
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorphism of Au11(PR3)7Cl3 clusters: understanding C–H⋯π interaction and C–H⋯Cl–C van der Waals interaction on cluster assembly by surface modification†
Chenwanli Qinab,
Qianqin Yuanab,
Peng Li *ab,
Shuxin Wangab,
Shuang Chen*abc and
Manzhou Zhu*ab
*ab,
Shuxin Wangab,
Shuang Chen*abc and
Manzhou Zhu*ab
aDepartment of Chemistry and Centre for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, P. R. China. E-mail: peng-li@ahu.edu.cn; chenshuang@ahu.edu.cn; zmz@ahu.edu.cn
bKey Laboratory of Structure and Functional Regulation of Hybrid Materials, Anhui University, Ministry of Education, Hefei, 230601, P. R. China
cInstitutes of Physical Science and Information Technology, Anhui University, Hefei, Anhui 230601, P. R. China
First published on 20th March 2020
Abstract
The C–H⋯π interaction and the C–H⋯Cl–C van der Waals interaction play a crucial role in the crystallization of nanoclusters. In this paper, we present an example of a crystal system transformation of Au11(PR3)7Cl3 from monoclinic (M) to trigonal (T) by surface modification. Atomically-resolved gold nanoclusters containing tris(4-chlorophenyl)phosphine and chloride ligands were synthesized and determined to be Au11(p-ClPPh3)7Cl3 (p-ClPPh3 = tris(4-chlorophenyl)phosphine) by X-ray crystallography. Crystal data demonstrated that the C–H⋯Cl–C interaction is dominant in a trigonal crystal system of Au11(p-ClPPh3)7Cl3 with a R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. However, the C–H⋯π interaction is the major driving force to form a monoclinic crystal system of Au11(PPh3)7Cl3 (PPh3 = triphenylphosphine) with a P2(1)/n space group. Moreover, UV-vis absorption spectra and X-ray photoelectron spectra reveal that the electronic structure of the Au11(p-ClPPh3)7Cl3 nanocluster is greatly influenced by p-ClPPh3. This work provides critical implications for the crystallization of metal nanoclusters, as well as a better understanding of the non-covalent interaction on the nanocluster assembly and the crystal engineering by surface modification.
space group. However, the C–H⋯π interaction is the major driving force to form a monoclinic crystal system of Au11(PPh3)7Cl3 (PPh3 = triphenylphosphine) with a P2(1)/n space group. Moreover, UV-vis absorption spectra and X-ray photoelectron spectra reveal that the electronic structure of the Au11(p-ClPPh3)7Cl3 nanocluster is greatly influenced by p-ClPPh3. This work provides critical implications for the crystallization of metal nanoclusters, as well as a better understanding of the non-covalent interaction on the nanocluster assembly and the crystal engineering by surface modification.
Introduction
Atomically precise noble metal nanoclusters have received widespread attention due to their peculiar applications, such as catalysis, energy conversion, and biomedicine.1–6 Recently, experimental and theoretical studies of metal nanoclusters have witnessed tremendous progress. More importantly, the structure of these nanoclusters can be identified by single-crystal X-ray diffraction (SC-XRD).7–10 Based on this advantage, it is possible to probe the structure details and the structure-related properties at the atomic level, which is highly desirable to further practical applications.11–13Ligands play a crucial role in the stabilization of nanoclusters.14–17 The non-covalent interactions between surface ligands not only alter properties of clusters but also have significant effects on nanocluster assembly and self-assembly.18–30 For instance, Wu and co-workers demonstrated that C–H⋯π interactions facilitated the chiral arrangement of ligands, and the assembled ligands presented rotational and parallel patterns on the surface of the nanoparticles.14 Pradeep and colleagues reported that C–H⋯π interactions are dominant in a cubic lattice and result in a higher luminescence efficiency.16 Crudden et al. showed that multiple C–H⋯π and π⋯π interactions rigidify the ligands and contribute to the high photoluminescent quantum yields in the Au13 nanocluster.31 Zheng et al. reported that π⋯π interactions play a crucial role in stabilizing silver nanoclusters.32 Xie et al. found that the assembly behaviour of clusters can be regulated by metal ions (i.e., Ag+/Cs+).33 Non-covalent interactions play an important role in nanocluster chemistry.34,35 To date, a series of Au11(PR3)Cl3 nanoclusters with adjustable ligands have been extensively studied comparing with other nanoclusters.15,36–39 Therefore, Au11(PR3)Cl3 clusters are chosen as models to investigate non-covalent interactions in the crystallization of nanoclusters by surface modification.
In the following, we synthesized Au11(p-ClPPh3)7Cl3 (Au11-Cl for short) and Au11(PPh3)7Cl3 (Au11-H for short) nanoclusters by using tris(4-chlorophenyl)phosphine and triphenylphosphine as the protecting ligand. The composition of the Au11-Cl was characterized by electron spin ionization (ESI-MS), and the structure of the Au11-Cl was determined by X-ray crystallography. We are engaged in illuminating the crystal system transformation ascribed to C–H⋯Cl–C interactions and C–H⋯π interactions. The different patterns of non-covalent interactions can alter the packing of nanoclusters into monoclinic or trigonal lattice. The distance between H and H atom (DHH for short) reveals the non-covalent interactions of the intra-molecular can also be changed through surface modification. Furthermore, UV-vis absorption spectra and X-ray photoelectron spectra reveal the difference in the electronic structure due to surface modification.
Experimental
Chemicals
All chemicals are commercial and were used without further purification. Hydrogen tetrachloroaurate tetrahydrate (HAuCl4·4H2O, 99.95%), triphenylphosphine (PPh3, 98%), tris(4-chlorophenyl)phosphine (p-ClPPh3, 98%), sodium borohydride (NaBH4, 99.99%), methanol (HPLC grade, 99.9%) methylene chloride (HPLC grade, 99.9%), hexane (HPLC grade, 99.9%), ethanol (HPLC grade, 99.9%). Ultrapure water was purchased from Wahaha Co. Ltd. All glassware was cleaned with aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of hydrochloric acid and nitric acid), rinsed with ultrapure water, and then dried before use.
:1 mix of hydrochloric acid and nitric acid), rinsed with ultrapure water, and then dried before use.
Synthesis of Au11(p-ClPPh3)7Cl3 (Au11-Cl) and crystallization
First, HAuCl4·4H2O (157 mg, 0.4 mmol) was dissolved in 15 mL ethanol, tris(4-chlorophenyl)phosphine (0.15 g, 0.5 mmol) was dissolved in the solution. The solution was stirred (∼1200 rpm) in a 50 mL round bottom flask. After stirring for 20 min, the solution colour turned turbid. Then, 5 mL ethanol solution of NaBH4 (40 mg) was added, and the colour of solution changed from turbid to dark immediately. After stirring for 8 h, the organic solution was evaporated and washed several times with hexane to remove the redundant phosphine and other by-products. Finally, The Au11-Cl nanocluster was crystallized in vapour diffusion of hexane/CH2Cl2 at room temperature.Synthesis of Au11(PPh3)7Cl3 (Au11-H)
The Au11(PPh3)7Cl3 was synthesized according to the method in the literature by Hutchison et al.36 First, AuCl(PPh3) (500 mg) was dissolved in 15 mL ethanol solution and NaBH4 (190 mg, 5 mmol dissolved) was slowly added to the solution. After stirring for 8 h, the organic solution was evaporated and washed several times with hexane to remove the excess PPh3 and other by-products. The pure Au11-H was red solid and crystallized in vapour diffusion of hexane/CH2Cl2 at room temperature.Characterization
UV-vis spectra of nanoclusters in CH2Cl2 were performed on the UV-6000PC instrument.Red-brown crystals were collected, and the structure of Au11-Cl was determined by X-ray crystallography. Single crystal X-ray diffraction data of the Au11-Cl was collected on a Bruker Smart 1000 CCD. The area detector used graphite-monochromatized Mo Kα radiation (λ = 0.71069 Å). A piece of red-brown block-shaped crystal (0.25 × 0.24 × 0.23 mm) was mounted onto a MiTeGen capillary with Fluorolube, and all structures were solved by direct methods using SHELXS-97. Data collection was performed under room temperature (296 K).
The electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) measurement was recorded using UPLC H-Class XEVO G2 XS Qtof (Waters. Crop.). The Au11-Cl was recorded in the positive ion mode when dissolving in CH3OH, containing 50 mM CsOAc, and centrifugation for 5 min (12000 rpm). The sample was infused at 180 μL h−1. The source temperature was kept at 50 °C with the spray voltage keeping at 4 kV. The mass spectra were processed using Masslynx 4.1 software (Waters Corp.).
The Thermo ESCALAB 250 instrument was used to measure X-ray photoelectron spectroscopy (XPS), which configured with a monochromated Al Kα (1486.8 eV) 150 W X-ray source, 0.5 mm circular spot size, and the analysis chamber base pressure lower than 1 × 10−9 mbar. Data were collected with FAT = 20 eV.
Results and discussions
As shown in Fig. 1a, the ultraviolet-visible (UV-vis) absorption spectrum of Au11-H (black) shows two prominent peaks at 325 nm and 416 nm. While the UV-vis absorption spectrum of the Au11-Cl (red) shows three peaks at 325 nm, 390 nm and 426 nm, respectively. The differences of absorption spectra possibly indicate that the electronic structure is distinctive, resulting from the replacement of PPh3 by p-ClPPh3. More, the thermodynamic stability of the Au11 was explored under room temperature (Fig. S1†). These results imply the Au11-H is more stable than Au11-Cl. The formula of Au11-Cl was confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS). As shown in Fig. 1b, the ESI-TOF-MS (positive ion mode) revealed a single intense peak at m/z = 5230.02 Da, and the 1 gap between isotopic peaks proves that z = 1. The formula is attributed to [Au11(C18H12Cl3P)7Cl3Cs3–2H]+ (Au11-Cl). The experimental data matched well with the theoretical simulation indicates the purity of Au11-Cl. X-ray photoelectron spectroscopy (XPS) was further performed to reveal the electronic structure of Au11-Cl and Au11-H. Fig. S2† shows the total spectra of XPS for two nanoclusters. The Au 4f peak (84.53 eV) of Au11-Cl nanocluster is on the higher energy side compared to that of Au11-H (84.05 eV), which means the Au atoms in the Au11-Cl are more oxidized than those in the Au11-H.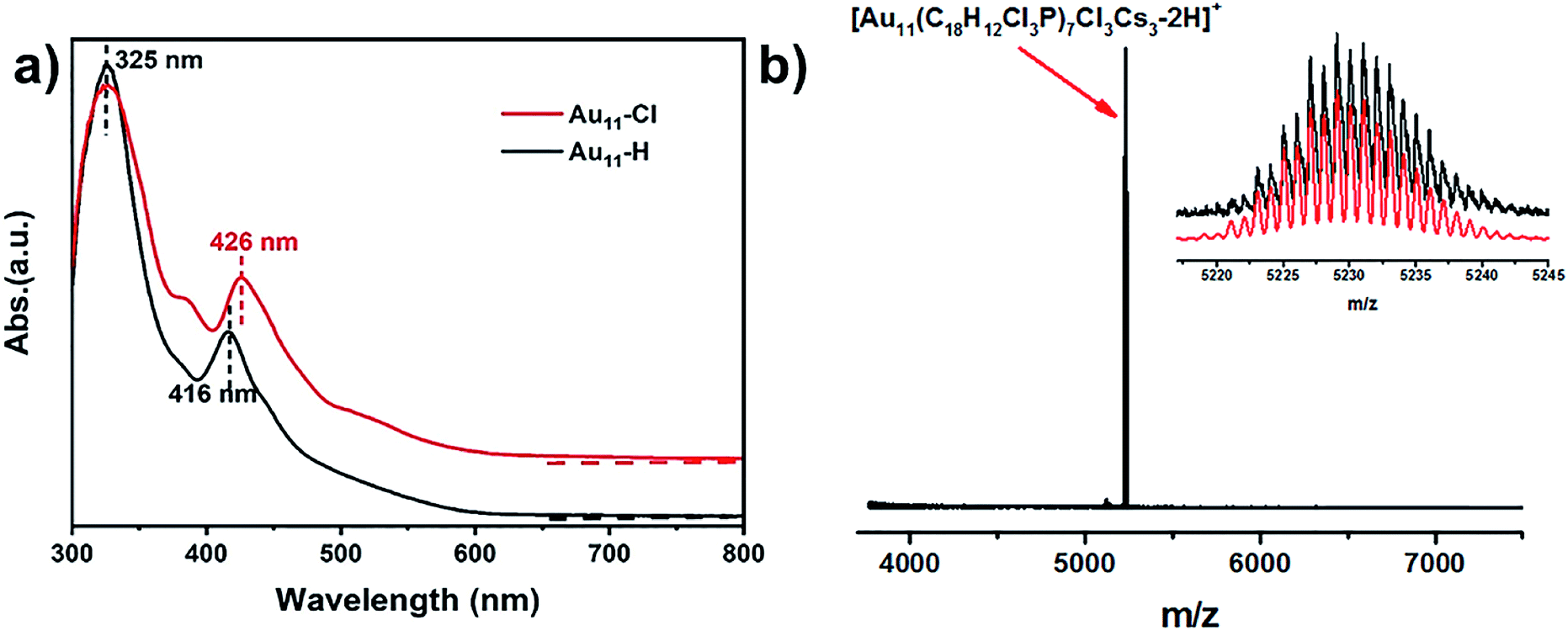 | ||
| Fig. 1 (a) The UV-vis absorption spectra of Au11-Cl and Au11-H. (b) Positive ion mode ESI-TOF-MS of Au11-Cl clusters in CH3OH, inset: overlap of the experimental data (black) and the simulated spectrum (red) for Au11-Cl. | ||
The structure of Au11-Cl was determined by the single crystal X-ray crystallography (Fig. 2). The molecular structure of the Au11-Cl is similar to that of the Au11-H. Ten gold atoms (yellow) form an incomplete icosahedral structure with one gold atom in the centre. And the ligands of six p-ClPPh3 (pink) and three Cl (green) atoms cap the metal core along the C3 axis with one p-ClPPh3 ligand on the C3 axis. In comparison with the Au11-H, the Au11-Cl crystal possesses a larger cell volume (23297.7 Å3 versus 12398 Å3) and less density (2.066 g cm−3 versus 2.243 g cm−3), which is possibly attributed to distinct non-covalent interactions in the unit cell. More comparisons of the cell details are given in Table S1.†
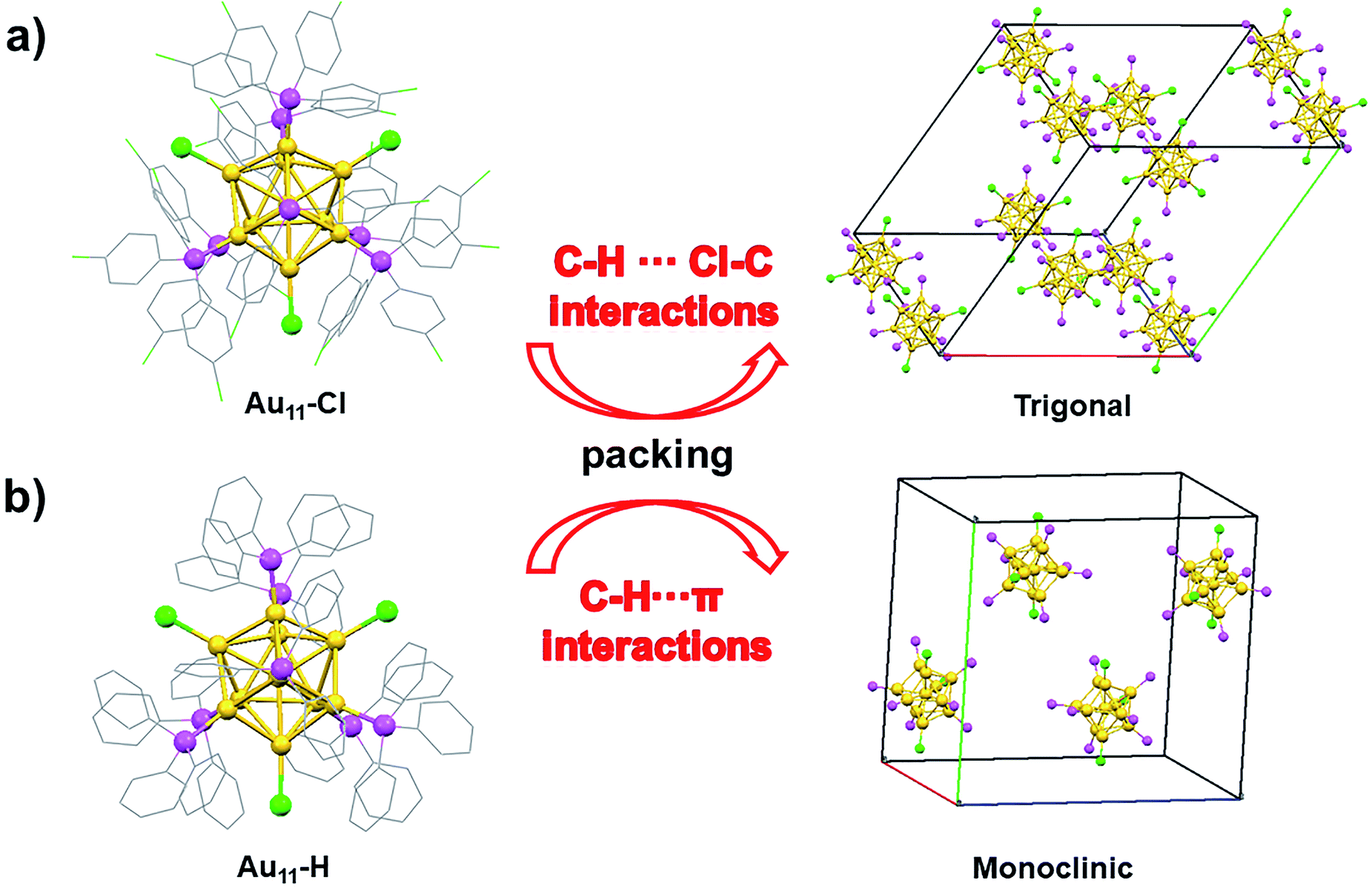 | ||
| Fig. 2 (a) Crystal structure and trigonal unit cell of the Au11-Cl. (b) Crystal structure and monoclinic unit of the Au11-H. (colour labels: yellow = Au, pink = P, gray = C, green = Cl, for clarity all H atoms are not shown). | ||
The packing of these two nanoclusters along (100), (010), and (001) planes is shown in Fig. 3, respectively. For the T system of Au11-Cl, the nanoclusters are organized layer by layer with an fcc-like ABCABC style viewing along a-axis, b-axis, and c-axis (Fig. 3a–c). Especially, viewing along the c-axis, the Au11-Cl gets organized into a hexagonal lattice, while the Au11-H gets decorated into a rectangular lattice. In the case of the Au11-H, the nanoclusters are packed layer by layer with an ABAB style viewing along the a-axis, b-axis, and c-axis (Fig. 3d–f). Although the Au11-Cl and the Au11-H are constructed with the same core structure, they display totally different lattices. The obvious packing difference of the Au11 nanoclusters is related to the difference of non-covalent interactions between nanoclusters.14
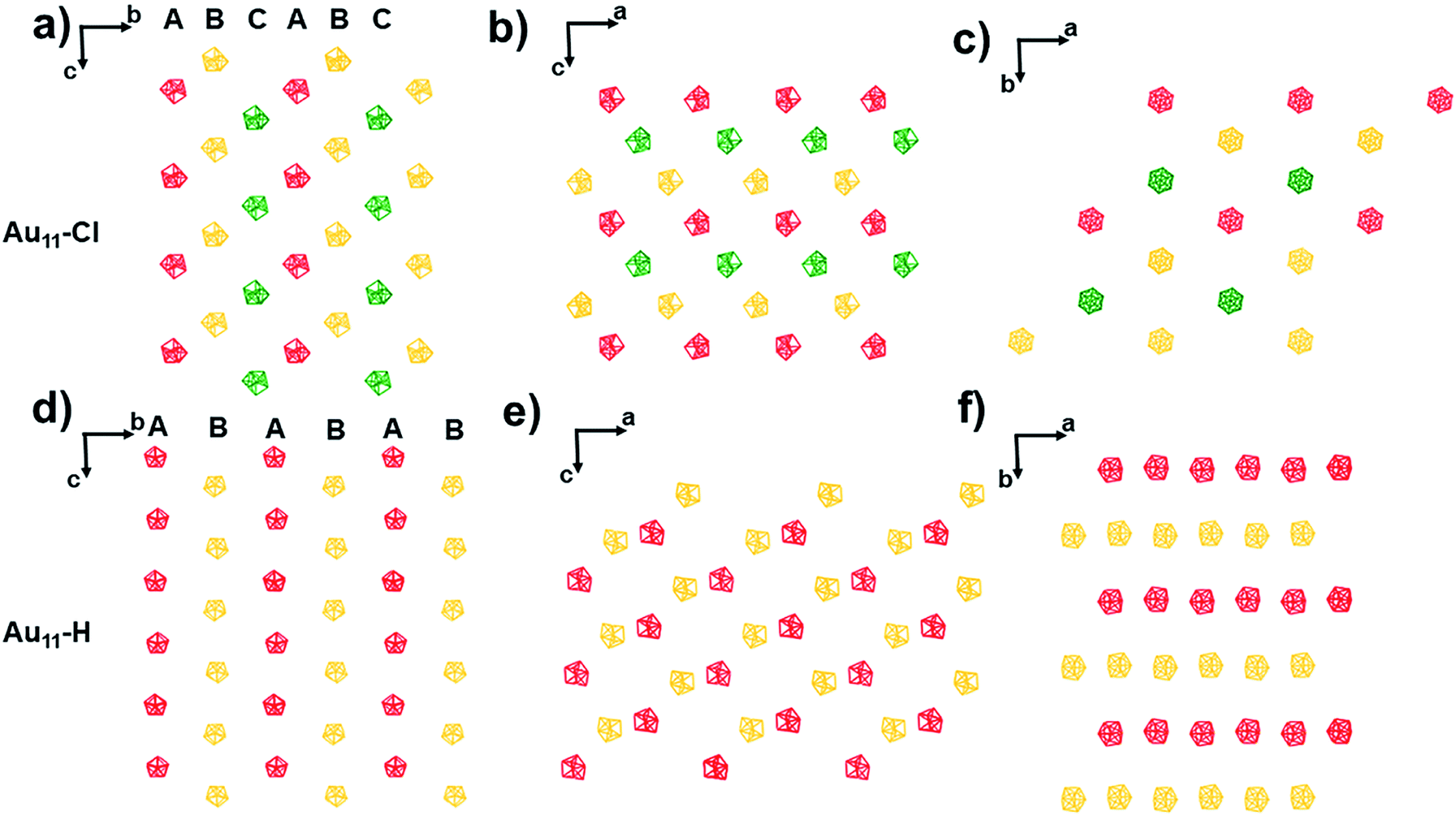 | ||
| Fig. 3 Packing of the Au11-H and the Au11-Cl view from the (a and d) a-axis, (b and e) b-axis and (c and f) c-axis. | ||
C–H⋯π interactions play a crucial role in the crystal packing of the Au11-H, which are the major driving force to form monoclinic. In the M system, each unit contains four Au11-H nanoclusters. Fig. 4 shows the C–H⋯π interactions (red/blue dashed line) of the Au11-H. C–H⋯π interactions exist between the C–H groups (blue) of the PPh3 ligands and adjacent benzenes (green) of the adjacent cluster, evidenced by their distance. The distance of C–H⋯π ranges from 2.80 to 2.89 Å, with an average of 2.835 Å (Fig. S3†). Strong C–H⋯π interactions form a chain to hold each Au11-H nanocluster and restrict the motion of the entire nanocluster.
 | ||
| Fig. 4 The C–H⋯π interactions in the Au11-H. Yellow = Au; pink = P; green = C; blue = H; bright green = Cl; blue red dashed, C–H⋯π interactions. | ||
Interestingly, most H atoms form C–H⋯π interactions are located on the para position of the benzene rings in the Au11-H, which inspires us to speculate whether these interactions could be destroyed by introducing an atom with a larger atomic radius in the para position. As we expected, by replacing PPh3 with p-ClPPh3 ligands that contain Cl atoms on the para position of aromatic rings, C–H⋯π interactions are not found in the unit cell of the as-obtained Au11-Cl. In order to discuss the non-covalent interactions of Au11-Cl, four Au11-Cl clusters are marked by the green quadrilateral (Fig. S7†).
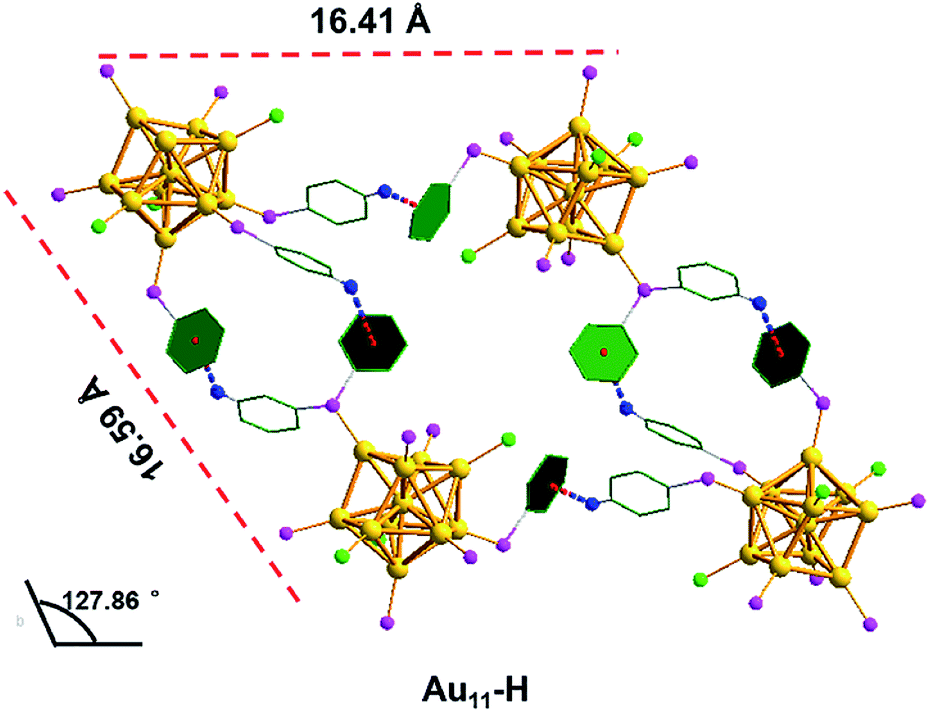
As shown in Fig. 5, the inter-cluster distances are 19.80 Å and 17.42 Å, respectively, and the corresponding obtuse is 125.42°. However, the lengths (16.41 Å and 16.59 Å) and obtuse (127.86°) of Au11-H are different from those of Au11-Cl. When zooming into the surfaces between nanoclusters, C–H⋯Cl–C interactions (bright green/blue dashed line) of the Au11-Cl lead to form T systems in the unit. In the T lattice of Au11-Cl, the average distance of the C–H⋯Cl–C interactions is 2.94 Å, with a range from 2.85 to 2.99 Å (Fig. S4†). This distance is consistent with the reported value.40 Overall, comparing the C–H⋯Cl–C interactions and the C–H⋯π interactions suggests that the insertion of chlorine atoms breaks C–H⋯π interactions and changes the crystal system from M to T.
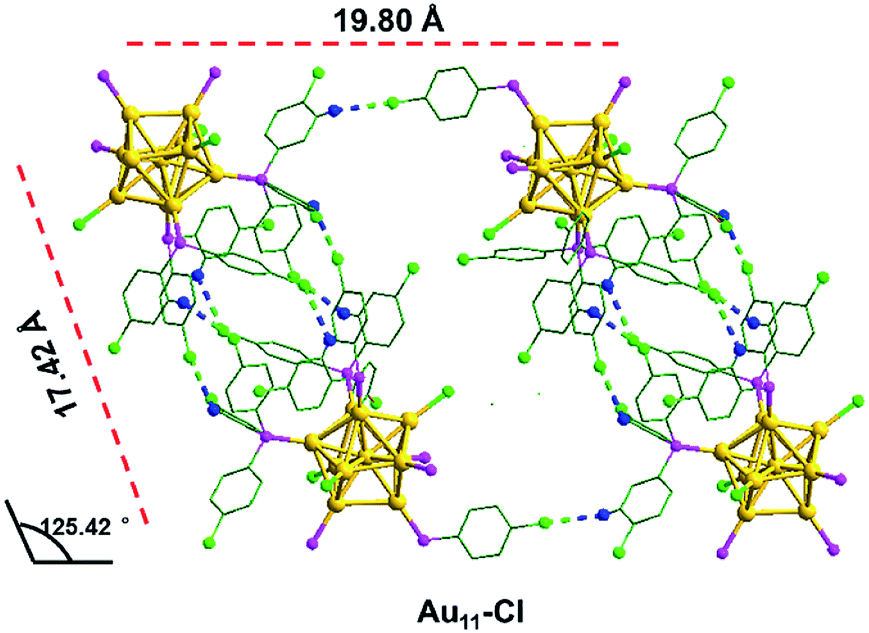 | ||
| Fig. 5 The C–H⋯Cl–C interactions in the Au11-Cl. Yellow = Au; pink = P; green = C; blue = H; bright green = Cl; bright green blue dashed, C–H⋯Cl–C interaction. | ||
Another significant aspect is that the non-covalent interactions of the intra-molecular also changed because of the introduction of chlorine atoms in the surface ligands. The average distance of H and H (DHH for short) in the Au11-Cl is 2.72 Å (Fig. 6a and S5†). In contrast, the DHH in the Au11-H is 2.80 Å, varying from 2.53 to 2.99 Å (Fig. 6b and S6†), which is slightly larger than that of the Au11-Cl. Viewing along the C3 axis, the triangle constituted by nearest H atoms is slightly rotating compared with the one in Au11-Cl, due to the introduction of the Cl atoms. The total numbers and average distances of the non-covalent interactions and the DHH were shown in Table 1. All these results verify that non-covalent interactions of intra-cluster induce the rotation and arrangement of ligands, which further enable every single nanocluster to be compacted and ordered with reduced intra-cluster vibration, contributing to the formation of high-quality single crystals.
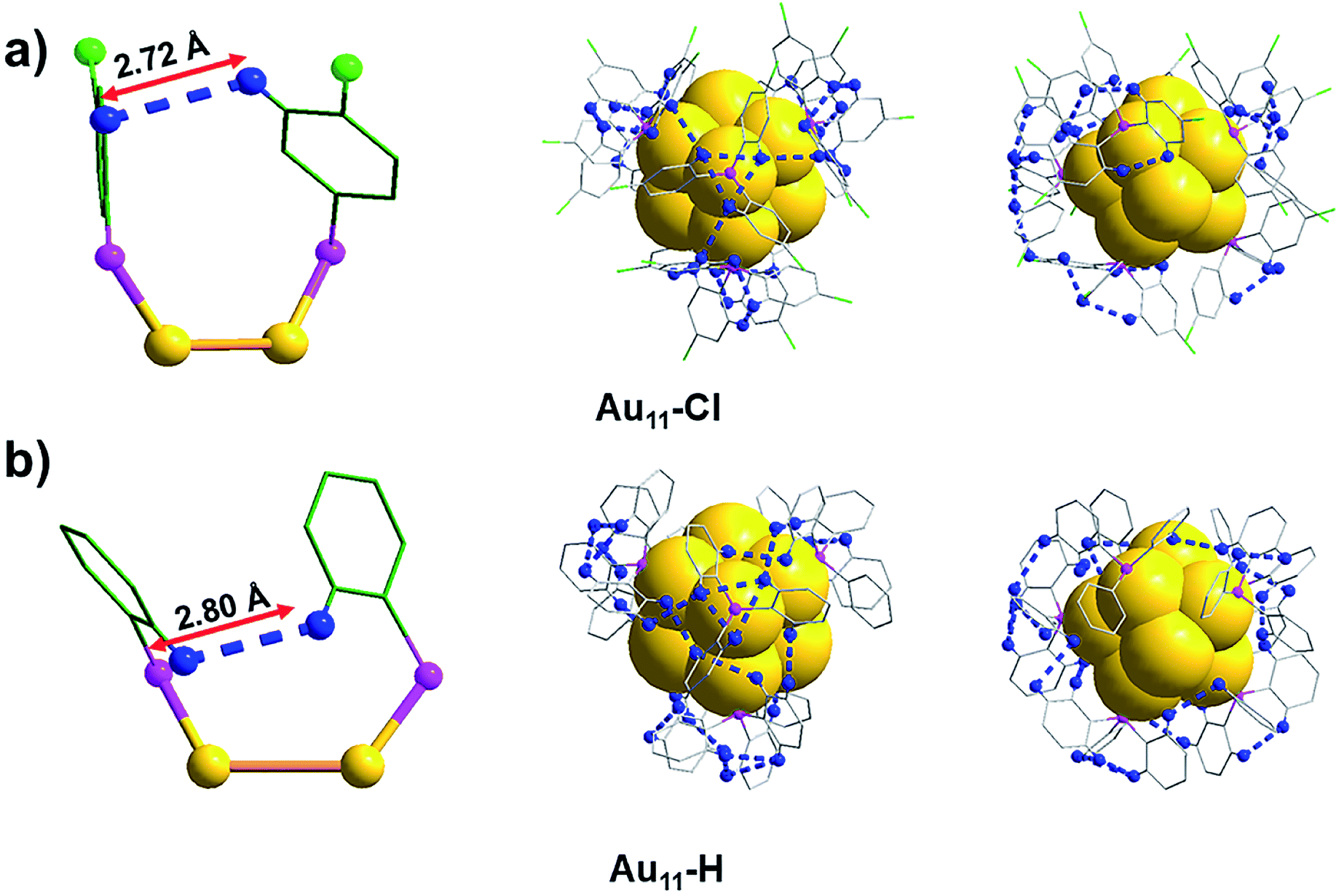 | ||
| Fig. 6 The nearest neighbour H and H atoms in the intra-Au11-Cl and intra-Au11-H. (a) The nearest neighbour H and H atoms in the Au11-Cl viewed from different angles. (b) The nearest neighbour H and H atoms in the Au11-H viewed from different angles. Yellow = Au; pink = P; green = C; blue = H; bright green = Cl; blue dashed, intra-DHH; for clarity other atoms are not shown. | ||
| Interaction (distance) | Au11-H | Au11-Cl |
|---|---|---|
| CH⋯π | 6 (2.84 Å) | × |
| C–H⋯Cl–C | × | 20 (2.94 Å) |
| Inter-DHH | 46 (2.80 Å) | 83 (2.81 Å) |
| Intra-DHH | 24 (2.79 Å) | 21 (2.72 Å) |
Conclusions
In summary, we have chosen the Au11(p-ClPPh3)7Cl3 (Au11-Cl) and the Au11(PPh3)7Cl3 (Au11-H) to investigate non-covalent interactions in the crystallization by surface modification. The chlorine atoms occupy the para of benzene rings in phosphine ligands, resulting in the breaking of C–H⋯π interactions and crystal system transformation. C–H⋯Cl–C interactions of Au11(p-PPh3)7Cl3 lead to the formation of trigonal systems, revealing the importance of the ligands on the non-covalent interaction modes. Moreover, UV-vis spectra and XPS spectra reveal the change of the electronic structure caused by surface modification. This work provides new insights into the effects of inter- and intra-interactions on nanocluster crystallization, making the assembly and self-assembly of metal nanoclusters more readily.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support by NSFC (No. 21571001, 21372006, 21631001, and U1532141), the Ministry of Education, the Education Department of Anhui Province, and 211 Project of Anhui University.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS.
- H. Zhu, N. Goswami, Q. Yao, T. Chen, Y. Liu, Q. Xu, D. Chen, J. Lu and J. Xie, J. Mater. Chem. A, 2018, 6, 1102–1108 RSC.
- C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 99–104 CrossRef CAS PubMed.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- Y. Wang, X. K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Hakkinen, B. K. Teo, Q. M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, S. Zang and J. Xie, ACS Mater. Lett., 2019, 1, 237–248 CrossRef CAS.
- M. Zhu, S. Jin and S. Wang, Chem.–Asian J., 2019, 14, 3222–3231 CrossRef PubMed.
- R. W. Huang, Y. S. Wei, X. Y. Dong, X. H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- X. Kang, Y. Song, H. Deng, J. Zhang, B. Liu, C. Pan and M. Zhu, RSC Adv., 2015, 5, 66879–66885 RSC.
- A. Nag, P. Chakraborty, M. Bodiuzzaman, T. Ahuja, S. Antharjanam and T. Pradeep, Nanoscale, 2018, 10, 9851–9855 RSC.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- P. Chakraborty, A. Nag, A. Chakraborty and T. Pradeep, Acc. Chem. Res., 2019, 52, 2–11 CrossRef CAS PubMed.
- P. Sun, Z. Wang, Y. Bi, D. Sun, T. Zhao, F. Zhao, W. Wang and X. Xin, ACS Appl. Nano Mater., 2020, 3, 2038–2046 CrossRef CAS.
- Z. Wu, J. Liu, Y. Gao, H. Liu, T. Li, H. Zou, Z. Wang, K. Zhang, Y. Wang, H. Zhang and B. Yang, J. Am. Chem. Soc., 2015, 137, 12906–12913 CrossRef CAS PubMed.
- Z. Wu, C. Dong, Y. Li, H. Hao, H. Zhang, Z. Lu and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 9952–9955 CrossRef CAS PubMed.
- J. Fontana, W. J. Dressick, J. Phelps, J. E. Johnson, R. W. Rendell, T. Sampson, B. R. Ratna and C. M. Soto, Small, 2014, 10, 3058–3063 CrossRef CAS PubMed.
- M. De Nardi, S. Antonello, D.-e. Jiang, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, ACS Nano, 2014, 8, 8505–8512 CrossRef CAS PubMed.
- P. Chakraborty, A. Nag, G. Paramasivam, G. Natarajan and T. Pradeep, ACS Nano, 2018, 12, 2415–2425 CrossRef CAS PubMed.
- V. Berry and R. F. Saraf, Angew. Chem., Int. Ed., 2005, 44, 6668–6673 CrossRef CAS PubMed.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef PubMed.
- Y. Song, S. Weng, H. Li, H. Yu and M. Zhu, Inorg. Chem., 2019, 58, 7136–7140 CrossRef CAS PubMed.
- S. Zhang, X. Lu, J. Sun, Y. Zhao and X. Shao, CrystEngComm, 2015, 17, 4110–4116 RSC.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- Z. Gan, J. Chen, J. Wang, C. Wang, M.-B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Hakkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Hakkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, D. Zhao, J. Xie and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 184–189 CrossRef CAS PubMed.
- J. Liang, X.-S. Wu, X.-L. Wang, C. Qin, K.-Z. Shao, Z.-M. Su and R. Cao, CrystEngComm, 2016, 18, 2327–2336 RSC.
- J. Shen, Z. Wang, D. Sun, G. Liu, S. Yuan, M. Kurmoo and X. Xin, Nanoscale, 2017, 9, 19191–19200 RSC.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- Z. Qin, D. Zhao, L. Zhao, Q. Xiao, T. Wu, J. Zhang, C. Wan and G. Li, Nanoscale Adv., 2019, 1, 2529–2536 RSC.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C. T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Hakkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- P. K. Thallapally and A. Nangia, CrystEngComm, 2001, 3, 114–119 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1966633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01288b |
| This journal is © The Royal Society of Chemistry 2020 |