
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of highly dispersed ultrafine Pd nanoparticles on a porous organic polymer for hydrogenation of CO2 to formate†
Xianzhao Shao*,
Xinyi Miao,
Xiaohu Yu,
Wei Wang and
Xiaohui Ji
and
Xiaohui Ji
Shaanxi Key Laboratory of Catalysis, School of Chemistry and Environment Science, Shaanxi University of Technology, Hanzhong 723001, Shaanxi, China. E-mail: xianzhaoshao@snut.edu.cn
First published on 4th March 2020
Abstract
Precise design of catalytic supports is an encouraging technique for simultaneously improving the activity and stability of the catalyst. However, development of efficient heterogeneous catalysts for transforming CO2 into formic acid (FA) is still a big challenge. Herein, we report that Pd nanoparticles (NPs) based on a porous organic polymeric support containing amide and pyridine functional groups (AP-POP) can be an efficient catalyst for selective hydrogenation of CO2 to form formate with high efficiency even under mild reaction conditions (6.0 MPa, 80 °C). Electron density of the active Pd species modulated via the interaction between pyridine nitrogen and Pd play important roles in dramatic enhancement of catalytic activity and was indicated by X-ray photoelectron spectroscopy (XPS) along with CO chemisorption. This work provides an interesting and effective strategy for precise support design to improve the catalytic performance of nanoparticles.
Introduction
The catalytic conversion of inorganic carbon dioxide (CO2) to organic matter plays a vital role in the carbon cycle and energy metabolism in nature, and thus provides new avenues for solving the problem of CO2 emission and energy conversion/storage in the modern lifestyle of human beings.1–3 Among the various reduction products, formic acid (FA) is one of the most significant due to its direct applications as a chemical component or as a renewable fuel and it is present in the form of formate during practical production for shifting the thermodynamic equilibrium forward.4–7 FA has been proposed as an efficient and safe delivery vehicle for fuel cell technologies with special emphasis in biomass degradation.8 Additionally, the formation of FA via hydrogenation of CO2 is an important and promising means in order to realize carbon-neutral hydrogen energy cycle, where CO2 is consumed to produce FA and is recycled from H2 release.9–15 There has been tremendous achievement in the recent few years on the application of Ir and Ru-based mono-nuclear complexes as catalysts in homogenous conversion of CO2 to formate.16–21 However, practical applications of homogenous catalysts in this field have been difficult to achieve due to various problems including the high-cost of molecular Ir complex based catalysts, limitation of separability as well as the reverse decomposition of the generated formate. The concept of insoluble matrix based immobilization of homogenous catalysts for conversion of FA by hydrogenation of CO2 is based on the maintenance of activity of the catalyst as well as easy separation.22–25 However, these catalysts suffer from various problems including deactivation of the catalyst, low turnover numbers and leaching of the metallic catalysts.Heterogeneous catalysts, in spite of its merit in separation, have been scarcely reported on the formation of formic acid/formate by hydrogenation of CO2 due to poor catalytic performance.26–31 Therefore, development of heterogeneous catalyst with well-defined properties is required in order to promote catalytic activity for the CO2 hydrogenation and to enhance long-term stability performance. The metal-support interactions, between specific functional groups introduced into the support structures which enable tailoring the electronic and geometric properties, is promising design strategy for metal nanoparticles based catalysts. Among all, nitrogen-doped carbon material is the preferred choice due to combined effects of its large surface area, variety of N chemical configurations and interactions with immobilized metal nanoparticles. Several recent studies have demonstrated that metal nanoparticles binding on the surface of N-doped carbon supports is an efficient method for improving the catalytic activity.32–34 However, the relation between metal nanoparticles and the support materials is still uncertain due to the inhomogeneous surface morphology and chemical properties of the carbon supports.
Porous organic polymers (POPs), in recent years have been recognized as versatile supports for multifunctional solid catalysts owing to large surface area, high stability and well-designed organic framework.35–38 Porous organic polymers containing nitrogen functionalities in the framework would not only provide binding sites for stabilizing the metal nanoparticles, but also provide the option for selective CO2 uptake via interactions which are Lewis acid–Lewis base types,39 leading to designing of multifunctional solid catalysts with the possibilities introduced by the organic ligands. Herein, a porous organic polymer with pore surface containing the amide and pyridine functional groups (AP-POP) was designed and it was envisaged that the central nitrogen neutral donor atoms of pyridine groups along with the amide groups will not only stabilize, but also tune the electronic structure of Pd NPs by the interaction between pyridine nitrogen and Pd. As a result, the Pd NPs catalyst supported on AP-POP exhibits efficient catalytic hydrogenation of CO2 to formate with high efficiency. The interaction of support with Pd results in electronically modified Pd nanoparticles resulting to high catalytic efficiency.
Experimental
Materials and characterization
The chemicals required in this study were availed from various suppliers and were used without further purification. 1,3,5-Benzenetricarbonyl chloride, 2,6-diaminopyridine and triethlyamine were obtained from Sigma-Aldrich. Palladium(II) chloride was purchased from Acros Organics whereas CO2 (99.99%) and H2 (99.99%) were obtained from Date Gas Industries. Various instrumental techniques like elemental analysis, solid state NMR, FTIR, TG-DTA, XRD, BET, XPS, SEM were used to characterize the synthesized catalysts. The details of all the above mentioned instrumental methods can be found in a recent literature report.40 High resolution transmission electron microscopy and scanning transmission electron microscopy (STEM) images were obtained using a JEM-2100F electron microscope at 200 kV. CO pulse adsorption method performed by Micromeritics AutoChem II 2920 automated catalyst characterization system was performed to measure metal adsorption.Synthesis of Pd/AP-POP catalyst
The methodology for preparation of the porous organic polymer (AP-POP) based on amino pyridine is described in an earlier report.40 A 0.5 g quantity of the AP-POP was added to 100 mL of a methanol solution of PdCl2 with 1 mol L−1 HCl solution containing 25 mg of Pd component after which the mixture was stirred vigorously at 70 °C for 12 h. A 20-fold excess of aqueous NaBH4 was added, followed by stirring for 1 h. The product was collected by filtration, washed several times with water then ethanol, and dried under vacuum at 50 °C for 12 h. Pd/AC and Pd/C3N4 catalysts were also prepared using the same route. The schematic procedure for the preparation of Pd/AP-POP catalyst is shown in Scheme S1.† The chemical composition and textural properties of selected materials were compared in Table S1.†General procedure for CO2 hydrogenation to formic acid/formate
The catalytic activity of the catalyst was followed according to the procedure described earlier using 20 mg of the Pd catalyst.40 For HPLC determination of formic acid, the mobile phase was 0.005 M H2SO4. The other parameters for HPLC measurement were identical to the earlier report.40 Turnover number (TON) based on all employed Pd atoms were determined according to TON = [produced moles of FA after 12 h]/[moles of total Pd], TON based on Pd atoms which were exposed in the surface were calculated by TON = [produced moles of formate after 12 h]/[moles of total Pd]/D, where D is the dispersion of metal atoms on the support surface. D value is roughly calculated by D = 1.12/d (mean diameter of Pd nanoparticles, nm).Results and discussion
As illustratively shown in Scheme S1,† in a solution mixture of dichloromethane and triethylamine (TEA), the porous organic polymers were reproducibly synthesized by the covalent coupling of 1,3,5-benzenetricarbonyl chloride (TMC) with 2,6-diaminopyridine (DAP) to reticulate aminopyridine functional groups into the POP, which was denoted as AP-POP. The as-prepared polymer was then characterized and confirmed by FTIR, solid-state 13C/CPMAS NMR, and XPS.For AP-POP, the disappearance of C–Cl stretching at 700 cm−1 along with the presence of stretching vibrational frequencies of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1680 cm−1) and N–H (1528 cm−1) of amide bond indicated the complete condensation of TMC and DAP. The peaks at 1514 cm−1 and 1395 cm−1 corresponds to N–H in plane bending and C–N stretching vibrations of amide groups (Fig. S1†). Further, the structure of AP-POP was also analyzed by 13C cross-polarization magic-angle-spinning (CP-MAS) solid state NMR (Fig. S2†). The presence of peaks at 164 ppm corresponds to the amide carbonyl, and all other peaks between 109 and 150 ppm correspond to the aromatic carbons from phenyl and pyridyl moieties.41 In terms of physical texture, the morphology of the particles of AP-POP material was spherical (SEM, Fig. S3†) and amorphous in nature (PXRD, Fig. S4†). The distribution of the mesopores was around 7.6 nm with a corresponding surface area of 43 m2 g−1 (Fig. S5†), with the thermal decomposition study indicating thermal stability below 300 °C (Fig. S6†).
O (1680 cm−1) and N–H (1528 cm−1) of amide bond indicated the complete condensation of TMC and DAP. The peaks at 1514 cm−1 and 1395 cm−1 corresponds to N–H in plane bending and C–N stretching vibrations of amide groups (Fig. S1†). Further, the structure of AP-POP was also analyzed by 13C cross-polarization magic-angle-spinning (CP-MAS) solid state NMR (Fig. S2†). The presence of peaks at 164 ppm corresponds to the amide carbonyl, and all other peaks between 109 and 150 ppm correspond to the aromatic carbons from phenyl and pyridyl moieties.41 In terms of physical texture, the morphology of the particles of AP-POP material was spherical (SEM, Fig. S3†) and amorphous in nature (PXRD, Fig. S4†). The distribution of the mesopores was around 7.6 nm with a corresponding surface area of 43 m2 g−1 (Fig. S5†), with the thermal decomposition study indicating thermal stability below 300 °C (Fig. S6†).
Supported Pd catalysts were synthesized using a conventional impregnation methodology with PdCl2 as precursor and methanol as solvent. The sample was subsequently reduced by NaBH4 without calcination. With the aim of comparison, different supports, activated carbon (AC) and carbon nitride (C3N4), were also employed for depositing Pd using the identical procedure. The chemical composition and textural properties of selected materials were compared in Table S1.† The actual Pd loading determined by ICP-OES analysis was found to be 3.2, 3.3 and 3.5 wt% on Pd/AC, Pd/C3N4 and Pd/AP-POP, respectively. XRD pattern of all the supports suggested high Pd dispersion as corroborated by absence of Pd nanoparticles on all of them (Fig. S7†). On the basis of indexed diffraction patterns, the average size of the Pd nanoparticles were 1.4 nm for Pd/AP-POP, 2.4 nm for Pd/AC and 1.9 nm on Pd/C3N4 and, respectively, as evident from Fig. 1 and S8.† The HRTEM images of Pd/AP-POP catalyst reveal lattice fringe of 2.20 Å for the nanoparticles corresponding to the spacing of metallic Pd{111} crystal plane.42 The scanning transmission electron microscopy (STEM)-energy dispersive spectroscopy (EDS) mapping further showed that C, N, O and Pd were all uniformly distributed over the Pd/AP-POP catalyst (Fig. 1c–g).
 | ||
| Fig. 1 STEM, HRTEM images (a and b) and the corresponding EDS mapping (c–g) of Pd/AP-POP. | ||
XPS measurements were carried out to investigate the chemical states of surface elements in the three samples. InPd/AP-POP (Fig. 2a), the two peaks located at 335.4 and 340.7 eV can be assigned to the metallic Pd species, whereas the other doublet peaks at 337.2 and 342.5 eV are assigned to Pd2+.43 Interestingly, these Pd0 signals for Pd/C3N4 are located at 335.6 and 340.9 eV and for Pd/AC, they are located at 335.8 and 341.1 eV, with the reason can be charge transfer from the support to Pd. In order to understand this interaction, the XPS spectra were also analyzed for N (Fig. 2b). The N 1s XPS spectra results indicated that the pyridinic N for Pd/AP-POP is located at 398.8 eV, about 0.2 eV higher than that for AP-POP and thus verifying the charge redistribution and the change of Pd state. It should be noted that there are two types of N species having atomic ratio of nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and they were ascribed to the pyridinic and amide (399.8 eV) groups in AP-POP and Pd/AP-POP. We also noticed that the binding energy of O 1s showed no visible changes (Fig. S9†). In brief, XPS revealed that Pd NPs in the Pd/AP-POP were having higher electron density due to charge transfer from the support.
:2, and they were ascribed to the pyridinic and amide (399.8 eV) groups in AP-POP and Pd/AP-POP. We also noticed that the binding energy of O 1s showed no visible changes (Fig. S9†). In brief, XPS revealed that Pd NPs in the Pd/AP-POP were having higher electron density due to charge transfer from the support.
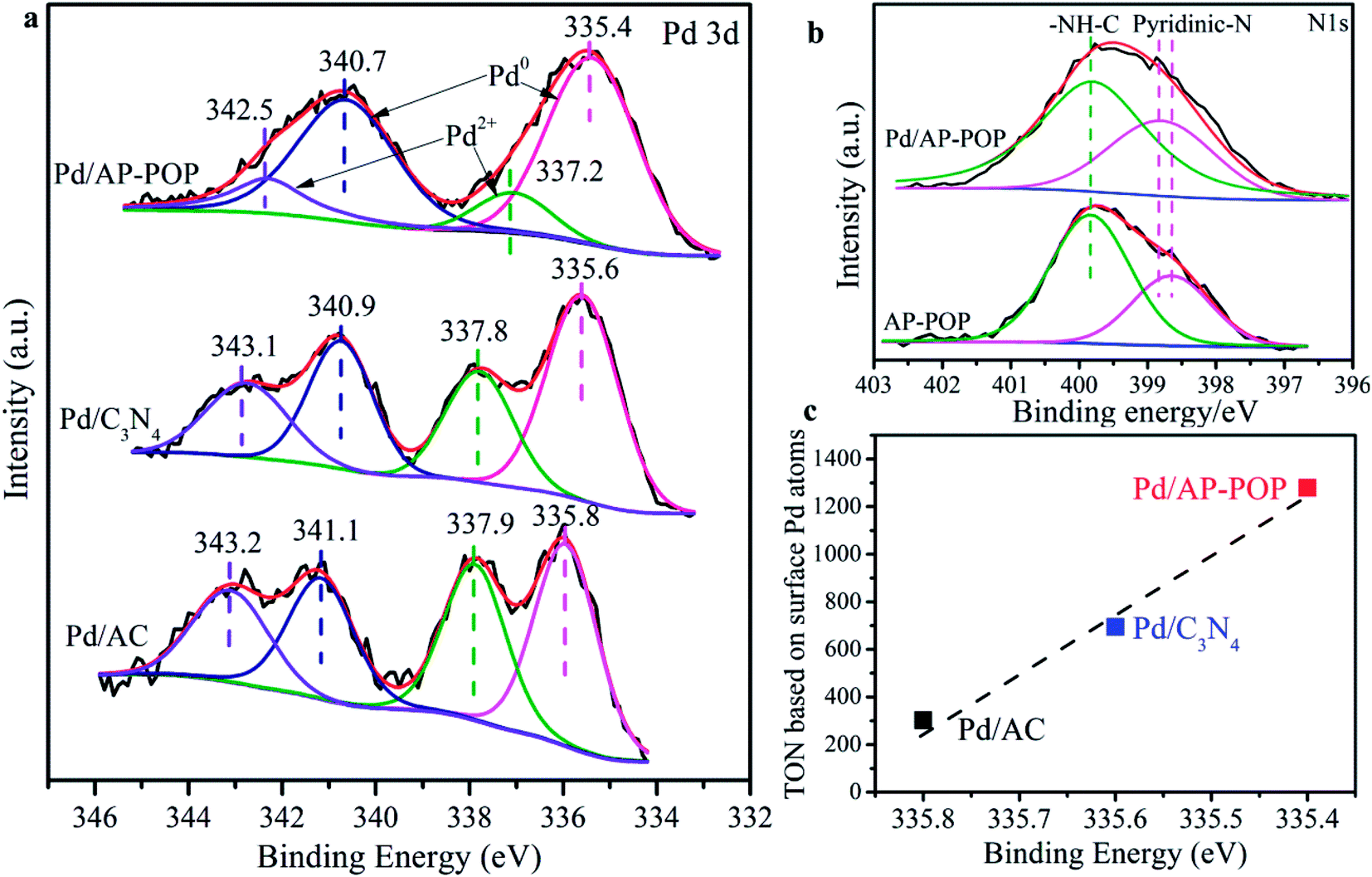 | ||
| Fig. 2 (a) High-resolution XPS spectra of different samples in the Pd 3d region. (b) XPS spectra of AP-POP and Pd/AP-POP in the N 1s region. (c) Relationship between the TON for CO2 hydrogenation based on surface exposed Pd atoms (as determined by STEM) and the Pd 3d binding energy (as determined by XPS). | ||
The activity of Pd based catalysts for the conversion of formate by hydrogenation of CO2 is shown in Fig. 3 and Table 1. The hydrogenation was carried out at 80 °C temperature over a period of 12 h in a stainless steel reactor (30 mL) when the pressure was maintained at 6.0 MPa (H2:CO2 = 1:1). The reaction medium was consisting of an aqueous solution containing 2.0 M triethylamine (NEt3). As per Fig. 3a (entries 1–3), the catalytic activity of Pd/AP-POP catalyst is much higher compared to the Pd/C3N4 and Pd/AC which indicates the important role played by AP-POP in the hydrogenation of CO2. The nature of the support for immobilizing the metal NPs was crucial due to the possibility of providing additional heterogeneous catalytic sites by the support and simultaneously may modify the particle sizes and electronic properties of the NPs as discussed later. Additionally, at higher pressure the formate yield was increased to 83% (entry 5) which was found to be higher than the well-known homogeneous catalyst of Ir(PNP) (70%).16 As expected, the catalytic activity of the hydrogenation increased with an increase of total pressure (entries 4 and 5). This is understandable because at higher pressure the equilibrium shifts towards the product side. Furthermore, raising the H2/CO2 ratio from 1:1 to 2:1, at the same total initial pressure, did not notably affect the formate yield and turnover number (TON) of the reaction (entries 3 and 6). When the H2 pressure was lower but CO2 pressure was higher (entry 7), the catalytic activity decreased which might be due to the enhancement of CO2 solubility by the added Et3N. The formation of formate was also influenced by temperature with the formate yield increased significantly when the CO2 hydrogenation was carried out at 100 °C (entry 3 vs. 8). As depicted in Fig. 3b, TON was increased to 1023 after 12 h of reaction, while further prolonged the reaction time to 16 h led to a slight increase. It is worth noting that the Pd/AP-POP can direct hydrogenation of CO2 into formic acid even in water (entry 9) with a TON of 128, which is close to the catalyst of Ru complexes (TON 159, 60 °C, 10 MPa),19 and is greater than those recently reported for other heterogeneous catalyst systems (Table S2†). Basic nature of the amide functional groups present in the polymeric structure is the reason behind this. No catalytic activity was observed for AP-POP material in absence of Pd (entry 10). In addition, HPLC and 1H NMR spectra (Fig. S10†) proved selective formation of formate without any byproduct in all the cases.
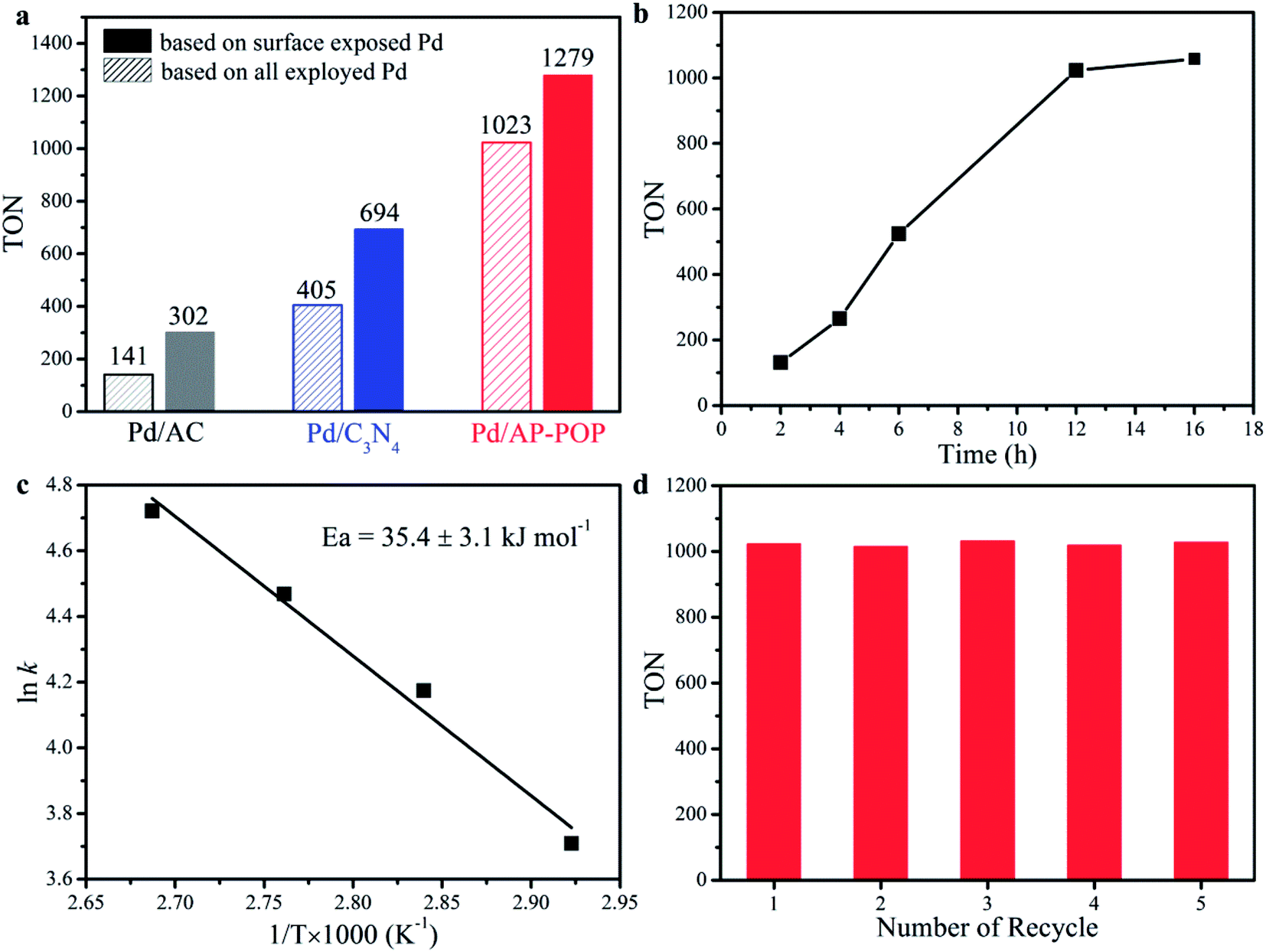 | ||
| Fig. 3 (a) Comparison of the catalytic activities of different supported Pd catalysts. (b) Times-dependent hydrogenation reaction catalyzed by Pd/AP-POP. (c) Arrhenius plots of Pd/AP-POP. (d) Recyclability of Pd/AP-POP in the CO2 hydrogenation reaction. Reaction conditions: catalyst (20 mg), 2.0 M aqueous Et3N solution (5 mL), H2:CO2 (1:1, total 6.0 MPa). TON based on all employed Pd atoms, unless stated otherwise. | ||
| Entry | Catalyst | p(H2)/p(CO2) (MPa) | Formic acid yieldb (%) | TONc@12 h |
|---|---|---|---|---|
| a Reaction conditions: catalyst (20 mg), 2.0 M aqueous Et3N solution (5 mL), 80 °C, 12 h.b Yields were calculated with respect to the base.c TON based on all employed Pd atoms.d 100 °C.e No base. | ||||
| 1 | Pd/AC | 3.0/3.0 | 8.5 | 141 |
| 2 | Pd/C3N4 | 3.0/3.0 | 25.1 | 405 |
| 3 | Pd/AP-POP | 3.0/3.0 | 67.3 | 1023 |
| 4 | Pd/AP-POP | 2.0/2.0 | 49.9 | 759 |
| 5 | Pd/AP-POP | 4.0/4.0 | 83.0 | 1262 |
| 6 | Pd/AP-POP | 4.0/2.0 | 68.4 | 1039 |
| 7 | Pd/AP-POP | 2.0/4.0 | 55.9 | 850 |
| 8d | Pd/AP-POP | 3.0/3.0 | 75.4 | 1146 |
| 9e | Pd/AP-POP | 3.0/3.0 | — | 128 |
| 10 | AP-POP | 3.0/3.0 | 0 | — |
The apparent activation energy (Ea) based on the Arrhenius plot was calculated to be 35 kJ mol−1 for Pd/AP-POP (Fig. 3c), which was comparable to the reported Pd based catalyst.31,42 Moreover, the recovered catalyst after participating in a reaction was found to have identical particle size without any significant agglomeration of the Pd NPs with the average diameter being determined to be 1.6 nm which was proved from the STEM image (Fig. S11†). Furthermore, the original activity was maintained even after fifth reaction for the spent catalyst (Fig. 3d).
The control experiments indicated that high catalytic activity for hydrogenation of CO2 is contributed both by the highly dispersed Pd and AP-POP support. CO2, a well-known linear, non-polar molecule, has two Lewis base sites (oxygen atoms) and a Lewis acid site (carbon atom) and the initial linear geometry of the molecule usually results in interaction with the electron-rich sites of AP-POP which are the surface nitrogen species as Lewis-basic sites. Notably, although BET surface area is low, the CO2 adsorption capacity in AP-POP at 273 K is 23.6 mg g−1 (Fig. S12†), meanwhile desorption curve is higher than adsorption curve, which is also confirmed interaction between CO2 and adsorbent. There is also evidently a correlation between the TON based on surface Pd atoms and the Pd 3d5/2 binding energy determined by XPS analysis (Fig. 2c). Therefore, the high catalytic activity of the Pd/AP-POP can be attributed to the modified electronic state of the Pd tuned by the electron-donating ability of the pyridinic N support. This phenomenon has been reported previously and was proved to be efficient for the hydrogenation of CO2 to formate.44–46 For instance, the strong electron-donating ability of nitrogen and phosphorus ligands has a positive significant effect for promoting the reaction.47 In earlier work, our group had proved that the AP-POP can stabilize Ir single-atom via strong metal-support interaction and endowed it with outstanding catalytic performance for CO2 hydrogenation reaction.48
Moreover, the order of decrease for metal dispersion was found to be Pd/AP-POP (10.3%) < Pd/C3N4 (16.6%) < Pd/AC (21.8%) measured by CO pulse chemisorption, suggesting interactions between Pd and pyridinic N in Pd/AP-POP, since the average diameters estimated by STEM revealed no significant differences in NP size, which is consistent with the previous results.45,48 Obviously, the presence of AP-POP, as a promoter, facilitated metal-support interactions and thereby changing the electronic properties of Pd, which resulted in enhanced catalytic activity.
Conclusions
In summary, we have successfully synthesized highly efficient Pd NPs on porous organic polymer support for hydrogenation of CO2 to formate. The introduction of an electron-donating element on the support induced a considerable charge transfer from nitrogen to Pd, leading to a significantly higher activity in CO2 hydrogenation. The enhanced metal-support interaction was found to facilitate carbon dioxide reduction, as evidenced by CO chemisorption and XPS. Additionally, the Pd/AP-POP exhibited a stable cycling performance even after five cycles. This work provides the importance of tuning electronic state of the active center with precisely designed support for promoting CO2 hydrogenation to FA. We believed that electron-rich support effect to design an advanced nanoparticles catalysts with high activity for CO2 hydrogenation reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21503125), and the Natural Science Basic Research Plan in Shaanxi Province of China (2018JM2048) and the Open Project of Shaanxi Key Laboratory of Catalysis (SLGPT2019KF01-21).References
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- T. P. Senftle and E. A. Carter, Acc. Chem. Res., 2017, 50, 472–475 CrossRef CAS PubMed.
- G. H. Gunasekar, K. Park, K. D. Jung and S. Yoon, Inorg. Chem. Front., 2016, 3, 882–895 RSC.
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- N. Onishi, M. Iguchi, X. Yang, R. Kanega, H. Kawanami, Q. Xu and Y. Himeda, Adv. Energy Mater., 2018, 1801275 Search PubMed.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- J. H. Lee, J. Ryu, J. Y. Kim, S. W. Nam, J. H. Han, T. H. Lim, S. Gautam, K. H. Chae and C. W. Yoon, J. Mater. Chem. A, 2014, 2, 9490–9495 RSC.
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Chem. Commun., 2017, 53, 4677–4680 RSC.
- D. A. Bulushev and J. R. H. Ross, Catal. Rev., 2018, 60, 566–593 CrossRef CAS.
- N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Coord. Chem. Rev., 2018, 373, 317–332 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- M. Caiti, D. Padovan and C. Hammond, ACS Catal., 2019, 9, 9188–9198 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- S. M. Lu, Z. Wang, J. Wang, J. Li and C. Li, Green Chem., 2018, 20, 1835–1840 RSC.
- H. Horváth, G. Papp, H. Kovács, Á. Kathó and F. Joó, Int. J. Hydrogen Energy, 2019, 44, 28527–28532 CrossRef.
- N. D. McNamara and J. C. Hicks, ChemSusChem, 2014, 7, 1114–1124 CrossRef CAS PubMed.
- A. Kann, H. Hartmann, A. Besmehn, P. J. C. Hausoul and R. Palkovits, ChemSusChem, 2018, 11, 1857–1865 CrossRef CAS PubMed.
- K. Park, G. H. Gunasekar, N. Prakash, K. D. Jung and S. Yoon, ChemSusChem, 2015, 8, 3410–3413 CrossRef CAS PubMed.
- G. H. Gunasekar and S. Yoon, J. Mater. Chem. A, 2019, 7, 14019–14026 RSC.
- D. Preti, C. Resta, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2011, 50, 12551–12554 CrossRef CAS PubMed.
- G. A. Filonenko, W. L. Vrijburg, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 343, 97–105 CrossRef CAS.
- Q. Liu, X. Yang, L. Li, S. Miao, Y. Li, Y. Li, X. Wang, Y. Huang and T. Zhang, Nat. Commun., 2017, 8, 1407 CrossRef PubMed.
- L. C. Lee, X. Y. Xing and Y. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 38436–38444 CrossRef CAS PubMed.
- C. Mondelli, B. Purtolas, M. Ackermann, Z. Chen and J. P. Ramírez, ChemSusChem, 2018, 11, 2859–2869 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, M. J. Hülsey and N. Yan, Mol. Catal., 2019, 475, 110461 CrossRef CAS.
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, ACS Catal., 2018, 8, 2277–2285 CrossRef CAS.
- K. Koh, M. Jeon, D. M. Chevrier, P. Zhang, C. W. Yoon and T. Asefa, Appl. Catal., B, 2017, 203, 820–828 CrossRef CAS.
- O. Y. Podyacheva, D. A. Bulushev, A. N. Suboch, D. A. Svintsitskiy, A. S. Lisitsyn, E. Modin, A. Chuvilin, E. Y. Gerasimov, V. I. Sobolev and V. N. Parmon, ChemSusChem, 2018, 11, 3724–3727 CrossRef CAS PubMed.
- Q. Sun, Z. F. Dai, X. J. Meng, L. Wang and F. S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- W. Zhang, B. Aguila and S. Ma, J. Mater. Chem. A, 2017, 5, 8795–8824 RSC.
- G. Kupgan, L. J. Abbott, K. E. Hart and C. M. Colina, Chem. Rev., 2018, 118, 5488–5538 CrossRef CAS PubMed.
- P. Bhanja, A. Modak and A. Bhaumik, ChemCatChem, 2019, 11, 244–257 CrossRef CAS.
- V. M. Suresh, S. Bonakala, H. S. Atreya, S. Balasubramanian and T. K. Maji, ACS Appl. Mater. Interfaces, 2014, 6, 4630–4637 CrossRef CAS PubMed.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
- L. Rajput and R. Banerjee, Cryst. Growth Des., 2014, 14, 2729–2732 CrossRef CAS.
- Z. Zhang, L. Zhang, S. Yao, X. Song, W. Huang, M. J. Hülsey and N. Yan, J. Catal., 2019, 376, 57–67 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- R. Arrigo, M. E. Schuster, Z. L. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- F. Wang, J. Xu, X. Shao, X. Su, Y. Huang and T. Zhang, ChemSusChem, 2016, 9, 246–251 CrossRef CAS PubMed.
- S. Masuda, K. Mori, Y. Kuwahara and H. Yamashita, J. Mater. Chem. A, 2019, 7, 16356–16363 RSC.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- K. Mori, T. Sano, H. Kobayashi and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 8902–8909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01324b |
| This journal is © The Royal Society of Chemistry 2020 |