
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLC-MS/MS determination of avanafil and its metabolites in rat plasma and brain: pharmacokinetic study after oral administration and transdermal film application
Ahmed K. Kammoun *a,
Alaa Khedra and
Osama A. A. Ahmedbc
*a,
Alaa Khedra and
Osama A. A. Ahmedbc
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah 21589, P. O. Box 80260, Saudi Arabia. E-mail: akammoun@kau.edu.sa
bDepartment of Pharmaceutics, Faculty of Pharmacy, King Abdulaziz University, Jeddah 21589, P. O. Box 80260, Saudi Arabia
cDepartment of Pharmaceutics and Industrial, Faculty of Pharmacy, Minia University, Minia, Egypt
First published on 4th March 2020
Abstract
Avanafil (AVA) has been FDA approved in 2012 as a phosphodiesterase-type five inhibitor drug (PDE-5), for the treatment of erectile dysfunction (ED). It was necessary to study the pharmacokinetics and bioavailability parameters of AVA since it exhibits side effects, a long time from drug administration. As a result of this, we described a sensitive high-performance-liquid chromatography-triple quad-mass spectrometric method (LC-QqQ-MS) for the analysis of AVA in rat plasma and brain. Furthermore, the concentrations of AVA and its primary metabolites were determined in rat brain since it is known that PDE-5 inhibitor drugs are capable of crossing the blood–brain barrier (BBB). The liquid–liquid extraction method was developed, optimized, and applied for maximum recovery of AVA from plasma and brain homogenates. The percentage of recovery was 96.60 ± 2.44% and 94.50 ± 1.86%, in rat plasma and brain homogenate, respectively. The separation was performed on a Nucleodur C18 column, with mobile phase composed of 0.1% formic acid and acetonitrile (29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :71, v/v), at flow rate 0.5 mL min−1, and monitored with QqQ-MS applying positive multiple reaction monitoring (MRM) mode. The calculated pharmacokinetic parameters, noncompartmental model, were: Cmax 1503.82 ± 354.11 ng mL−1 with a t1/2 value of 4.87 ± 0.42 h and Cmax 141.94 ± 22.57 ng mL−1 with a t1/2 value of 7.05 ± 1.59 h, for oral AVA suspension and transdermal film, respectively. The average percentage of total metabolites in plasma and brain was 27.1 ± 2.2% and 7.0 ± 1.0%, respectively.
:71, v/v), at flow rate 0.5 mL min−1, and monitored with QqQ-MS applying positive multiple reaction monitoring (MRM) mode. The calculated pharmacokinetic parameters, noncompartmental model, were: Cmax 1503.82 ± 354.11 ng mL−1 with a t1/2 value of 4.87 ± 0.42 h and Cmax 141.94 ± 22.57 ng mL−1 with a t1/2 value of 7.05 ± 1.59 h, for oral AVA suspension and transdermal film, respectively. The average percentage of total metabolites in plasma and brain was 27.1 ± 2.2% and 7.0 ± 1.0%, respectively.
1. Introduction
Avanafil (AVA); (S)-4-((3-chloro-4-methoxybenzyl)amino)-2-(2-(hydroxymethyl)pyrrolidin-1-yl)-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide1 is a phosphodiesterase type 5 (PDE-5) inhibitor and used for treatment of erectile dysfunction (ED).2 ED is one of the most common diseases worldwide and referred to as impotence.3,4 Stendra®, 200 mg tablets, has been recently FDA approved, in April 2012, for the treatment of ED.5,6 AVA has a vasodilation effect and increases the blood flow via its highly selective inhibition of cGMP-specific PDE-5 enzyme compared to other marketed PDE-5 inhibitors.7A few analytical methods were found in the literature that describe the measurement of AVA in the biological matrix or pharmaceutical dosage forms. An HPLC-UV and UV spectrometric methods have been reported for monitoring of AVA in the new formulation8 and in combination with dapoxetine.9 Also, Patel M. N. K. et al. (2016) described a multivariate UV-spectrophotometric method for the analysis of AVA in a binary mixture with dapoxetine.10 Mans D. J. et al. (2013) analyzed AVA in combination with acetildenafil and sildenafil using ion mobility spectrometry.11 Jung S. et al. (2010) studied the tolerability and pharmacokinetics of AVA in Korean male subjects using ion mobility spectrometry and monitoring with mass spectrometry.12 Recently, Can, N. O. (2018) published a stability-indicating LC method for the analysis of AVA in its dosage forms with the characterization of some related substances.13
It has been reported that PDE-5 inhibitors are capable of crossing the blood–brain barrier (BBB).14 Many pharmacological side effects may be attributed to this critical information. Subsequently, it has been reported that tadalafil showed to improve the experimental stroke due to the vasodilatation effect.15 Furthermore, it has been reported that AVA is primarily and extensively metabolized by hepatic cytochrome P450 to eleven or twenty-one metabolites. Some of the identified metabolites were pharmacologically inactive, including metabolite M-16, and some have shown side effects, including headache and nausea.16,17 The pharmacokinetic studies of such pharmaceuticals utilize a robust and sensitive LC-MS method to enable both quantitative measurements of drug entity and structural confirmation of the monitored compounds, including some metabolites.18 For such newly FDA-approved drugs, a pharmacokinetic profile is helpful to optimize the bioavailability criteria including, half-life, Cmax, clearance rate, and the route of administration. In the present study, we developed a pioneer bioanalytical LC-MS/MS method for monitoring AVA in rat plasma and brain tissue after oral administration and transdermal application. Simple liquid–liquid extraction (LLE) technique was optimized and utilized for measurement and characterization of AVA and its primary metabolites in rat plasma and brain homogenate. The results revealed that AVA and its metabolites have existed in brain tissues, as proved by MS2 spectral data.
2. Experimental
2.1. Chemicals and reagents
Avanafil, ≥99.0%, and valsartan, ≥97.0%, w/w, were purchased from Sigma-Aldrich (GmbH, Steinheim, Germany). Acetonitrile, formic acid, and other chemicals were of HPLC grade.The animals were obtained from king Fahd animal house, King Abdulaziz University. The Research Ethics Committee, Faculty of Pharmacy, King Abdulaziz University, was approved the animal study protocol. The committee ensures animal use complied with the EU Directive 2010/63/EU on the protection of animals used for scientific purposes and the Guiding Principle in Care and Use of Animals (DHEW publication NIH 80-23).
2.2. Chromatographic conditions
Agilent 6460 liquid chromatography – triple quad mass spectrometer (LC-QqQ-MS) (Agilent Technologies, USA) was used for the quantification of AVA using valsartan as internal standard (IS). The system was controlled by MassHunter software (version B.03.01, Build 3.1.346.0). The MS conditions were as follows: gas temperature, 330 °C; gas flow rate, 11 L min−1; nebulizer pressure; 35 psi, and capillary voltage, 4100 V. The MS settings were optimized for each compound separately, including the fragmentor voltage (at Q1), dwell time, and collision energy voltage (at Q2). Two MRM transitions were applied applying positive ionization polarity at corresponding time segment; AVA, m/z 485.0–375.0, fragmentor 135 eV, collision energy 28 eV, and valsartan (IS), m/z 436.0–306.0, fragmentor 135 eV, collision energy 20 eV. The dwell time for both AVA and IS was 200 milliseconds. The chromatographic separation was performed on Nucleodur C18 column, 5 μm, 4.6 × 250 mm (Macherey Nagel, Duren, Germany). The mobile phase composition was water containing 0.1% formic acid (w/v), and acetonitrile, 29:71, v/v, and pumped at a flow rate of 0.5 mL min−1. Sample injection volume, 5 μL.
2.3. Preparation of solutions
AVA and IS stock solutions were prepared separately in methanol to obtain a concentration of 250 μg mL−1. The AVA working solutions were prepared in methanol spanning the range of 0.5 to 8.0 μg mL−1. The serial calibration concentration of AVA was prepared in plasma and brain homogenate at a concentration of 50.0, 100.0, 200.0, 400.0, 600.0, 800.0, and 3200.0 ng mL−1. The concentrations of quality control (QC) samples were prepared at a concentration of 50.0, 200.0, and 800.0 ng mL−1. The concentration of IS in all samples was 10 μg mL−1.2.4. Sample preparation
The plasma and brain samples were extracted and prepared by utilizing the liquid–liquid extraction method. Plasma or brain homogenate samples, 100 μL, were mixed with 50 μL IS, 250 μg mL−1, and 1 mL acetonitrile. Samples were agitated by vortex for 10 s and centrifuged at 5000 rpm for 7 min. The supernatant was filtered, and a volume of 5 μL was injected for LC-MS analysis.2.5. Method performance
The method performance for the determination of AVA was optimized and tested in both plasma and brain tissue. All validation parameters were studied, including; selectivity, linearity, precision, accuracy, percentage recovery, matrix effect, and sample stability. The selectivity of the assay was evaluated by analyzing blank samples with AVA and IS versus control samples. Linearity of AVA in rat plasma and brain tissue homogenate were constructed at the range; 50.0–3200.0 ng mL−1, and the regression coefficient value was calculated. The accuracy was evaluated by calculation of the actual concentration in comparative with the estimated value for each QC sample. The precision was assessed by analysis of QC samples five times over one day, and analysis on each of 7 days to evaluate both intra-day and inter-day precision, respectively. The lowest concentration in the calibration curve was defined as the lower limit of quantification (LLOQ). The recovery of extraction was determined by comparing the area under the peak of post-spiked standards with those of pre-spiked standards. Also, the effect of the matrix was assessed by comparing pretreated blank samples spiked at a concentration of QC samples with the direct injection of corresponding standard solutions. Finally, sample stability, including; long-term, short-term, and freeze–thaw, were all investigated at three levels of concentrations applying the same chromatographic conditions.2.6. Pharmacokinetic study
Male Wistar rats, weighing 200–250 g each, were selected for this study. Animals were fasted for 12 h before dose administration. The orally administered group (20 animals) was given AVA orally (1 mL) 0.5% w/v sodium carboxymethyl cellulose aqueous suspension in a dose of 60 mg mL−1. Each group was divided into two sub-groups (10 each) for blood sampling by an alternative method to avoid blood volume depletion from animals (hypovolemia) and as advised by the local ethical committee. Blood samples, 250 μL each, were collected at 0, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8 and 24 h post-oral administration of AVA dose. Collected blood samples were centrifuged at 3500 rpm for 10 min, and the plasma layer was transferred to a new Eppendorf's tubes. Rat brain samples were collected from animals at six or 24 h post-dose administration. The collected brains were weighed and then homogenized in saline solution (150 mg mL−1). The collected plasma and brain samples were stored at −80 °C until analysis.Raw AVA transdermal films were prepared according to previously reported work.19 Briefly, hydroxypropyl methylcellulose (HPMC), 2% w/v, as a matrix-forming polymer and propylene glycol (2% w/v) as a plasticizer were dispersed in distilled water. Accurately weighed amount of AVA was incorporated with stirring with the HPMC solution and then kept overnight at 4 °C for the gel to clear. The gel was poured into 9 cm diameter Petri dishes and subjected to drying at 40 °C. After that, 4–7.5 cm2 film areas that contain the required AVA dose for each tested rat was cut and used as a transdermal film for the in vivo assessment on rat skin. Pharmacokinetic assessment of raw AVA transdermal film, following application, was carried out in male Wistar rats (n = 20), at a dose of 60 mg kg−1, with the same sampling time intervals used for AVA oral suspension. The applied transdermal films were occluded on adhesive patches.
2.7. Statistics of pharmacokinetic parameters
Pharmacokinetic parameters were calculated by applying a noncompartmental model. The results are presented as mean ± standard deviation (SD). The studied pharmacokinetic parameters were; maximum AVA plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), half-life (t1/2), elimination rate constant (Ke), area under AVA plasma concentration–time curve (AUC), the area under the first moment curve (AUMC), mean AVA residence time (MRT). Kinetica® software was used to evaluate the data by means of noncompartmental analysis. Any significant difference in drug plasma time concentration curve between the two studied groups was evaluated with repeated measure two-way ANOVA followed by Sidak's multi-comparison test using GraphPad Prism 6. Pharmacokinetic parameters were investigated for the significance of data difference using an unpaired t-test (two-tailed). Results were considered significant at P < 0.05.3. Results and discussion
3.1. Method development
One of the main advantages of the developed method is the simplicity and efficiency of the applied LLE procedure. This procedure showed a high percentage recovery with minimal sample loss because of skipped steps of evaporation or reconstitution. A standard solution mixture of AVA and IS was analyzed repetitively applying interchangeable chromatographic separation parameters and MS settings. The optimal separation was achieved on a Nucleodur C18 column. Besides, the optimal MS detectability of stable MS precursor ion was achieved upon applying MS-scan mode at alternative fragmentor and collision energy voltages. The optimal mass transitions were achieved by applying different and alternative values of collision energy voltages (CE), and the most stable and abundant product ions were selected. Fig. 1 showed the MRM transition – chromatograms of standard AVA, 0.2 ng μL−1 at 11.65 min, and IS, 10 ng μL−1 at 4.48 min.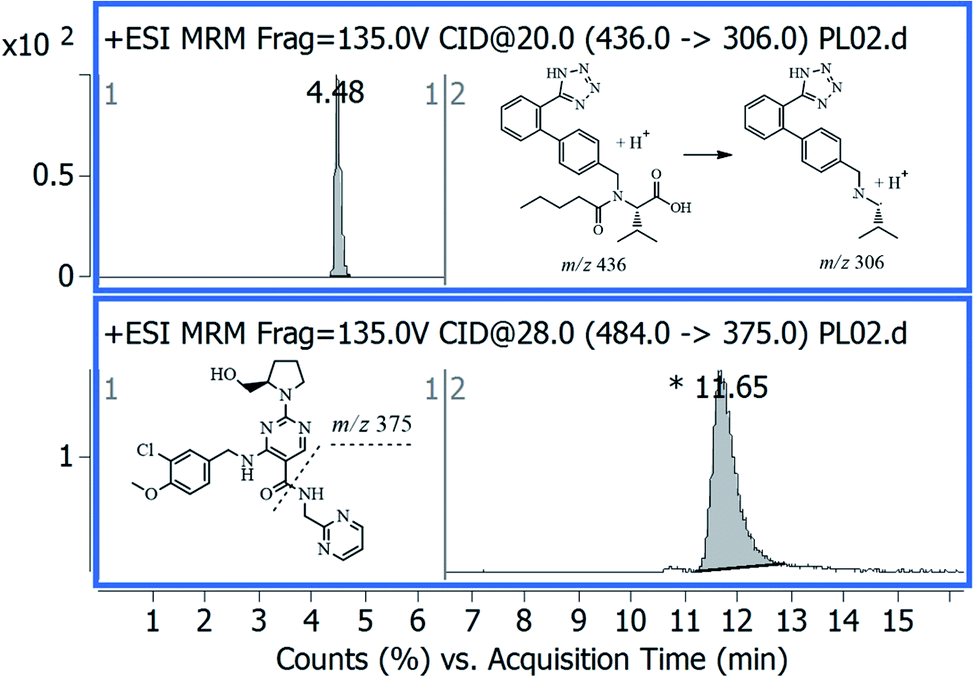 | ||
| Fig. 1 Representative MRM chromatogram of standard avanafil, 0.2 ng μL−1 at 11.65 min, and internal standard, 10 ng μL−1 at 4.48 min. | ||
3.2. Method performance
The developed method showed excellent specificity and selectivity as there is no interference from co-extracted plasma or biogenic brain material, with low background noise. The method performance was validated according to the ICH guidelines.20 The calibration curve for AVA showed excellent linearity at the range; 50.0–3200.0 ng mL−1 with squared regression coefficient (r2) value close to unity in both matrixes, plasma, and brain samples, and equal to 0.9979 and 0.9921, respectively. The response factor, slope, in both matrixes, was 0.00175 MS count per each picogram. The precision was tested, using spiked rat plasma. The intra-day and inter-day precision, using QC samples, expressed as RSD value, were within a range of 0.82 to 1.05% and 1.03 to 1.75%, respectively. The calculated percentage error (Er%) of recovered concentrations, relative to nominated values, for intra-day and inter-day, were in the range −0.85 to −1.02% and −1.10 to −2.15%, respectively (Table 1). The calculated percentage recoveries, in rat plasma and brain tissues, were 96.60 ± 2.44% and 94.50 ± 1.86%, respectively. These results indicated minimal matrix effect and minimal ionization suppression with acceptable accuracy and precision. The relative standard deviation value of recovered concentrations was less than 15% after three freeze–thaw cycles (Table 1). The relative errors for three levels of concentration of QC samples were within ±3.25% in other stability tests. Consequently, it was anticipated that the developed method would be successfully applied to AVA pharmacokinetic studies in rat's plasma and brain tissue.| Concentration in plasma, ng mL−1 | Average recovery (%) ± SD | Intra-day | Inter-day | |||
|---|---|---|---|---|---|---|
| Autosampler (25 °C/24 h) | Freeze–thaw (−20 °C/3 cycles) | Recovery% (RSD)a | Erb (%) | Recovery% (RSD)a | Erb (%) | |
| a Mean recovery% (RSD) of five determinations.b Percentage relative error. | ||||||
| 50.0 | 97.98 ± 1.35 | 94.04 ± 3.95 | 98.98 (0.82) | −1.02 | 97.85 (1.03) | −2.15 |
| 200.0 | 98.23 ± 0.98 | 96.68 ± 2.92 | 99.23 (0.93) | −0.77 | 98.61 (1.25) | −1.39 |
| 800.0 | 99.15 ± 2.20 | 98.23 ± 4.20 | 99.15 (1.05) | −0.85 | 98.90 (1.75) | −1.10 |
3.3. Pharmacokinetic parameters
AVA plasma and brain concentration data after oral and transdermal applications are shown in Fig. 2 and 3, respectively.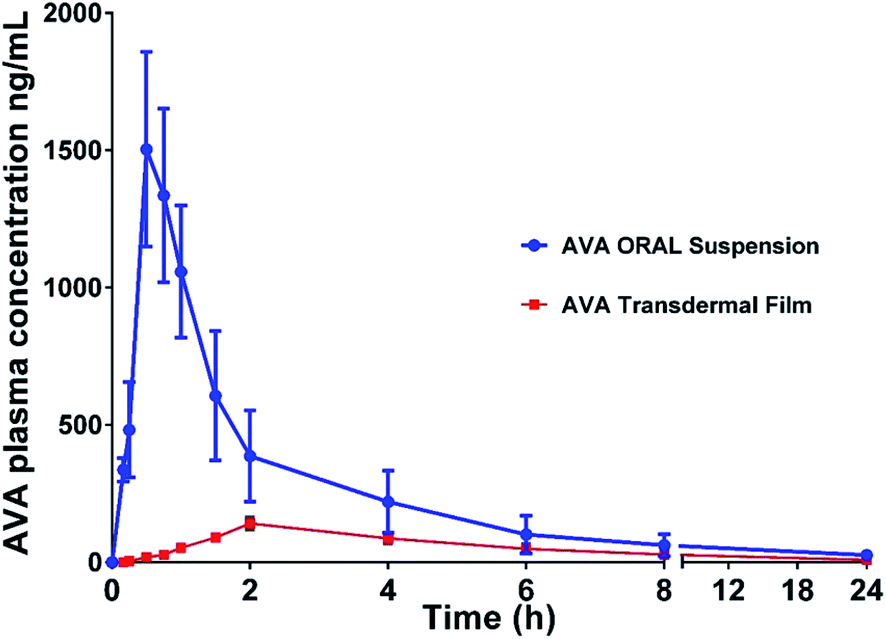 | ||
| Fig. 2 Plasma concentration–time profile of avanafil after oral administration and transdermal-film application. Data represent the mean value ± standard deviation (n = 6). | ||
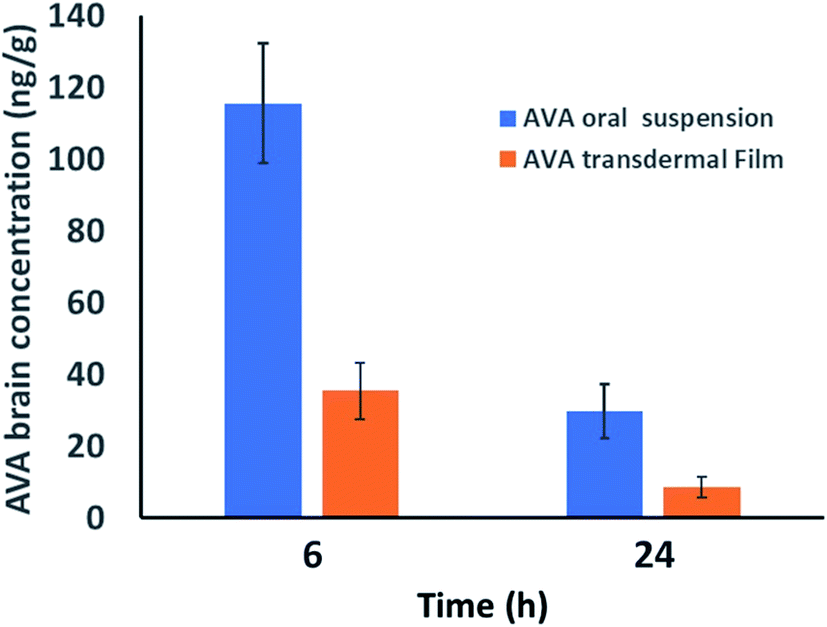 | ||
| Fig. 3 Brain concentration of avanafil in rat brain at 6 and 24 h after 60 mg kg−1 oral administration and transdermal application, respectively. Data represent the mean value ± standard deviation (n = 6). | ||
The pharmacokinetic parameters (noncompartmental model) calculated from the rat plasma data are presented in Table 2. The results showed a t1/2 value of 4.87 ± 0.42 and 7.05 ± 1.59 h, for oral AVA suspension and AVA transdermal film, respectively. The elimination rate constant was 0.14 ± 0.01 and 0.10 ± 0.02 (Ke) for oral and transdermal AVA, respectively. The results revealed a significant (p < 0.05) increase in Cmax, AUC, and AUMC of oral AVA-suspension when compared with AVA-transdermal film (Table 2).
| Pharmacokinetic parameter | Unit | Transdermal film ± SD | Oral administration ± SD |
|---|---|---|---|
| a Abbreviations: Ke; elimination rate constant, t1/2; half-life, Tmax; time to reach AVA maximum plasma concentration, Cmax; maximum AVA plasma concentration, AUC; area under AVA plasma concentration–time curve, AUMC; the area under the first moment curve, MRT; mean AVA residence time. | |||
| Ke | h−1 | 0.10 ± 0.02 | 0.14 ± 0.01 |
| t1/2 | h | 7.05 ± 1.59 | 4.87 ± 0.42 |
| Tmax | h | 2.00 (0.50 to 2.10 h) | 0.5 (0.45 to 0.55 h) |
| Cmax | ng mL−1 | 141.94 ± 22.57 | 1503.83 ± 354.11 |
| AUC0–t | ng mL−1 h−1 | 861.96 ± 187.98 | 3437.94 ± 1424.32 |
| AUC(24-end) | ng mL−1 h−1 | 101.10 ± 66.04 | 191.74 ± 129.45 |
| AUC0–inf_obs | ng mL−1 h−1 | 963.06 ± 227.77 | 3629.68 ± 1550.67 |
| AUMC(0–24) | ng mL−1 h−2 | 5596.27 ± 1553.57 | 14864.13 ± 8322.42 |
| AUMC(24-end) | ng mL−1 h−2 | 2426.35 ± 1584.86 | 4601.87 ± 3106.83 |
| AUMC(0-end) | ng mL−1 h−2 | 8022.62 ± 2895.18 | 19466.00 ± 11408.13 |
| MRT0–inf_obs | h | 8.21 ± 1.44 | 5.156 ± 0.78 |
| Cmax/AUC(0–24) | h−1 | 0.17 ± 0.02 | 0.46 ± 0.08 |
| Total clearance rate | mL min−1 | 1.12 ± 0.23 | 0.31 ± 0.09 |
On the other hand, AVA transdermal film showed longer t1/2, Tmax, and MRT results compared with oral AVA-suspension. These results could be attributed to the enhanced AVA absorption from the oral suspension form compared to the reduced absorption of AVA from transdermal film through the barrier skin layers. AVA–brain data showed that oral AVA-suspension reaches a concentration of 115.48 ± 16.68 and 29.73 ± 7.47 ng g−1 brain after 6 and 24 h of dose administration. However, AVA transdermal film showed lower AVA-levels in the brain with concentrations of 35.41 ± 7.84 and 8.571 ± 3.02 ng g−1 in the brain after 6 and 24 h of dose administration, respectively. These results indicate that AVA penetrates the blood–brain barrier in lower levels compared to plasma levels (Fig. 3).
3.4. Confirmation of extracted AVA-metabolites in rat plasma and brain
Rat plasma and brain samples, collected at 30 and 120 min after dosing, respectively, were extracted. The extracted samples were analyzed by LC-QqQ-MS applying scan mode (100–650 m/z) to allocate the metabolites that previously reported (Fig. 4).16,17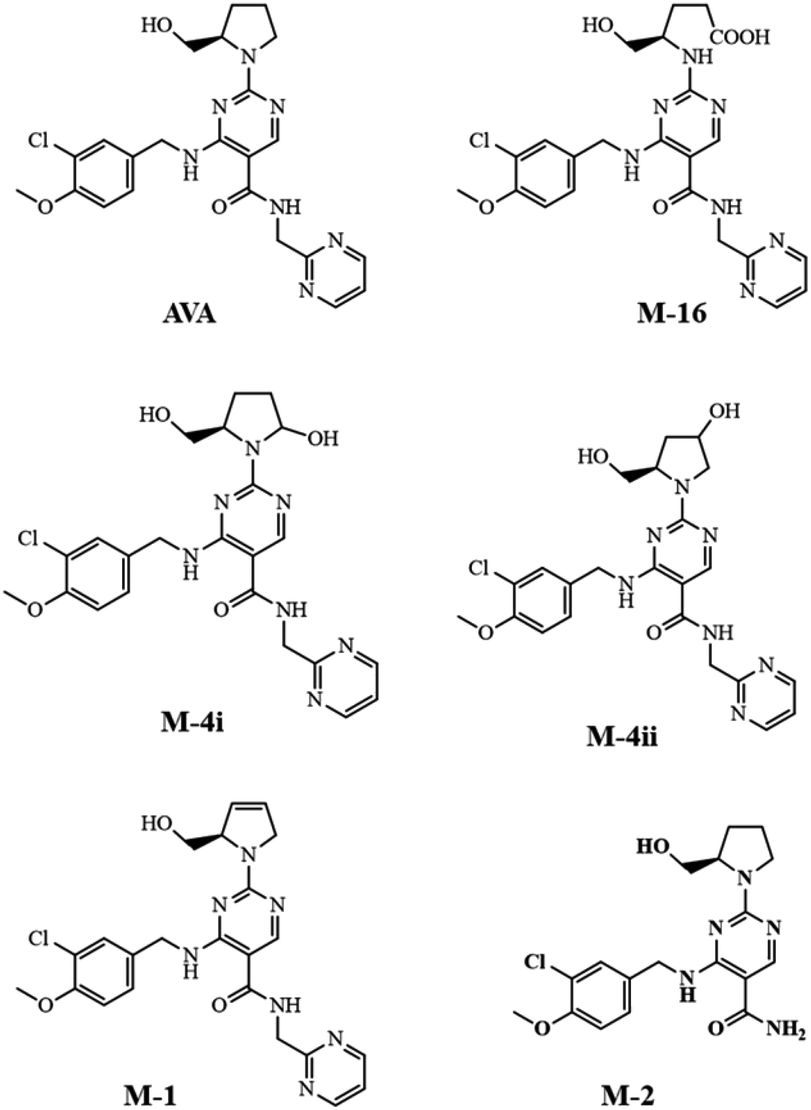 | ||
| Fig. 4 Chemical structure of AVA and detected major metabolites in rat plasma and brain. | ||
The extracted ion chromatographic peaks correspond to the primary metabolites were further re-analyzed applying fragmentation mode using collision energies ranged from 32 to 22 V. Furthermore, the MS2 spectrum of each metabolite found in plasma and brain, were interpreted and confirmed as shown in Fig. 5.
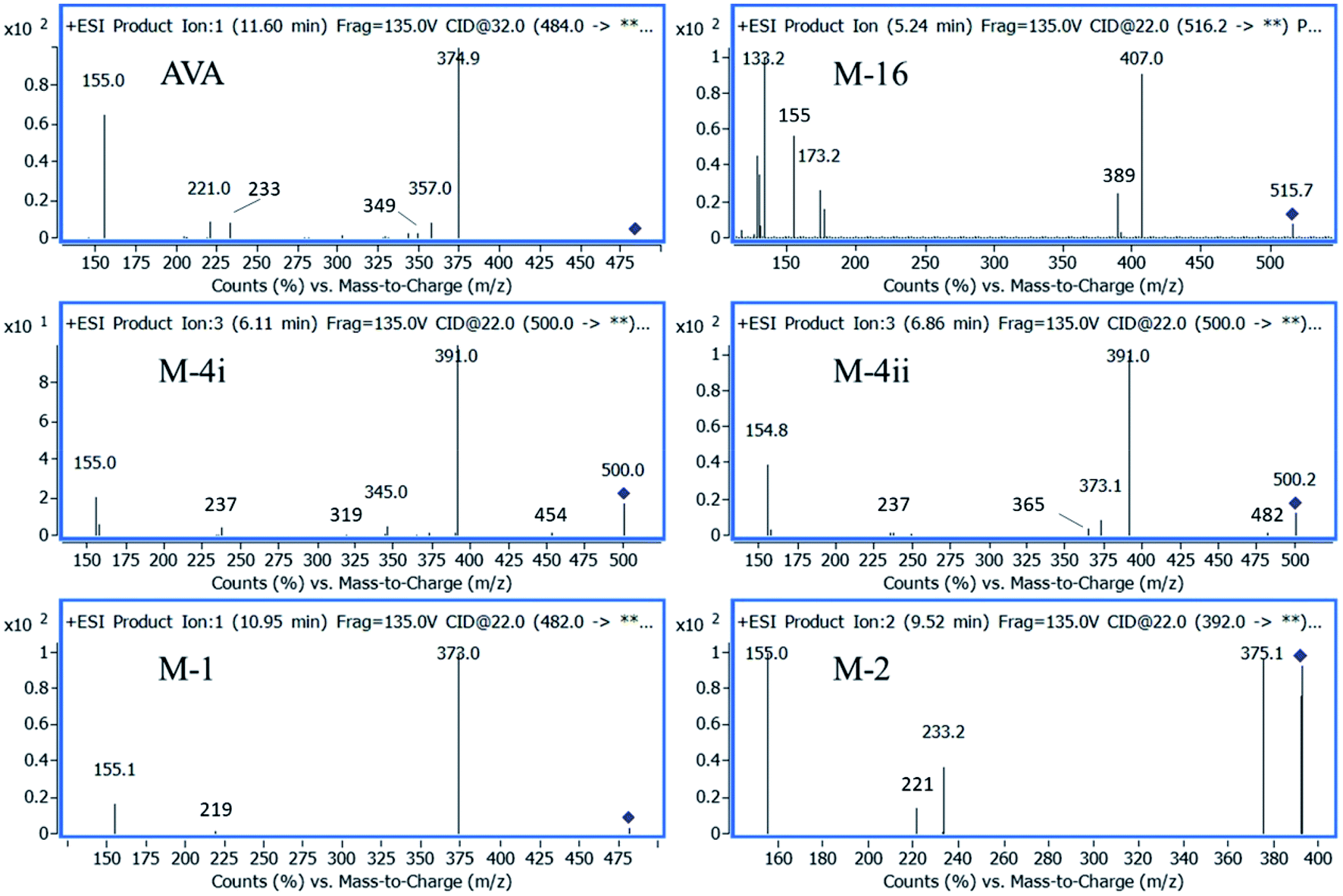 | ||
| Fig. 5 QqQ-MS2 spectra of AVA and its metabolites. | ||
The fragmentation pathway of AVA and its identified five metabolites, preferentially proceed via formation of; m/z 155 ion “2-chloro-1-methoxy-4-methylbenzene”, (fragment 1), and [M – neutral loss of “pyrimidin-2-ylmethanamine”]+ ion. The neutral cleaved part was assigned as “fragment 2” and equal to m/z 108. These two favored cleavage pathways resulted in the production of the two most abundant MS2 peaks. Table 3 showed the MS-product ions of each compound with average values of relative abundance percentages. The MS2 spectrum of metabolite M-16 showed fragment ions at m/z 133 (100%), corresponds to “4-amino-5-hydroxypentanoic acid” moiety, and at m/z 398, corresponds to [M – “5-hydroxypentanoic acid” + H]+. Metabolites M-4i and M-4ii were verified by MS2 spectra and elution order, as previously reported.16,17 Metabolite M-2 showed a fragment ions at m/z 375, which corresponds to [M – “NH2”]+, and m/z 221, corresponds to [M – fragment 1 – “NH2” + H]+. Fig. 5 showed the MS2 of AVA and identified metabolites at a corresponding retention time.
| Name | [M + H]+, m/z | CE, V | Product ions, m/z (% abundance) | |||||
|---|---|---|---|---|---|---|---|---|
| a Fragment 1, m/z 155.b [M – fragment 2]+.c [M – fragment 2 – H2O]+.d [M – fragment 1 – fragment 2 + H]+.e [M – fragment 1 + H]+. | ||||||||
| AVA | 484 | 32 | 375 (100)b | 155 (75.0)a | 357 (14.0)c | 233 (12.0) | 221 (1.8)d | 105 (1.5) |
| M-4i | 500 | 22 | 391 (100)b | 155 (17.0)a | 237 (2.8)d | 345 (1.8)e | 454 (1.2) | 319 (0.3) |
| M-4ii | 500 | 22 | 391 (100)b | 155 (35.0)a | 373 (4.5) | 365 (2.4) | 237 (0.9)d | 345 (0.3)e |
| M-16 | 516 | 22 | 133 (100) | 407 (70.0)b | 129 (50.0) | 155 (35.0)a | 389 (22.0)c | 498 (3.7) |
| M-1 | 482 | 22 | 373 (100)b | 155 (36.0) a | 219 (2.2)d | 231 (1.5) | — | |
| M-2 | 392 | 22 | 155 (100)a | 375 (85.0) | 233 (28) | 221 (10.0) | 346 (0.8) | — |
3.5. Analysis of AVA and its metabolites in rat brain
AVA and its metabolites, in rat brain, were identified, quantified, and the results were compared with plasma data, carried parallel together. A plasma sample, n = 5, collected at 30 min after oral administration, was extracted and analyzed as described under the experimental section. Parallel, five rat brain samples, collected after 120 min after oral administration of the same dose, were homogenized. The brain extraction procedure was repeated five times, and a volume of 100 μL of the supernatant, obtained from each extraction step, was combined, 500 μL, dried, reconstituted in 100 μL acetonitrile, and a volume of 5 μL was analyzed by LC-QqQ-MS applying + MRM mode, as shown in Fig. 6.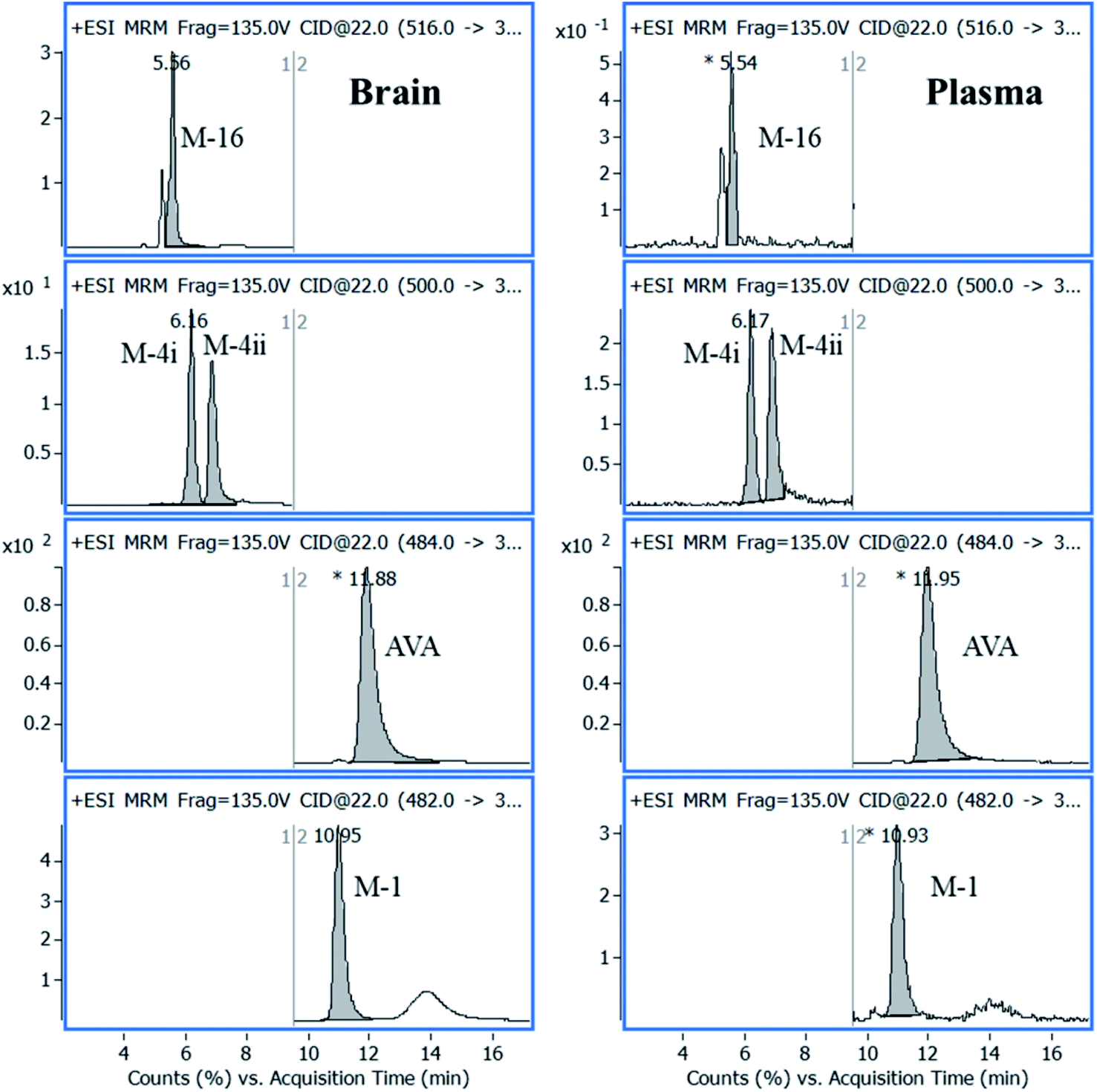 | ||
| Fig. 6 Representative MRM chromatograms of AVA in its characterized metabolites in plasma and brain samples analyzed after 30 min, and 120 min from drug administration, respectively. | ||
The concentration of AVA in plasma and brain was 350.00 and 50.00 ng mL−1, respectively. The concentration of metabolites was calculated from the calibration curve of AVA in plasma and brain (Table 4). The average percentage of total metabolites in plasma and brain was 27.1 ± 2.2% and 7.0 ± 1.0%, respectively.
| Name | tR, min | m/z | Product ion, m/z | CE, V | Concentration, ng mL−1 | |
|---|---|---|---|---|---|---|
| Plasmaa | Brainb | |||||
| a Sample collected at 30 min after drug administration.b Sample collected at 120 min after drug administration. | ||||||
| AVA | 11.87 | 484 | 375 | 32 | 350.00 | 50.00 |
| M-4i | 6.16 | 500 | 391 | 22 | 29.05 | 0.56 |
| M-4ii | 6.84 | 500 | 391 | 22 | 31.48 | 0.78 |
| M-16 | 5.62 | 516 | 389 | 22 | 24.11 | 0.49 |
| M-1 | 10.96 | 482 | 373 | 22 | 10.14 | 1.66 |
4. Conclusions
A robust LC-MS/MS method for a PHD-5 inhibitor (AVA) in rat plasma and brain tissue was developed and validated. The method showed satisfactory performance and applied successfully for qualitative and quantitative analysis of AVA and related metabolites in rat plasma and brain tissue homogenate. The pharmacokinetic parameters of AVA in rat plasma and brain tissue were determined following single-dose oral administration and transdermal application. Five major metabolites of AVA were confirmed by LC-MS2 and quantified in rat plasma and brain tissue. The obtained results confirmed that AVA and five of its metabolites go to the brain and would be the reason for AVA-side effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. D-076-166-1440. The authors, therefore, gratefully acknowledge the DSR technical and financial support.References
- T. Sakamoto, Y. Koga, M. Hikota, K. Matsuki, H. Mochida, K. Kikkawa, K. Fujishige, J. Kotera, K. Omori, H. Morimoto and K. Yamada, Bioorg. Med. Chem. Lett., 2015, 25, 1431–1435 CrossRef CAS PubMed.
- C. N. McMahon, C. J. Smith and R. Shabsigh, BMJ, 2006, 332, 589–592 CrossRef PubMed.
- G. Jackson, Arab J. Urol., 2013, 11, 212–216 CrossRef PubMed.
- C. B. Johannes, A. B. Araujo, H. A. Feldman, C. A. Derby, K. P. Kleinman and J. B. McKinlay, J. Urol., 2000, 163, 460–463 CrossRef CAS PubMed.
- J. M. Belavic, Nurse Pract., 2013, 38, 24–42 CrossRef PubMed , quiz 42-23.
- J. P. Mulhall, A. L. Burnett, R. Wang, K. T. McVary, J. W. Moul, C. H. Bowden, K. DiDonato, W. Shih and W. W. Day, J. Urol., 2013, 189, 2229–2236 CrossRef CAS PubMed.
- J. A. Kyle, D. A. Brown and J. K. Hill, Ann. Pharmacother., 2013, 47, 1312–1320 CrossRef CAS PubMed.
- K. A. Soliman, H. K. Ibrahim and M. M. Ghorab, Int. J. Pharm., 2017, 517, 148–156 CrossRef CAS PubMed.
- M. Hegazy, A. Kessiba, M. Abdelkawy and A. E. El-Gindy, J. Liq. Chromatogr. Relat. Technol., 2015, 38, 1660–1665 CrossRef CAS.
- M. N. K. Patel and S. Charmy, J. AOAC Int., 2016, 3(15), 649–663 CrossRef PubMed.
- D. J. Mans, R. J. Callahan, J. D. Dunn and C. M. Gryniewicz-Ruzicka, J. Pharm. Biomed. Anal., 2013, 75, 153–157 CrossRef CAS PubMed.
- J. Jung, S. Choi, S. H. Cho, J. L. Ghim, A. Hwang, U. Kim, B. S. Kim, A. Koguchi, S. Miyoshi, H. Okabe, K. S. Bae and H. S. Lim, Clin. Ther., 2010, 32, 1178–1187 CrossRef CAS PubMed.
- N. O. Can, Molecules, 2018, 23(7), 1771–1784 CrossRef PubMed.
- C. A. Peixoto, A. K. Nunes and A. Garcia-Osta, Mediators Inflammation, 2015, 2015, 940207 Search PubMed.
- L. Zhang, Z. Zhang, R. L. Zhang, Y. Cui, M. C. LaPointe, B. Silver and M. Chopp, Brain Res., 2006, 1118, 192–198 CrossRef CAS PubMed.
- C. T. Broman and E. Sheu, US Pat., 2016/0331687A1, 2016.
- M. Limin, N. Johnsen and W. J. Hellstrom, Expert Opin. Invest. Drugs, 2010, 19, 1427–1437 CrossRef PubMed.
- Q. A. Xu and T. L. Madden, LC-MS in Drug Bioanalysis, Springer US, 2012 Search PubMed.
- O. A. A. Ahmed and S. M. Badr-Eldin, Int. J. Pharm., 2019, 570, 118657 CrossRef CAS PubMed.
- International Conference on Harmonization (ICH) Guidelines M10 on bioanalytical method validation, 13 March, 2019.
| This journal is © The Royal Society of Chemistry 2020 |