
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium on phosphorous-modified alumina as an effective and stable catalyst for catalytic transfer hydrogenation of furfural†
Thibault Fovanna‡
 ab,
Sebastiano Campisi‡c,
Alberto Villa*c,
Anastasios Kambolis§
a,
Gael Peng¶
a,
Daniel Rentschd,
Oliver Kröcherab,
Maarten Nachtegaala and
Davide Ferri*a
ab,
Sebastiano Campisi‡c,
Alberto Villa*c,
Anastasios Kambolis§
a,
Gael Peng¶
a,
Daniel Rentschd,
Oliver Kröcherab,
Maarten Nachtegaala and
Davide Ferri*a
aPaul Scherrer Institut, CH-5232 Villigen PSI, Switzerland. E-mail: davide.ferri@psi.ch; Tel: +41 56 310 2781
bÉcole polytechnique fédérale de Lausanne (EPFL), Institute of Chemical Sciences and Engineering, CH-1015 Lausanne, Switzerland
cDipartimento di Chimica, Università degli Studi di Milano, I-20133 Milano, Italy. E-mail: alberto.villa@unimi.it; Tel: +39 02 503 14361
dSwiss Federal Laboratories for Materials Science and Technology (Empa), Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
First published on 20th March 2020
Abstract
Supported ruthenium was used in the liquid phase catalytic transfer hydrogenation of furfural. To improve the stability of Ru against leaching, phosphorous was introduced on a Ru/Al2O3 based catalyst upon impregnation with ammonium hypophosphite followed by either reduction or calcination to study the effect of phosphorous on the physico-chemical properties of the active phase. Characterization using X-ray diffraction, solid state 31P nuclear magnetic resonance spectroscopy, X-ray absorption spectroscopy, temperature programmed reduction with H2, infrared spectroscopy of pyridine adsorption from the liquid phase and transmission electron microscopy indicated that phosphorous induces a high dispersion of Ru, promotes Ru reducibility and is responsible for the formation of acid species of Brønsted character. As a result, the phosphorous-based catalyst obtained after reduction was more active for catalytic transfer hydrogenation of furfural and more stable against Ru leaching under these conditions than a benchmark Ru catalyst supported on activated carbon.
1 Introduction
Biomass can be converted into a pool of highly functionalized, platform molecules1,2 to enable the synthesis of fine chemicals, pharmaceuticals, intermediates, polymers and additives. The production of value-added compounds from biomass involves catalytic transformations, where the catalyst design represents a key factor for achieving the desired efficiency and selectivity.3Furfural (furan-2-carbaldehyde) is an example of a building block from hemicellulose-derived pentoses, which can be transformed into various compounds of industrial interest.4 The hydrogenation of furfural can proceed along several pathways leading to various useful chemicals such as tetrahydrofuran, furan, 2-methylfuran and furfuryl alcohol (Scheme 1).5 The latter is the result of a selective single-step reduction of the carbonyl group of furfural and is a key intermediate in the production of lysine, ascorbic acid (vitamin C), polymers, synthetic fibers, rubbers, resins and lubricants.6 The conventional catalytic hydrogenation process currently used for producing furfuryl alcohol requires high temperature (130–200 °C) and pressure (up to 30 bar) and toxic elements, i.e. copper chromite.7 Therefore, the development of heterogeneous catalytic processes operating under mild conditions and with less-toxic metal catalysts remains a priority to achieve a more sustainable industrial process. In this view, the catalytic transfer hydrogenation (CTH), consisting in the use of organic molecules as sources of hydrogen, represents a valid alternative, which minimizes safety hazards deriving from handling high pressure hydrogen gas.8
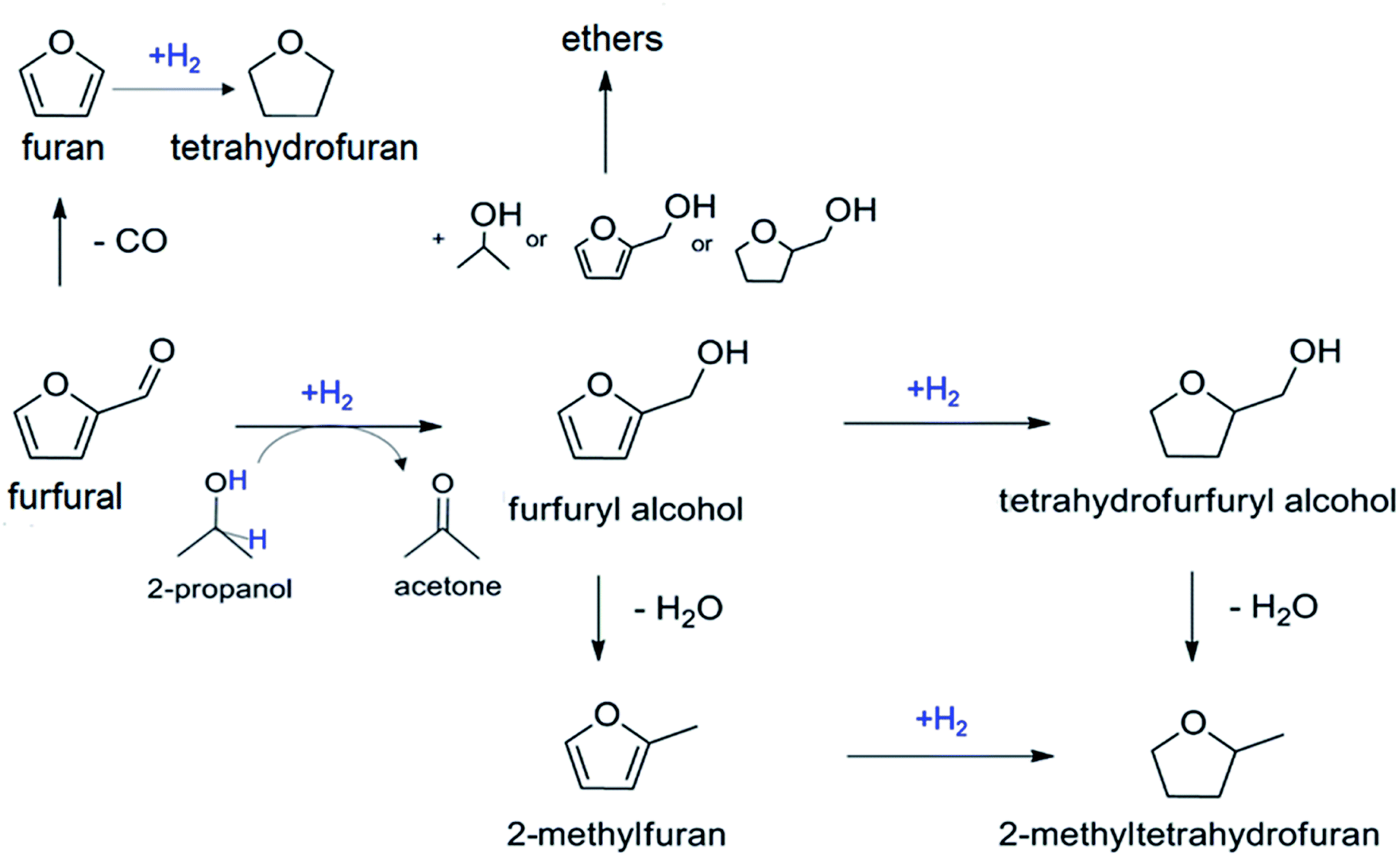 | ||
| Scheme 1 Reaction pathways for the catalytic hydrogenation of furfural. | ||
A large variety of metal-based catalysts have been proposed in the literature as alternatives for CTH9 and include cobalt,10 nickel,11 copper,12 platinum,13 palladium14 and ruthenium.15 Ruthenium emerged by its superior activity and efficiency under liquid phase conditions and by its lower cost compared to other precious metals.16 Ru/Al2O3 (ref. 17–20) and Ru on carbonaceous supports21 are well studied hydrogenation catalysts. Interest in Ru as an effective metal for the hydrogenation of biomass-derived carbonyl compounds in the aqueous phase22–25 has probably increased due to its unique oxophilicity and ability to dissociatively adsorb water molecules.26 However, Ru-based catalysts often suffer from the lack of stability, due to passivation, poisoning or leaching of Ru species.27 Besides the possibility to alloy Ru with Au to improve the stability for hydrogenation reactions,28 modifiers and promoters can be used to increase the efficiency and the stability of the catalysts.29 Organic linkers,30 metal oxides31 as well as heteroatoms introduced on the support32 can commonly act as promoters by anchoring metal species, introducing defects or stabilizing particular oxidation states of the active metal.33 P-containing functionalities (e.g. phosphates, phosphides) form a broad class of promoters, which are experiencing a growing interest in the field of heterogeneous catalysis for biomass valorization.34–45 Besides acting as ligands for the stabilization of metal species, phosphorous introduces Brønsted acid sites on the support surface and promotes electron transfer phenomena resulting in enhanced activity.36,40
In this work, we investigate the effect of the modification of alumina with phosphorous on the catalytic performance of Ru towards catalytic transfer hydrogenation of furfural in the presence of isopropanol as hydrogen donor. A suite of characterization techniques (X-ray powder diffraction, X-ray absorption spectroscopy, attenuated total reflectance infrared spectroscopy, temperature programmed reduction, solid-state nuclear magnetic resonance spectroscopy, transmission electron microscopy) was used to clarify the nature, structure, speciation and coordination of Ru species as well as of P functionalities on the alumina surface. This information allowed rationalizing the catalytic results in the frame of appropriate structure–activity relationships.
2 Experimental
2.1 Catalyst synthesis
The catalyst precursor (Ru_dry) was obtained by impregnation of γ-Al2O3 (PURALOX NWa155, Sasol; 132 m2 g−1) with a solution of ruthenium(III) nitrosyl nitrate (1.5 wt% Ru(NO)(NO3)3 in diluted HNO3, Sigma-Aldrich) in order to achieve 5 wt% metal loading. After drying at 60 °C/170 mbar in a rotary evaporator, a portion of Ru_dry was calcined in air in a muffle oven at 400 °C for 3 h to obtain Ru_O2. A second aliquot of Ru_dry was reduced at 500 °C in 30 vol% H2 (Ru_H2). A third portion was further impregnated with a solution of ammonium hypophosphite (NH4H2PO2, Sigma-Aldrich) to a P/Ru ratio of 1.4. After drying in a rotary evaporator at 60 °C/170 mbar, an aliquot of the material (RuP_dry) was reduced in 30 vol% H2 at 500 °C for 4 h, cooled to 180 °C in the same atmosphere and then under N2 to ambient temperature. Finally, the powder was exposed to 1 vol% O2 at room temperature for 30 min (sample labelled as RuP_H2). For comparison, a portion of RuP_dry was calcined at 400 °C (RuP_O2) for 3 h. An additional sample (P_Al2O3) was prepared by impregnation of γ-Al2O3 with NH4H2PO2 to achieve 5 wt% P loading. The material was dried as described above then calcined at 400 °C for 3 h. The overall sample preparation is summarized in Scheme 2.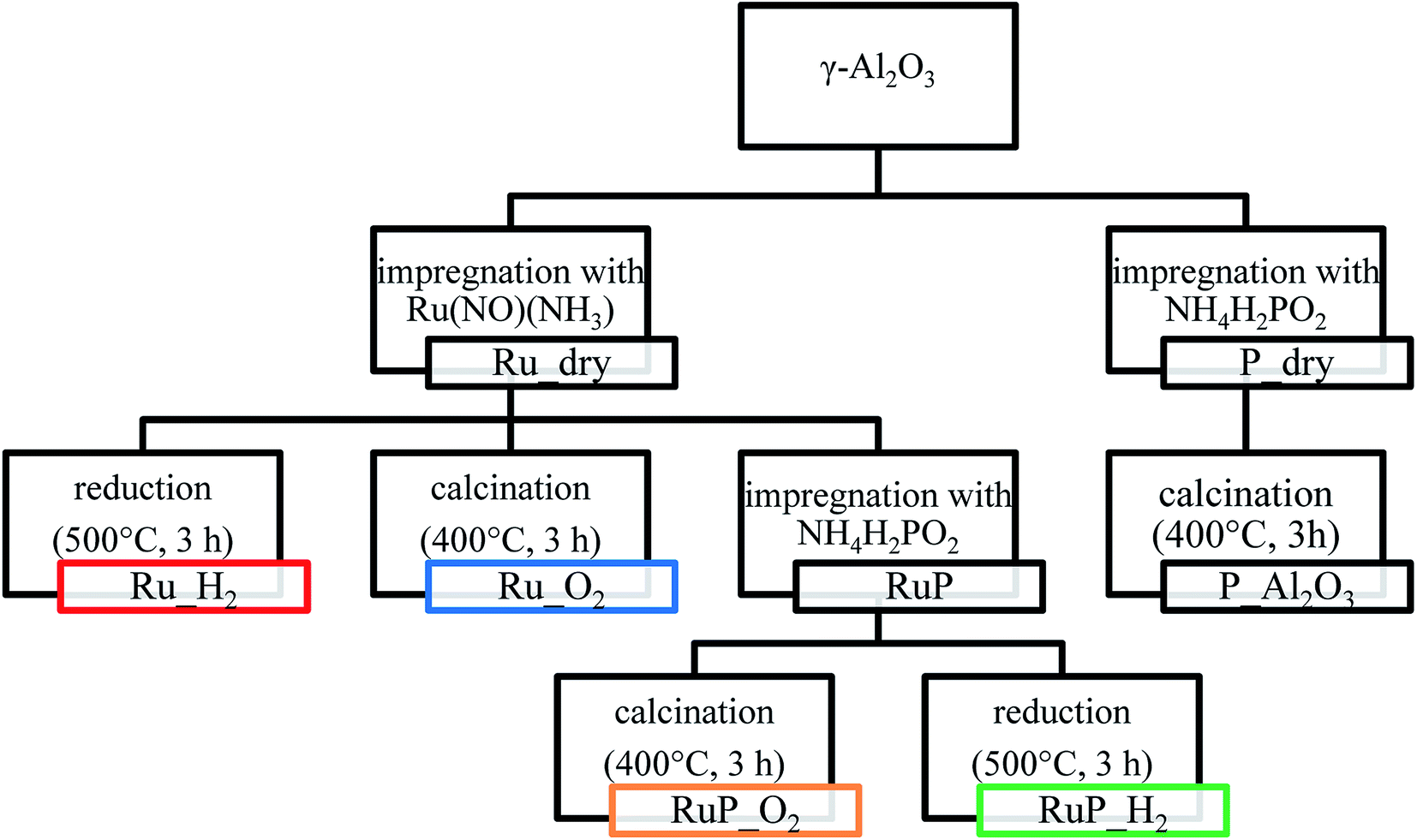 | ||
| Scheme 2 Synthesis approach for the preparation of the various materials. | ||
2.2 Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 eV) at the Swiss Norwegian beamline (SNBL) of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and the SuperXAS beamline of the Swiss Light Source (SLS, Paul Scherrer Institute, Switzerland) using 15 cm long ionization chambers filled with 1 bar N2 and 1 bar Ar. A reference spectrum of a Ru metal mesh was measured simultaneously with the sample between the second and the third ionization chamber for energy calibration. At SNBL, the white beam of a 0.8 T ESRF bending magnet was monochromatized by a Si(111) double crystal monochromator. Harmonic suppression was obtained by detuning the second crystal to 65% throughput. The beam size on the sample was set to 5 to 0.5 mm (l × h). At the SuperXAS beamline, the polychromatic beam from the 2.9 T bending magnet was collimated by a Pt coated mirror at 2.5 mrad, monochromatized by a continuous scanning Si(111) channel cut monochromator and subsequently focused using a Pt coated toroidal mirror to a spot size of 100 × 100 μm at the sample position. XAS spectra were background corrected, normalized and energy corrected using the Demeter software package.46 Spectra of metallic Ru (mesh) and RuO2 were collected for reference purposes.
117 eV) at the Swiss Norwegian beamline (SNBL) of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and the SuperXAS beamline of the Swiss Light Source (SLS, Paul Scherrer Institute, Switzerland) using 15 cm long ionization chambers filled with 1 bar N2 and 1 bar Ar. A reference spectrum of a Ru metal mesh was measured simultaneously with the sample between the second and the third ionization chamber for energy calibration. At SNBL, the white beam of a 0.8 T ESRF bending magnet was monochromatized by a Si(111) double crystal monochromator. Harmonic suppression was obtained by detuning the second crystal to 65% throughput. The beam size on the sample was set to 5 to 0.5 mm (l × h). At the SuperXAS beamline, the polychromatic beam from the 2.9 T bending magnet was collimated by a Pt coated mirror at 2.5 mrad, monochromatized by a continuous scanning Si(111) channel cut monochromator and subsequently focused using a Pt coated toroidal mirror to a spot size of 100 × 100 μm at the sample position. XAS spectra were background corrected, normalized and energy corrected using the Demeter software package.46 Spectra of metallic Ru (mesh) and RuO2 were collected for reference purposes.Operando experiments were carried out in a flow cell adapted for liquid phase experiments.47 The catalyst (35 mg) was loaded between two quartz wool plugs. The cell was connected to a HPLC pump (Azura P 4.1S 10 mL min−1 head; Knauer) and set to a flow of 0.2 mL min−1. At the outlet of the cell a backpressure regulator (KCB series, Swagelok) maintained the reactive environment in liquid phase at 16 bar. The catalyst bed was recorded dry at room temperature before allowing the Ar-saturated 2-propanol solution of furfural (5 mM; Sigma-Aldrich, 99.9%) to enter the cell. Quick EXAFS spectra were recorded for 1 min every 30 °C until the reactor reached 180 °C. Once the desired reaction temperature was attained, the catalyst bed was left under reaction conditions for 30 min while collecting samples for analysis by gas chromatography (see below) every 15 min. In order to improve the signal to noise ratio, the time-resolved spectra were averaged over the whole acquisition period (1 min, 60 spectra). Fourier transform was performed in the k-range of 3–15 Å−1 for Ru_H2 and 3–11 Å−1 for RuP_H2. The Fourier transformed spectra were not phase-shift corrected. A curve fitting analysis was carried out on the data in the R-range of 1.9–3.0 Å. Only the first coordination shell was considered and metallic Ru was used as reference. The amplitude reduction factor was set to 0.78 after fitting the EXAFS spectra of the Ru mesh.
:Al mol ratio 11.3, ABCR, 379 m2 g−1, calcined at 550 °C in air prior to use).2.3 Catalytic activity
Furfural hydrogenation was performed at 180 °C, using a stainless steel reactor (30 mL capacity), equipped with heater, mechanical stirrer, gas supply system and thermometer. The furfural solution (15 mL; 0.3 M in 2-propanol) was added into the reactor at room temperature and the desired amount of catalyst (F/Ru ratio = 100 mol mol−1) was suspended in the solution. The N2 pressure was 5 bar. The mixture was heated to the reaction temperature and was then mechanically stirred (1250 rpm). At the end of the reaction, the autoclave was cooled to room temperature, N2 flow stopped and the autoclave purged with flowing N2. Samples were removed periodically (0.2 mL) using a micro-syringe through a withdrawal valve and were analyzed by gas chromatography (HP 7820A gas chromatograph equipped with a capillary column HP-5 30 m × 0.32 mm, 0.25 μm film; Agilent Technologies). Standard solutions of reactants and products were analyzed to determine separation times. Quantitative analysis was performed using an external standard (n-octanol). Identification of the products was performed using a Thermo Scientific Trace ISQ QD Single Quadrupole GC-MS equipped with a capillary column (HP-5 30 m × 0.32 mm, 0.25 μm film; Agilent Technologies).For catalyst recycling tests, each run was carried out under the same conditions (furfural = 0.3 M; F/Ru mol ratio, 100; 180 °C, 5 bar N2). The catalyst was recycled to be used in the subsequent run (reaction times of 3 h each) after filtration without any further treatment.
3 Results and discussion
3.1 Catalyst preparation and characterization
The various catalyst materials that are compared in this study were prepared according to Scheme 2. Ruthenium was deposited on γ-Al2O3 by wet impregnation from ruthenium(III) nitrosyl nitrate solution to attain a final metal loading of 5 wt%. After drying, Ru_dry underwent nitrosyl nitrate precursor decomposition either by calcination in air (Ru_O2) or by reduction in H2 (Ru_H2). For both samples, specific surface areas of 169 and 163 m2 g−1 were determined, respectively.The P-promoted catalysts were prepared similarly by sequential wet impregnation with ammonium hypophosphite. After impregnation with the ruthenium precursor, Ru_dry was contacted with the phosphite containing solution. The material containing 7.5 wt% P (RuP_dry) was subsequently calcined (RuP_O2; 131 m2 g−1) or reduced (RuP_H2; 126 m2 g−1). The specific surface area of the resulting samples decreased to approximately the value obtained for the support (Al2O3, 132 m2 g−1).
The structural analysis by X-ray powder diffraction (XRPD, Fig. 1) helped to identify the composition and structure of Ru containing crystallites. Calcination of the Ru precursors on P-free Al2O3 (Ru_O2) and after impregnation with the P precursor (RuP_O2) produced large crystallites of RuO2 as suggested by the presence of sharp reflections of this phase. Conversely, reduction (Ru_H2) produced metallic Ru crystallites of ca. 11 nm in diameter as estimated using the Scherrer equation (Table S1†). The XRPD pattern of RuP_H2 displayed only the reflections of the Al2O3 support and no specific reflection of RuO2 nor metallic Ru could be detected. Transmission electron micrographs of Ru_H2 and RuP_H2 (Fig. S1†) confirmed the absence of well-defined particles in RuP_H2. This observation suggests that the presence of phosphorous allowed Ru to disperse uniformly on Al2O3 and prevented the growth of Ru or RuO2 particles. It also provides a possible explanation for the observed lower increase of surface area after Ru wet impregnation compared to the corresponding P-free materials. On the other hand, phosphorous did not prevent the formation of large RuO2 crystallites when RuP_dry was subjected to calcination at 400 °C (sample RuP_O2). Hence, reduction is required to disperse Ru in the presence of phosphorous.
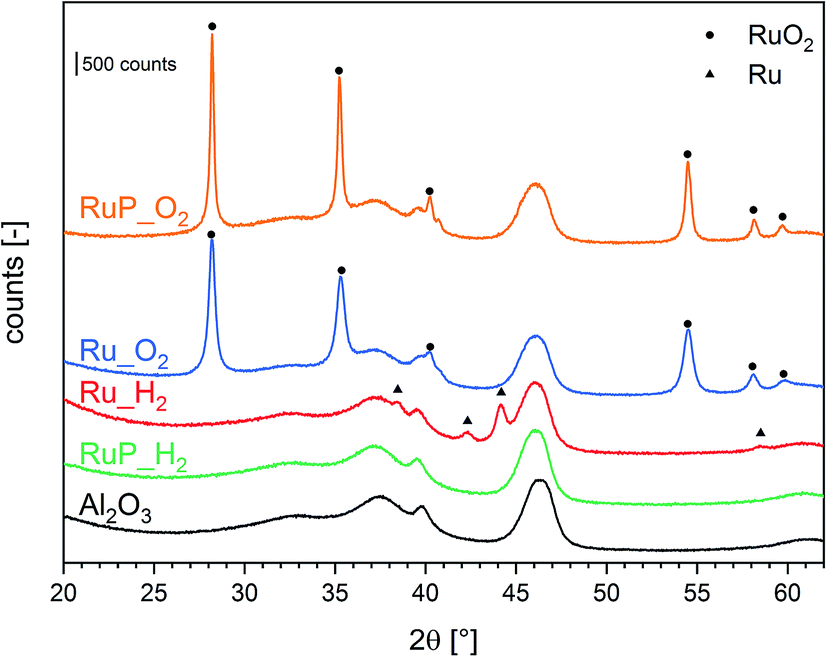 | ||
| Fig. 1 XRD patterns of Ru catalysts and of the aluminum oxide reference material. | ||
Information on the local environment of Ru was determined by analysis of the Ru K-edge X-ray absorption near edge structure (XANES, Fig. S2†) and extended X-ray absorption fine structure (EXAFS, Fig. 2) spectra. The XANES spectra of Ru_O2 and RuP_O2 exhibited the typical white line of RuO2 (Fig. S2†) confirming the presence of RuO2 already identified by XRPD. The edge energy position of Ru_H2 was close to that of metallic Ru while the edge of RuP_H2 was shifted to higher energy suggesting the presence of partially oxidized Ru species. The attenuation of the features of the white line intensity of RuP_H2 compared to the intensity of the Ru reference suggests the presence of small Ru domains. The FT-EXAFS spectra of the two reduced samples (Fig. 2) are very different. Ru_H2 presented coordination shells up to 10 Å, suggesting the presence of large Ru metal particles, while RuP_H2 did not exhibit further coordination shells past the first Ru–O coordination shell (at ca. 1.6 Å). Hence, RuP_H2 possessed smaller particles or clusters, thus justifying the absence of a Ru-containing phase in XRPD and of evident particles in TEM.
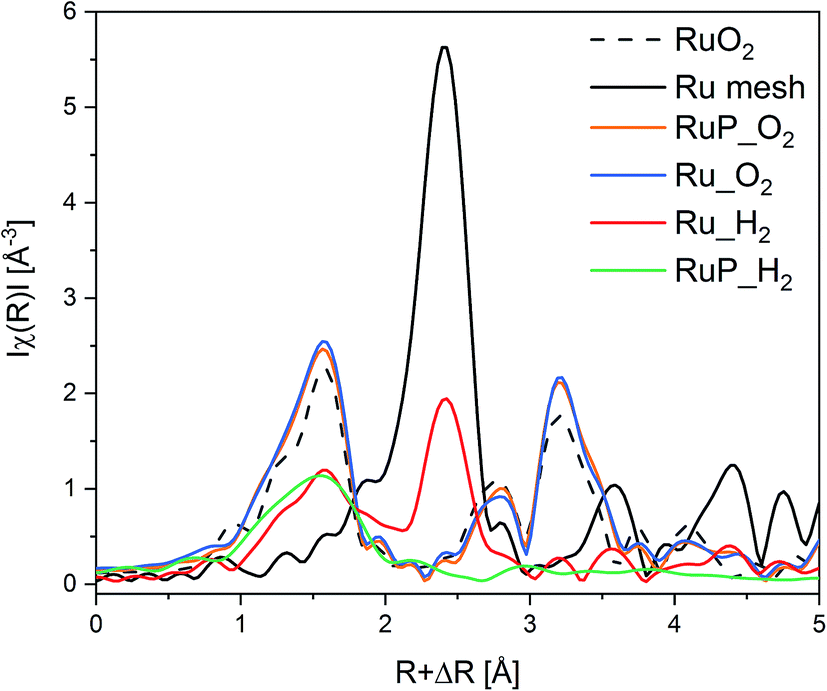 | ||
| Fig. 2 Ru K-edge FT-EXAFS of Ru catalysts and of the Ru0 and RuO2 references. | ||
The differences in the extent of aggregation, the oxidation state and the coordination environment of Ru are expected to influence also the reactivity of Ru. Hence, the reducibility of the materials was investigated by temperature programmed reduction (H2-TPR). The H2-TPR profiles of Ru_H2 and RuP_H2 shown in Fig. 3 confirmed that the different Ru dispersion and aggregation also affected the reducibility of Ru. The P-free sample presented a reduction peak at 195 °C, while highly dispersed Ru in sample RuP_H2 was already reduced at 98 °C, in agreement with the increased difficulty to reduce small oxide particles than large particles. The opposite behavior displayed by the small Ru domains present on RuP_H2 reveals that phosphorous decreases the activation barrier for hydrogen dissociation on small Ru clusters.
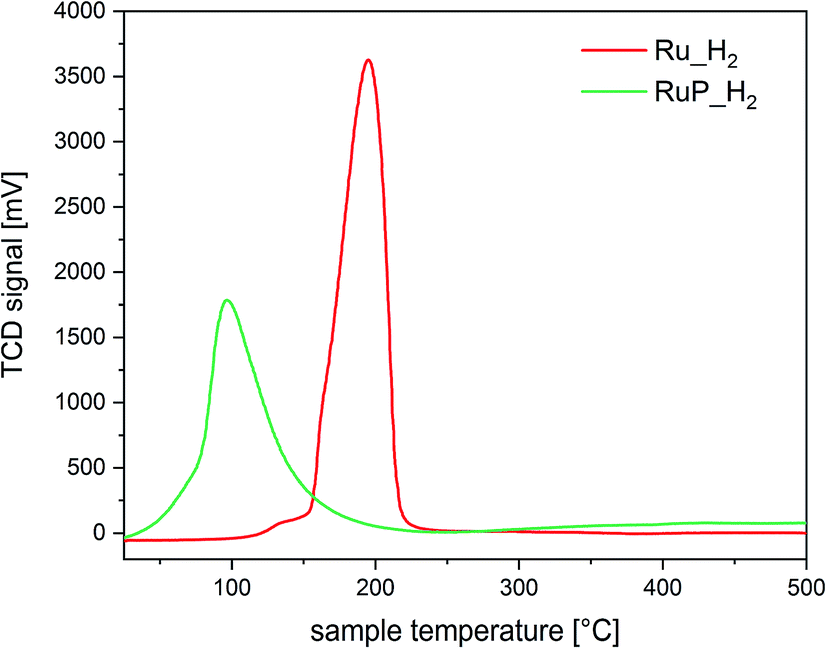 | ||
| Fig. 3 H2-TPR of Ru_H2 and RuP_H2. | ||
Based on the characterization focused on Ru species, it appears clear that phosphorus induces an improvement of the dispersion of Ru on alumina. Phosphorus groups stabilize Ru species of low nuclearity, which are characterized by a more pronounced tendency to split hydrogen compared to larger Ru species immobilized on P-free alumina.
Understanding the role of phosphorous includes also analysis of possible changes in the acidity of the samples. The type of acidity (Brønsted or Lewis) of Ru_H2 and RuP_H2 was investigated by adsorption of pyridine in cyclohexane solvent using ATR-IR (Fig. 4a). The spectra of both samples (Fig. S3†) exhibited signals of pyridine coordinated to Lewis acid sites (LAS) at 1448 cm−1 (ν19b) and 1610 cm−1 (ν8a).48 The vibrations at 1540 cm−1 (ν19b mode) and 1637 cm−1 (ν8a) corresponding to the pyridinium ion coordinated to Brønsted acid sites (BAS) were negligible. Despite the similarities, significant information was obtained from the inspection of the band at ca. 1490 cm−1, which is attributed to the interaction of pyridine with both BAS and LAS. The intensity of this signal is typically higher than that of the signal at 1540 cm−1 (BAS). It was centered at 1491 cm−1 in the case of pyridine adsorbed on RuP_H2, while the position changed to 1485 cm−1 in the case of Ru_H2 (Fig. 4a). While the signal was asymmetric in both samples, the spectra obtained after pyridine desorption clearly confirmed the existence of two signals close in energy. Pyridine adsorption on H-ZSM-5, which contains mainly BAS (Fig. S3†), shows the characteristic signals of BAS at 1540 and 1490 cm−1.49 In the absence of the former signal, which is likely too weak to be detected in the spectrum of pyridine adsorbed on RuP_H2, the signal at 1490 cm−1 can be taken as evidence of a contribution from pyridine coordinated to BAS. Thus, phosphorous modified the acidity of alumina by increasing the tendency of the surface to donate protons.
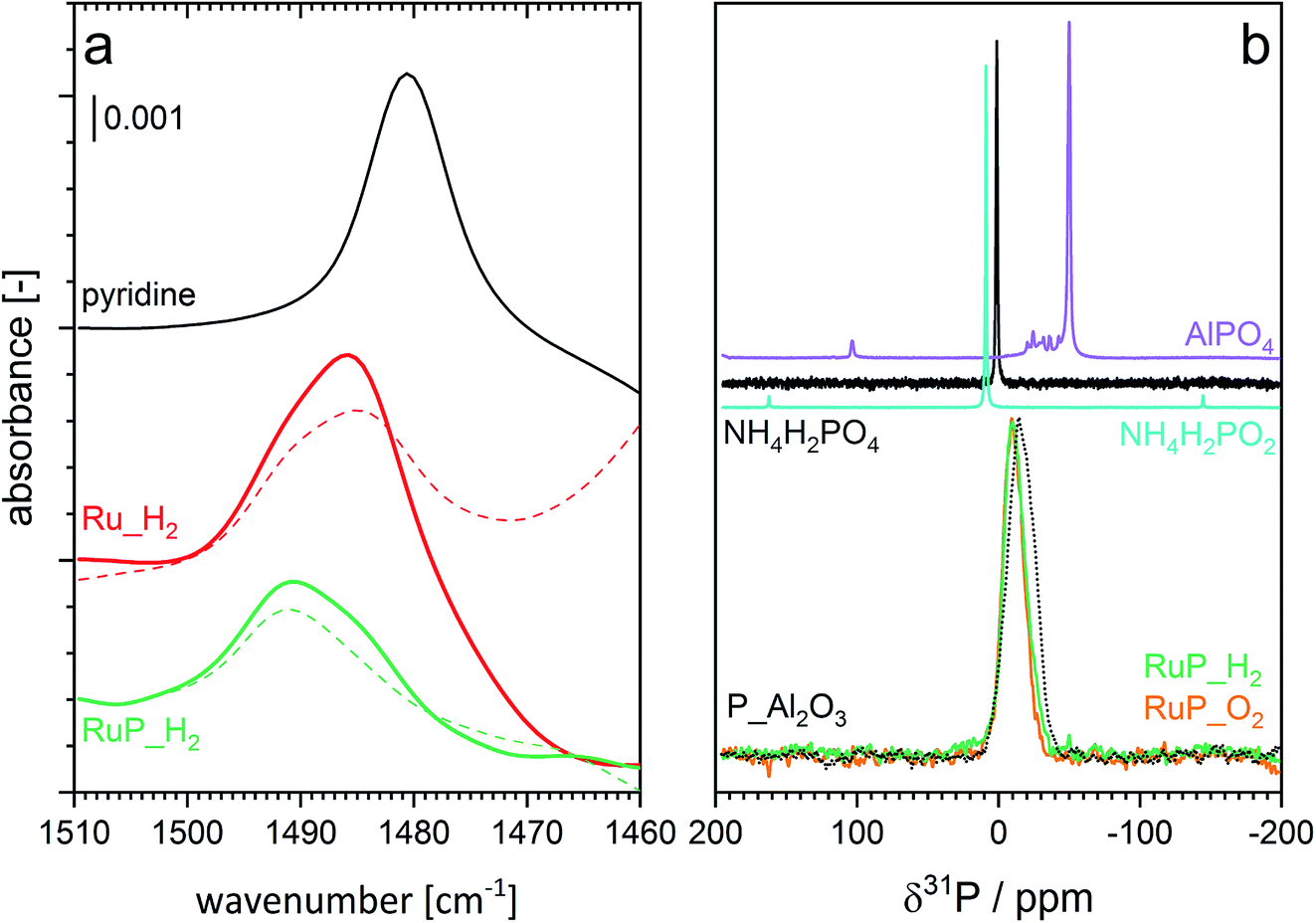 | ||
| Fig. 4 (a) Spectral region of interest of ATR-IR spectra of pyridine adsorption (solid) and desorption (dashed) on Ru_H2 and RuP_H2. Enlarged region of the spectra are provided in Fig. S3.† (b) 31P MAS NMR spectra of RuP_H2, RuP_O2 and of reference materials. | ||
The interaction between phosphorus and alumina and thus the nature of phosphorous was studied by magic angle spinning nuclear magnetic resonance (MAS-NMR). The broad resonance at −10 ppm observed in the 31P MAS-NMR spectra of RuP_H2 (Fig. 4b) can be assigned to surface phosphate species.50,51 The same line shape was noticed in the spectrum of RuP_O2, indicating that the same phosphate species must be present in both cases irrespective of the synthesis conditions. The shift from the value of 0 ppm of the NH4H2PO2 precursor provides an additional proof of the interaction of phosphorous with alumina by the formation of an amorphous aluminum phosphate species that is not visible by XRPD because it is not yet a well crystallized AlPO4 phase. The similarity of the spectrum of P_Al2O3 to those of RuP_H2 and RuP_O2 confirmed that no specific interaction with Ru could be detected. No additional information was obtained from the 27Al MAS-NMR data (Fig. S4†).
In summary, the characterization data indicate that phosphorous is present in the form of phosphate groups on the surface of alumina, likely anchoring sites for Ru species, thus ensuring the observed high metal dispersion and finally, it changes the acid properties of the surface by providing more BAS.
3.2 Catalytic activity
Table 1 shows the selectivity and activity data of the prepared catalysts compared to a commercial 5 wt% Ru supported on activated carbon (Ru/AC) as benchmark catalyst for the liquid phase catalytic transfer hydrogenation of furfural in batch conditions. We identified furfuryl alcohol as the major product for all materials.| Samplea | Activityb | Selectivityf (%) | |||
|---|---|---|---|---|---|
| Furfuryl alcohol | Tetrahydro furfuryl alcohol | 2-Methyl furan | Ethers | ||
| a Reaction conditions: F0 = 0.3 M; F/Ru ratio = 100 mol mol−1, 180 °C, 5 bar N2. F0, initial concentration of furfural.b (molF0–molFi) (molRu)−1 h−1; molF0, initial mol of F; molFi, mol of F at time i (i = 15 min).c At 30% conversion.d At 50% conversion.e At 80% conversion.f Selectivity is calculated as Sj = 100 molj (molF0–molFk)−1; molF0, initial mol of F; molFk, mol of F at conversion value k (k = 30 and 80%). | |||||
| Ru/AC | 38 | 84c | — | 12c | 3c |
| 80d | 2d | 16d | 2d | ||
| Ru_H2 | 40 | 98c | 2c | — | — |
| 96e | 1e | — | 3e | ||
| RuP_H2 | 58 | 93c | 2c | — | 5c |
| 81e | 3e | 2e | 14e | ||
| Ru_O2 | 14 | 86c | 3c | 1c | 10c |
| RuP_O2 | 18 | 70c | 18c | 1c | 9c |
Ru_O2 and RuP_O2 demonstrated the lowest activity, likely due to the fact that reduction of RuO2 needs to take place first (Fig. 5a). The hydrogen-treated sample (RuP_H2) exhibited the highest activity, 93% selectivity to furfuryl alcohol after 15 min of reaction and 58 (molF0–molFi) molRu−1 h−1 compared to 84% for the benchmark catalyst (38 (molF0–molFi) molRu−1 h−1). This improved performance can be attributed to the superior reducibility and the presence of Ru metal. Ru/Al2O3-based catalysts did not produce 2-methylfuran, which was observed in the case of Ru/AC.
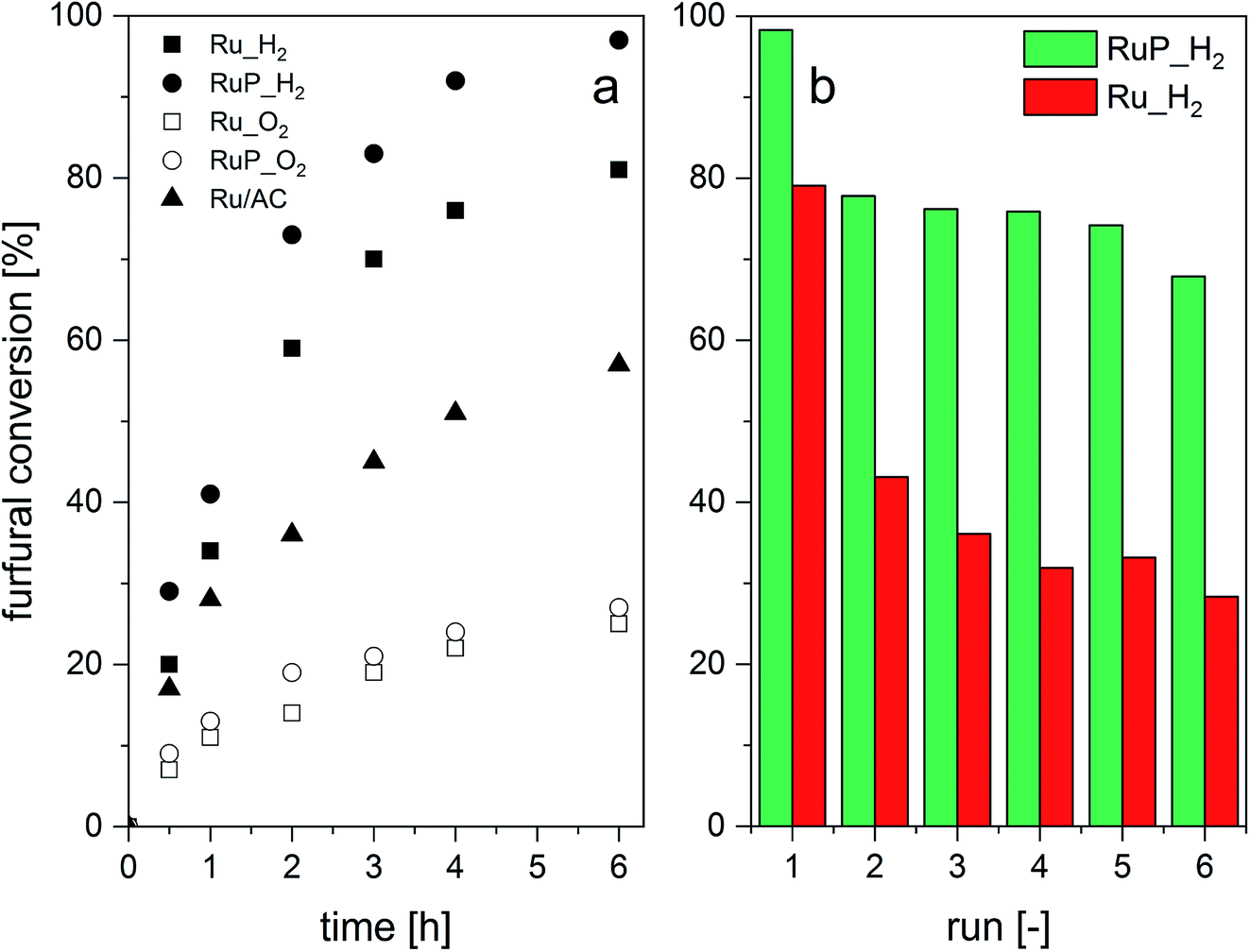 | ||
| Fig. 5 Catalytic transfer hydrogenation of furfural: (a) furfural conversion without catalyst pre-reduction and (b) consecutive leaching tests. | ||
Whether the additional acidity determined by pyridine adsorption impacts catalytic activity and selectivity cannot be completely excluded from the present data. Brønsted acidity was shown to promote etherification reactions52 and Table 1 shows that RuP_H2 promoted the production of ethers at 80% conversion of furfural (14% selectivity towards ethers) compared to the P-free catalysts but at the expense of selectivity to furfuryl alcohol. This may indicate that the presence of Brønsted acidity at the surface of the solid catalyst may be a strategy to direct the reaction towards etherification (Fig. S5†).
The stability of the catalysts to the reaction conditions and the liquid phase environment was evaluated by performing six consecutive recycling tests (Fig. 5b). Both Ru_H2 and RuP_H2 experienced a decrease in furfural conversion between the first and the second run. However, the extent of activity loss was significantly higher for the P-free catalyst. Elemental analysis of the filtered solution evidenced 4% of Ru leaching in the case of RuP_H2 between the first and the second run and no further leaching in the subsequent runs. On the contrary, Ru_H2 leached 10% of Ru between the first and the second run, and another 5% in the subsequent catalytic test. For comparison, 7% of Ru leached into the solution in the case of Ru/AC in the same sequence.
These results indicate that besides increasing the dispersion and the reducibility of Ru, phosphorus strengthens the interaction between Ru and alumina. We consider this behavior a result of the increased dispersion of the active phase and the presence of phosphate groups.
The effect of reduction of oxidized Ru species on the activity was investigated by reducing the catalysts (7 bar H2, 150 °C, 1 h) in situ prior to reaction. The catalytic activity and product selectivity are summarized in Table 2. The beneficial effect of the pre-reduction is evident from comparison with the data of Table 1. RuP_O2, in particular, reproduced the same conversion profile of the reduced counterpart RuP_H2 (Fig. S6†), thus confirming the hypothesis that metallic ruthenium favors the hydrogenation reaction. In situ reduction produced a slight decrease in the activity of RuP_H2, likely in agreement with the observation that the co-existence of RuOx species and Ru metal has a positive impact on the performance of Ru-based catalysts.53 Therefore, the lower activity of pre-reduced RuP_H2 is ascribable to the complete reduction of the dispersed RuOx species (most likely Ru–OH species as indicated by EXAFS) present in the pristine catalyst. Whether remainders of oxidized Ru were present on Ru_O2 and RuP_O2 after reduction was not ascertained.
| Samplea | Activityb | Selectivitye (%) | |||
|---|---|---|---|---|---|
| Furfuryl alcohol | Tetrahydro-furfuryl alcohol | 2-Methyl-furan | Ethers | ||
| a Reaction conditions: F0 = 0.3 M; F/Ru ratio = 100 mol mol−1, 180 °C, 5 bar N2. F0, initial concentration of furfural.b (molF0–molFi) (molRu)−1 h−1; molF0, initial mol of F; molFi, mol of F at time i (i = 15 min).c At 30% conversion.d At 80% conversion.e Selectivity is calculated as Sj = 100 molj (molF0–molFk)−1; molF0, initial mol of F; molFk, mol of F at conversion value k (k = 30 and 80%). | |||||
| Ru/AC | 42 | 80c | — | 18c | 1c |
| 76d | 2d | 20d | 1d | ||
| Ru_H2 | 38 | 84c | 4c | — | 10c |
| RuP_H2 | 48 | 88c | 1c | — | 11c |
| Ru_O2 | 30 | 70c | 18c | 1c | 9c |
| RuP_O2 | 42 | 62c | 17c | 2c | 17c |
Operando XAS experiments were performed on the most active of the pristine catalysts, i.e. Ru_H2 and RuP_H2 to monitor the Ru speciation under reaction in flow. Fig. 6 shows the FT-EXAFS spectra obtained while heating an Ar-saturated 2-propanol solution of furfural to 180 °C. While the XANES region (Fig. S7†) demonstrates the reduction of both samples to Ru metal by a weakening of the white line at higher temperature, Fig. 6a shows the appearance of a Ru–Ru shell of the metal phase in RuP_H2 at ca. 2.5 Å with a simultaneous gradual disappearance of the Ru–O shell (at ca. 1.5 Å). This process also occurs on Ru_H2, but the reduction of the oxidized phase contributes to the increase in the fraction of Ru metal already present in the catalyst (at ca. 2.5 Å, Fig. 6b). The less intense and broader contribution of the first Ru–Ru shell observed for RuP_H2 is an indication that the Ru particles formed upon reduction are smaller than in Ru_H2, which is confirmed by the values of the Ru coordination number obtained from the fit of the first Ru–Ru shell (Table 3) of both catalysts (EXAFS fits are shown in Fig. S8†). Moreover, it is evident that the Ru–O species do not completely disappear during reaction in both catalysts, which can be related to the need of mixed RuOx/Ru interfaces.54 After 30 min at 180 °C, furfural conversion was 98% on both RuP_H2 and Ru_H2.
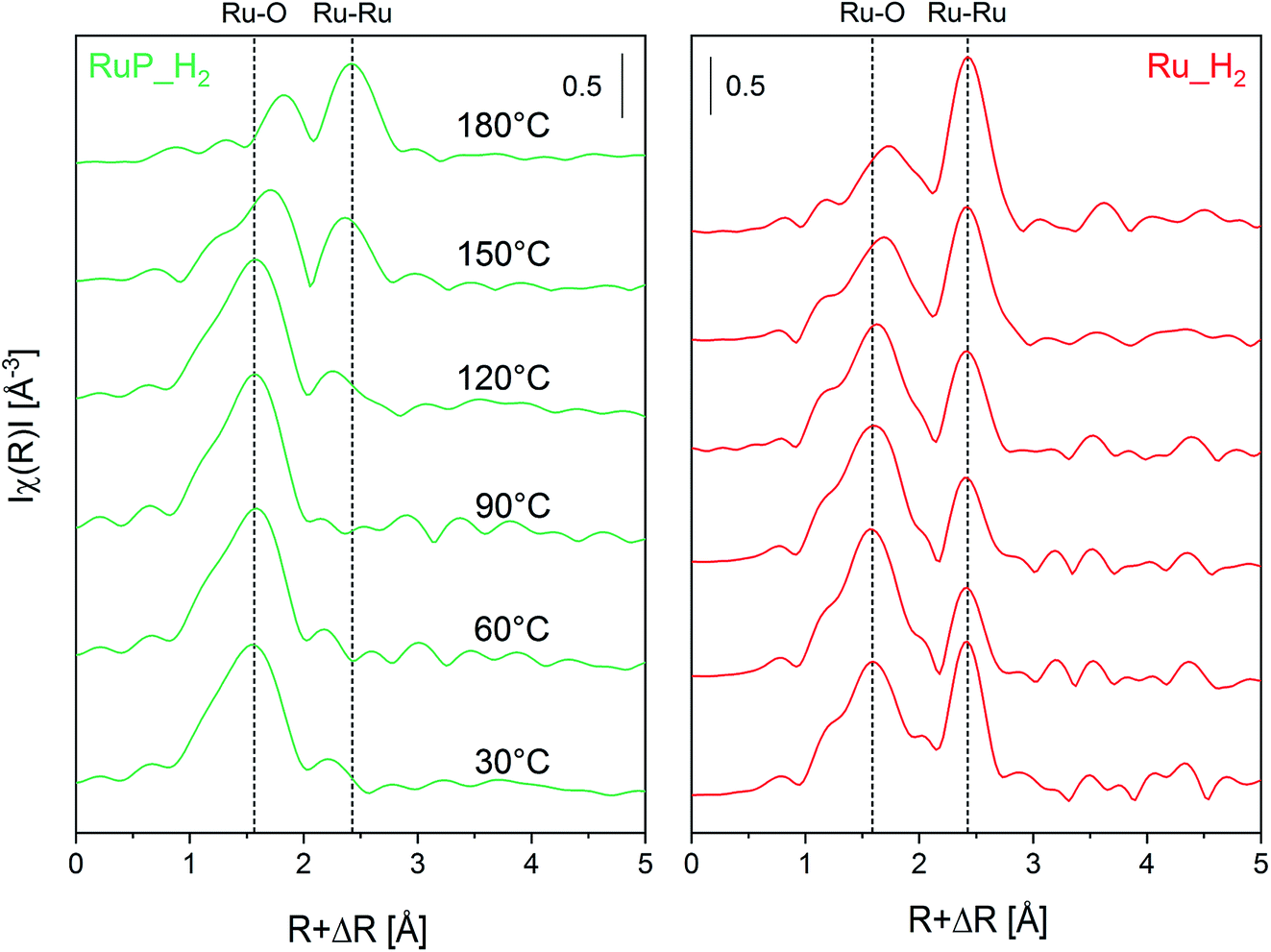 | ||
| Fig. 6 Operando non-phase shift-corrected FT-EXAFS spectra at the Ru K-edge under reaction conditions of Ru_H2 and RuP_H2 determined from 30 °C to 180 °C. Conditions: 5 mM furfural in Ar-saturated 2-propanol at a flow rate of 0.2 mL min−1, under 16 bar and with 35 mg of material. | ||
| Sample | CNa | Rb (Å) | DWc (Å2) | ΔEd (eV) | R factore | Average sizef (nm) |
|---|---|---|---|---|---|---|
a Coordination number.b Interatomic distance.c Debye–Waller factor.d Shift in the edge energy.e Defined as 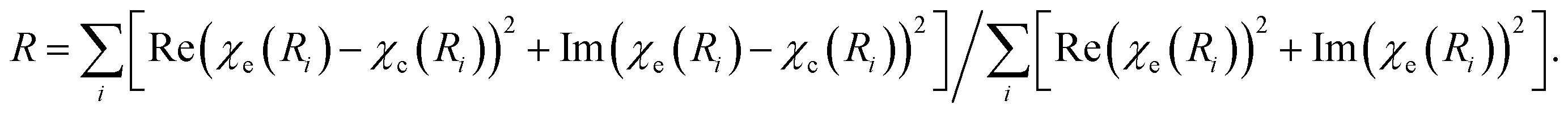 f The average size was calculated using the hcp model as described in ref. 55. f The average size was calculated using the hcp model as described in ref. 55. |
||||||
| Ru_H2 | 8.12 ± 1.1 | 2.67 ± 0.01 | 0.006 ± 0.001 | 3.7 ± 1.0 | 0.024 | 1.2 |
| RuP_H2 | 4.92 ± 1.0 | 2.67 ± 0.01 | 0.009 ± 0.002 | 3.6 ± 1.4 | 0.019 | 0.7 |
4 Conclusion
In conclusion, the results from the characterization and from catalytic activity data indicate that addition of phosphorous changed significantly the dispersion and reducibility of Ru. Also, the type of acid site of the Al2O3 support was modified from a Lewis-based material towards a more Brønsted-based material. While it is difficult to disentangle which effect is more critical for catalytic activity and selectivity because of the vastly different Ru particle size in these catalysts, these combined effects improved the activity of Ru/Al2O3 towards catalytic transfer hydrogenation of furfural to furfuryl alcohol. Further systematic study is needed to disentangle the role of the various parameters involved, such as particle size and P loading effects. Moreover, the nature of the binding mode of P to Al2O3 and possibly to Ru should also be addressed in future studies. Phosphorous improved also the stability of Ru by reducing Ru leaching into the solution significantly.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are grateful to the Paul Scherrer Institut (CROSS project) and the Competence Center for Energy and Mobility (CCEM, project ARRMAT-PLUS) for financial support. Beamtime allocation at beamlines SNBL (ESRF, France) and SuperXAS (SLS, Switzerland) is kindly acknowledged. The NMR hardware was partially granted by the Swiss National Science Foundation (SNSF, grant no. 206021_150638/1). D. F. thanks Dr M. Schöneborn for providing the alumina sample and Dr C. Proff for the TEM measurements. Dr D. Kuzmenko is thanked for support with the EXFAS fit.References
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- G. Centi and R. A. van Santen, Catalysis for Renewables, Wiley, 2007 Search PubMed.
- H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2007 Search PubMed.
- G. M. Lopez and A. D. Martin, Furfural: An Entry Point of Lignocellulose in Biorefineries to Produce Renewable Chemicals, Polymers, and Biofuels, World Scientific Publishing Company, 2018 Search PubMed.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- Ashland Oil Inc OP, US 1557479 A 19790226, 1981.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- Á. O'Driscoll, J. J. Leahy and T. Curtin, Catal. Today, 2017, 279, 194–201 CrossRef.
- M. Audemar, C. Ciotonea, K. De Oliveira Vigier, S. Royer, A. Ungureanu, B. Dragoi, E. Dumitriu and F. Jérôme, ChemSusChem, 2015, 8, 1885–1891 CrossRef CAS PubMed.
- B.-J. Liaw, S.-J. Chiang, C.-H. Tsai and Y.-Z. Chen, Appl. Catal., A, 2005, 284, 239–246 CrossRef CAS.
- W. Gong, C. Chen, R. Fan, H. Zhang, G. Wang and H. Zhao, Fuel, 2018, 231, 165–171 CrossRef CAS.
- M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef CAS.
- R. E. Palmer, N. Jian, A. Villa, N. Dimitratos, C. R. A. Catlow, A. Chutia, P. P. Wells, C. E. Chan-Thaw, S. M. Rogers, M. Perdjon and A. Thetford, ACS Catal., 2017, 7, 2266–2274 CrossRef.
- C. Ramirez-Barria, M. Isaacs, K. Wilson, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 2018, 563, 177–184 CrossRef CAS.
- J. Su, Y. Yang, G. Xia, J. Chen, P. Jiang and Q. Chen, Nat. Commun., 2017, 8, 14969 CrossRef PubMed.
- M. Hoang, C. Evangelisti, P. Pertici, G. Vitulli, A. Maria Caporusso, G. Marconi, T. W. Turney and G. Capannelli, J. Organomet. Chem., 2004, 689, 639–646 CrossRef.
- C. Kellner, J. Catal., 2004, 75, 251–261 CrossRef.
- L. Kępiński, L. Krajczyk, J. Okal, M. Zawadzki and W. Tylus, Appl. Catal., A, 2006, 319, 202–209 Search PubMed.
- P. Betancourt, A. Rives, R. Hubaut, C. E. Scott and J. Goldwasser, Appl. Catal., A, 1998, 170, 307–314 CrossRef CAS.
- M. Oubenali, G. Vanucci, B. Machado, M. Kacimi, M. Ziyad, J. Faria, A. Raspolli-Galetti and P. Serp, ChemSusChem, 2011, 4, 950–956 CrossRef CAS PubMed.
- P. Panagiotopoulou, N. Martin and D. G. Vlachos, ChemSusChem, 2015, 8, 2046–2054 CrossRef CAS PubMed.
- J. M. Tukacs, M. Bohus, G. Dibó and L. T. Mika, RSC Adv., 2017, 7, 3331–3335 RSC.
- J. Xu, F. Liu, A. Wang, B. Qiao, S. Miao, Y. Su, H. Sun, L. Li, T. Zhang, Y.-T. Cui, L. Li, R. Lang, F. Jerome and Q. Liu, Green Chem., 2018, 20, 1770–1776 Search PubMed.
- J. Jae, W. Zheng, R. F. Lobo and D. G. Vlachos, ChemSusChem, 2013, 6, 1158–1162 CrossRef CAS PubMed.
- C. Michel and P. Gallezot, ACS Catal., 2015, 5, 4130–4132 CrossRef CAS.
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321–329 CrossRef CAS.
- A. Villa, C. E. Chan-Thaw, S. Campisi, C. L. Bianchi, D. Wang, P. G. Kotula, C. Kübel and L. Prati, Phys. Chem. Chem. Phys., 2015, 17, 28171–28176 RSC.
- G. J. Hutchings, Catal. Lett., 2001, 75, 1–12 CrossRef CAS.
- J. Yang, J. Ma, Q. Yuan, P. Zhang and Y. Guan, RSC Adv., 2016, 6, 92299–92304 RSC.
- C. P. Jiménez-Gómez, J. A. Cecilia, F. I. Franco-Duro, M. Pozo, R. Moreno-Tost and P. Maireles-Torres, Mol. Catal., 2018, 455, 121–131 CrossRef.
- J. Zhang, X. Mu, B. Zhang, B. Fei, X. Liu and X. Chen, Faraday Discuss., 2017, 202, 79–98 RSC.
- Z. Gao, L. Yang, G. Fan and F. Li, ChemCatChem, 2016, 8, 3769–3779 CrossRef CAS.
- J. Xi, Y. Zhang, Q. Xia, X. Liu, J. Ren, G. Lu and Y. Wang, Appl. Catal., A, 2013, 459, 52–58 CrossRef CAS.
- M. Lou, R. Wang, J. Zhang, X. Tang, L. Wang, Y. Guo, D. Jia, H. Shi, L. Yang, X. Wang, Z. Sun and T. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6431–6441 CrossRef CAS PubMed.
- Z. C. Ding, C. Y. Li, J. J. Chen, J. H. Zeng, H. T. Tang, Y. J. Ding and Z. P. Zhan, Adv. Synth. Catal., 2017, 359, 2280–2287 CrossRef CAS.
- J. Tuteja, H. Choudhary, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 96–100 CrossRef CAS PubMed.
- Z. Zhang, J. Sun, M. Dou, J. Ji and F. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16236–16242 CrossRef CAS PubMed.
- F. J. L. Heutz, C. Erken, M. J. B. Aguila, L. Lefort and P. C. J. Kamer, ChemCatChem, 2016, 8, 1896–1900 CrossRef CAS.
- S. Campisi, S. Bennici, A. Auroux, P. Carniti and A. Gervasini, Top. Catal., 2018, 61, 1939–1948 CrossRef CAS.
- Q. Liang, F. Shi, X. Xiao, X. Wu, K. Huang and S. Feng, ChemCatChem, 2018, 10, 2179–2183 CrossRef CAS.
- M. Martinelli, M. Kumaran Gnanamani, S. D. Hopps, D. E. Sparks, A. MacLennan, Y. Hu, B. H. Davis and G. Jacobs, ChemCatChem, 2018, 10, 3709–3716 CrossRef CAS.
- R. Radhakrishan, D. M. Do, S. Jaenicke, Y. Sasson and G. K. Chuah, ACS Catal., 2011, 1, 1631–1636 CrossRef CAS.
- M. Machida, Chem. Rec., 2016, 16, 2219–2231 CrossRef CAS PubMed.
- Y.-M. Chai, K. Yan, C.-G. Liu, J. Lin, J. Qin, B. Liu, J. Chi, B. Dong and W. Gao, ChemSusChem, 2017, 11, 743–752 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- G. L. Chiarello, M. Nachtegaal, V. Marchionni, L. Quaroni and D. Ferri, Rev. Sci. Instrum., 2014, 85, 074102 CrossRef PubMed.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- N. S. Gould and B. Xu, Chem. Sci., 2018, 9, 281–287 RSC.
- S. K. Matam, E. H. Otal, M. H. Aguirre, A. Winkler, A. Ulrich, D. Rentsch, A. Weidenkaff and D. Ferri, Catal. Today, 2012, 184, 237–244 CrossRef CAS.
- E. W. Shin, J. S. Han, M. Jang, S. H. Min, J. K. Park and R. M. Rowell, Environ. Sci. Technol., 2004, 38, 912–917 CrossRef CAS PubMed.
- H. Nguyen, N. Xiao, S. Daniels, N. Marcella, J. Timoshenko, A. Frenkel and D. G. Vlachos, ACS Catal., 2017, 7, 7363–7370 CrossRef CAS.
- A. V. Mironenko and D. G. Vlachos, J. Am. Chem. Soc., 2016, 138, 8104–8113 CrossRef CAS PubMed.
- M. J. Gilkey, P. Panagiotopoulou, A. V. Mironenko, G. R. Jenness, D. G. Vlachos and B. Xu, ACS Catal., 2015, 5, 3988–3994 CrossRef CAS.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00415d |
| ‡ These authors contributed equally to the work. |
| § Present address: CheMa Laboratories, Xenofontos 2, GR-Korinthos 201 31, Greece. |
| ¶ Present address: École polytechnique fédérale de Lausanne (EPFL), Institute of Chemical Sciences and Engineering, CH-1015 Lausanne, Switzerland. |
| This journal is © The Royal Society of Chemistry 2020 |