatalytic systems.
Utilising only natural residues with no chemical modification needed, the N-terminal proline approach is arguably the simplest in establishing a biocompatible organocatalytic system. As a range of reactions have already been established, 4-OT is an attractive system for performing organic reactions in biological contexts. However, a major limitation is that it is only able to catalyse secondary amine organocatalysis. Other useful organocatalytic transformations (based on e.g. thiourea or counterion based catalysis) are unavailable and thus other approaches must be employed.
Genetic code expansion
Fundamentals of genetic code expansion
Genetic code expansion enables site-specific incorporation of unnatural amino acids, which can be used to mediate bioorthogonal chemical reactions. To achieve this goal, a pair of orthogonal aminoacyl-tRNA synthetase/tRNA pair is needed. Specifically, the orthogonal tRNA decodes a blank codon, commonly the amber stop codon (TAG) as it is often the least used codons in most organisms. To produce recombinant proteins that contain unnatural amino acids in E. coli, pyrrolysyl-tRNA synthetase/tRNA and tyrosyl-tRNA synthetase/tRNA pairs from archaea are the most versatile and popular choices.120 The pyrrolysyl-tRNA synthetase/tRNA pair is particularly useful as it naturally decodes the amber codon.121 To incorporate the unnatural amino acid, a TAG codon is then introduced into the gene of interest at the position of choice. Double transformation of E. coli with plasmids containing the gene of interest and the synthetase are conducted. By including the unnatural amino acid in the medium, the orthogonal synthetase specifically charges the orthogonal tRNA with the unnatural amino acid, which will allow for production of full-length protein with the unnatural amino acid at the desired position. To date, over 200 unnatural amino acids can be genetically incorporated into a protein of interest using this technique, so there exists a vast opportunity to exploit these unnatural amino acids for organocatalytic transformations (Fig. 9).91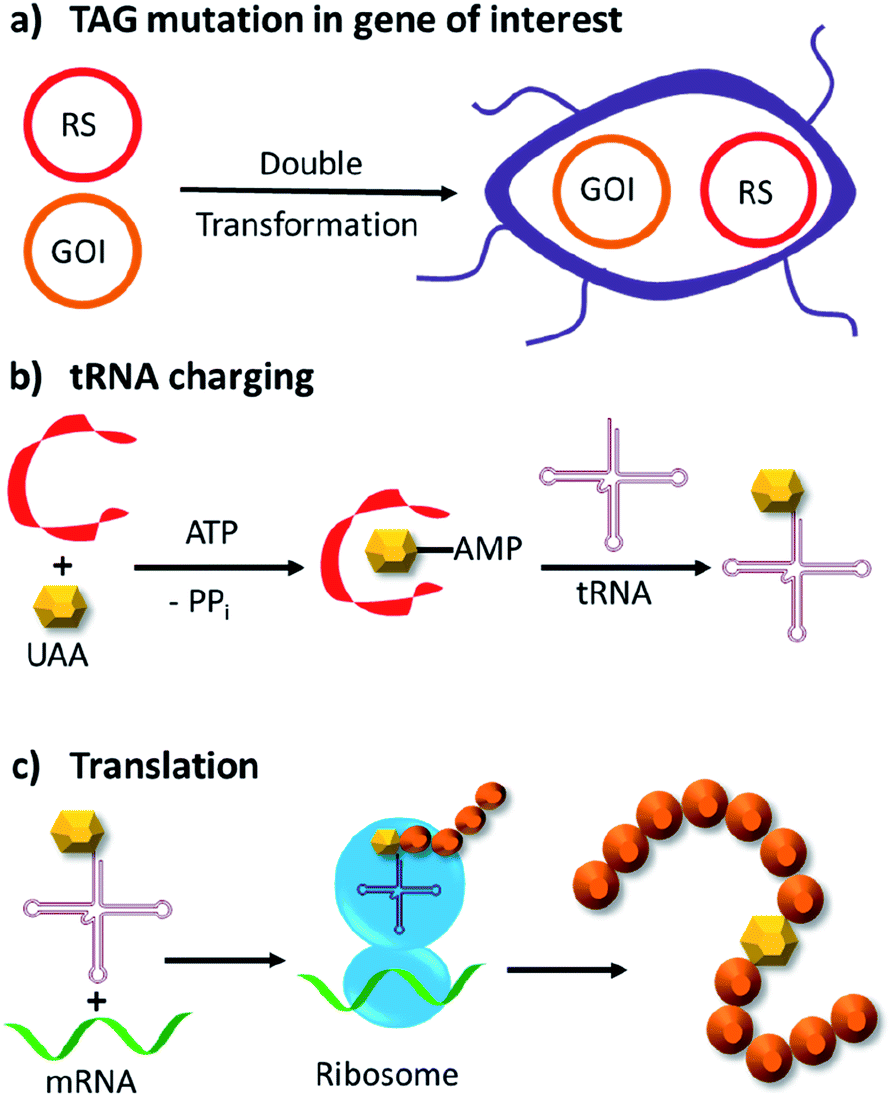 | ||
| Fig. 9 Incorporation of unnatural amino acids by genetic code expansion in E. coli. (a) Double transformation of two plasmids, in which one bears an exogenous amino acid tRNA synthetase (RS) and cognate tRNA, whereas the other contains the gene of interest (GOI) with a site-specific TAG mutation. (b) Expression of the tRNA synthetase and addition of the unnatural amino acid (UAA) allows the tRNA to be charged with the UAA. (c) Ribosomal translation of the GOI with the unnatural amino acid incorporated site specifically into the protein. ATP = adenosine triphosphate, PPi = inorganic pyrophosphate. | ||
The multidrug regulator protein LmrR
LmrR is a dimeric protein isolated from Lactococcus lactis that has a hydrophobic pore in the centre, allowing for the recruitment of organic molecules (Fig. 10a). Four positions located within the hydrophobic pore (Val15, Asn19, Met89 and Phe93) were individually mutated to a TAG codon and tested for the incorporation of the unnatural amino acid p-azidophenylalanine under the action of an evolved tyrosyl-tRNA synthetase from Methanococcus jannaschii. The azido group was chosen and subsequently reduced to the catalytically active aniline, because direct incorporation of p-aminophenylalanine proved to be challenging.75 The designer enzyme was then tested for hydrazone and oxime formation. It was found that unnatural amino acid replacement at the Val15 position yielded the most promising result (Fig. 10b).75 Laboratory evolution was used to screen the library variants in 96 well plates by measuring the loss of the UV absorbance from the substrate.50 The resulting variant which carries additional mutations, including A11L, N19M, A92R and F93H, showed a 74-fold increase in catalytic efficiency. Based on the knowledge of these positions from previous structures, Leu11 and Met19 are thought to help position the aniline in a more “reaction-ready” position. Furthermore, Arg92 was reasoned to stabilise the build-up of negative charge that appears during the condensation of the aniline with the carbonyl group. Lastly, His93 was proposed to serve as proton shuttle assisting in the formation of iminium ion intermediates and promoting the transamination processes.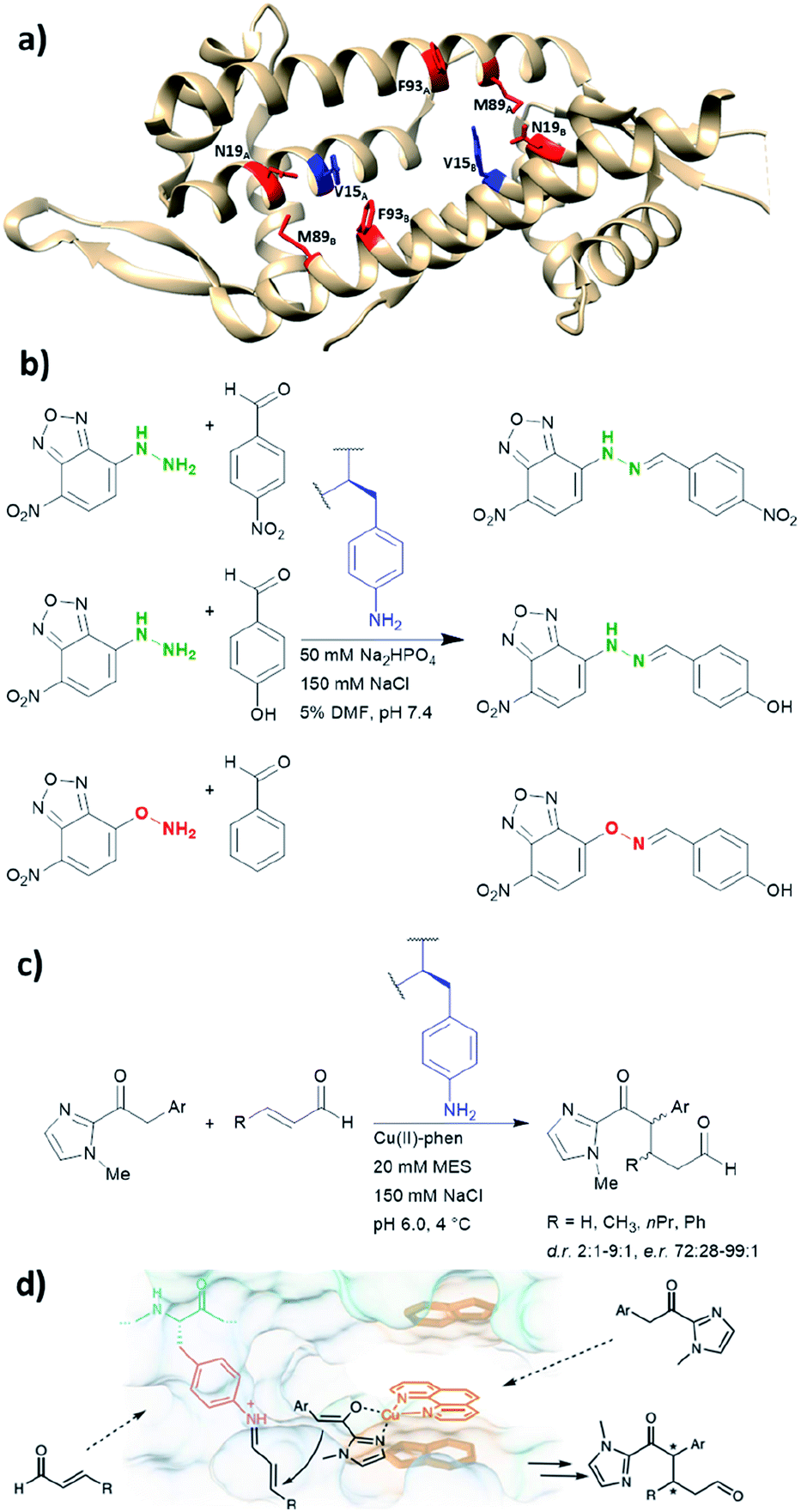 | ||
| Fig. 10 (a) Crystal structure of the homodimeric protein LmrR (PDB: 3F8F). Residues in red are those targeted for unnatural amino acid incorporation (Asn19, Met89, Phe93). Residues in blue (Val15) show the most promising result, when they are replaced with the unnatural amino acid p-amino-phenylalanine. (b) Hydrazone and oxime ligation performed by the unnatural amino acid. (c) Conjugate addition catalysed by dual substrate activation using LmrR V15pAF and a copper complex. (d) Postulated mechanism for the conjugate addition via dual activation from (c). (d) Adapted from ref. 122, https://www.nature.com/articles/s41929-019-0420-6, with permission of Springer Nature, copyright 2020. Further permissions related to the material excerpted should be directed to Springer Nature. DMF = dimethylformamide, phen = phenanthroline, pAF = p-aminophenylalanine. | ||
Recently, the p-aminophenylalanine/LmrR system has been further modified for a novel dual substrate activation strategy.122 Through combination with a supramolecularly bound Lewis acidic Cu(II) complex, the resulting artificial enzyme was able to mediate a Michael reaction that involves both formation of a Cu-enolate and an organocatalytic iminium intermediate. Yields of this novel reaction mode were up to 90%, with d.r. and ee up to 9:1 and >99% respectively. This work highlights that importance of developing different approaches to artificial enzyme design (e.g. genetic code expansion and supramolecular approach), as proteins can be used to host multiple catalytic centres for coupled reaction cascades.
De novo designed BH32
BH32 is an enzyme originally created by Rosetta to perform the Morita–Baylis–Hillman reaction,101 and this protein has been further re-engineered into a potent hydrolase through the combined use of genetic code expansion and laboratory evolution.74 Substitution of the catalytic His23 with methyl-histidine was achieved by using an evolved variant of the pyrrolysyl-tRNA synthetase and its cognate tRNA (Fig. 11a). The resulting enzyme was able to perform ester hydrolysis for a range of compounds that fluoresce upon reaction (Fig. 11b). Screening for variants with improved activity was performed using 96 well plates on a plate reader where formation of the fluorescein product could be monitored. Six mutations resulted in a 15-fold increase in enzyme activity. Mutations resulting from the evolution were L10P, A19H, S22M, E46N, P63G and D125G. Based on the data derived from crystallography and kinetic investigations, the authors concluded that the aromatic ester formed between the substrate and Me-His was significantly more prone to hydrolysis (Fig. 11c). In contrast, the neutral acyl enzyme intermediate formed from the natural amino acid histidine hydrolyses slowly under the same condition.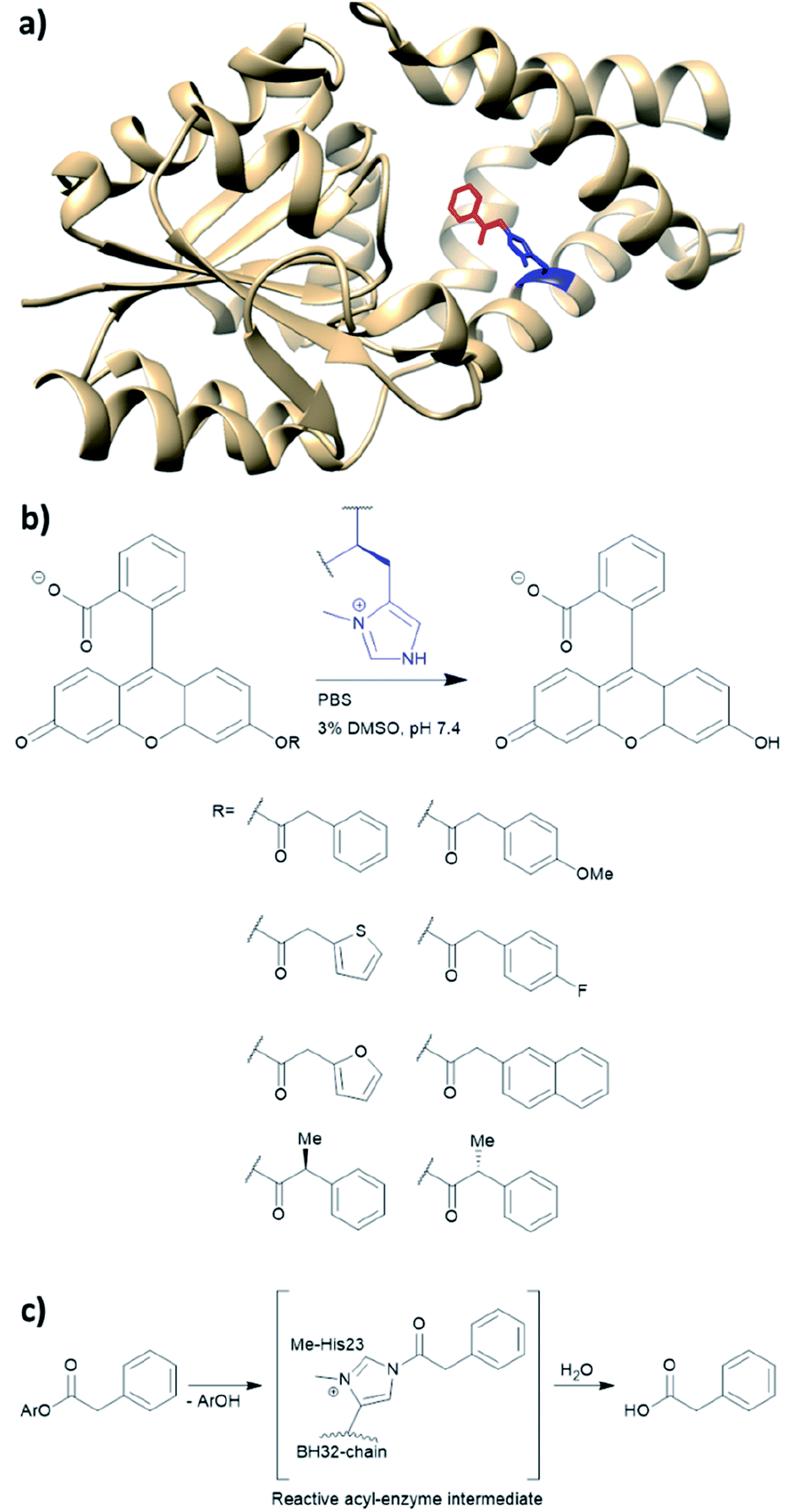 | ||
| Fig. 11 (a) Crystal structure of the protein BH32 (PDB: 6Q7N). Residue in blue is the unnatural amino acid methyl-histidine bound to acetophenone (red). (b) Substrate scope of the ester hydrolysis performed by BH32 with the unnatural amino acid Me-His. (c) Shortened mechanism of the acyl enzyme intermediate formation of BH32 Me-His23 with aromatic esters. PBS = phosphate-buffered saline, DMSO = dimethyl sulfoxide. | ||
The technique of genetic code expansion allows exploration beyond the limit of what natural amino acids offer, thus holding great promise in contemporary enzymology. Incorporation of unnatural amino acids in vivo enables laboratory evolution in a fashion similar to those of natural enzymes. Consequently, artificial enzymes made by this fashion can also be applied to whole cell catalysis or synthetic biological pathways. However, the efficiency of incorporation greatly depends on the unnatural amino acid used. The choice of protein to harbour the amino acid also needs to be considered carefully. Both LmrR and BH32 have been previously used in artificial enzyme design (LmrR for artificial metallo-enzymes and BH32 was computationally designed for carbon–carbon bond forming reactions).47,92 Both examples have shown promise in performing biocompatible organocatalysis. As genetic code expansion has become more readily available, this technique will likely gain increasing popularity in the future of enzyme design.
Non-covalent supramolecular systems
Harnessing non-covalent interactions
Non-covalent but strong protein–ligand interactions have been exploited to generate organocatalytic artificial enzymes. In these systems, a catalytic moiety is covalently attached to a section of a ligand that is only weakly involved in protein binding and introduced to the protein partner. Consequently, the resulting protein–ligand complex is converted into a potential catalytic entity (Fig. 12).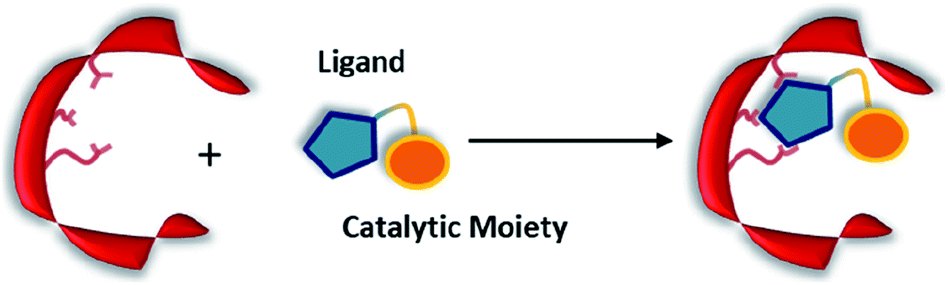 | ||
| Fig. 12 Combination of a catalytically inactive protein scaffold with a ligand–catalyst conjugate leads to a catalytically active supramolecularly assembled protein complex. | ||
When compared to other approaches, a supramolecular complex has little restrictions on the choice of the catalytic motifs. Preparation of the modified ligands tends to be straightforward, involving simple synthetic techniques such as amide bond coupling and click chemistry.35,39,123–126 Hence, the supramolecular approach enables researchers to screen activity for a broad range of candidates within a short period of time. In addition, the protein hosts can still be engineered via rational design or laboratory evolution.46,48,127–129 To this end, the supramolecular approach is an important technique for creating artificial enzymes. As a rule of thumb, the supramolecular catalytic complexes are built based on protein–ligand interactions that have dissociation constants (KD) ranging from low μM to pM.123–125,130
The ligand needs to possess a site for easy functionalisation while causing minimal effect on protein–ligand interaction. One such pair is the (strept)avidin and biotin, whose KD value is approximately 10−14 M−1.130 The (strept)avidin–biotin system has already been exploited in the late 1970s to tether a rhodium catalyst to the valeric motif of biotin for asymmetric hydrogenations.131 Subsequently, a variety of streptavidin based artificial metallo-enzymes operated by iridium, rhodium, ruthenium and palladium have been reported.35,48,123,126,127,132,133 Below we describe two different types of organocatalytic artificial enzymes based on biotin–streptavidin.
Anion–π-catalysis has become a contemporary topic in organocatalysis.9,134–139 In this catalytic mode, anion intermediates formed during the reaction can be stabilised by π-acidic molecules such as naphthalenediimides (NDIs, bold blue core in 11, Fig. 13a), which possess a positive quadrupole moment. This consequently facilitates organic transformations such as conjugate additions (Fig. 13b). Whereas all natural aromatic amino acids are π-basic and interact with cations, the streptavidin–biotin technology was recently used to create an organocatalytic artificial enzyme that drives catalysis by anion–π interactions.36,76
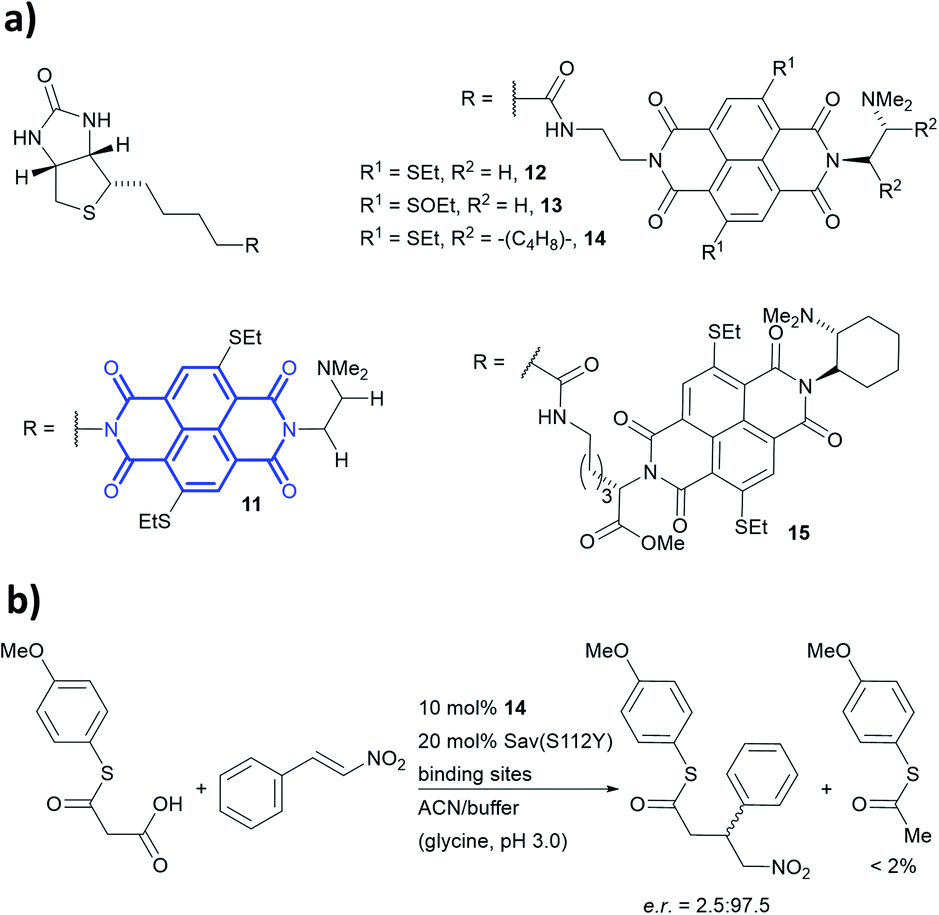 | ||
| Fig. 13 (a) Reported biotin–NDI–amine organocatalyst conjugates, NDI fragment in bold and blue.36 (b) Decarboxylative Michael addition reaction catalysed by 14 within S112Y mutant streptavidin. Sav = streptavidin. | ||
To add anion–π interactions into the repertoire of enzyme catalysis, a combined chemical and genetic screening approach was used. A library of five bifunctional catalytic moieties were attached to biotin (compounds 11–15, Fig. 13a) that contain both an NDI motif and a tertiary amine connected through a linker of different length. The π-acidic surface of the NDI motif was proposed to be able stabilise the enolate intermediate formed in the reaction, whereas the tertiary amine acts as a base and localises the enolate intermediate over the NDI moiety.140 Hence, their ability to mediate a decarboxylative alkylation between thioester malonates and nitrostyrenes was evaluated (Fig. 13b).36
Ligand 14 was identified to be most reactive, and the activity was screened using a streptavidin library of 20 variants. The combination of ligand 14 and S112Y variant yields an organocatalytic artificial enzyme that selects for product formation over the decarboxylated starting material at a ratio >30:1. The conversion in ACN:glycine buffer at pH 3.0 was found to be 90% with e.r. up to 97.5:2.5.
Based on the site-directed mutagenesis studies and docking simulations, a plausible mechanism operated by ligand 14/Sav-S112Y was proposed. A medium sized linker between biotin and NDI (i.e. ligand 14) is essential to accommodate the catalytic unit close to the biotin-binding vestibule, whilst not causing any steric clash. Large electron-withdrawing substituents at the NDI motif were found to weaken the binding (13 vs. 14, Fig. 13a), while a flexible dimethylene bridge instead of a rigid one (12 vs. 14, Fig. 13a) hampers both the conversion and selectivity. The tertiary amine/NDI motif locates in close proximity to the intersubunit interface of the homotetrameric streptavidin, which has a C2 symmetry (Fig. 14a). Hence, residues from both monomers can interact with the catalyst and substrates,141 and the docking studies revealed that the S112Y mutation from each monomer, namely S112YA and S112YB, is essential to the catalysis by ligand 14.
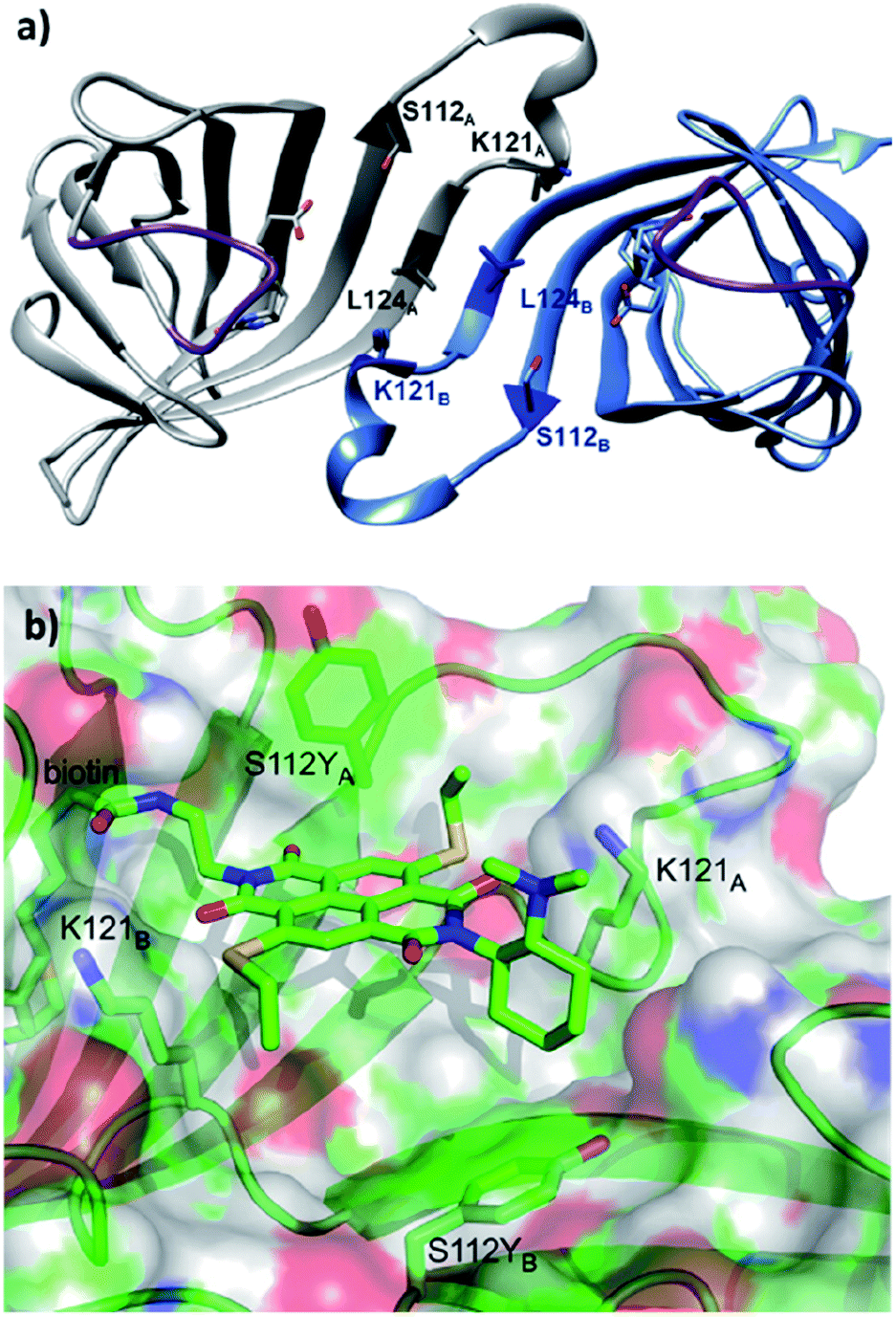 | ||
| Fig. 14 (a) Cartoon overview of the assembly of streptavidin with full binding-site occupation with D-biotin in both binding sites, monomeric unit in light blue. The C2 symmetric interface of two streptavidin subunits is illustrated (PDB: 1MK5). Amino acids Ser112, Lys121, and Leu124 are highlighted. Purple loop regions represent amino acids 46–52. The other two subunits of the homotetrameric streptavidin are omitted for illustration purpose. (b) Cartoon overview of a single binding-site of anion–π catalyst 14 in Sav S112Y. (b) Adapted from ref. 36, https://pubs.acs.org/doi/abs/10.1021/acscentsci.6b00097, with permission of ACS, copyright 2016. Further permissions related to the material excerpted should be directed to the ACS. | ||
When the ligand is bound to monomer A, the NDI motif forms π–π-interactions with S112YA; this is supported by the observation that the mutant S112E and S112W resulted in decreased activity, while the S112F mutant showed similar reactivity to that of S112Y (Fig. 14b). In contrast, S112YB shielded its own biotin binding site. Accordingly, optimal activity was obtained when the catalyst to free binding-sites ratio was kept at 1:2. The wild type lysine residues Lys121A and Lys121B anchor the NDI at the designated location. Furthermore, Lys121A helped maintaining a low pKa value for the tertiary amine of 14, keeping it in its deprotonated form for reaction (even at pH 3.0). Mutation of Lys121 led to a detrimental effect on the activity and selectivity. This study revealed the intricate interactions between the residues and catalytic motif, thereby highlighting that screening of both ligands and variants is critical to obtain an efficient and selective supramolecular system.
The hybrid catalyst system of streptavidin and conjugate 14 was further employed to perform a bioorthogonal domino-Michael–aldol reactions between diketones and nitrostyrenes (Fig. 15).76 With 1–5 mol% catalyst loading, the bicyclic products were obtained in moderate yields (≈50%), decent enantioselectivities (0–80% ee) and significant diastereoselectivity (>20:1) after screening with four streptavidin mutants. Interestingly, the protein–ligand assembly lead to an inversion of stereoselectivity when compared to the nascent biotin-catalyst conjugate.
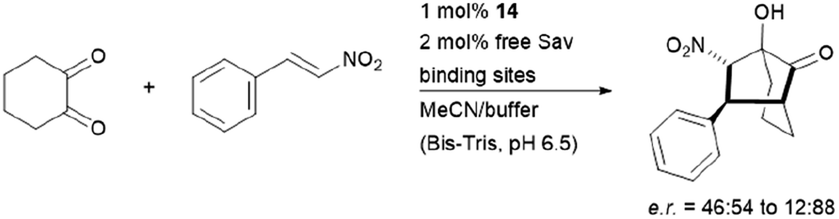 | ||
| Fig. 15 Domino-Michael–Aldol-reaction catalysed by 14. Bis-Tris = bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane. | ||
The biotin-binding-sites in wild-type Sav are rather shallow, exposing a good portion of the catalytic moiety to the solvent. The lack of amino acid side chains in proximity makes mutational optimisation difficult to achieve (Fig. 14b). This led to the development of chimeric Sav variants, which contain insertions of amino acid loops around the biotin-binding sites of Sav like naturally occurring random loops or α-helices.55 Eight chimeric Sav variants containing random coils and alpha helix motifs inserted between residues 46–52 (purple region, Fig. 14a) and one with an addition at the C-terminus have been tested as host for the decarboxylative alkylation catalysed by ligand 14 (Fig. 13b). Though initially thought to increase stereoselectivity and reactivity, three of these chimeric protein hosts were completely inactive and the rest showed lower yields and enantioselectivities than the previously optimised mutant S112Y. Nonetheless, there are similar levels of selectivity for product formation over the decarboxylated starting material (>30:1).
Secondary amine organocatalysis
We have recently employed the streptavidin–biotin technology to create protein-based secondary amine organocatalytic systems. Seven biotinylated secondary amines (ligands 16–22, Fig. 16a) were prepared via either copper-catalysed azide–alkyne cycloaddition or amide bond coupling reactions.39 These catalysts can be broadly segregated into three types: MacMillan-like imidazolidinones (16–19), prolines (20, 21) and pyrrolidines (22, 23), and their ability to catalyse the Michael addition of nitromethane to aromatic α,β-unsaturated aldehydes was tested (Fig. 16b).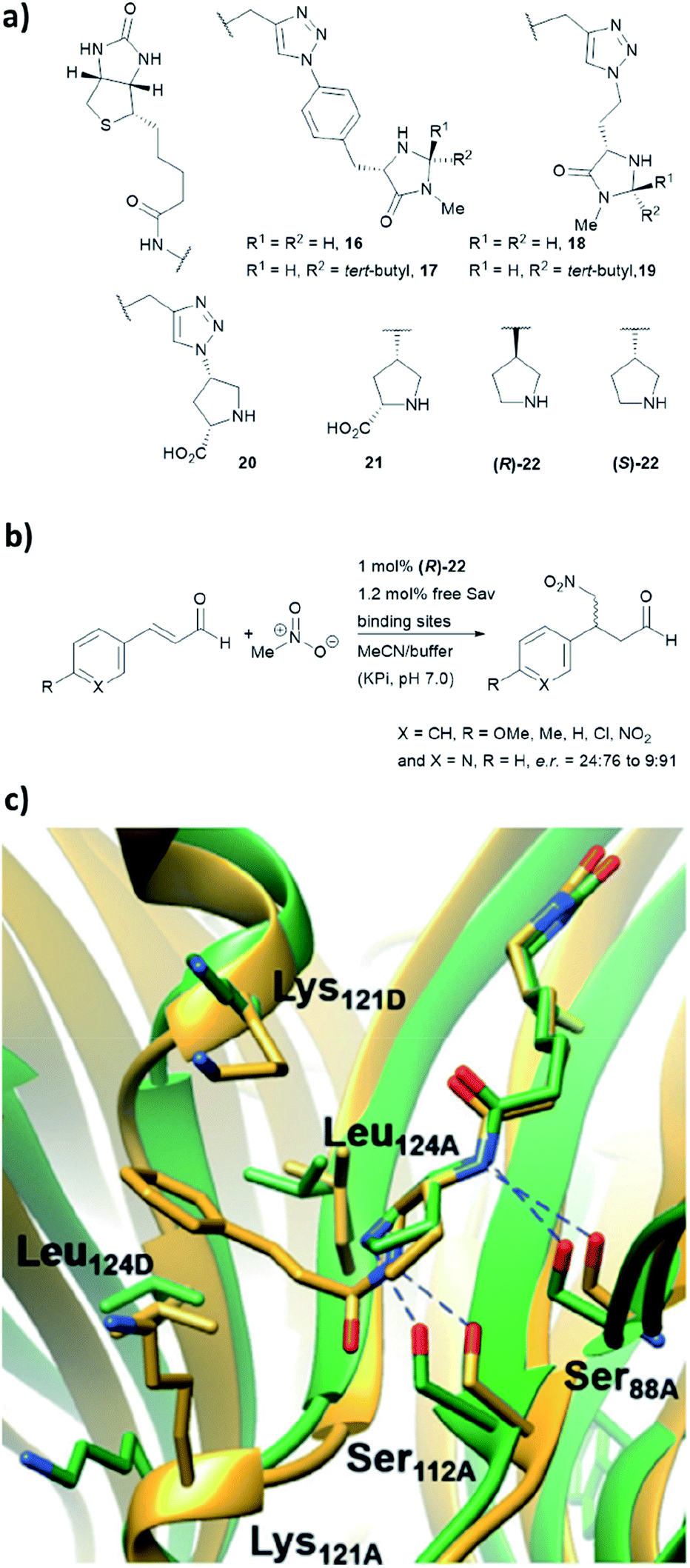 | ||
| Fig. 16 (a) Reported biotin–secondary amine organocatalyst conjugates.39 (b) Set of Michael addition reactions catalysed by 22 within a recombinant core streptavidin. (c) Cartoon overview of Sav:(R)-22 crystal structure (green, PDB: 6GH7) and aminol adduct obtained from QM/MM simulations (golden), and it was adapted from ref. 39, which is an open access article under the terms of the Creative Commons Attribution License, published by Wiley-VCH, copyright 2018, the authors. | ||
Both (R)- and (S)-22 alone are not enantioselective. However, when introduced to the tetrameric streptavidin, they were found to be able to mediate the model reaction with high reactivity and stereoselectivity.39 Moderate to good yields (37–80%) were obtained using only 1 mol% of protein catalyst and 1:1 MeOH/potassium phosphate buffer as reaction medium. Notably, these two protein complexes, namely Sav:(R)-22 and Sav:(S)-22, differed by only one chiral centre, but their stereoselectivity was opposite, with the former favouring for the S enantiomer and latter for the R enantiomer. Through crystallographic and computational structural studies, the position of the secondary amine motif was found to be in proximity to Ser112A. The lysine and leucine residues located at the dimer interface (Lys121A/B and Leu124A/B) dictate the face for which the intermediate was exposed for nucleophilic addition, thereby dictating both regio- (1,2 vs. 1,4-addition) and enantioselectivity (R and S) of the reaction (Fig. 16c).39
Based on the precedence of metallo-enzyme development and supramolecular capsules,142,143 two organocatalytic artificial enzymes that operate distinctively different mechanisms have been designed. Nevertheless, the conditions developed so far are not completely biocompatible, as acidic conditions (pH 3.0) and/or a large volume of organic co-solvents are needed (though the latter was mostly due to the use of substrate with poor solubility in aqueous environments).23 Furthermore, activity of these non-covalent complexes could be potentially optimised via laboratory evolution, as demonstrated by the existing Sav-based catalytic systems.46,48,127,128 Though being a robust and reliable system, issues related to the C2 symmetry of the intersubunit interface of Sav was only recently addressed by the creation of “dimeric” Sav.141 This “dimeric” Sav variant will facilitate mechanistic studies and the design of tailored and asymmetric scaffolds for chemical catalysis. In addition to the streptavidin:biotin system, other protein–ligand systems should serve as inspiration for novel protein-based organocatalytic systems, including the siderophore binding proteins124 or coumarin binding albumins.144
Conclusions and outlook
In this review, we summarised five approaches which are currently employed to perform organocatalysis within proteins (Table 1). The supramolecular tethering and N-terminal proline approaches have proven to be successful, and chemical catalysis with improved biocompatibility has been exploited in different applications including chemo-enzymatic synthesis116 and gene switches.145 In turn, covalent modification, computational de novo design and genetic code expansion are anticipated to excel, as related technologies have vastly improved and gained popularity in recent years.146–148 Importantly, a much-improved system can be achieved by combining different approaches. This can be exemplified by the recent development of LmrR modified with dual catalytic groups.122 Genetic code expansion can also be used to introduce novel catalytic functionalities into a de novo designed enzyme active site. This has been demonstrated in a recently reported artificial metalloenzyme, in which the designed active site includes the unnatural amino acid bipyridylalanine for metal binding.149Despite all these exciting opportunities, there are aspects that need to be immediately addressed in the area of organocatalytic artificial enzyme design. Notably, many of the current systems suffer from poor reactivity, with enzyme loadings up to 20 mol% needed for reaction. However, the development of RA95, aniline/LmrR and methylated histidine/BH32 have demonstrated that laboratory evolution is a feasible approach for activity improvement. The choice of protein scaffold and screening system likely play critical roles during the design. Furthermore, most protein-based organocatalytic systems are based on enamine and iminium catalysis that have similar counterparts in Nature. Useful bioorthogonal reactions that are frequently used in small molecule synthesis have not been tested, including α-fluorinations, aziridinations and Diels–Alder reactions.4 Also, sophisticated catalytic modes such as singly occupied molecular orbital (SOMO)150 activation or photo-radical chemistry151 can also be explored. In turn, other useful catalysts, including hydrogen bonding activators (thioureas and squaramides),152,153 N-heterocyclic carbene32,154 and ion pairing catalysis155 have yet been explored. Additionally, repurposing flavin-dependent enzymes for novel photo-redox reactions represent a valuable avenue for artificial enzyme design.156–158 In summary, this review illustrated that the design of artificial organocatalytic enzymes has become an exciting area of research and it will play critical roles in both chemical and synthetic biology research in future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank for the financial support provided by Leverhulme Trust (RPG-2017-195), Royal Society (RG170187) and Wellcome Seed Trust (202056/Z/16/Z).Notes and references
- M. Tavakolian, S. Vahdati-Khajeh and S. Asgari, ChemCatChem, 2019, 11, 2943 CrossRef CAS.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- Y. B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed.
- D. W. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed.
- T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS PubMed.
- S. G. Ouellet, J. B. Tuttle and D. W. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS PubMed.
- W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed.
- O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC.
- Y. Zhao, Y. Cotelle, L. Liu, J. Lopez-Andarias, A. B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255 CrossRef CAS PubMed.
- J. Lou, F. Liu, C. D. Lindsay, O. Chaudhuri, S. C. Heilshorn and Y. Xia, Adv. Mater., 2018, 30, 1705215 CrossRef PubMed.
- R. J. Spears, R. L. Brabham, D. Budhadev, T. Keenan, S. McKenna, J. Walton, J. A. Brannigan, A. M. Brzozowski, A. J. Wilkinson, M. Plevin and M. A. Fascione, Chem. Sci., 2018, 9, 5585 RSC.
- R. J. Spears and M. A. Fascione, Org. Biomol. Chem., 2016, 14, 7622 RSC.
- F. R. Bisogno, M. G. López-Vidal and G. de Gonzalo, Adv. Synth. Catal., 2017, 359, 2026 CrossRef CAS.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099 CrossRef CAS.
- G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944 CrossRef CAS PubMed.
- K. Baer, M. Krausser, E. Burda, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2009, 48, 9355 CrossRef CAS PubMed.
- M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491 CrossRef CAS.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952 CrossRef CAS PubMed.
- L.-H. Qin, W. Hu and Y.-Q. Long, Tetrahedron Lett., 2018, 59, 2214 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- A. Mondal, S. Bhowmick, A. Ghosh, T. Chanda and K. C. Bhowmick, Tetrahedron: Asymmetry, 2017, 28, 849 CrossRef CAS.
- S. Bhowmick, A. Mondal, A. Ghosh and K. C. Bhowmick, Tetrahedron: Asymmetry, 2015, 26, 1215 CrossRef CAS.
- C. Jimeno, Org. Biomol. Chem., 2016, 14, 6147 RSC.
- Y.-Q. Yu and Z.-L. Wang, J. Chin. Chem. Soc., 2013, 60, 288 CrossRef CAS.
- L. Burroughs, M. E. Vale, J. A. R. Gilks, H. Forintos, C. J. Hayes and P. A. Clarke, Chem. Commun., 2010, 46, 4776 RSC.
- A. Seingeot, Y. Charmasson, M. Attolini and M. Maffei, Heteroat. Chem., 2017, 28, e21352 CrossRef.
- W. W. Y. Leong, X. Chen and Y. R. Chi, Green Chem., 2013, 15, 1505 RSC.
- J. Yan, R. Sun, K. Shi, K. Li, L. Yang and G. Zhong, J. Org. Chem., 2018, 83, 7547 CrossRef CAS PubMed.
- N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS PubMed.
- G. Zhong, R. A. Lerner and C. F. Barbas III, Angew. Chem., Int. Ed., 1999, 38, 3738 CrossRef CAS PubMed.
- B. S. Vachan, M. Karuppasamy, P. Vinoth, S. V. Kumar, S. Perumal, V. Sridharan and J. C. Menendez, Adv. Synth. Catal., 2020, 362, 87 CrossRef CAS.
- C. K. Prier and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13992 CrossRef CAS PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12 CrossRef.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Kohler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142 CrossRef CAS PubMed.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711 CrossRef CAS PubMed.
- Y. Cotelle, V. Lebrun, N. Sakai, T. R. Ward and S. Matile, ACS Cent. Sci., 2016, 2, 388 CrossRef CAS PubMed.
- M. Rahimi, E. M. Geertsema, Y. Miao, J. Y. van der Meer, T. van den Bosch, P. de Haan, E. Zandvoort and G. J. Poelarends, Org. Biomol. Chem., 2017, 15, 2809 RSC.
- E. Zandvoort, E. M. Geertsema, B. J. Baas, W. J. Quax and G. J. Poelarends, Angew. Chem., Int. Ed., 2012, 51, 1240 CrossRef CAS PubMed.
- A. R. Nödling, K. Świderek, R. Castillo, J. W. Hall, A. Angelastro, L. C. Morrill, Y. Jin, Y.-H. Tsai, V. Moliner and L. Y. P. Luk, Angew. Chem., Int. Ed., 2018, 57, 12478 CrossRef PubMed.
- X. Garrabou, D. S. Macdonald, B. I. M. Wicky and D. Hilvert, Angew. Chem., Int. Ed., 2018, 57, 5288 CrossRef CAS PubMed.
- X. Garrabou, D. S. Macdonald and D. Hilvert, Chem.–Eur. J., 2017, 23, 6001 CrossRef CAS PubMed.
- X. Garrabou, R. Verez and D. Hilvert, J. Am. Chem. Soc., 2017, 139, 103 CrossRef CAS PubMed.
- X. Garrabou, B. I. Wicky and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 6972 CrossRef CAS PubMed.
- X. Garrabou, T. Beck and D. Hilvert, Angew. Chem., Int. Ed., 2015, 54, 5609 CrossRef CAS PubMed.
- L. Li, C. Li, Z. Zhang and E. Alexov, J. Chem. Theory Comput., 2013, 9, 2126 CrossRef CAS PubMed.
- M. Hestericová, T. Heinisch, L. Alonso-Cotchico, J. D. Maréchal, P. Vidossich and T. R. Ward, Angew. Chem., Int. Ed., 2018, 57, 1863 CrossRef PubMed.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545 CrossRef CAS PubMed.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585 CrossRef CAS PubMed.
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131 CrossRef CAS PubMed.
- C. Mayer, C. Dulson, E. Reddem, A. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2019, 58, 2083 CrossRef CAS PubMed.
- Y. Wang, J. Chen and Z. Kang, Biochemistry, 2019, 58, 1451 CrossRef CAS PubMed.
- V. Vaissier Welborn and T. Head-Gordon, Chem. Rev., 2019, 119, 6613 CrossRef CAS PubMed.
- H. Kries, R. Blomberg and D. Hilvert, Curr. Opin. Chem. Biol., 2013, 17, 221 CrossRef CAS PubMed.
- C. Trindler and T. R. Ward, in Effects of Nanoconfinement on Catalysis, ed. R. Poli, Springer International Publishing, Cham, 2017, ch. 49, pp. 49–82, DOI:10.1007/978-3-319-50207-6_3.
- M. M. Pellizzoni, F. Schwizer, C. W. Wood, V. Sabatino, Y. Cotelle, S. Matile, D. N. Woolfson and T. R. Ward, ACS Catal., 2018, 8, 1476 CrossRef CAS.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120 CrossRef CAS PubMed.
- Z. P. Wu and D. Hilvert, J. Am. Chem. Soc., 1990, 112, 5647 CrossRef CAS.
- D. Häring, E. Schüler, W. Adam, C. R. Saha-Möller and P. Schreier, J. Org. Chem., 1999, 64, 832 CrossRef PubMed.
- C. J. Suckling and L.-M. Zhu, Bioorg. Med. Chem. Lett., 1993, 3, 531 CrossRef CAS.
- H. Kuang, M. L. Brown, R. R. Davies, E. C. Young and M. D. Distefano, J. Am. Chem. Soc., 1996, 118, 10702 CrossRef CAS.
- T. L. Williams, A. R. Nödling, Y. H. Tsai and L. Y. P. Luk, Wellcome Open Res., 2018, 3, 107 Search PubMed.
- H. A. Bunzel, H. Kries, L. Marchetti, C. Zeymer, P. R. E. Mittl, A. J. Mulholland and D. Hilvert, J. Am. Chem. Soc., 2019, 141, 11745 CrossRef CAS PubMed.
- C. Zeymer, R. Zschoche and D. Hilvert, J. Am. Chem. Soc., 2017, 139, 12541 CrossRef CAS PubMed.
- R. Obexer, A. Godina, X. Garrabou, P. R. E. Mittl, D. Baker, A. D. Griffiths and D. Hilvert, Nat. Chem., 2017, 9, 50 CrossRef CAS PubMed.
- R. Obexer, S. Studer, L. Giger, D. M. Pinkas, M. G. Grütter, D. Baker and D. Hilvert, ChemCatChem, 2014, 6, 1043 CrossRef CAS.
- L. Giger, S. Caner, R. Obexer, P. Kast, D. Baker, N. Ban and D. Hilvert, Nat. Chem. Biol., 2013, 9, 494 CrossRef CAS PubMed.
- E. A. Althoff, L. Wang, L. Jiang, L. Giger, J. K. Lassila, Z. Wang, M. Smith, S. Hari, P. Kast, D. Herschlag, D. Hilvert and D. Baker, Protein Sci., 2012, 21, 717 CrossRef CAS PubMed.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas III, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387 CrossRef CAS PubMed.
- J.-Y. van der Meer, H. Poddar, B.-J. Baas, Y. Miao, M. Rahimi, A. Kunzendorf, R. van Merkerk, P. G. Tepper, E. M. Geertsema, A.-M. W. H. Thunnissen, W. J. Quax and G. J. Poelarends, Nat. Commun., 2016, 7, 10911 CrossRef CAS PubMed.
- E. M. Geertsema, Y. Miao, P. G. Tepper, P. de Haan, E. Zandvoort and G. J. Poelarends, Chem.–Eur. J., 2013, 19, 14407 CrossRef CAS PubMed.
- Y. Miao, E. M. Geertsema, P. G. Tepper, E. Zandvoort and G. J. Poelarends, ChemBioChem, 2013, 14, 191 CrossRef CAS PubMed.
- E. Zandvoort, E. M. Geertsema, W. J. Quax and G. J. Poelarends, ChemBioChem, 2012, 13, 1274 CrossRef CAS PubMed.
- E. Zandvoort, B.-J. Baas, W. J. Quax and G. J. Poelarends, ChemBioChem, 2011, 12, 602 CrossRef CAS PubMed.
- A. J. Burke, S. L. Lovelock, A. Frese, R. Crawshaw, M. Ortmayer, M. Dunstan, C. Levy and A. P. Green, Nature, 2019, 570, 219 CrossRef CAS PubMed.
- I. Drienovská, C. Mayer, C. Dulson and G. Roelfes, Nat. Chem., 2018, 10, 946 CrossRef PubMed.
- L. Liu, Y. Cotelle, J. Klehr, N. Sakai, T. R. Ward and S. Matile, Chem. Sci., 2017, 8, 3770 RSC.
- M. D. Toscano, K. J. Woycechowsky and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 3212 CrossRef CAS PubMed.
- E. T. Kaiser and D. S. Lawrence, Science, 1984, 226, 505 CrossRef CAS PubMed.
- D. Qi, C. M. Tann, D. Haring and M. D. Distefano, Chem. Rev., 2001, 101, 3081 CrossRef CAS PubMed.
- L. Polgar and M. L. Bender, J. Am. Chem. Soc., 1966, 88, 3153 CrossRef CAS.
- L. Polgar and M. L. Bender, Biochemistry, 1967, 6, 610–620 CrossRef CAS PubMed.
- K. E. Neet, A. Nanci and D. E. Koshland Jr, J. Biol. Chem., 1968, 243, 6392 CAS.
- Z. P. Wu and D. Hilvert, J. Am. Chem. Soc., 1989, 111, 4513 CrossRef CAS.
- H. L. Levine, Y. Nakagawa and E. T. Kaiser, Biochem. Biophys. Res. Commun., 1977, 76, 64 CrossRef CAS PubMed.
- S. Mao, Z. Dong, J. Liu, X. Li, X. Liu, G. Luo and J. Shen, J. Am. Chem. Soc., 2005, 127, 11588 CrossRef CAS PubMed.
- J. C. Maza, D. L. V. Bader, L. Xiao, A. M. Marmelstein, D. D. Brauer, A. M. ElSohly, M. J. Smith, S. W. Krska, C. A. Parish and M. B. Francis, J. Am. Chem. Soc., 2019, 141, 3885 CrossRef CAS PubMed.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697 CrossRef CAS PubMed.
- P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005 CAS.
- B. Bhushan, Y. A. Lin, M. Bak, A. Phanumartwiwath, N. Yang, M. K. Bilyard, T. Tanaka, K. L. Hudson, L. Lercher, M. Stegmann, S. Mohammed and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 14599 CrossRef CAS PubMed.
- S. R. G. Galan, J. R. Wickens, J. Dadova, W.-L. Ng, X. Zhang, R. A. Simion, R. Quinlan, E. Pires, R. S. Paton, S. Caddick, V. Chudasama and B. G. Davis, Nat. Chem. Biol., 2018, 14, 955 CrossRef CAS PubMed.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50 RSC.
- M. Morais, N. Forte, V. Chudasama and J. R. Baker, in Bioconjugation: Methods and Protocols, ed. S. Massa and N. Devoogdt, Springer, New York, NY, 2019, ch. 15, pp. 15–24, DOI:10.1007/978-1-4939-9654-4_2.
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS PubMed.
- O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115, 2174 CrossRef CAS PubMed.
- D. Hilvert, Annu. Rev. Biochem., 2013, 82, 447 CrossRef CAS PubMed.
- A. Zanghellini, L. Jiang, A. M. Wollacott, G. Cheng, J. Meiler, E. A. Althoff, D. Röthlisberger and D. Baker, Protein Sci., 2006, 15, 2785 CrossRef CAS PubMed.
- A. Leaver-Fay, M. Tyka, S. M. Lewis, O. F. Lange, J. Thompson, R. Jacak, K. W. Kaufman, P. D. Renfrew, C. A. Smith, W. Sheffler, I. W. Davis, S. Cooper, A. Treuille, D. J. Mandell, F. Richter, Y.-E. A. Ban, S. J. Fleishman, J. E. Corn, D. E. Kim, S. Lyskov, M. Berrondo, S. Mentzer, Z. Popović, J. J. Havranek, J. Karanicolas, R. Das, J. Meiler, T. Kortemme, J. J. Gray, B. Kuhlman, D. Baker and P. Bradley, in Methods in Enzymology, ed. M. L. Johnson and L. Brand, Academic Press, 2011, vol. 487, ch. 545, pp. 545–574 Search PubMed.
- D. J. Tantillo, C. Jiangang and K. N. Houk, Curr. Opin. Chem. Biol., 1998, 2, 743 CrossRef CAS PubMed.
- M. Merski and B. K. Shoichet, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16179 CrossRef CAS PubMed.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190 CrossRef PubMed.
- S. Bjelic, L. G. Nivón, N. Çelebi-Ölçüm, G. Kiss, C. F. Rosewall, H. M. Lovick, E. L. Ingalls, J. L. Gallaher, J. Seetharaman, S. Lew, G. T. Montelione, J. F. Hunt, F. E. Michael, K. N. Houk and D. Baker, ACS Chem. Biol., 2013, 8, 749 CrossRef CAS PubMed.
- M. Hennig, B. D. Darimont, J. N. Jansonius and K. Kirschner, J. Mol. Biol., 2002, 319, 757 CrossRef CAS PubMed.
- B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard and D. Baker, Science, 2003, 302, 1364 CrossRef CAS PubMed.
- T. A. Kunkel, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 488 CrossRef CAS PubMed.
- A. R. Buller, P. van Roye, J. K. B. Cahn, R. A. Scheele, M. Herger and F. H. Arnold, J. Am. Chem. Soc., 2018, 140, 7256 CrossRef CAS PubMed.
- Y. Zhang, P. Doruker, B. Kaynak, S. Zhang, J. Krieger, H. Li and I. Bahar, Curr. Opin. Chem. Biol., 2019, 62, 14 Search PubMed.
- D. L. Trudeau and D. S. Tawfik, Curr. Opin. Chem. Biol., 2019, 60, 46 CrossRef CAS PubMed.
- M. A. Maria-Solano, E. Serrano-Hervas, A. Romero-Rivera, J. Iglesias-Fernandez and S. Osuna, Chem. Commun., 2018, 54, 6622 RSC.
- R. K. Wierenga, FEBS Lett., 2001, 492, 193 CrossRef CAS PubMed.
- R. K. Wierenga, E. G. Kapetaniou and R. Venkatesan, Cell. Mol. Life Sci., 2010, 67, 3961–3982 CrossRef CAS PubMed.
- A. D. Goldman, J. T. Beatty and L. F. Landweber, J. Mol. Evol., 2016, 82, 17 CrossRef CAS PubMed.
- J. Lazic, J. Spasic, D. Francuski, Z. Tokic-Vujosevic, J. Nikodinovic-Runic, V. Maslak and L. Djokic, J. Serb. Chem. Soc., 2016, 81, 871–881 CrossRef CAS.
- T. K. Harris, R. M. Czerwinski, W. H. Johnson Jr, P. M. Legler, C. Abeygunawardana, M. A. Massiah, J. T. Stivers, C. P. Whitman and A. S. Mildvan, Biochemistry, 1999, 38, 12343 CrossRef CAS PubMed.
- E. A. Burks, C. D. Fleming, A. D. Mesecar, C. P. Whitman and S. D. Pegan, Biochemistry, 2010, 49, 5016 CrossRef CAS PubMed.
- C. Guo, M. Saifuddin, T. Saravanan, M. Sharifi and G. J. Poelarends, ACS Catal., 2019, 9, 4369 CrossRef CAS PubMed.
- L. Biewenga, T. Saravanan, A. Kunzendorf, J. Y. van der Meer, T. Pijning, P. G. Tepper, R. van Merkerk, S. J. Charnock, A. W. H. Thunnissen and G. J. Poelarends, ACS Catal., 2019, 9, 1503 CrossRef CAS PubMed.
- T. Narancic, J. Radivojevic, P. Jovanovic, D. Francuski, M. Bigovic, V. Maslak, V. Savic, B. Vasiljevic, K. E. O'Connor and J. Nikodinovic-Runic, Bioresour. Technol., 2013, 142, 462 CrossRef CAS PubMed.
- L. Djokic, J. Spasic, S. Jeremic, B. Vasiljevic, O. Prodanovic, R. Prodanovic and J. Nikodinovic-Runic, Bioprocess Biosyst. Eng., 2015, 38, 2389 CrossRef CAS PubMed.
- J. Radivojevic, G. Minovska, L. Senerovic, K. O'Connor, P. Jovanovic, V. Savic, Z. Tokic-Vujosevic, J. Nikodinovic-Runic and V. Maslak, RSC Adv., 2014, 4, 60502 RSC.
- A. R. Nödling, L. A. Spear, T. L. Williams, L. Y. P. Luk and Y.-H. Tsai, Essays Biochem., 2019, 63, 237 CrossRef PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026 CrossRef CAS PubMed.
- Z. Zhou and G. Roelfes, Nat. Catal., 2020, 3, 289 CrossRef CAS.
- I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, J. Am. Chem. Soc., 2019, 141, 4815 CrossRef CAS PubMed.
- D. J. Raines, J. E. Clarke, E. V. Blagova, E. J. Dodson, K. S. Wilson and A.-K. Duhme-Klair, Nat. Catal., 2018, 1, 680 CrossRef CAS.
- F. W. Monnard, E. S. Nogueira, T. Heinisch, T. Schirmer and T. R. Ward, Chem. Sci., 2013, 4, 3269 RSC.
- T. R. Ward, Acc. Chem. Res., 2011, 44, 47 CrossRef CAS PubMed.
- M. T. Reetz, J. J. Peyralans, A. Maichele, Y. Fu and M. Maywald, Chem. Commun., 2006, 4318 RSC.
- T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marliere, S. Panke and T. R. Ward, Chem. Sci., 2018, 9, 5383 RSC.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661 CrossRef CAS PubMed.
- A. Zocchi, N. Humbert, T. Berta and T. R. Ward, Chimia, 2003, 57, 589 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- L. Olshansky, R. Huerta-Lavorie, A. I. Nguyen, J. Vallapurackal, A. Furst, T. D. Tilley and A. S. Borovik, J. Am. Chem. Soc., 2018, 140, 2739 CrossRef CAS PubMed.
- C.-C. Lin, C.-W. Lin and A. S. C. Chan, Tetrahedron: Asymmetry, 1999, 10, 1887 CrossRef CAS.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed.
- Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582 CrossRef CAS PubMed.
- Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101 CrossRef CAS PubMed.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778 RSC.
- Y. Cotelle, S. Benz, A.-J. Avestro, T. R. Ward, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2016, 55, 4275 CrossRef CAS PubMed.
- S. Wu, Y. Zhou, J. G. Rebelein, M. Kuhn, H. Mallin, J. Zhao, N. V. Igareta and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 15869 CrossRef CAS PubMed.
- T. M. Brauer, Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2017, 139, 17500 CrossRef CAS PubMed.
- T. M. Brauer, Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2016, 55, 7698 CrossRef PubMed.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780 CrossRef CAS.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef PubMed.
- B. D. Weitzner, Y. Kipnis, A. G. Daniel, D. Hilvert and D. Baker, Protein Sci., 2019, 28, 2036 CrossRef CAS PubMed.
- I. Drienovská and G. Roelfes, Nat. Catal., 2020, 3, 193 CrossRef.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203 CrossRef CAS.
- J. H. Mills, S. D. Khare, J. M. Bolduc, F. Forouhar, V. K. Mulligan, S. Lew, J. Seetharaman, L. Tong, B. L. Stoddard and D. Baker, J. Am. Chem. Soc., 2013, 135, 13393 CrossRef CAS PubMed.
- T. D. Beeson, A. Mastracchio, J. B. Hong, K. Ashton and D. W. Macmillan, Science, 2007, 316, 582 CrossRef CAS PubMed.
- M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678 CrossRef CAS PubMed.
- B. L. Zhao, J. H. Li and D. M. Du, Chem. Rec., 2017, 17, 994 CrossRef CAS PubMed.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166 CrossRef CAS PubMed.
- M. J. Black, K. F. Biegasiewicz, A. J. Meichan, D. G. Oblinsky, B. Kudisch, G. D. Scholes and T. K. Hyster, Nat. Chem., 2020, 12, 71 CrossRef PubMed.
- B. A. Sandoval, A. J. Meichan and T. K. Hyster, J. Am. Chem. Soc., 2017, 139, 11313 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |