
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct use of 1,3-dienes for the allylation of ketones via catalytic hydroindation†
Itaru Suzuki a,
Kensuke Yagib,
Shinji Miyamotoa and
Ikuya Shibata*a
a,
Kensuke Yagib,
Shinji Miyamotoa and
Ikuya Shibata*a
aResearch Center for Environmental Preservation, Osaka University, 2-4 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: shibata@epc.osaka-u.ac.jp
bDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1, Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 6th February 2020
Abstract
In this study, in situ catalytically generated allylic indium from 1,3 dienes and InCl2H was developed for use in the allylation of ketones. This protocol resulted in the unprecedented establishment of a successive combining of quaternary C–C bonds, which could then be applied to many types of ketones. Other branched 1,3 dienes and vinyl cyclopropanes, could also be coupled with ketones in a reaction where CuH would not be applicable.
Homoallylic alcohols are useful building blocks in the synthesis of bioactive natural compounds and pharmaceuticals. For these syntheses, the preparation of tertiary compounds has remained challenging regardless of whether or not they are given asymmetrically.1 The allylation of ketones with allylic reagents is a typical method for the preparation of these compounds (Scheme 1a).2 This method, however, cannot avoid wasteful steps such as the transmetalation between Grignard reagents and B, Si or Sn sources and the reductive generation between allylic halides and low-valent metals. Although this method can be applied to highly stereocontrolled reactions, the wasteful steps are cumbersome and more practical reaction methods are required.
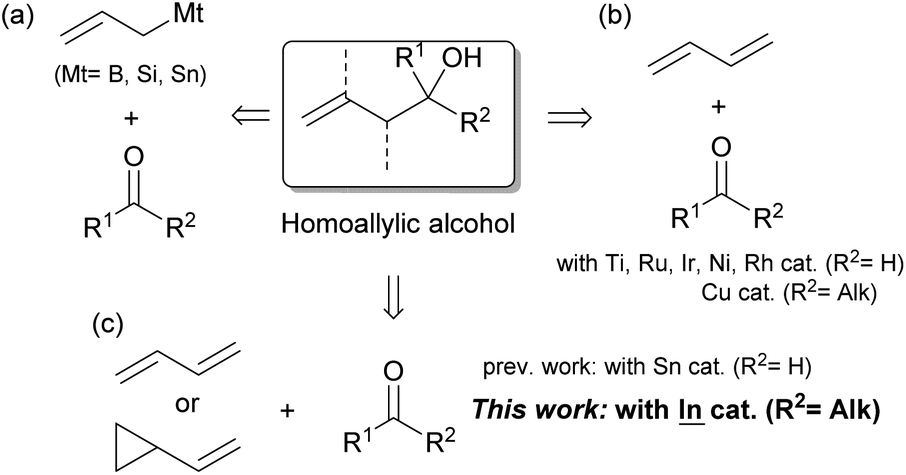 | ||
| Scheme 1 Synthesis of homoallylic alcohols from allylation reagents with ketones. | ||
For this process, 1,3-dienes are an important industrial feedstock that is produced on a massive scale via either the cracking of ethylenes or the transformation of biomass. Easily available dienes have recently replaced the conventional allylation of aldehydes with the aid of transition metal catalysts (Scheme 1b).3 After the first application using a Ti catalyst by Gendre and Moïse,4 Krische has expanded the field with the introduction of Ru-catalyzed stereocontrolled reactions.5 Other transition metals such as Ni,6 Ir,7 and Rh8 have contributed to improvements in coupling. A recent adoption of ketones as viable substrates was achieved by Liu and Buchwald via proficient Cu–H chemistry.9 The scope of possible substrates could be expanded even further,10 however, particularly with the use of 1,3-dienes and ketones that possess a variety of functional groups.
Our group has explored the hydrostannylation11 or indation12 of unsaturated bonds in the preparation of reactive organostannanes or indiums that could be applied to further transformations, although stoichiometric amounts of Sn or In sources must be added to the reaction systems. Recently, a transition metal-free reductive coupling of 1,3-dienes,13 or their derivatives such as vinyl cyclopropanes,14 with aldehydes catalyzed by Bu2SnXH has been developed, but the method would not allow the use of ketones due to the low reactivity of the reaction intermediate, allylic stannanes (Scheme 1c). On the other hand, our group has already developed a process for the hydroindation of 1,3-dienes with a stoichiometric amount of InX2H to give allylic indiums followed by the allylation of ketones.12b,12d Herein, we report the catalytic coupling of 1,3-dienes or vinyl cyclopropanes with ketones through the generation of allylic indiums via the hydroindation of 1,3-dienes with a catalytic amount of InX2H (Scheme 1c).
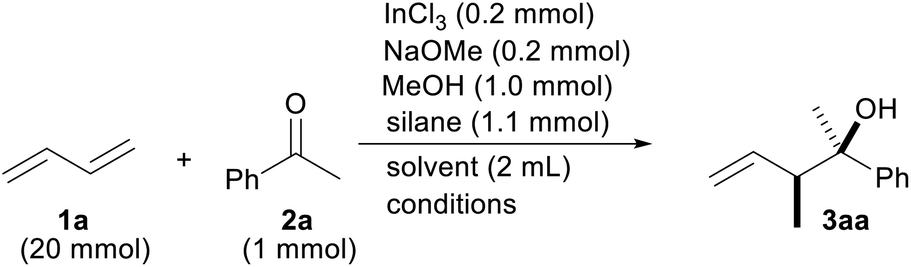
We initiated the optimization of the reaction conditions by combining 1,3-butadiene (1a) and acetophenone (2a) in a sealed test tube (Table 1). The gaseous diene 1a was liquefied and weighed before addition to the reaction. Based on our previous reports, InCl2OMe generated from InCl3/NaOMe and a silane were chosen as indium and hydride sources, respectively.13 The desired product 3aa was obtained in a 92% yield as a mixture of the diastereomers (entry 1). The yield was lowered either when no MeOH was used or when the reaction time was cut by half (entries 2 and 3). A screening of the silanes showed that MePhSiH2 was the optimal hydride source (entries 4–7). We found that the reaction was finished in 3 h when the reaction temperature was raised to 60 °C (entry 8). Replacing the solvent with MeCN, Et2O or toluene did not improve the reaction yield (entries 9–11). It was necessary to add NaOMe to the reaction system for a facile generation of InCl2H (entry 12).16 It was important to add InCl3 to the reaction (entry 13). A radical scavenger, TEMPO, suppressed the progress of the reaction, which implied that this reaction contains a radical process (entry 14). The reaction yield was decreased when the lower amount of the catalyst was employed (entry 15).
|
||||
|---|---|---|---|---|
| Entry | Silane | Solvent | Conditions | Yield (%)3aa (syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti)b :anti)b |
| a The yields were determined by 1H NMR.b Stereochemistry, see: ref. 15.c dr could not be determined because of complex of the reaction mixture. | ||||
| 1 | MePhSiH2 | THF | 25 °C, 48 h | 92 (80:20) |
| 2 | 25 °C, 48 h w/o MeOH | 42 (88:12) |
||
| 3 | 25 °C, 24 h w/o MeOH | 54 (76:24) |
||
| 4 | Et3SiH | THF | 25 °C, 24 h | 12c |
| 5 | Ph3SiH | THF | 25 °C, 24 h | 0 |
| 6 | Ph2SiH2 | THF | 25 °C, 24 h | 32 (83:17) |
| 7 | PhSiH3 | THF | 25 °C, 24 h | 54 (83:17) |
| 8 | MePhSiH2 | THF | 60 °C, 3 h | 92 (80:20) |
| 9 | MeCN | 38 (80:20) |
||
| 10 | Et2O | 71 (76:42) |
||
| 11 | Toluene | 9c | ||
| 12 | THF | w/o NaOMe | Trace | |
| 13 | w/o InCl3 | 0 | ||
| 14 | With TEMPO (0.2 mmol) | 0 | ||
| 15 | With InCl2OMe (0.1 mmol) | 51 (78:22) |
||
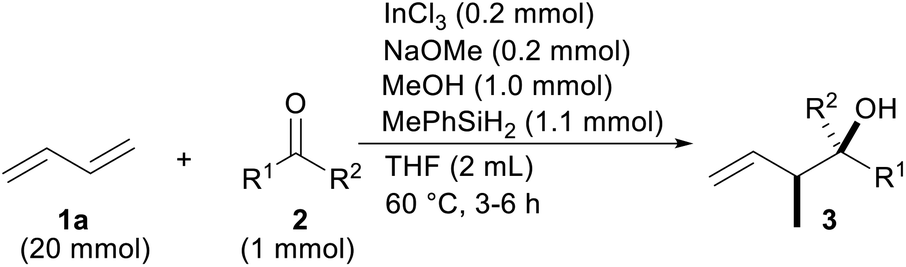
With the optimal reaction conditions in hand (Table 1, entry 8), the scope of the ketones was investigated (Table 2). Electron-deficient substitution on the phenyl ring had a small effect on the reaction yield, but an electron-rich substitution decreased it appreciably (entries 2–6). The efficiency was also attenuated by steric hindrance around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of propiophenone (2g) and butyrophenone (2h) (entries 7 and 8). α-Cyano and -bromo acetophenone 2i and 2j, respectively, reacted sufficiently (entries 9 and 10). The tolerance to reduction of the C–Br bond by InX2H under reductive conditions is a characteristic of coupling (entries 3 and 10).17 On the other hand, α-methoxy one was unsatisfactory as a reactant probably due to chelation between the OMe and CO groups with the catalyst that would have promoted a reduction in the ketone 2k (entry 11). β-Keto ester 2l was a good partner even though a similar chelation involving two CO groups could have happened (entry 12). Both acyclic and cyclic aliphatic ketones were allylated (entries 13–14). Other aromatic rings such as naphthalenes were introduced into the products 3ao and 3ap (entries 15 and 16).
O moiety of propiophenone (2g) and butyrophenone (2h) (entries 7 and 8). α-Cyano and -bromo acetophenone 2i and 2j, respectively, reacted sufficiently (entries 9 and 10). The tolerance to reduction of the C–Br bond by InX2H under reductive conditions is a characteristic of coupling (entries 3 and 10).17 On the other hand, α-methoxy one was unsatisfactory as a reactant probably due to chelation between the OMe and CO groups with the catalyst that would have promoted a reduction in the ketone 2k (entry 11). β-Keto ester 2l was a good partner even though a similar chelation involving two CO groups could have happened (entry 12). Both acyclic and cyclic aliphatic ketones were allylated (entries 13–14). Other aromatic rings such as naphthalenes were introduced into the products 3ao and 3ap (entries 15 and 16).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Ketone | Product | Yield (%) (dr) |
| a The yields were determined by 1H NMR.b Reaction time was 24 h. | |||||
| 1 | C6H5 | Me | 2a | 3aa | 92 (80:20) |
| 2 | p-ClC6H4 | 2b | 3ab | 83 (79:21) |
|
| 3 | p-BrC6H4 | 2c | 3ac | 88 (80:20) |
|
| 4 | p-CNC6H4 | 2d | 3ad | 79 (79:21) |
|
| 5 | p-MeC6H4 | 2e | 3ae | 53 (75:25) |
|
| 6 | p-OMeC6H4 | 2f | 3af | 58 (75:25) |
|
| 7 | C6H5 | Et | 2g | 3ag | 69 (84:16) |
| 8 | nPr | 2h | 3ah | 66 (84:16) |
|
| 9 | CH2CN | 2i | 3ai | 75 (82:18) |
|
| 10 | CH2Br | 2j | 3aj | 74 (84:16) |
|
| 11 | CH2OMe | 2k | 3ak | 22 (80:20) |
|
| 12 | CH2CO2Et | 2l | 3al | 59 (80:20) |
|
| 13b | PhCH2CH2–(CH2)5– | Me | 2m | 3am | 73 (57:43) |
| 14 | 2n | 3an | 75 | ||
| 15 | 1-Np | Me | 2o | 3ao | 77 (75:25) |
| 16 | 2-Np | Me | 2p | 3ap | 80 (76:24) |
Reductive coupling was then applied to other dienes (Scheme 2). In the case of isoprene (1b), two different products, 3ba and 3ba′, were formed even though the reaction was very slow (eqn (1)). The regioselectivity derived from the different structures of the allylic indiums. To our delight, diene 1c made it possible to establish contiguous quaternary C–C bonds with ketones 2a–2c (eqn (2)). To date, construction of contiguous quaternary C–C bonds with a catalyst remains a challenging task in organic synthesis,18 and the task has never been realized by the same type of reductive coupling that is catalyzed by transition metal catalysts.
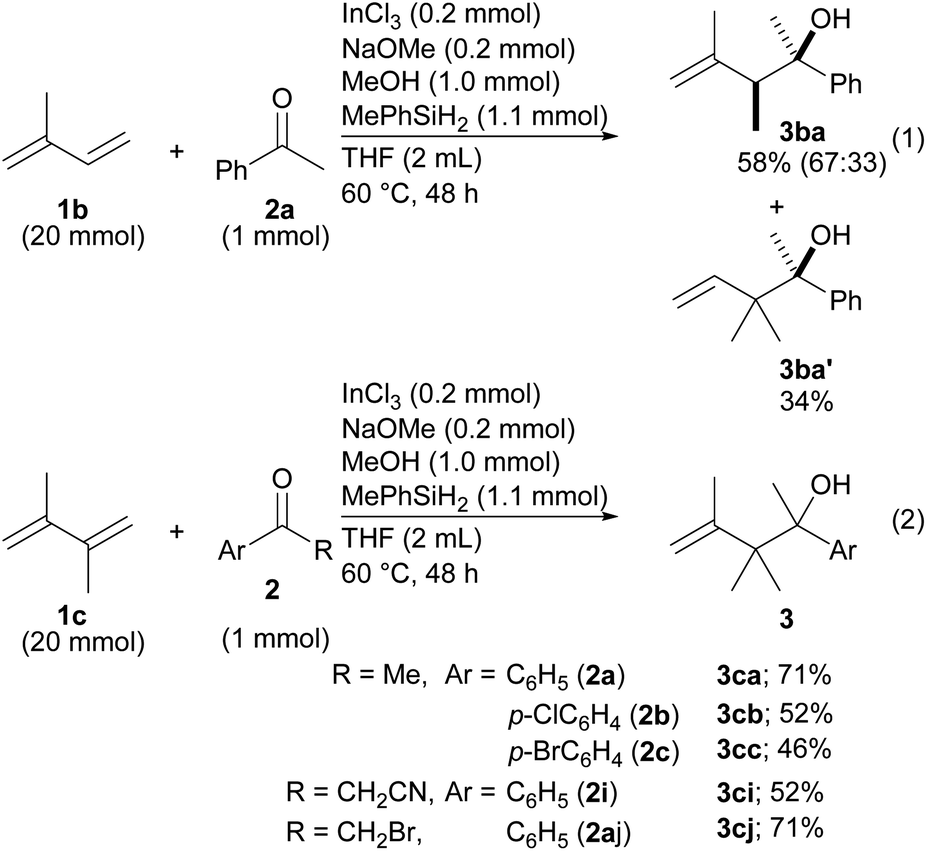 | ||
| Scheme 2 Application of substituted 1,3-butadienes to the coupling. | ||
Our proposal of the reaction mechanism is described in Scheme 3. Initially, prepared InCl2(OMe) is reduced by MePhSiH2 to give HInCl2. The indium radical is formed in the presence of tiny amounts of O2 and adds to diene 1a.17 The stable allylic radical A extracts hydrogen from InCl2H to afford allylic indium B, which regenerates the indium radical. Following the allylation of ketone 2, the generated indium alkoxide 3′ is protonated by CH3OH to give the product 3 and InCl2(OMe). The reaction mechanism was investigated using a deuterated silane, Ph2SiD2 (Scheme 4). We found that the product 3aa-CH2D was afforded without any other deuterated compound. The product 3aa-CH2D is formed from δ-deuterated intermediate B-d1 as shown in Scheme 4. Further investigation on the reaction mechanism is ongoing in our laboratory.
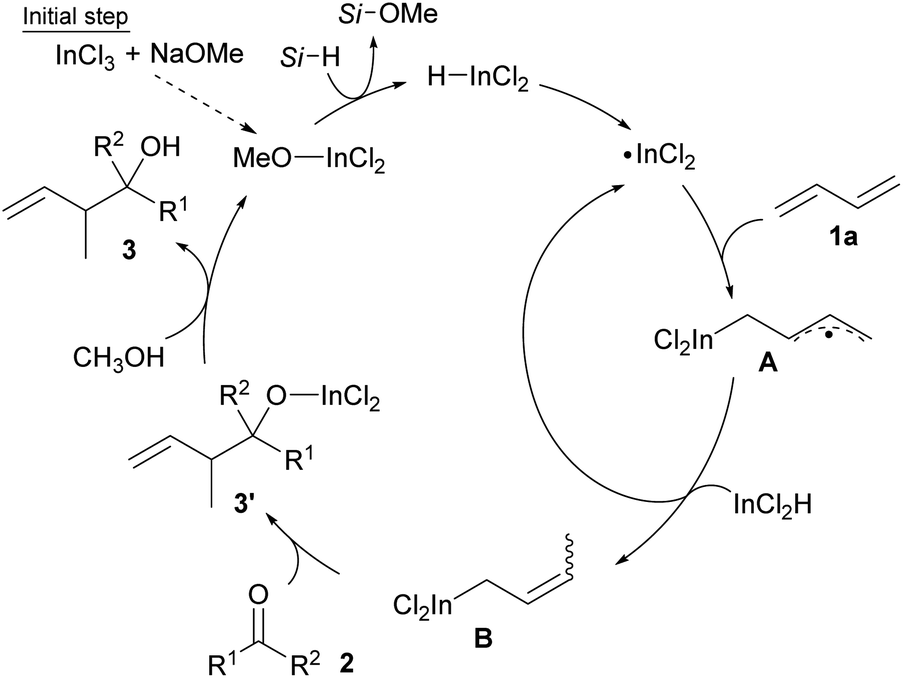 | ||
| Scheme 3 A plausible catalytic cycle. | ||
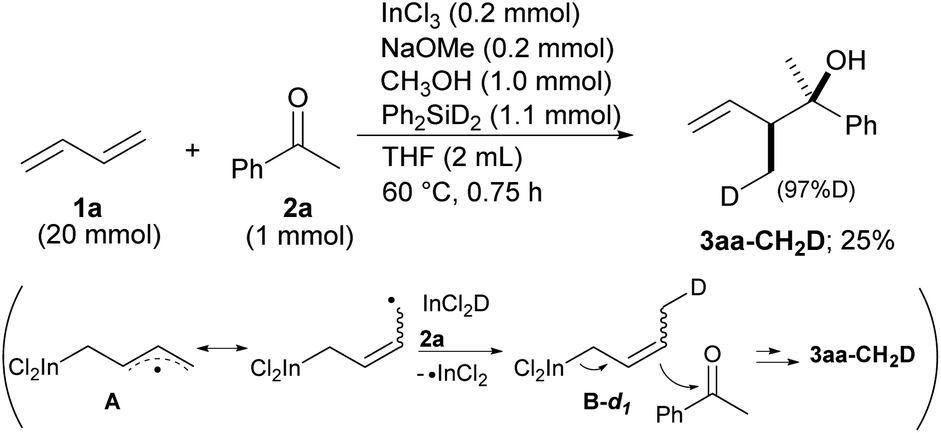 | ||
| Scheme 4 Deuterated experiments of allylation of ketone 2a. | ||
Finally, vinyl cyclopropane 4, a diene derivative, was tested for reductive coupling with ketones.19 The desired product 5a was produced in the presence of a radical initiator, V-70L, even though diastereoselectivity was not achieved (Scheme 5, eqn (1)). This could have been caused by low E/Z selectivity of the allylic indiums. The seminal work developed by Buchwald that was related to a Cu–H catalyzed reaction did not allow the use of vinyl cyclopropane as a reactant because their method has no process for a radical opening of the cyclopropane ring (Scheme 5, eqn (2)).9 Our method expands the scope of the dienes by allowing use of their derivatives.
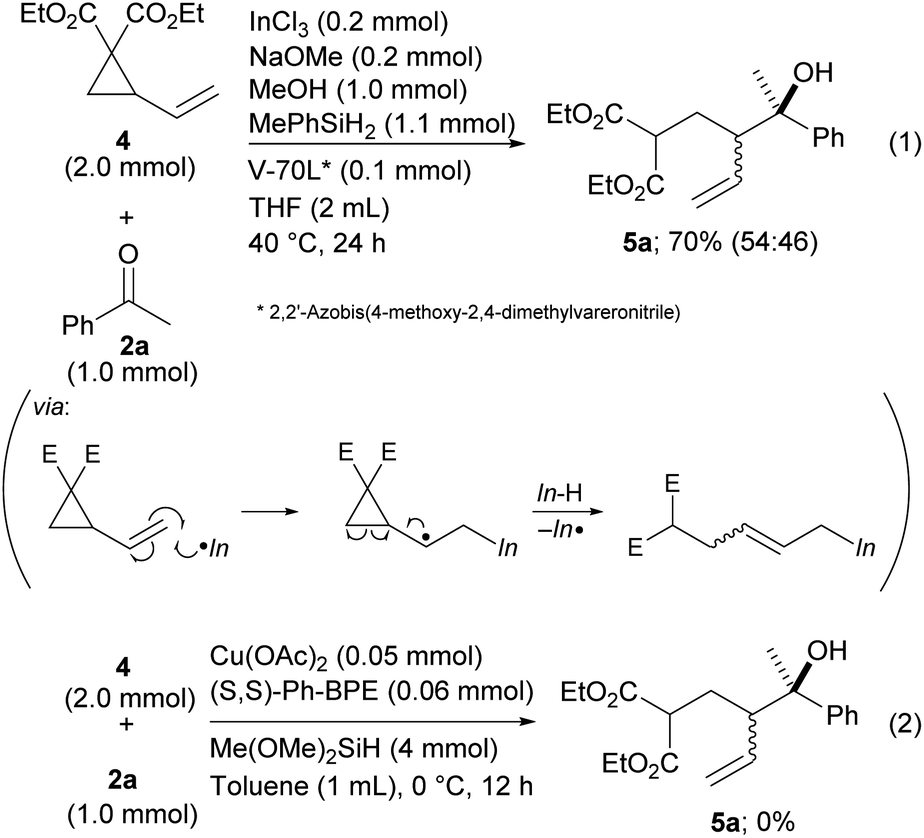 | ||
| Scheme 5 Coupling of vinyl cyclopropanes with ketones. | ||
In summary, we developed a process whereby the reductive coupling of 1,3-dienes with various ketones could be sufficiently catalyzed by HInCl2. This approach allowed the introduction of functional groups into homoallylic alcohols, which generated sequential Ctert–Ctert bonds with expansion to vinyl cyclopropane 4. Application to an asymmetric version of the coupling and improvement of the diastereoselectivity are underway.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed.
- (a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed; (b) M. Hatano and K. Ishihara, Synthesis, 2008, 1647–1675 CAS; (c) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS.
- M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed.
- L. Bareille, P. Le Gendre and C. Moïse, Chem. Commun., 2005, 775–777 RSC.
- (a) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338–6339 CrossRef CAS PubMed; (b) J. R. Zbieg, J. Moran and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 10582–10586 CrossRef CAS PubMed; (c) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324–327 CrossRef CAS PubMed; (d) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628–20631 CrossRef CAS; (e) B. Y. Park, T. P. Montgomery, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320–16323 CrossRef CAS PubMed; (f) T.-Y. Chen and M. J. Krische, Org. Lett., 2013, 15, 2994–2997 CrossRef CAS PubMed; (g) J. C. Leung, L. M. Geary, T.-Y. Chen, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 15700–15703 CrossRef CAS PubMed.
- (a) K. M. Miller and T. F. Jamison, Org. Lett., 2005, 7, 3077–3080 CrossRef CAS PubMed; (b) M. Kimura, A. Ezoe, M. Mori, K. Iwata and Y. Tamaru, J. Am. Chem. Soc., 2006, 128, 8559–8568 CrossRef CAS PubMed; (c) S. Ogoshi, K.-i. Tonomori, M.-a. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077–7086 CrossRef CAS PubMed; (d) A. Köpfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1876–1880 RSC.
- (a) J. F. Bower, R. L. Patman and M. J. Krische, Org. Lett., 2008, 10, 1033–1035 CrossRef CAS; (b) J. R. Zbieg, T. Fukuzumi and M. J. Krische, Adv. Synth. Catal., 2010, 352, 2416–2420 CrossRef CAS PubMed; (c) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14210–14213 CrossRef CAS.
- (a) M. Kimura, D. Nojiri, M. Fukushima, S. Oi, Y. Sonoda and Y. Inoue, Org. Lett., 2009, 11, 3794–3797 CrossRef CAS PubMed; (b) H.-Y. Jang, R. R. Huddleston and M. J. Krische, Angew. Chem., Int. Ed., 2003, 42, 4074–4077 CrossRef CAS PubMed.
- C. Li, R. Y. Liu, L. T. Jesikiewicz, Y. Yang, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 5062–5070 CrossRef CAS PubMed.
- B. Fu, X. Yuan, Y. Li, Y. Wang, Q. Zhang and T. Xiong, Org. Lett., 2019, 21, 3576–3580 CrossRef CAS PubMed.
- (a) I. Shibata, T. Suwa, K. Ryu and A. Baba, J. Am. Chem. Soc., 2001, 123, 4101–4102 CrossRef CAS PubMed; (b) N. Hayashi, K. Kusano, S. Sekizawa, I. Shibata, M. Yasuda and A. Baba, Chem. Commun., 2007, 4913–4915 RSC; (c) N. Hayashi, Y. Hirokawa, I. Shibata, M. Yasuda and A. Baba, J. Am. Chem. Soc., 2008, 130, 2912–2913 CrossRef CAS PubMed; (d) I. Shibata, T. Suwa, K. Ryu and A. Baba, J. Org. Chem., 2001, 66, 8690–8692 CrossRef CAS PubMed.
- (a) A. Baba and I. Shibata, Chem. Rec., 2005, 5, 323–335 CrossRef CAS PubMed; (b) N. Hayashi, H. Honda, M. Yasuda, I. Shibata and A. Baba, Org. Lett., 2006, 8, 4553–4556 CrossRef CAS PubMed; (c) N. Hayashi, Y. Hirokawa, I. Shibata, M. Yasuda and A. Baba, Org. Biomol. Chem., 2008, 6, 1949–1954 RSC; (d) N. Hayashi, H. Honda, I. Shibata, M. Yasuda and A. Baba, Synlett, 2008, 1407–1411 CAS.
- I. Suzuki, Y. Uji, S. Kanaya, R. Ieki, S. Tsunoi and I. Shibata, Org. Lett., 2017, 19, 5392–5394 CrossRef CAS PubMed.
- S. Tsunoi, Y. Maruoka, I. Suzuki and I. Shibata, Org. Lett., 2015, 17, 4010–4013 CrossRef CAS.
- P. Dey, M. Koli, D. Goswami, A. Sharma and S. Chattopadhyay, Eur. J. Org. Chem., 2018, 1333–1341 CrossRef CAS.
- N. Hayashi, I. Shibata and A. Baba, Org. Lett., 2005, 7, 3093–3096 CrossRef CAS PubMed.
- K. Inoue, A. Sawada, I. Shibata and A. Baba, Tetrahedron Lett., 2001, 42, 4661–4663 CrossRef CAS.
- (a) K. Ohmatsu, N. Imagawa and T. Ooi, Nat. Chem., 2013, 6, 47 CrossRef; (b) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed; (c) X. Liu, J. Zhang, L. Zhao, S. Ma, D. Yang, W. Yan and R. Wang, J. Org. Chem., 2015, 80, 12651–12658 CrossRef CAS PubMed; (d) X. Huang, S. Wu, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2016, 7, 12382 CrossRef PubMed; (e) F. I. Amr, C. Vila, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2016, 358, 1583–1588 CrossRef CAS; (f) S. Nakamura, R. Yamaji and M. Iwanaga, Chem. Commun., 2016, 52, 7462–7465 RSC; (g) L. Ye, Q.-S. Gu, Y. Tian, X. Meng, G.-C. Chen and X.-Y. Liu, Nat. Commun., 2018, 9, 227 CrossRef PubMed; (h) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, Angew. Chem., Int. Ed., 2018, 57, 8664–8667 CrossRef CAS PubMed; (i) W.-L. Chan, X. Tang, F. Zhang, G. Quek, G.-J. Mei and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 6260–6264 CrossRef CAS PubMed; (j) M. Sun, J.-F. Chen, S. Chen and C. Li, Org. Lett., 2019, 21, 1278–1282 CrossRef CAS PubMed.
- Ir-catalyzed coupling of vinylcyclopropanes with aldehydes was developed. See: J. Moran, A. G. Smith, R. M. Carris, J. S. Johnson and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 18618–18621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00853b |
| This journal is © The Royal Society of Chemistry 2020 |