
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemiluminescent organic nanophotosensitizer for a penetration depth independent photodynamic therapy†
Xiaomei Lu‡
a,
Xingwen Song‡a,
Qi Wanga,
Wenbo Hu*a,
Wei Shi*a,
Yufu Tanga,
Zizi Wua,
Quli Fan *b and
Wei Huangabc
*b and
Wei Huangabc
aKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Nanjing Tech University (Nanjing Tech), Nanjing 211816, China. E-mail: iamwbhu@njtech.edu.cn; iamwshi@njtech.edu.cn
bKey Laboratory for Organic Electronics and Information Displays, Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications (NUPT), Nanjing 210023, China. E-mail: iamqlfan@njupt.edu.cn
cShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), Xi'an 710072, China
First published on 24th March 2020
Abstract
Photodynamic therapy initiated by external photoexcitation is a clinically-approved therapeutic paradigm, but its practical application has been severely hindered by the shallow penetration of light. Here, we describe a penetration-independent PDT modality using a chemiluminescent organic nanophotosensitizer, which is activated by hydrogen peroxide instead of external photoexcitation.
Photodynamic therapy (PDT) performed with the cooperation of a photosensitizer, molecular oxygen and light has become a minimally non-invasive therapeutic paradigm in clinics for the treatment of various diseases such as psoriasis, vitiligo and cancer.1–4 In general, exposing photosensitizers to suitable light, generates very toxic reactive oxygen species (mainly, single oxygen, 1O2) that kill tumour cells. Near-infrared light is preferred as external light to activate PDT owing to its considerably deeper penetration into tissue as compared to ultraviolet or visible light.5,6 However, advances in PDT have been severely confined to superficial lesions for decades as all these external light-based phototherapies, including PDT, suffer from rapid attenuation of external light in tissue.7 Recently, Cerenkov radiation has been used as external light to break the depth dependency of PDT.7–9 Although conceptually impressive, the expensive radiation source and inevitable DNA damage induced by ionizing radiation remain major limitations for practical applications. Therefore, it is highly desirable to develop novel better PDT with the penetration depth-independent feature.
Unlike external light sources, internal light sources, such as fibre-optic light sources, could address the penetration issue and have thus been proposed as an alternative solution to activate PS, but it brings invasive problems.10 In this case, another intriguing internal light arising from chemiluminescence has been proposed for penetration depth independent PDT. In this paradigm, chemiluminescence, which occurs when a specific chemical (such as luminol) is mixed with an appropriate oxidizing agent (such as hydrogen peroxide, H2O2), could activate the adjacent photosensitizer to proceed to PDT. Moreover, elevated H2O2 levels have been found in several types of cancer cells compared to that in normal cells, potentially affording H2O2-activatable PDT with good selectivity. However, only few chemiluminescent PDT systems have been reported.11–14 Moreover, these chemiluminescent PDT systems usually require coupling with additional PS to activate PDT by energy transfer, complicating the system with reduced reproducibility. In addition, to match the absorption of near-infrared absorptive PS (such as chlorin e6) for efficient energy transfer, quantum dots were usually employed to red-shift bioluminescence,10,15 which not only complicates the system but also raises the potential of metal-induced toxicity issues.
Herein, we fabricated a novel chemiluminescent NPs (C NPs) as a nanophotosensitizer for penetration depth independent PDT in tumour cells and bacteria (Scheme 1). This concise chemiluminescent NPs consisted of luminol and horseradish peroxidase (HRP), which is activated by hydrogen peroxide instead by external photoexcitation. In H2O2-rich conditions, C NPs exhibited remarkably enhanced 1O2 production compared to the luminol/HRP mixture. Finally, C NPs was demonstrated as a powerful nanophotosensitizer for efficient PDT in tumour cells and bacteria without external photoexcitation, becoming a promising platform for the future design of efficient PDT at high tissue depth.
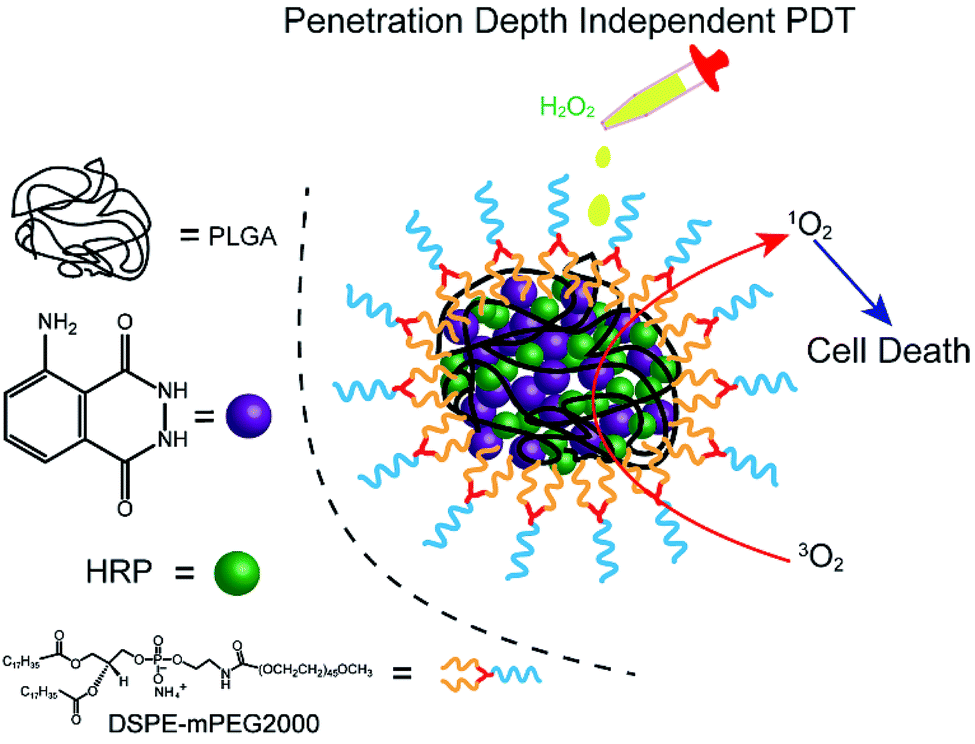 | ||
| Scheme 1 Schematic illustration of the NPs structure and penetration depth-independent PDT. | ||
C NPs were prepared by the self-assembly of PLGA, luminol, HRP, and DSPE-mPEG2000 via a nanoprecipitation method (Scheme 1).16 The detailed preparation process is presented in the ESI.† Briefly, the PLGA was dissolved in acetonitrile. Luminol and HRP were dissolved in an aqueous solution, which was then added into the previous PLGA acetonitrile solution. This mixed solution was added dropwise to an aqueous solution of DSPE-PEG2000. After gently stirring for 4 h at room temperature, the remaining organic solvent and free molecules were removed by ultrafiltration. The designed C NPs could be stored in an aqueous solution up to 2 months without any eye-observed precipitation (Fig. 1a), indicating their excellent stability. Transmission electron microscopy (TEM) results reveal the spherical morphology of the C NPs with high monodispersity (Fig. 1b), while dynamic light scattering (DLS) indicated their average hydrodynamic diameter with a value of about 70 nm (Fig. 1c).
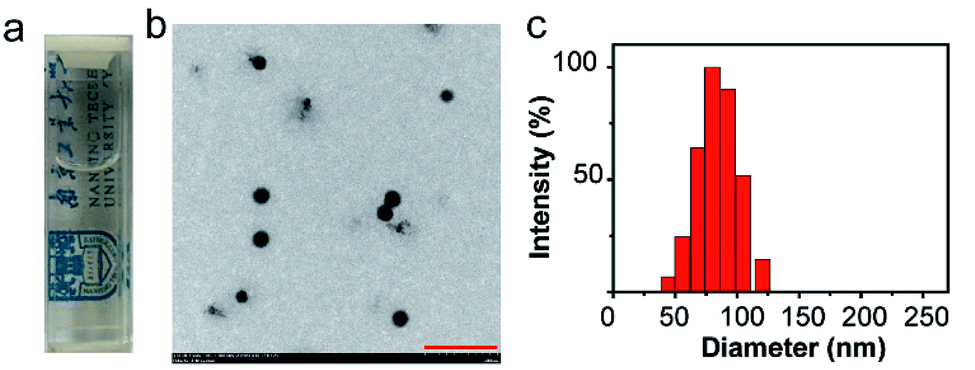 | ||
| Fig. 1 (a) Photograph of C NPs in an aqueous solution. (b) TEM image of C NPs. Scale bar: 500 nm. (c) Representative DLS of C NPs. | ||
The production of reactive oxygen species (ROS, e.g., 1O2) in NPs or luminol (L) + HRP was experimentally confirmed by the photodegradation of anthracene-9,10-diyl-bis-methylmalonate (ADMA) in the presence of H2O2 and molecular oxygen.17,18 After the addition of H2O2, the characteristic absorbances (260, 358, 378 and 399 nm) of ADMA dispersed in L + HRP solutions gradually decreased with prolonged time (Fig. 2a), indicating the inefficient production of 1O2. In sharp contrast, the absorbance of ADMA decreased remarkably in a mixture of ADMA and NPs after the addition of H2O2 (Fig. 2b). This is a solid evidence to illustrate that C NPs can be activated by H2O2 rather than external photoexcitation to perform PDT. Notably, within 2 min, the NPs almost totally consumed the AMDA, while the L + HRP only showed negligible consumption (Fig. 2c), which unambiguously demonstrated much more efficient production of 1O2 by C NPs. This is reasonable because L and HRP within NPs are close together, which is favourable for chemiluminescence. Despite the origin of 1O2 production during the C NP-induced chemiluminescence, the above-mentioned results clearly indicate that C NPs can efficiently produce 1O2 under H2O2 activation, which shows tremendous potential in vivo PDT in high tissue depth.
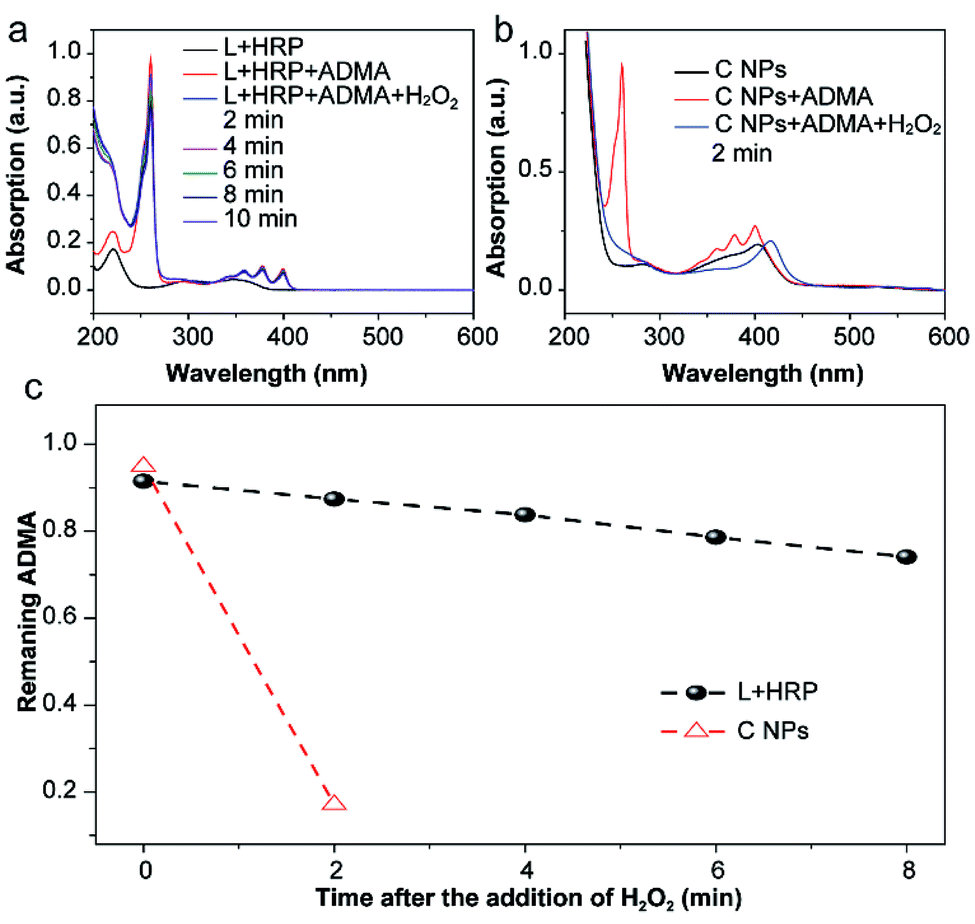 | ||
| Fig. 2 The absorption spectra of a C NPs (a) or L + HRP (b) and ADMA mixture before and after addition of H2O2. The spectra were obtained every 2 min after addition of H2O2. Rapidly decreased characteristic absorbance at 260 nm of ADMA within 2 min confirms the efficient 1O2 production of C NPs in the presence of H2O2. (c) Kinetic curves of the ADMA consumption of L + HRP and C NPs. | ||
To verify the PDT effects, we utilized calcein-AM (living cell) and propidium iodide (PI, dead cell) cell viability kits to distinguish the dead cells from living ones (Fig. 3).19,20 After incubation with H2O2, H2O2 + HRP, and H2O2 + HRP alone, HeLa cells showed comparable cellular viability to the blank group (control) without any treatments, which demonstrate the resistance of HeLa cells toward H2O2, H2O2 + HRP, and H2O2 + HRP. Without the addition of H2O2, both L + HRP and C NP-incubated HeLa cells exhibited negligible cytotoxicity, suggesting low dark-cytotoxicity of L + HRP and C NPs. After the addition of H2O2, C NP-incubated HeLa cells exhibited strong cytotoxicity relative to the mixed solution of L + HRP.
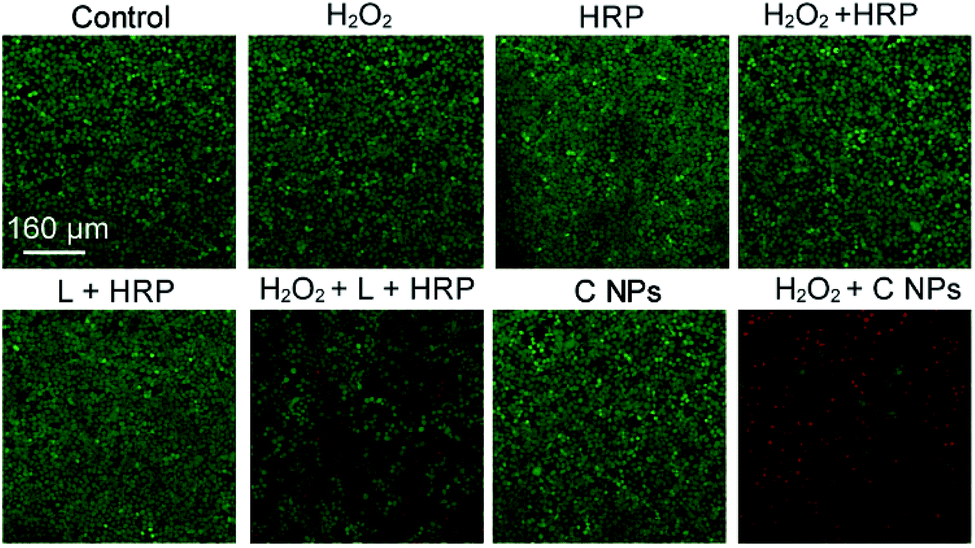 | ||
| Fig. 3 Live/dead assay of HeLa cells. Green colour represents live cells, and red colour represents dead cells. | ||
Furthermore, we evaluated the antibacterial ability of NPs using Gram-negative bacteria, namely E. coli. As shown in Fig. 4a, all groups treated with C NPs, L + HRP, and H2O2 alone showed a tiny difference as compared to the blank group (control) without any treatment, which means that C NPs, L + HRP, and H2O2 have no obvious influence on E. coli. After the addition of H2O2, a huge decrease in C NPs + H2O2 was observed, while only a small decrease in L + HRP + H2O2, indicating a stronger antibacterial ability of C NPs. The antibacterial efficiency of NPs + H2O2 was determined to be 70% (Fig. 4b), which is approximately 10-fold stronger than L + HRP + H2O2 (7%). These results clearly demonstrated the strong antibacterial ability of C NPs.
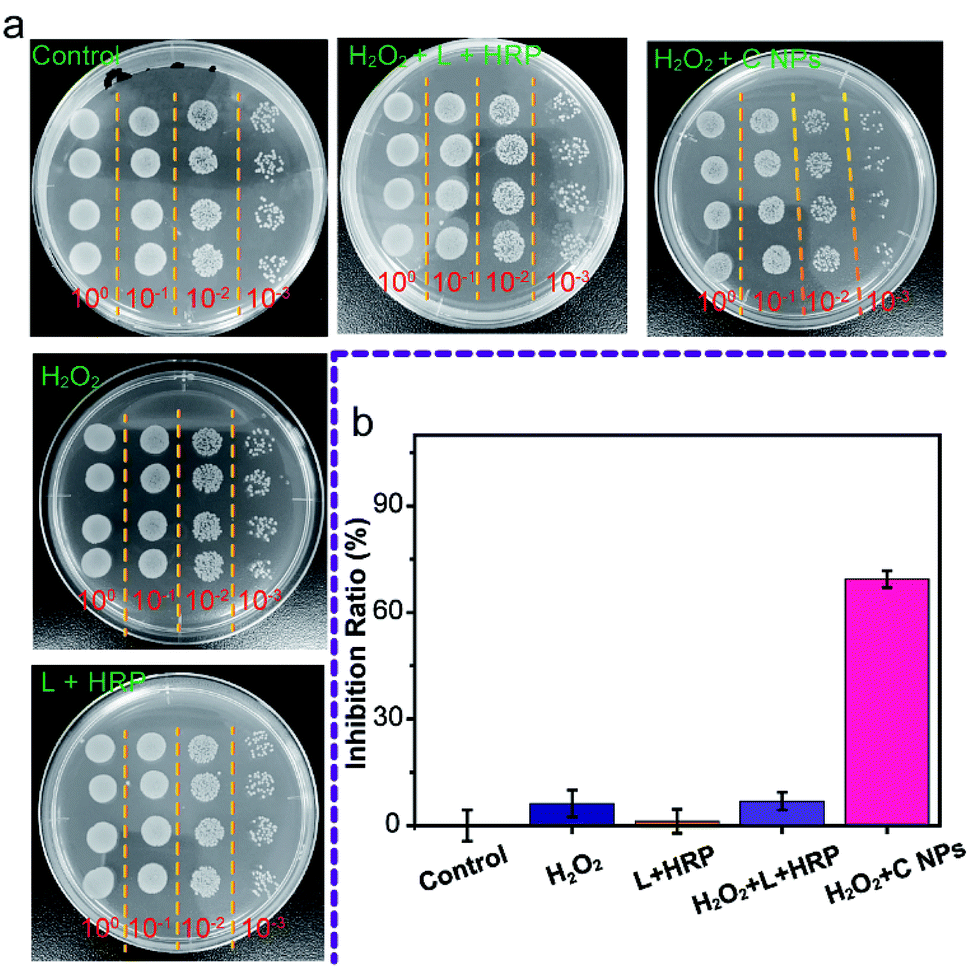 | ||
| Fig. 4 (a) Colony-forming units (CFU) for E. coli treated with NPs before and after addition of H2O2 on LB agar plate. The treated E. coli diluted 100 to 10−3, 6 microliters of each dilution were inoculated to solid LB media, the E. coli were grown at 37 °C for 12 h. (b) Antibacterial activity of NPs before and after addition of H2O2. | ||
In summary, we have fabricated a chemiluminescent organic nanophotosensitizer, namely C NPs, which could be activated by H2O2 instead by external photoexcitation. The C NPs show a very strong 1O2 generation ability in the presence of H2O2. The tumour cells and bacteria explanation clearly demonstrate that C NPs could be used as a powerful nanophotosensitizer for potential penetration depth-independent PDT. Our results provide an attractive platform for the future design of a powerful photosensitizer, which can expand the application scope of PDT.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 61805118 and 21674048), the Natural Science Foundation of Jiangsu Province of China (No. BK20171020), the China Postdoctoral Science Foundation (No. 2018T110488), and open research fund of Key Laboratory for Organic Electronics and Information Displays.Notes and references
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- H. Yuan, B. Wang, F. Lv, L. Liu and S. Wang, Adv. Mater., 2014, 26, 6978–6982 CrossRef CAS PubMed.
- W. Hu, H. Ma, B. Hou, H. Zhao, Y. Ji, R. Jiang, X. Hu, X. Lu, L. Zhang, Y. Tang, Q. Fan and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 12039–12047 CrossRef CAS PubMed.
- J. D. Bhawalkar, N. D. Kumar, C. F. Zhao and P. N. Prasad, Photomed. Laser Surg., 1997, 15, 201–204 CAS.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS PubMed.
- D. Ni, C. A. Ferreira, T. E. Barnhart, V. Quach, B. Yu, D. Jiang, W. Wei, H. Liu, J. W. Engle, P. Hu and W. Cai, J. Am. Chem. Soc., 2018, 140, 14971–14979 CrossRef PubMed.
- A. Kamkaew, L. Cheng, S. Goel, H. F. Valdovinos, T. E. Barnhart, Z. Liu and W. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 26630–26637 CrossRef CAS PubMed.
- Y. R. Kim, S. Kim, J. W. Choi, S. Y. Choi, S. H. Lee, H. Kim, S. K. Hahn, G. Y. Koh and S. H. Yun, Theranostics, 2015, 5, 805–817 CrossRef CAS PubMed.
- H. Yuan, H. Chong, B. Wang, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, J. Am. Chem. Soc., 2012, 134, 13184–13187 CrossRef CAS PubMed.
- D. Mao, W. Wu, S. Ji, C. Chen, F. Hu, D. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991–1007 CAS.
- Y. Zhang, L. Pang, C. Ma, Q. Tu, R. Zhang, E. Saeed, A. E. Mahmoud and J. Wang, Anal. Chem., 2014, 86, 3092–3099 CrossRef CAS PubMed.
- X. Xu, H. An, D. Zhang, H. Tao, Y. Dou, X. Li, J. Huang and J. Zhang, Sci. Adv., 2019, 5, eaat2953 CrossRef PubMed.
- C. Y. Hsu, C. W. Chen, H. P. Yu, Y. F. Lin and P. S. Lai, Biomaterials, 2013, 34, 1204–1212 CrossRef CAS PubMed.
- C. Zheng, M. Zheng, P. Gong, D. Jia, P. Zhang, B. Shi, Z. Sheng, Y. Ma and L. Cai, Biomaterials, 2012, 33, 5603–5609 CrossRef CAS PubMed.
- W. Hu, T. He, R. Jiang, J. Yin, L. Li, X. Lu, H. Zhao, L. Zhang, L. Huang, H. Sun, W. Huang and Q. L. Fan, Chem. Commun., 2017, 53, 1680–1683 RSC.
- W. Hu, Q. Wang, X. Miao, L. Bai, L. Li, G. S. He, J. Li, S. Yao, T. He, X. Lu, W. Huang, P. N. Prasad and Q. Fan, J. Phys. Chem. C, 2018, 122, 20945–20951 CrossRef CAS.
- W. Hu, T. He, H. Zhao, H. Tao, R. Chen, L. Jin, J. Li, Q. Fan, W. Huang, A. Baev and P. N. Prasad, Angew. Chem., Int. Ed., 2019, 58, 11105–11111 CrossRef CAS PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R.-F. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01477j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |