
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase junction enhanced photocatalytic activity of Ga2O3 nanorod arrays on flexible glass fiber fabric†
Hanlin Suna,
Liying Zhanga,
Jingyan Yua,
Shunli Wang*a,
Daoyou Guo a,
Chaorong Lia,
Fengmin Wua,
Aiping Liua,
Peigang Lib and
Weihua Tangb
a,
Chaorong Lia,
Fengmin Wua,
Aiping Liua,
Peigang Lib and
Weihua Tangb
aKey Laboratory of Optical Field Manipulation of Zhejiang Province, Center for Optoelectronics Materials and Devices, Department of Physics, Zhejiang Sci-Tech University Hangzhou, 310018, China. E-mail: slwang@zstu.edu.cn
bState Key Laboratory of Information Photonics and Optical Communications, Information Functional Materials and Devices, School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, China
First published on 20th March 2020
Abstract
Ga2O3 nanostructures hold great potential applications in photocatalytic fields due to their stability, high efficiency and environmental friendliness. The construction of phase junction has been proved to be one of the most effective strategies for enhancing Ga2O3 photocatalytic activity. However, the influence of the formation process at the interface of the phase junction on the photocatalytic activity of Ga2O3 nanostructures is far less well understood. In this work, for the first time, large-area Ga2O3 nanorod arrays (NRAs) with controllable α/β phase junction were prepared in situ on a flexible glass fiber fabric by a facile and environmentally friendly three-step method. The α/β-Ga2O3 phase junction NRAs exhibit an ultra-high photocatalytic degradation rate of 97% during Ultraviolet (UV) irradiation for 60 min, which is attributed to a unique phase junction promoting efficient charge separation. However, the photocatalytic activity of α/β-Ga2O3 phase junction NRAs is not evident in the early phase transition, possibly due to the presence of defects acting as charge recombination centers.
1. Introduction
With the continuous development of the social economy, environmental pollution has become an increasingly serious problem, prompting humans to continuously explore new solutions.1–6 Photocatalytic reaction, as a simple, efficient and cost-effective method, has promising applications in the removal of environmental pollutants and attracted wide attention.7–13 Recently, various metal oxides with d10 (In3+, Ga3+, Ge4+, Sn4+) configurations have been reported as effective photocatalysts for photodegradation of various organic pollutants.14,15 Ga2O3 is a typical representative among them.16With a wide bandgap (4.2–4.9 eV) and excellent physical and chemical properties, Ga2O3 is recognized as one of the most promising semiconductors of this century.17–22 It has extensively been applied to power devices,23–25 solar-blind ultraviolet (UV) photodetectors,26–29 gas sensors,30 solar cells31 and photocatalysis.32,33 For photocatalysis applications, related studies claim that Ga2O3 can theoretically exhibit better and more stable photocatalytic activity than commercial TiO2 and realize the degradation of refractory pollutants.34,35 This is attributed to the extraordinary redox capability of photogenerated electron–hole pairs.1,36,37 Furthermore, Ga2O3 is also widely accepted as an environmentally friendly material with low cost and high chemical stability.38 Many methods have been investigated to further improve the photocatalytic activity of Ga2O3, including morphology controlling, doping, surface modification and semiconductor coupling.39–44 Nitu Syed et al. reported a two-step method for the synthesis of porous α-Ga2O3 nanosheets from liquid metal gallium, explaining that the excellent photocatalytic activity of α-Ga2O3 originated from the narrowed bandgap caused by trap states.1 Han et al. revealed that the modification of in situ Ag nanoparticles can effectively improve the photocatalytic property of Ga2O3 for hydrogen evolution.39 Zhang et al. incorporated solvothermally synthesized Ga2O3 nanoparticles into liquid metal/metal oxide frameworks to form enhanced photocatalytic systems.41 Xu et al. fabricated two-dimensional TiO2–Ga2O3 p–n heterostructures, demonstrating the contribution of heterostructures in enhancing photocatalytic activity.45
Furthermore, the construction of appropriate phase junction structure in Ga2O3 can also significantly enhance photocatalytic activity.46–48 Liu et al. demonstrated that the mesopores and heterojunction in the mixed-phase Ga2O3 are responsible for enhancing photocatalytic activity.7 However, the influence of the formation process at the interface of the phase junction on the photocatalytic activity of Ga2O3 nanostructures has not been fully understood. For example, phase transformation is a process from the surface to the bulk, and different thicknesses of phase interface may result in various photocatalytic activities.49 Therefore, an in-depth understanding of junction-related issues will aid in the design and preparation of efficient Ga2O3 nanostructured photocatalysts. On the other hand, almost all of the reported Ga2O3 nanostructured photocatalysts are currently applied in suspension systems. The disadvantages of photocatalysts, such as agglomeration, inadequate illumination and difficulty in recovery, restrict their large-scale practical applications. Glass fiber fabric as a support for in situ growth of Ga2O3 nanostructures is expected to effectively overcome this difficulty.18 To the best of our knowledge, there are no reports of in situ preparation of Ga2O3 nanostructures on glass fiber fabric for the application of photocatalytic degradation.
Herein, for the first time, we reported a facile and environmentally friendly three-step method for in situ preparation of large-area Ga2O3 nanorod arrays (NRAs) with controllable α/β phase junction on a flexible glass fiber fabric. The as-prepared α/β-Ga2O3 phase junction NRAs exhibited excellent photocatalytic activity for the degradation of Rhodamine B (RhB) aqueous solution. In addition, the mechanism of photocatalytic activity enhancement was discussed and compared with related literature.
2. Experimental
2.1. Materials
Glass fiber fabric, model TS-BXB, specification 0.06 MM × 1.20 M, obtained from Hangzhou Gaojing Fine Chemical Industry Co., Ltd. The glass fiber fabrics were cut into a size of approximately 20 × 20 mm2 as the substrate of Ga2O3 NRAs. Rhodamine B (RhB), gallium nitrate hydrate (Ga(NO3)3·nH2O) were purchased from Shanghai Saen Chemical Technology Co., Ltd. Fluorine doped tin oxide (FTO) conductive glass (14 Ω cm−2, size: 10 × 20 × 2.2 mm3) was made by Japan Nippon Sheet Glass Co., Ltd. Sodium sulphate (Na2SO4) was got from Tianjin Yongda Chemical Regent Co., Ltd. All chemicals are analytical grade.2.2. Sample preparation
The preparation of Ga2O3 NRAs involves the following three steps. In the first step, a SnO2 thin film was fabricated by radio frequency magnetron sputtering on the surface of the cleaned glass fiber fabric, which was used as a growth seed layer of Ga2O3. The growth temperature and Ar gas pressure were fixed at 550 °C and 0.8 Pa, respectively. The second step is to prepare GaOOH nanorod precursor by hydrothermal method. Here, 0.20 g of Ga(NO3)3·nH2O was dissolved in 30 mL of DI water to prepare a growth solution. Then the substrates glass fiber fabric completed in the first step was placed in the growth solution and transferred separately into a 50 mL Teflon-lined stainless steel autoclave for hydrothermal treatment at 150 °C for 12 h. After the solution was naturally cooled down to room temperature, the precipitates were filtered and washed with DI water, then dried in air at 80 °C for 2 h to obtain GaOOH NRAs precursors. The last step, the as-prepared precursors were annealed at 400 °C for 4 h in air to obtain α-Ga2O3 NRAs. The detailed synthesis is schematically demonstrated in Fig. 1. Further, other samples were obtained by annealing α-Ga2O3 NRAs in air at 700 °C for different times from 20 min to 120 min.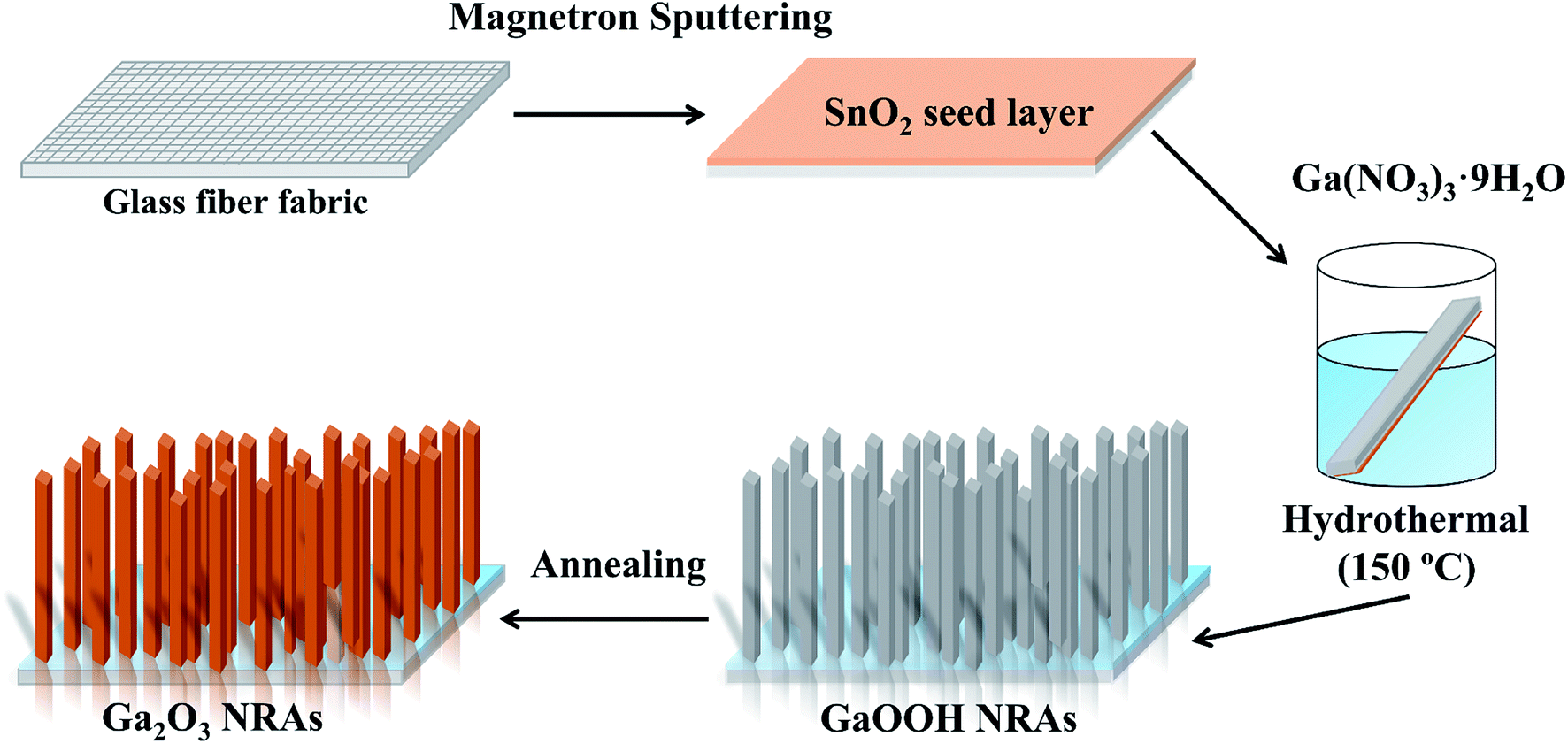 | ||
| Fig. 1 Schematic illustration of the preparation process of α-Ga2O3 NRAs. | ||
2.3. Characterization
The crystal structure of samples was analyzed by a Bruker D8 DISCOVER X-ray diffractometer (XRD). UV-Raman spectra were recorded on a Jobin-Yvon T64000 triple-stage spectrograph with spectral resolution of 2 cm−1. The thermal behavior of the GaOOH nanorod was investigated by thermal gravimetric analyzer (Pyris1 TGA). For the morphological and microstructural analysis, a Hitachi S-4800 field-emission scanning electron microscope (SEM) equipped and a JEOL JEM-2100 transmission electron microscopy (TEM) were utilized. The ultraviolet-visible (UV-vis) absorption spectra were taken using a Hitachi U-3900 UV-vis spectrophotometer. The chemical composition of samples was characterized by a Thermo Scientific K-Alpha X-ray photoelectron spectroscopy (XPS).2.4. Photocatalytic experiments
In this experiment, the glass fiber fabric with Ga2O3 NRAs were dropped into 50 mL of RhB aqueous solution (2 × 10−5 M) and placed in the dark for 30 min to ensure adsorption–desorption equilibrium was reached. Then irradiated reaction solution with a 10 W UV light lamp (λ = 254 nm). The light intensity of the UV lamp was always maintained at 1.0 mW cm−2. During the process, about 3 mL of solution was withdrawn from the reaction system at a given time interval (10 min) for absorbance testing by UV-vis spectrophotometry.2.5. Mott–Schottky measurement
For Mott–Schottky measurements, 5 mg Ga2O3 NRAs powder was scraped from the glass fiber fabric and dispersed in 2 mL of absolute ethanol, followed by the addition of 20 μL of 0.5% Nafion. After the mixed solution was sonicated for 1 h, 0.5 mL was transferred onto a FTO conductive glass. The resulting electrodes were dried in air and further heated at 150 °C for 1 h under a N2 gas flow. The electrochemical measurements were performed in a three-electrode configuration system using a CHI 760E electrochemical workstation (CH Instruments, China), including the as-prepared FTO working electrodes (with an active area of 1.0 cm2), Pt foil as the counter electrode and saturated calomel electrode (SCE) as the reference electrode. 0.5 M Na2SO4 aqueous solution was used as the electrolyte.3. Results and discussion
Fig. 2(a) shows the XRD patterns of as-synthesized GaOOH and α-Ga2O3 NRAs. All the peaks can be indexed to the orthorhombic GaOOH phase (JCPDS no. 06-0180) except the diffraction peak of the SnO2 seed layer. After annealing GaOOH at 400 °C for 4 h, the observed diffraction peaks occur at new locations, indicating that the GaOOH is completely converted into α-Ga2O3 of the corundum structure (JCPDS no. 06-0503).50 The phase transition of α-Ga2O3 at 700 °C for various times was also analyzed by XRD, and the corresponding results are shown in Fig. 2(b). With α-Ga2O3 annealed at 700 °C for 30 min, a diffraction peak corresponding to the (111) plane attributed to monoclinic β-Ga2O3 is detected, and it becomes stronger with the further increase of annealing time. Ga2O3 with different phase structures can be obtained during annealing for 30–90 min. No diffraction peak assigned to α-Ga2O3 is observed through annealing for 120 min, suggesting that the α-Ga2O3 is totally transformed into β-Ga2O3 at this point.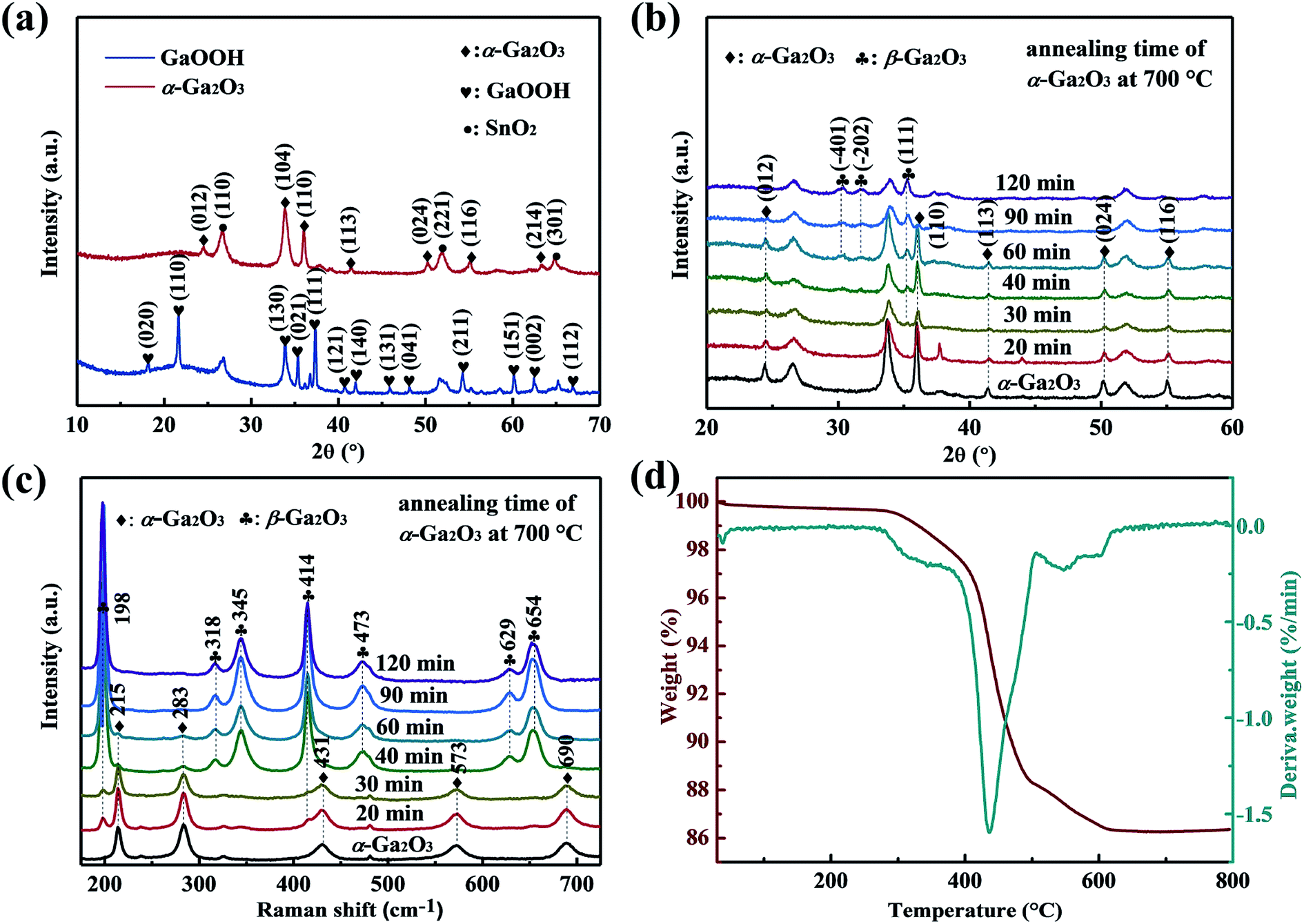 | ||
| Fig. 2 (a) XRD patterns of as-synthesized GaOOH and α-Ga2O3 NRAs. (b) XRD patterns and (c) UV Raman spectra of α-Ga2O3 NRAs annealed in air at 700 °C for various times. (d) TG/DTG curve of the as-prepared GaOOH NRAs precursor by heating from room temperature to 800 °C in air. | ||
UV Raman spectroscopy was also used to monitor the α to β phase transformation of Ga2O3. As shown in Fig. 2(c), it is worth noting that the typical characteristic Raman bands of β-Ga2O3 at 198 cm−1 and 414 cm−1 can be clearly observed after annealing α-Ga2O3 at 700 °C for 20 min, in addition to the existing Raman bands of α-Ga2O3, indicating the formation of β-Ga2O3. However, only Raman bands at 215 cm−1 and 283 cm−1 attributed to α-Ga2O3 are detected with annealing time to 60 min, and both disappeared at 90 min. The result is unsynchronized with that revealed by the XRD patterns, which can be attributed to the strong sensitivity of UV Raman spectroscopy to the surface region, and XRD mainly reflects the bulk information of materials.47 Based on the above results, we suggest that the samples annealed for 20–90 min are α/β-Ga2O3 phase junction. In the following sections, the α-Ga2O3 NRAs annealed at 700 °C for various time will be labeled as Ga2O3-20 (annealed for 20 min), Ga2O3-30 (annealed for 30 min), Ga2O3-40 (annealed for 40 min), Ga2O3-60 (annealed for 60 min), Ga2O3-90 (annealed for 90 min) and β-Ga2O3 NRAs (annealed for 120 min), respectively.
The TG/DTG curve of the as-prepared GaOOH NRAs precursor by heating from room temperature to 800 °C in air atmosphere is shown in Fig. 2(d). A major weight loss of 11.2% can be noticed in the temperature range of approximately 260–480 °C, with the fastest weight loss rate occurring at 438 °C, which is attributed to transformation of GaOOH into α-Ga2O3 by thermal dehydration. A weak weight loss of 2% is also noted at the range of 500–630 °C, indicating the conversion of α-Ga2O3 to β-Ga2O3. With further prolonged heating up to 800 °C, there is no weight loss.
A typical SEM image of as-synthesized α-Ga2O3 NRAs, as presented in Fig. 3(a), which reveals the uniform and dense growth of the sample on each fiber rod. High-magnification SEM images of α-Ga2O3 NRAs and other Ga2O3 NRAs obtained by annealing (Ga2O3-60 and β-Ga2O3) are also shown in Fig. 3(b–d), respectively. Further revealing that the diameter of all nanorods ranged from 100 to 400 nm and the tips are all diamond-shaped. The annealing process has not significantly changed the morphology of Ga2O3 NRAs.51 Fig. 3(e and f) shows the low and high resolution TEM images of the Ga2O3-60 NRAs. Different lattice fringes are observed in here. The lattice-spacing value of 0.249 nm matches the (110) planes of α-Ga2O3, while the lattice-spacing values of 0.242 nm and 0.255 nm are ascribed to the (−310) and (111) planes of β-Ga2O3. This result clearly demonstrates the formation of α/β heterophase junctions in Ga2O3-60 NRAs, supporting the previous analysis of XRD and UV Raman spectroscopy.
 | ||
| Fig. 3 (a) SEM images of α-Ga2O3 NRAs. High-magnification SEM images of (b) α-Ga2O3 NRAs, (c) Ga2O3-60 NRAs, (d) β-Ga2O3 NRAs. (e) Low and high resolution TEM images of (f). | ||
The photocatalytic activities of Ga2O3 NRAs were evaluated by the degradation of RhB aqueous solution under UV light irradiation. A typical UV-vis absorption spectrum of RhB aqueous solution during photocatalytic degradation process in the presence of the Ga2O3 NRAs is shown in Fig. S1.† The decrease of the characteristic peak at 554 nm during illumination suggests RhB decomposition.1,43 Fig. 4(a) and S2† reveals the comparison of photocatalytic activities of different Ga2O3 NRAs. Among them, the Ga2O3-60 NRAs exhibits the best photocatalytic with a degradation rate of 97%, which can be attributed to the α/β-Ga2O3 phase junction promoting the separation of photogenerated electrons and holes.47 Furthermore, the photocatalytic degradation process of these Ga2O3 NRAs were fitted using the first-order kinetic curve according to the Langmuir–Hinshelwood model.32,52 As shown in Fig. 4(b), the value of the reaction rate constant (K) are estimated to be 0.0232, 0.0301, 0.0265, 0.0245, 0.0589, 0.0546 and 0.0418 min−1, corresponding to the α-Ga2O3, Ga2O3-20, Ga2O3-30, Ga2O3-40, Ga2O3-60, Ga2O3-90 and β-Ga2O3 NRAs, respectively.
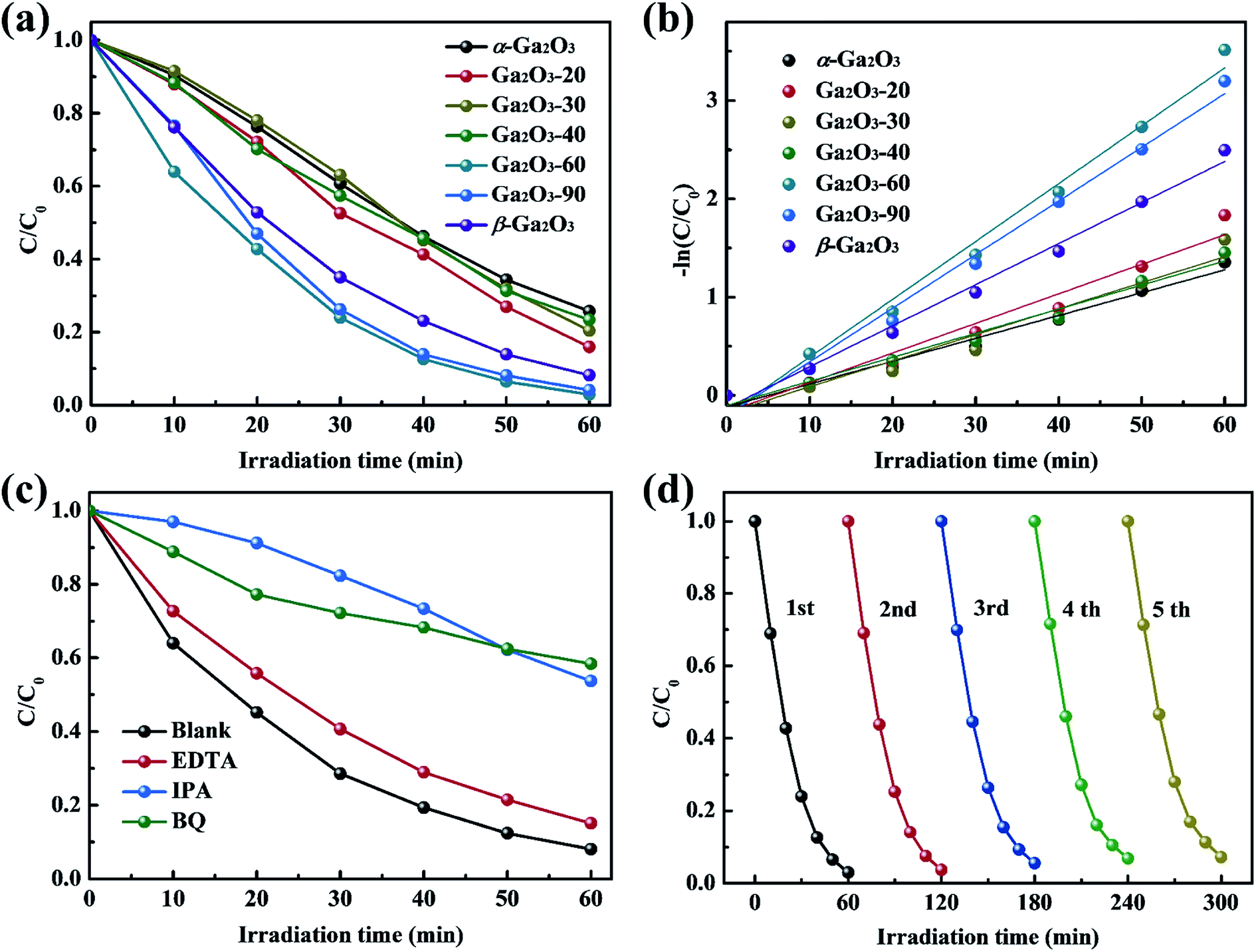 | ||
| Fig. 4 (a) Photocatalytic degradation of RhB in the presence of different Ga2O3 NRAs and (b) corresponding kinetic linear simulation curves. (c) Effect of trapping agents on the photocatalytic degradation of RhB over the Ga2O3-60 NRAs. (d) Photocatalytic stability test of the Ga2O3-60 NRAs in recycling reactions. | ||
Generally, a series of photogenerated reactive species, such as h+, ˙O2− and ˙OH, are involved in the photocatalytic process.4,53 To reveal the main reactive species responsible for the degradation of RhB solution by the Ga2O3-60 NRAs, radical trapping experiments were performed by adding EDTA (ethylenediamine tetraacetic acid, h+ trapping agents), IPA (isopropyl alcohol, ˙OH trapping agents) and BQ (benzoquinone, ˙O2− trapping agents), respectively. As shown in Fig. 4(c), the photocatalytic activity of the Ga2O3-60 NRAs is affected slightly with the addition of EDTA, indicating that h+ is not the main factor in this system. In contrast, the introduction of IPA or BQ greatly suppressed the photocatalytic activity of the Ga2O3-60 NRAs, indicating that ˙OH and ˙O2− acted as dominating reactive species in the reaction system.
Moreover, the cycling stability of the Ga2O3-60 NRAs was evaluated by conducting five consecutive cycle degradation experiments. As shown in Fig. 4(d), the degradation ratio of RhB is not obviously reduced during the repeated experiments, indicating the remarkable stability of the Ga2O3-60 NRAs. XRD patterns (Fig. S3†) also indicates that no structural difference can be observed between the Ga2O3-60 NRAs before and after photocatalytic degradation of RhB solution.
For the proposed photocatalytic degradation mechanism of the system, the band structures of α-Ga2O3 and β-Ga2O3 were characterized by Mott–Schottky measurements and XPS. As shown in Fig. 5(a and b), the flat band potential of α-Ga2O3 is calculated to be −1.26 eV (vs. SCE), which is more negative than the −0.96 eV (vs. SCE) of β-Ga2O3, and the valence band potential of β-Ga2O3 is 3.05 eV, which is more positive than the 2.92 eV of α-Ga2O3. Further combined with the band gap of Ga2O3 reported in our previous work,51 a schematic illustration of photocatalytic reaction process and charge separation transfer of α/β-Ga2O3 phase junction under UV light irradiation is shown in Fig. 5(c). Under UV light irradiation, the internal electric field of the α/β-Ga2O3 phase junction could drive the photogenerated charge transfer, promoting the photogenerated electrons transfer from the α phase to the β phase, and the photogenerated holes transfer from the β phase to the α phase. Following that, the photogenerated electrons react with O2 to generate ˙O2− and the photogenerated holes oxidize OH− to ˙OH, which together involve in RhB degradation. Efficient charge separation inhibits their recombination, resulting in improved photocatalytic degradation performance.47,49 In addition, it should be mentioned that phase junction can form on both the surface of Ga2O3 and in the bulk. Although almost no phase junctions were observed on the surface of the Ga2O3-60 NRAs, they still function as charge separation centers in the bulk. The separated carriers eventually diffuse to the surface of the sample to participate in the photocatalytic reaction.4 On the other hand, for Ga2O3 as a photocatalytic degradation material, the performance of the β phase is generally better than that of the α phase,7,43 which is confirmed in Fig. 4(a) of this research. Therefore, in addition to the efficient charge separation due to the phase junction in the bulk, the excellent photocatalytic activity of the Ga2O3-60 NRAs is also derived from the inherently high activity of the β surface phase. Interestingly, the Ga2O3-20, Ga2O3-30 and Ga2O3-40 NRAs did not exhibit excellent photocatalytic activity despite the formation of phase junctions on various surfaces. The phase transformation of Ga2O3 is a surface-preferred process, which is accompanied by the formation of defects.54 These defects may become the recombination center of photogenerated electron–hole pairs, reducing the number of efficient carriers on the surface.55 As a result, the photocatalytic activity of the initially annealed Ga2O3 phase junction NRAs was not satisfactory.
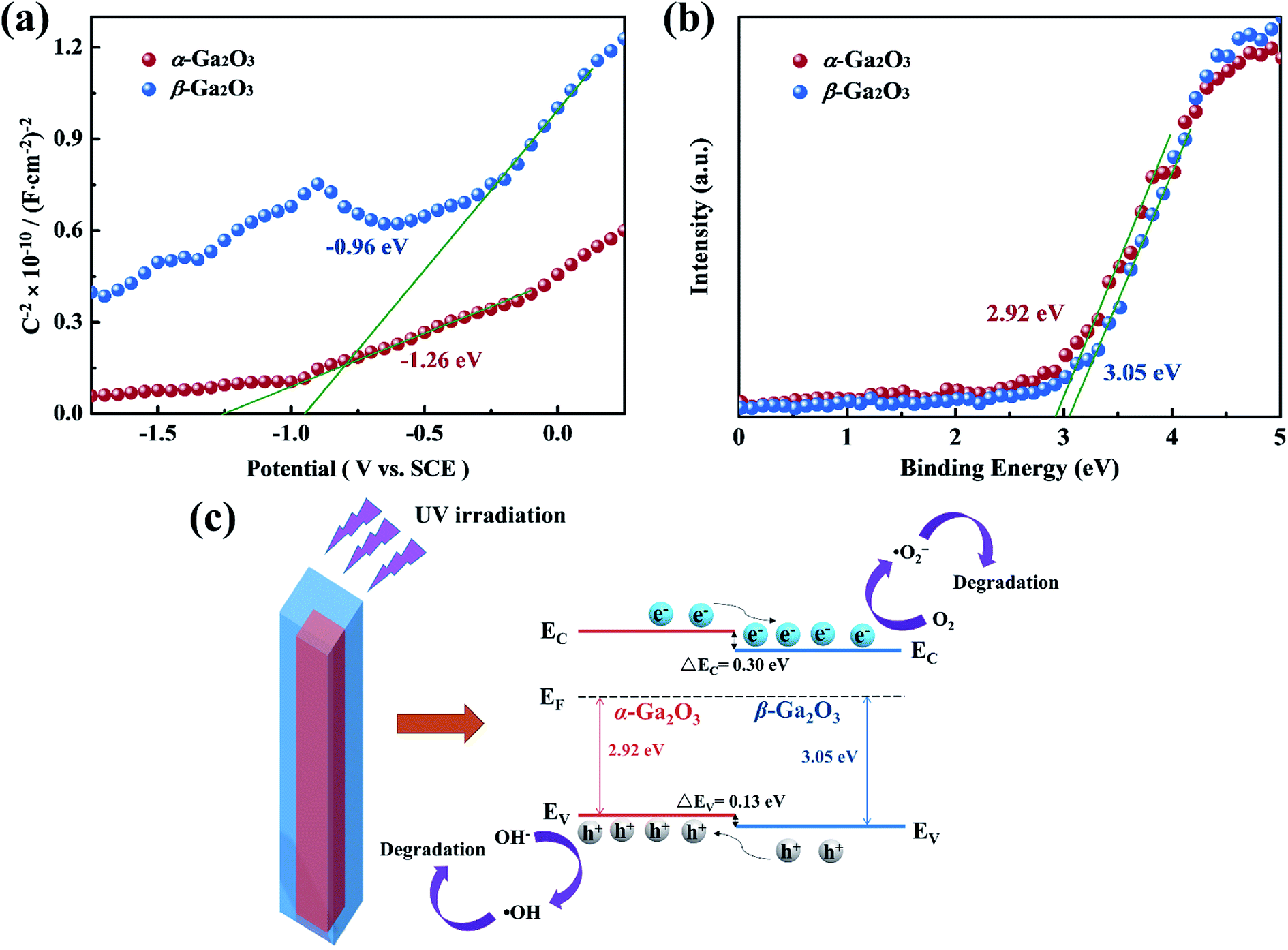 | ||
| Fig. 5 (a) Mott–Schottky curves of α-Ga2O3 NRAs and β-Ga2O3 NRAs electrodes measured in 0.5 M Na2SO4 solution. (b) XPS valence band spectra of α-Ga2O3 NRAs and β-Ga2O3 NRAs. (c) Schematic illustration of photocatalytic reaction process and charge separation transfer of α/β-Ga2O3 phase junction under UV light irradiation. | ||
To obtain more insight into the effect of defects on photocatalytic activity, the Ga2O3-20 NRAs and Ga2O3-60 NRAs were selected as typical samples for XPS analysis. As shown in Fig. 6, the O 1s spectra could be divided into two peaks: I and II, representing lattice oxygen ions and oxygen ions in the oxygen vacancies region, respectively.28 The peak ratio (II/I) of the Ga2O3-20 NRAs is 1/3, which is higher than that of the Ga2O3-60 NRAs (1/5), indicating the presence of more oxygen vacancies. Obviously, the Ga2O3-20 NRAs exhibits poor photocatalytic activity due to the existence of abundant oxygen vacancy defects.
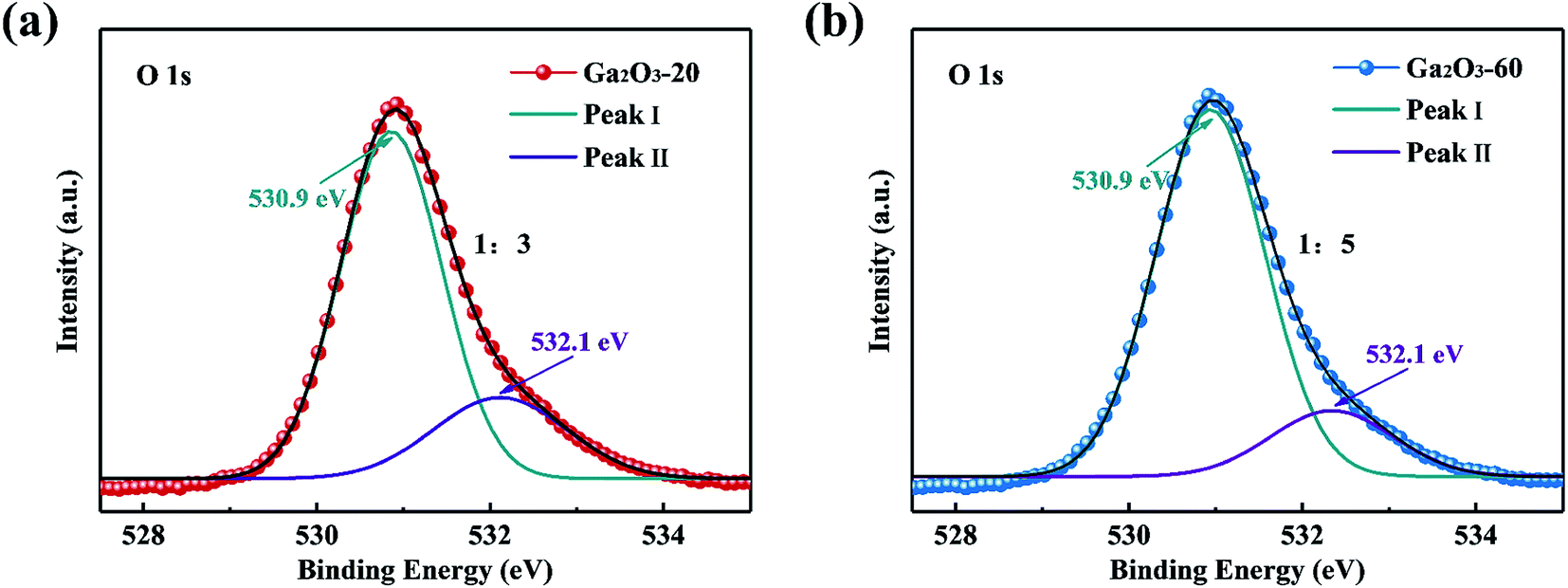 | ||
| Fig. 6 O 1s XPS spectra of (a) Ga2O3-20 NRAs and (b) Ga2O3-60 NRAs. | ||
The comparison of the photocatalytic degradation activity of α/β-Ga2O3 phase junction NRAs in this work and other previously reported Ga2O3 related materials is listed in Table 1. Although the comparison of photocatalytic activity is not absolutely reasonable due to the different light source conditions and pollutants used in each experiment, the photocatalytic activity of α/β-Ga2O3 phase junction NRAs in this work is significantly superior to almost all previous reports on Ga2O3 related materials. This method has realized the large-area growth of α/β-Ga2O3 phase junction NRAs on the flexible glass fiber fabric and obviously improved its photocatalytic performance, which is of great significance in the future research in the field of photocatalysis.
| Photocatalyst, concentration (mg L−1) | Pollutants, concentration (mol L−1) | Light source | Degradation after 60 min | Reference |
|---|---|---|---|---|
| α-Ga2O3 nanoplates, 90 | RhB, 0.45 × 10−5 | AM 1.5 solar simulator | 53% | 1 |
| α-Ga2O3 nanoparticles, 400 | TC, 5.6 × 10−5 | 30 W UV lamp | 85% | 5 |
| α-Ga2O3 nanorods, 1000 | RhB, 0.84 × 10−5 | 300 W Hg lamp | 62% | 33 |
| β-Ga2O3 nanorods, 1000 | RhB, 2 × 10−5 | 150 W xenon lamp | 39% | 36 |
| β-Ga2O3 microspheres, 1000 | RhB, 2 × 10−5 | 150 W xenon lamp | 60% | 37 |
| Ga2O3 sheet, 500 | CR, 2.15 × 10−5 | 30 W UV lamp | 33% | 38 |
| β-Ga2O3 nanorods, 1000 | RhB, 2 × 10−4 | 1000 W UV lamp | 38% | 43 |
| TiO2–Ga2O3 heterojunctions | MO, 1.8 × 10−5 | 30 W UV lamp | 83% | 45 |
| α/β-Ga2O3 NRAs, 200 | RhB, 2 × 10−5 | 10 W UV lamp | 97% | This work |
4. Conclusions
In summary, large-area Ga2O3 NRAs with controllable α/β phase junction were firstly prepared in situ on a flexible glass fiber fabric by a facile and environmentally friendly three-step method. Photocatalytic degradation experiments showed that the α/β-Ga2O3 phase junction NRAs synthesized by annealing α-Ga2O3 NRAs at 700 °C for 60 min exhibited remarkable performance for RhB, with a degradation rate of 97% in 60 min under UV light. The enhanced photocatalytic activity can be attributed to the unique phase junction promoting efficient charge separation and inhibiting the recombination of photogenerated electron–hole pairs. Additionally, the glass fiber fabric can realize large-area growth of the Ga2O3 NRAs, effectively solve the trouble of difficult recovery and reuse of photocatalysts, as well as the insufficient absorption of light. A facile environmentally friendly and inexpensive synthesis route will open new avenues for the development of efficient photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 61704153, 51572241, 61774019, 51572033), Natural Science Foundation of Zhejiang Province (No. LY20F040005), Zhejiang Public Service Technology Research Program/Analytical Test (LGC19F040001), and Fundamental Research Funds of Zhejiang Sci-Tech University (2019Q061, 2019Q067).References
- N. Syed, A. Zavabeti, M. Mohiuddin, B. Zhang, Y. Wang, R. S. Datta, P. Atkin, B. J. Carey, C. Tan, J. van Embden, A. S. R. Chesman, J. Z. Ou, T. Daeneke and K. Kalantar-zadeh, Adv. Funct. Mater., 2017, 27, 1702295 CrossRef.
- J. Wei, F. Guo, X. Wang, K. Xu, M. Lei, Y. Liang, Y. Zhao and D. Xu, Adv. Mater., 2018, 30, 1805153 CrossRef PubMed.
- W. Zhong, S. Shen, S. Feng, Z. Lin, Z. Wang and B. Fang, CrystEngComm, 2018, 20, 7851–7856 RSC.
- Y. Wang, W. Zhang, Z. Wang, Y. Cao, J. Feng, Z. Wang and Y. Ma, Chin. J. Catal., 2018, 39, 1500–1510 CrossRef CAS.
- J. Liu, W. Lu, Q. Zhong, H. Wu, Y. Li, L. Li and Z. Wang, J. Colloid Interface Sci., 2018, 519, 255–262 CrossRef CAS PubMed.
- X. He, S. Z. Luan, L. Wang, R. Y. Wang, P. Du, Y. Y. Xu, H. J. Yang, Y. G. Wang, K. Huang and M. Lei, Mater. Lett., 2019, 244, 78–82 CrossRef CAS.
- J. Liu and G. Zhang, Mater. Res. Bull., 2015, 68, 254–259 CrossRef CAS.
- L. C. Tien, W. T. Chen and C. H. Ho, J. Am. Ceram. Soc., 2011, 94, 3117–3122 CrossRef CAS.
- Z. Dong, J. Pan, B. Wang, Z. Jiang, C. Zhao, J. Wang, C. Song, Y. Zheng, C. Cui and C. Li, J. Alloys Compd., 2018, 747, 788–795 CrossRef CAS.
- W. Zhong, Y. Lou, S. Jin, W. Wang and L. Guo, Sci. Rep., 2016, 6, 23235 CrossRef CAS PubMed.
- K. Huang, J. Liu, L. Wang, G. Chang, R. Wang, M. Lei, Y. Wang and Y. He, Appl. Surf. Sci., 2019, 487, 1145–1151 CrossRef CAS.
- W. Zhong, J. Huang, S. Liang, J. Liu, Y. Li, G. Cai, Y. Jiang and J. Liu, ACS Energy Lett., 2019, 5, 31–38 CrossRef.
- S. Lin, X. Bai, H. Wang, H. Wang, J. Song, K. Huang, C. Wang, N. Wang, B. Li and M. Lei, Adv. Mater., 2017, 29, 1703238 CrossRef PubMed.
- A. A. Ismail, I. Abdelfattah, M. Faisal and A. Helal, J. Hazard. Mater., 2018, 342, 519–526 CrossRef CAS PubMed.
- W. Zhong, S. Shen, M. He, D. Wang, Z. Wang, Z. Lin, W. Tu and J. Yu, Appl. Catal., B, 2019, 258, 117967 CrossRef CAS.
- Y. Ma, X. Zhao, M. Niu, W. Li, X. Wang, C. Zhai, T. Wang, Y. Tang and X. Dai, RSC Adv., 2017, 7, 4124–4134 RSC.
- A. Kakoria, B. Devi, A. Anand, A. Halder, R. R. Koner and S. Sinha-Ray, ACS Appl. Nano Mater., 2018, 2, 64–74 CrossRef.
- S. Wang, H. Sun, Z. Wang, X. Zeng, G. Ungar, D. Guo, J. Shen, P. Li, A. Liu, C. Li and W. Tang, J. Alloys Compd., 2019, 787, 133–139 CrossRef CAS.
- J. Yu, Z. Nie, L. Dong, L. Yuan, D. Li, Y. Huang, L. Zhang, Y. Zhang and R. Jia, J. Alloys Compd., 2019, 798, 458–466 CrossRef CAS.
- J. Zhang, S. Jiao, Y. Wan, S. Gao, D. Wang and J. Wang, CrystEngComm, 2018, 20, 4329–4335 RSC.
- H. Lu, S. Jiao, Y. Nie, S. Liu, S. Gao, D. Wang, J. Wang, L. Li and X. Wang, J. Alloys Compd., 2020, 823, 153903 CrossRef CAS.
- C. Wu, D. Guo, L. Zhang, P. Li, F. Zhang, C. Tan, S. Wang, A. Liu, F. Wu and W. Tang, Appl. Phys. Lett., 2020, 116, 072102 CrossRef.
- M. Baldini, Z. Galazka and G. Wagner, Mater. Sci. Semicond. Process., 2018, 78, 132–146 CrossRef CAS.
- X. Wang, Y. Cui, T. Li, M. Lei, J. Li and Z. Wei, Adv. Opt. Mater., 2018, 7, 1801274 CrossRef.
- H. Zhang, L. Yuan, R. Jia, X. Tang, J. Hu, Y. Zhang, Y. Zhang and J. Sun, J. Phys. D: Appl. Phys., 2019, 52, 215104 CrossRef CAS.
- D. Guo, Y. Su, H. Shi, P. Li, N. Zhao, J. Ye, S. Wang, A. Liu, Z. Chen, C. Li and W. Tang, ACS Nano, 2018, 12, 12827–12835 CrossRef CAS PubMed.
- L. Dong, J. Yu, R. Jia, J. Hu, Y. Zhang and J. Sun, Opt. Mater. Express, 2019, 9, 1191 CrossRef CAS.
- D. Guo, H. Liu, P. Li, Z. Wu, S. Wang, C. Cui, C. Li and W. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 1619–1628 CrossRef CAS PubMed.
- J. Zhang, S. Jiao, D. Wang, S. Ni, S. Gao and J. Wang, J. Mater. Chem. C, 2019, 7, 6867–6871 RSC.
- H. J. Lin, H. Gao and P. X. Gao, Appl. Phys. Lett., 2017, 110, 043101 CrossRef.
- A. Pérez-Tomás, E. Chikoidze, Y. Dumont, M. R. Jennings, S. O. Russell, P. Vales-Castro, G. Catalan, M. Lira-Cantú, C. Ton-That, F. H. Teherani, V. E. Sandana, P. Bove and D. J. Rogers, Mater. Today Energy, 2019, 14, 100350 CrossRef.
- S. Jin, W. Lu, P. C. Stanish and P. V. Radovanovic, Chem. Phys. Lett., 2018, 706, 509–514 CrossRef CAS.
- D. Li, X. Duan, Q. Qin, H. Fan and W. Zheng, J. Mater. Chem. A, 2013, 1, 12417 RSC.
- X. Zhang, H. Huang, Y. Zhang, D. Liu, N. Tong, J. Lin, L. Chen, Z. Zhang and X. Wang, ACS Omega, 2018, 3, 14469–14476 CrossRef CAS PubMed.
- H. Liu, Z. Wang, H. Li, X. Zhang, X. Qin, Y. Dai, P. Wang, Y. Liu and B. Huang, RSC Adv., 2018, 8, 14328–14334 RSC.
- K. Girija, S. Thirumalairajan, A. K. Patra, D. Mangalaraj, N. Ponpandian and C. Viswanathan, Semicond. Sci. Technol., 2013, 28, 035015 CrossRef.
- K. Girija, S. Thirumalairajan, V. R. Mastelaro and D. Mangalaraj, J. Mater. Chem. A, 2015, 3, 2617–2627 RSC.
- M. Bagheri and A. R. Mahjoub, RSC Adv., 2016, 6, 87555–87563 RSC.
- C. Han, W. Mao, K. Bao, H. Xie, Z. Jia and L. Ye, Int. J. Hydrogen Energy, 2017, 42, 19913–19919 CrossRef CAS.
- S. Hong, C. K. Rhee and Y. Sohn, J. Alloys Compd., 2019, 774, 11–17 CrossRef CAS.
- W. Zhang, B. S. Naidu, J. Z. Ou, A. P. O'Mullane, A. F. Chrimes, B. J. Carey, Y. Wang, S. Y. Tang, V. Sivan, A. Mitchell, S. K. Bhargava and K. Kalantar-Zadeh, ACS Appl. Mater. Interfaces, 2015, 7, 1943–1948 CrossRef CAS PubMed.
- S. Kikkawa, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2018, 122, 21132–21139 CrossRef CAS.
- L. S. Reddy, Y. H. Ko and J. S. Yu, Nanoscale Res. Lett., 2015, 10, 364 CrossRef PubMed.
- S. Luan, L. Dong, X. Ma and R. Jia, J. Alloys Compd., 2020, 812, 152026 CrossRef CAS.
- H. Xu, F. Han, C. Xia, S. Wang, R. K. Ramachandran, C. Detavernier, M. Wei, L. Lin and S. Zhuiykov, Nanoscale Res. Lett., 2019, 14, 163 CrossRef PubMed.
- X. Wang and C. Li, J. Phys. Chem. C, 2018, 122, 21083–21096 CrossRef CAS.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089–13092 CrossRef CAS PubMed.
- D. Guo, K. Chen, S. Wang, F. Wu, A. Liu, C. Li, P. Li, C. Tan and W. Tang, Phys. Rev. Appl., 2020, 13, 24051 CrossRef.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- C. He, D. Guo, K. Chen, S. Wang, J. Shen, N. Zhao, A. Liu, Y. Zheng, P. Li, Z. Wu, C. Li, F. Wu and W. Tang, ACS Appl. Nano Mater., 2019, 2, 4095–4103 CrossRef CAS.
- K. Chen, S. Wang, C. He, H. Zhu, H. Zhao, D. Guo, Z. Chen, J. Shen, P. Li, A. Liu, C. Li, F. Wu and W. Tang, ACS Appl. Nano Mater., 2019, 2, 6169–6177 CrossRef CAS.
- B. Das, B. Das, N. S. Das, S. Sarkar and K. K. Chattopadhyay, Microporous Mesoporous Mater., 2019, 288, 109600 CrossRef CAS.
- S. Wang, D. Li, C. Sun, S. Yang, Y. Guan and H. He, Appl. Catal., B, 2014, 144, 885–892 CrossRef CAS.
- V. Ghodsi, S. Jin, J. C. Byers, Y. Pan and P. V. Radovanovic, J. Phys. Chem. C, 2017, 121, 9433–9441 CrossRef CAS.
- D. Y. Guo, Z. P. Wu, Y. H. An, X. C. Guo, X. L. Chu, C. L. Sun, L. H. Li, P. G. Li and W. H. Tang, Appl. Phys. Lett., 2014, 105, 023507 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01461c |
| This journal is © The Royal Society of Chemistry 2020 |