
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and thermal properties of Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O†
Hamdi Ben Yahia *a,
Alaa Alkhateeba and
Rachid Essehlib
*a,
Alaa Alkhateeba and
Rachid Essehlib
aQatar Environment and Energy Research Institute (QEERI 2.0), Hamad Bin Khalifa University, Qatar Foundation, P. O. Box 34110, Doha, Qatar. E-mail: Hyahia@hbku.edu.qa
bEnergy and Transportation Science Division, Oak Ridge National Laboratory, Oak Ridge, TN, USA
First published on 11th March 2020
Abstract
The title compounds were prepared via a wet chemistry route and their crystal structures were determined from single crystal X-ray diffraction data. Na2Mn(SO4)2·4H2O crystallizes with a monoclinic symmetry, space group P21/c, with a = 5.5415(2), b = 8.3447(3), c = 11.2281(3) Å, β = 100.172(1)°, V = 511.05(3) Å3 and Z = 2. Na2Ni(SO4)2·10H2O also crystallizes with a monoclinic symmetry, space group P21/c, with a = 12.5050(8), b = 6.4812(4), c = 10.0210(6) Å, β = 106.138(2)°, V = 780.17(8) Å3 and Z = 2. Na2Mn(SO4)2·4H2O is a new member of the blödite family of compounds, whereas Na2Ni(SO4)2·10H2O is isostructural with Na2Mg(SO4)2·10H2O. The structure of Na2Mn(SO4)2·4H2O is built up of [Mn(SO4)2(H2O)4]2− building blocks connected through moderate O–H⋯O hydrogen bonds with the sodium atoms occupying the large tunnels along the a axis and the manganese atom lying on an inversion center, whereas the structure of Na2Ni(SO4)2·10H2O is built up of [Ni(H2O)6]2+ and [Na2(SO4)2(H2O)4]2− layers. These layers which are parallel to the (100) plane are interconnected through moderate O–H⋯O hydrogen bonds. The thermal gravimetric- and the powder X-ray diffraction-analyzes showed that only the nickel phase was almost pure. At a temperature above 300 °C, all the water molecules evaporated and a structural phase transition from P21/c-Na2Ni(SO4)2·10H2O to C2/c-Na2Ni(SO4)2 was observed. C2/c-Na2Ni(SO4)2 is thermally more stable than Na2Fe(SO4)2 and therefore it would be suitable as the positive electrode for sodium ion batteries if a stable electrolyte at high voltage is developed.
1. Introduction
The sulfate salts of general formula Na2M(SO4)2·nH2O (n = 1, 2, 3, 4, 5, 6, 10, 16)1–14 have attracted much attention from mineralogists during the last century owing to their important role in desertification, soil contamination and surface and ground water salinization [ref. 13 and references therein]. These natural and synthetic sulfate salts crystallize with a wide range of crystal structure types depending on their degree of hydration. This parameter is at the origin of the presence of unique hydrogen bond features in these compounds. For this reason, numerous studies have focused on solving their crystal structures. In the system with magnesium five phases exist. The blödite-type Na2Mg(SO4)2·4H2O8 and the konyaite-type Na2Mg(SO4)2·5H2O9 are both minerals that form due to the evaporation of saline solutions, whereas Na2Mg(SO4)2·10H2O13 and Na2Mg(SO4)2·16H2O14 are synthetic phases that were obtained from the evaporation of solutions containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of MgSO4 and Na2SO4 salts. When the same mixture was heated to 650 °C and slow cooled at a rate of 1° min−1, Na2Mg(SO4)2 was obtained.15 The Na2M(SO4)2·nH2O phases are very sensitive to temperature, pressure and relative humidity. When heated at relatively high temperatures (T > 200 °C) these phases could be completely dehydrated to form the Na2M(SO4)2 phases which are of interest as positive electrodes for sodium- or lithium-ion batteries.
:1 molar ratio of MgSO4 and Na2SO4 salts. When the same mixture was heated to 650 °C and slow cooled at a rate of 1° min−1, Na2Mg(SO4)2 was obtained.15 The Na2M(SO4)2·nH2O phases are very sensitive to temperature, pressure and relative humidity. When heated at relatively high temperatures (T > 200 °C) these phases could be completely dehydrated to form the Na2M(SO4)2 phases which are of interest as positive electrodes for sodium- or lithium-ion batteries.
Among the Na2M(SO4)2 phases, Na2Fe(SO4)2 showed interesting electrochemical properties in Li- and Na-ion batteries. This phase enables the removal of nearly one sodium at potentials around ∼3.6 V vs. Li+/Li or ∼3.3 V vs. Na+/Na.16 Na2Fe(SO4)2 could also be obtained by intercalating one sodium into the structure of the eldfellite-type NaFe(SO4)2.17 At 0.1C, this material delivers a discharge capacity of 80 mA h g−1 with an operating potential around 3.25 V vs. Na+/Na. Even the hydrated phases such as the blödite-type Na2Fe(SO4)2·4H2O16 or the kröhnkite-type Na2Fe(SO4)2·2H2O18 were active at ∼3.3 V and ∼3.25 V vs. Na+/Na, respectively. On the other hand, the Na2Co(SO4)2 phase did not show any electrochemical activity up to 5 V.16 In the system Na2Mn(SO4)2·nH2O two phases were reported (n = 0 and 2). The thermal decomposition at 500 K of Na2Mn1.167(SO4)2S0.33O1.167·2H2O19 and the kröhnkite-type Na2Mn(SO4)2·2H2O20 led to the formation of two different Na2Mn(SO4)2 phases that crystallize with the glauberite- and alluaudite-type of structures, respectively. The glauberite-type Na2Mn(SO4)2 sample was not tested as positive electrode since it contains few MnS2O7 impurities and the alluaudite-type Na2Mn(SO4)2 has shown to be active in sodium ion batteries (NIBs), however the performance was worse than the iron analogues.21–23 In the system Na2Ni(SO4)2·nH2O three phases are known (n = 0, 4 and 6), however they were not tested as positive electrode materials for NIBs.24–26 For these reasons we prepared recently several Na2M(SO4)2·nH2O phases (M = Mn and Ni) in order to test their electrochemical performances in NIBs.
In this paper we report on the synthesis of the new phases Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O by wet chemistry route. The crystal structures of these phases were solved using single crystal X-ray diffraction (XRD) and their compositions were confirmed by the combination of thermal gravimetric analyzes (TGA) and energy-dispersive X-ray spectrometry analyzes (EDX). The fully dehydrated phases were analyzed by ex situ powder XRD. Our results are presented in the following sections.
2. Experimental section
2.1. Synthesis
Na2Mn(SO4)2·4H2O powder was synthesized via a wet chemistry route (super-saturation method) from a stoichiometric mixture of Na2SO4 (Aldrich, ≥99%) and MnSO4·H2O (Aldrich, ≥99%). The starting materials with a 1:1 molar ratio were dissolved in 20 ml of water (solution A). The solution A was stirred for few hours then left drying at room temperature during four weeks. This enabled the growth of a mixture of large colorless single crystals of Na12Mn7(SO4)13·15H2O, Na2Mn(SO4)2·2H2O and the new phase Na2Mn(SO4)2·4H2O. Powder sample of this sample was obtained by grinding few crystals.
Na2Ni(SO4)2·10H2O powder was also synthesized via a wet chemistry route (super-saturation method) from a stoichiometric mixture of Na2SO4 (Aldrich, ≥99%) and NiSO4·6H2O (Aldrich, ≥99%). The starting materials with a 1:1 molar ratio were dissolved in 20 ml of water (solution B). The solution B was stirred for few hours then left drying at room temperature during three weeks. This enabled the growth of large green single crystals of the new phase Na2Ni(SO4)2·10H2O. Powder sample of this material was obtained by grinding few crystals. The solution B was also dried at 80 °C for 12 hours then the resulting powder was grinded and fired at 350 °C for 32 hours. This led to the formation of the green powder of SS-Na2Ni(SO4)2. A similar powder of WC-Na2Ni(SO4)2 was obtained when heating under argon the crystals of Na2Ni(SO4)2·10H2O at 400 °C for 6 hours.
2.2. Powder X-ray diffraction measurements
To ensure the purity of the prepared powders, routine powder XRD measurements were performed. The data were collected at room temperature over the 2θ angle range of 5° ≤ 2θ ≤ 75° with a step size of 0.01° using a Bruker D8 advance diffractometer operating with CuKα radiation. Full pattern matching refinement was performed with the JANA2006 program package.27 The background was estimated by a Legendre function and the peak shapes were described by a pseudo-Voigt function.2.3. Single crystal X-ray diffraction measurements
Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O single crystals suitable for single crystal X-ray diffraction were selected on the basis of the size and the sharpness of the diffraction spots. The data collections were carried out on a Bruker D8 Venture diffractometer using MoKα radiation. Data processing and all refinements were performed with the APEX3 and JANA2006 program packages, respectively.27,28 For the data collection details, see Table 1. Further details on the structure refinements of Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O may be obtained from the Fachinformationszentrum Karlsruhe, D-76344 Eggenstein-Leopoldshafen (Germany), by quoting the Registry No. CSD – 1962424 and 1962425.| Crystal data | ||
| Chemical formula | Na2Mn(SO4)2·4H2O | Na2Ni(SO4)2·10H2O |
| Mr | 365.1 | 476.9 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 5.5415 (2), 8.3447 (3), 11.2281 (3) | 12.5050 (8), 6.4812 (4), 10.0210 (6) |
| β (°) | 100.172 (1) | 106.138 (2) |
| V (Å3) | 511.05 (3) | 780.17 (8) |
| Z | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 1.84 | 1.66 |
| Crystal size (mm) | 0.16 × 0.13 × 0.11 | 0.34 × 0.10 × 0.08 |
|
||
| Data collection | ||
| Diffractometer | Bruker D8 VENTURE | Bruker D8 VENTURE |
| Absorption correction | Multi-scan (SADABS) | Multi-scan SADABS |
| Tmin, Tmax | 0.88, 0.92 | 0.630, 0.747 |
| No. of measured, independent and observed [I > 3σ(I)] reflections | 9618, 1314, 1198 | 22158, 3819, 2781 |
| Rint | 0.022 | 0.029 |
| (sinθ/λ)max (Å−1) |
0.676 | 0.853 |
|
||
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.018, 0.060, 1.16 | 0.024, 0.079, 1.11 |
| No. of reflections | 1314 | 3819 |
| No. of parameters | 96 | 147 |
| No. of restraints | 6 | 15 |
| H-atom treatment | All H-atom parameters refined | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.19, −0.23 | 0.33, −0.33 |
2.4. Electron microprobe analyzes
Semi-quantitative energy-dispersive X-ray spectrometry (EDX) analyzes were carried out on the single crystals used for the data collections with a 7610F (JEOL) scanning electron microscope (SEM). The experimentally observed Na/M/S atomic ratios (M = Mn and Ni) were close to 2:1:2, as expected for Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O.
2.5. Thermal analyzes
Thermal gravimetric analyzes (TGA) were carried out on the prepared samples Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O using a TA-SDT 650 instrument. The measurements were conducted between 25 and 850 °C at a heating rate of 10 °C min−1. The experiments were performed in alumina crucible under N2 atmosphere.3. Results and discussion
3.1. Structure refinement
The systematic absences observed for Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O agree with the space group P21/c. Atomic positions of the majority of atoms were found by the Superflip program implemented in JANA2006 program package.27,29 The use of the difference Fourier synthesis allowed us to localize the remaining oxygen atomic positions. With anisotropic atomic displacement parameters (ADPs), the residual factors converged to the value R(F) = 0.0264 and wR(F2) = 0.0949 (G.O.F. = 1.92) for 79 refined parameters and 1198 observed reflections for the compound Na2Mn(SO4)2·4H2O and R(F) = 0.0345 and wR(F2) = 0.1165 (G.O.F. = 1.92) for 106 refined parameters and 2781 observed reflections for the compound Na2Ni(SO4)2·10H2O. At this stage of the refinements the chemical formulas were Na2Mn(SO4)2·4O and Na2Ni(SO4)2·10O. The H atomic positions were determined from the difference-Fourier maps. The O–H distances and H–O–H angles were set to 0.96 Å and 104.5°, respectively. The Uiso(H) were refined without constrains. This led to the final chemical formula Na2Mn(SO4)2·4H2O with the residual factors R(F) = 0.0198 and wR(F2) = 0.0629 (G.O.F. = 1.22 for 95 refined parameters) and the chemical formula Na2Ni(SO4)2·10H2O with the residual factors R(F) = 0.0251 and wR(F2) = 0.0707 (G.O.F. = 1.18 for 146 refined parameters). By refining the extinction parameters, the residual factors converged to the values given in Table 1. The refined atomic positions and anisotropic ADPs are given in Tables 2 and S1,† respectively. The EDX elemental analyzes performed on the crystals used for the data collection confirmed the compositions Na2Mn(SO4)2·xH2O and Na2Ni(SO4)2·xH2O (Fig. 1). The examination of the powder XRD pattern of the manganese sample revealed the presence of mainly Na12Mn7(SO4)13·15H2O30 besides a small amount of Na2Mn(SO4)2·2H2O2 and Na2Mn(SO4)2·4H2O (Fig. S1†), whereas the nickel sample was almost a pure Na2Ni(SO4)2·10H2O phase. Fig. 2 shows a good agreement between the experimental and calculated patterns of Na2Ni(SO4)2·10H2O. Evaluation of these data revealed the refined cell parameters a = 12.4926(5), b = 6.4763(2), c = 10.0153(4) Å and β = 106.070(2)° which are in good agreement with those from single crystal diffraction data (see Table 1). Only three tiny impurity peaks were detected. The composition of the Na2Ni(SO4)2·10H2O sample was confirmed by the combination of EDX (Fig. 1b) and TGA analyzes.| Atom | Wyck | x | y | z | Ueq/iso* |
|---|---|---|---|---|---|
| Na2Mn(SO4)2·4H2O | |||||
| Na1 | 4e | 0.12901(9) | 0.92766(6) | 0.36268(5) | 0.02082(16) |
| Mn1 | 2a | 0 | 0.5 | 0.5 | 0.01387(10) |
| S1 | 4e | 0.37292(5) | 0.79212(3) | 0.63561(2) | 0.01132(10) |
| O1 | 4e | 0.20851(18) | 0.91681(12) | 0.57815(8) | 0.0234(3) |
| O2 | 4e | 0.31892(17) | 0.63864(11) | 0.57209(9) | 0.0233(3) |
| O3 | 4e | 0.63145(16) | 0.83269(11) | 0.63209(9) | 0.0222(3) |
| O4 | 4e | 0.34724(18) | 0.77406(12) | 0.76397(8) | 0.0216(3) |
| O5 | 4e | 0.13017(16) | 0.45884(12) | 0.33232(8) | 0.0181(3) |
| H5b | 4e | 0.232(3) | 0.3650(13) | 0.3423(18) | 0.040(5)* |
| H5a | 4e | 0.235(3) | 0.5421(17) | 0.312(2) | 0.053(6)* |
| O6 | 4e | 0.18486(17) | 0.28225(10) | 0.58278(8) | 0.0192(3) |
| H6a | 4e | 0.223(3) | 0.210(2) | 0.5222(13) | 0.053(7)* |
| H6b | 4e | 0.3331(17) | 0.288(2) | 0.6416(12) | 0.039(5)* |
|
|||||
| Na2Ni(SO4)2·10H2O | |||||
| Na1 | 4e | 0.48096(4) | 0.25441(7) | 0.39586(5) | 0.02511(14) |
| Ni1 | 2c | 1.00000 | 0 | 1/2 | 0.01379(6) |
| S1 | 4e | 0.72260(2) | 0.51630(3) | 0.33017(3) | 0.01445(7) |
| O1 | 4e | 0.79184(6) | 0.42137(12) | 0.24800(9) | 0.0233(2) |
| O2 | 4e | 0.78000(8) | 0.49977(11) | 0.48001(9) | 0.0234(3) |
| O3 | 4e | 0.61500(7) | 0.41153(15) | 0.29742(9) | 0.0296(3) |
| O4 | 4e | 0.70453(7) | 0.73541(11) | 0.29108(9) | 0.0254(2) |
| O5 | 4e | 0.86644(7) | 0.03383(11) | 0.33044(10) | 0.0247(2) |
| H5a | 4e | 0.8331(9) | 0.1634(11) | 0.3001(15) | 0.036(4)* |
| H5b | 4e | 0.8058(7) | −0.0615(16) | 0.309(2) | 0.057(5)* |
| H6a | 4e | 0.9914(11) | 0.294(3) | 0.6835(6) | 0.064(6)* |
| H6b | 4e | 0.9025(7) | 0.342(2) | 0.5531(12) | 0.029(4)* |
| O6 | 4e | 0.97063(7) | 0.27252(11) | 0.58560(8) | 0.0239(2) |
| O7 | 4e | 0.60084(7) | 0.40809(12) | 0.60325(9) | 0.0250(2) |
| H7a | 4e | 0.6193(11) | 0.314(2) | 0.6794(13) | 0.059(5)* |
| H7b | 4e | 0.6698(7) | 0.435(2) | 0.5838(18) | 0.057(5)* |
| O8 | 4e | 0.38581(8) | 0.03475(14) | 0.52844(10) | 0.0287(3) |
| H8a | 4e | 0.3566(12) | 0.123(3) | 0.5863(16) | 0.068(6)* |
| H8b | 4e | 0.3216(8) | −0.023(2) | 0.4645(15) | 0.050(6)* |
| O9 | 4e | 0.90973(6) | −0.16068(11) | 0.61345(8) | 0.0219(2) |
| H9a | 4e | 0.8634(10) | −0.085(2) | 0.6567(16) | 0.044(5)* |
| H9b | 4e | 0.8662(11) | −0.2770(18) | 0.5734(19) | 0.059(5)* |
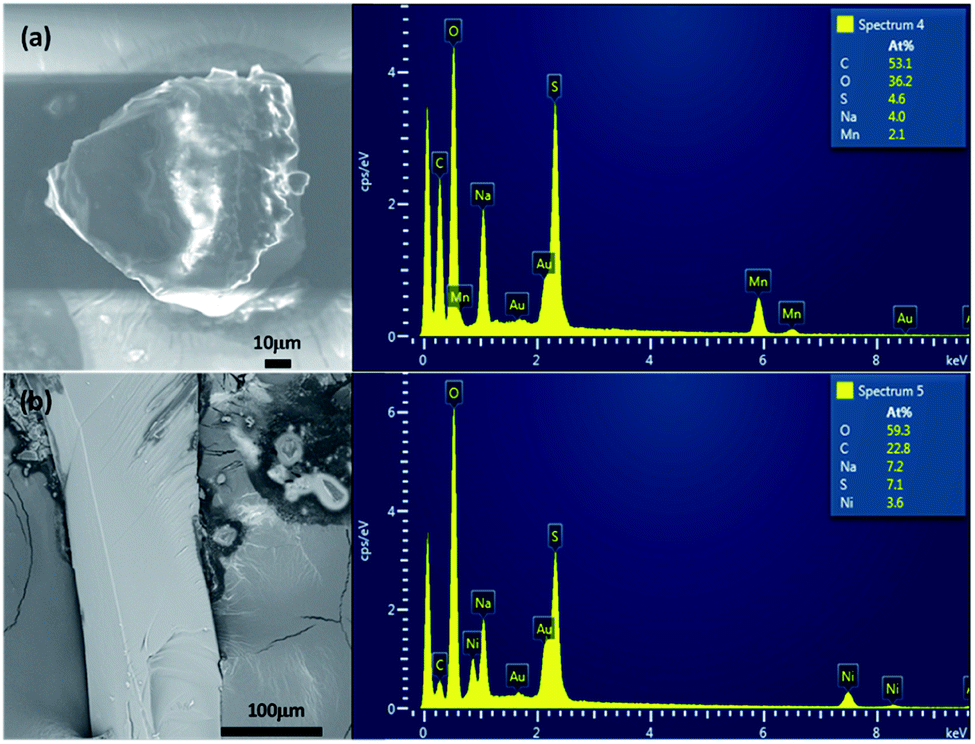 | ||
| Fig. 1 Images and EDX analyzes of Na2Mn(SO4)2·4H2O (a) and Na2Ni(SO4)2·10H2O (b). These are the single crystals used for the data collections. | ||
 | ||
| Fig. 2 Theoretical and experimental powder X-ray diffraction patterns of Na2Ni(SO4)2·10H2O (Cu-Kα radiation). Asterisk (*) corresponds to an unidentified impurity. | ||
Fig. 3 clearly indicates a 37.7% of weight loss, which corresponds exactly to the evaporation of ten water molecules. This confirms that the prepared sample Na2Ni(SO4)2·10H2O decomposes below 300 °C to form WC-Na2Ni(SO4)2 according to the following scheme:
| Na2Ni(SO4)2·10H2O(s) (476.95 g mol−1) → WC-Na2Ni(SO4)2(s) (296.8 g mol−1) + 10H2O(g) (180.15 g mol−1) |
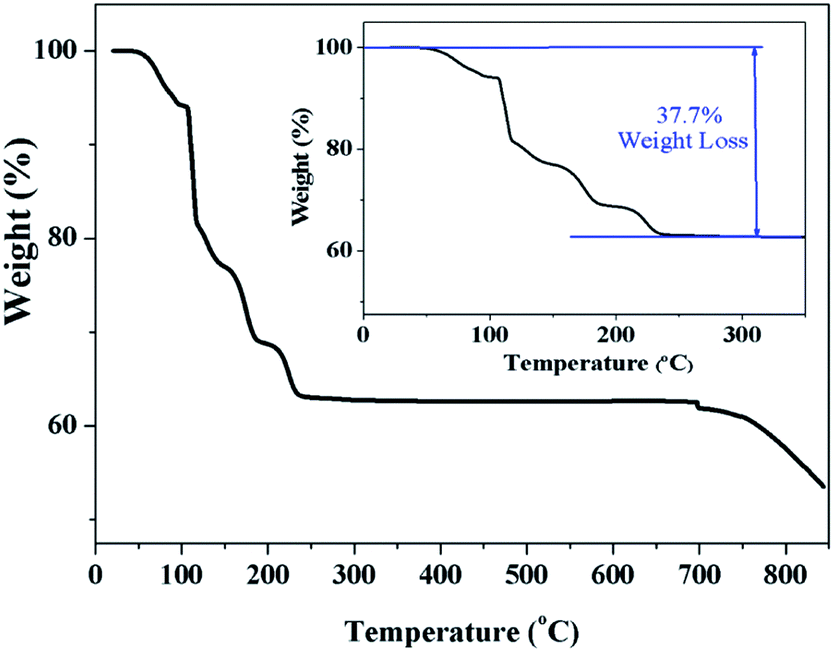 | ||
| Fig. 3 TGA thermal analysis of the Na2Ni(SO4)2·10H2O sample. | ||
This decomposition mechanism was confirmed by powder XRD, since the powder pattern of the dehydrated Na2Ni(SO4)2·10H2O phase is identical to the theoretical pattern of Na2Ni(SO4)2 and the experimental pattern of SS-Na2Ni(SO4)2 (Fig. 4). Above 700 °C, the sample decomposes by releasing SO2(g). It should be noted that the thermal behavior of the sample containing manganese was also performed (Fig. S2†). The weight loss was only 12.9% which is lower than the weight loss of 19.7% expected for Na2Mn(SO4)2·4H2O, however it is almost identical to the weight loss of 12.4% expected for Na12Mn7(SO4)13·15H2O. This is in good agreement with PXRD data which indicate that the prepared manganese phase contains mainly the Na12Mn7(SO4)13·15H2O phase.
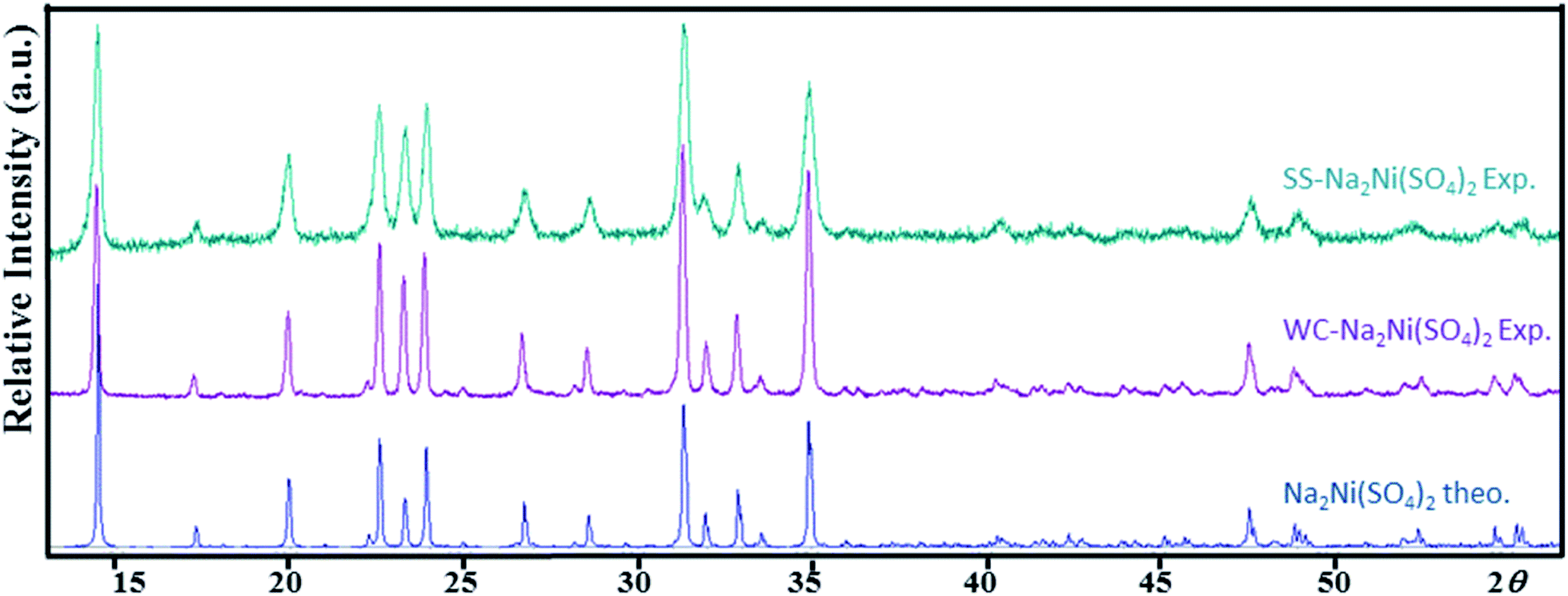 | ||
| Fig. 4 Theoretical and experimental powder X-ray diffraction patterns of Na2Ni(SO4)2 (Cu-Kα radiation). SS-Na2Ni(SO4)2 was obtained using solid state synthesis method, whereas WC-Na2Ni(SO4)2 corresponds to the dehydration of Na2Ni(SO4)2·10H2O obtained using wet chemistry route. | ||
3.2. Crystal structure of Na2Mn(SO4)2·4H2O
The Na2Mn(SO4)2·4H2O compound is isostructural with the blödite-type compounds Na2M(SO4)2·4H2O (M = Mg, Fe, Co, Ni, Zn, Cd).31–37 Conventionally, the blödite-type of structure is described as a stacking of layers parallel to the (010) plane and formed of MnO2(H2O)4 and NaO4(H2O)2 octahedra (Fig. 5a and b). These layers are interconnected through SO4 tetrahedra to form a 3d-framework (Fig. 5c). The interatomic distances are listed in Table 3. | ||
| Fig. 5 View along the a and b axes of the layers of MnO2(H2O)4 and NaO4(H2O)2 octahedra (a and b), and projection view of the crystal structure of Na2Mn(SO4)2·4H2O in the (010) plane (c). View along the a axis of the crystal structure of Na2Mn(SO4)2·4H2O (d). The atoms of the asymmetric unit are labeled (d). View of the building block [Mn(SO4)2(H2O)4]2− (e) and the [Na2O6(H2O)4]10− dimer units (f). | ||
| Distances (Å) | Distances (Å) | ||
|---|---|---|---|
| a Average distances are given in 〈 〉 and coordination numbers are given in [ ].b Bond valence sum, B.V. = e(r0−r)/b with the following parameters: b = 0.37, r0 (NaI–O) = 1.803, r0 (SVI–O) = 1.624, r0 (NiII–O) = 1.654 and r0 (MnII–O) = 1.624. | |||
| Na2Mn(SO4)2·4H2O | Na2Ni(SO4)2·10H2O | ||
| Na1–O1 | 2.3831(10) | Na1–O3 | 2.3956(11) |
| Na1–O3 | 2.3952(11) | Na1–O4 | 2.5474(9) |
| Na1–O5 | 2.4111(9) | Na1–O7 | 2.4138(9) |
| Na1–O4 | 2.4490(11) | Na1–O7 | 2.4159(9) |
| Na1–O1 | 2.4623(12) | Na1–O8 | 2.4671(12) |
| Na1–O6 | 2.6162(11) | Na1–O8 | 2.4821(10) |
| 〈dNa1–O〉 | 〈2.4528〉 | 〈dNa1–O〉 | 〈2.4537〉 |
| BVS | b1.06[6] | BVS | b1.04[6] |
| Mn1–O2 (×2) | 2.1475(9) | Ni1–O5 (×2) | 2.0365(8) |
| Mn1–O5 (×2) | 2.1593(9) | Ni1–O6 (×2) | 2.0409(8) |
| Mn1–O6 (×2) | 2.2088(9) | Ni1–O9 (×2) | 2.0892(9) |
| 〈dMn1–O〉 | 〈2.1718〉 | 〈dNi1–O〉 | 〈2.0555〉 |
| BVS | b2.14[6] | BVS | b2.03[6] |
| S1–O1 | 1.4565(10) | S1–O1 | 1.4842(10) |
| S1–O2 | 1.4707(10) | S1–O2 | 1.4772(9) |
| S1–O3 | 1.4794(9) | S1–O3 | 1.4607(9) |
| S1–O4 | 1.4807(10) | S1–O4 | 1.4738(7) |
| 〈dS1–O〉 | 〈1.4718〉 | 〈dS1–O〉 | 〈1.4740〉 |
| BVS | b6.03[4] | BVS | b6.00[4] |
The manganese atom is lying on an inversion center and is coordinated to two oxygen atoms and four water molecules forming [MnO2(H2O)4]2− octahedron. This octahedron share corners with two SO4 tetrahedra forming the building block [Mn(SO4)2(H2O)4]2− (Fig. 5e). The [MnO2(H2O)4]2− octahedron share also corners with four [Na2O6(H2O)4]10− dimer units leading to layers in the (010) plane (Fig. 5a). The d(Mn1–O) distances range from 2.1475 to 2.2088 Å with an average distance of 2.1718 Å which is slightly shorter than the sum of the effective ionic radii of the six-coordinated Mn2+ and O2− {IR(Mn2+) + IR(O2−) = 0.83 + 1.4 Å}.38 The BVS value of 2.14 is in good agreement with the oxidation state 2+ expected for the Mn atoms.39
The slightly distorted SO4 tetrahedra share only corners with the MnO2(H2O)4 and NaO4(H2O)2 octahedra (Fig. 5c). The d(S1–O) distances range from 1.4565 to 1.4807 Å with the average value of 1.4718 Å. This value is shorter than the sum of the effective ionic radii of the four-coordinated S6+ and O2−{IR(S6+) + IR(O2−) = 0.12 + 1.38 Å}. The BVS value of 6.03 is in good agreement with the oxidation state 6+ expected for sulfur.
The sodium atom is coordinated to four oxygen atoms and two water molecules forming [NaO4(H2O)2]7− octahedra. These octahedra share edges and form [Na2O6(H2O)4]10− dimer units (Fig. 5f). The average Na1–O distance of 2.4528 Å is consistent with the sum of the effective ionic radii of the six-coordinated Na+ and O2−{IR(Na+) + IR(O2−) = 1.02 + 1.40 Å}. The BVS value of 1.06 is in good agreement with the oxidation state 1+ expected for the Na atoms.
In the crystal structure of Na2Mn(SO4)2·4H2O, the H2O molecules play the role of hydrogen-bond donors whereas the oxygen atoms O3 and O4 are the hydrogen bond acceptors (Table 4 and Fig. 6). The O–H⋯O hydrogen bonds connect the [Mn(SO4)2(H2O)4]2− building blocks (Fig. 5e) forming a tunnel-like structure with large voids along the a axis (Fig. 6a). In these voids, the sodium atoms are located (Fig. 5d and 6a). This feature indicates that the compounds of the blödite-family might be good ionic conductors. This may explain the electrochemical activity of the Na2Fe(SO4)2·4H2O phase in NIBs and LIBs.16 Based on the classification of Jeffrey, all the O–H⋯O hydrogen bonds are moderate (Table 4 and Fig. 6b and c).40,41
| Donor | Hydrogen | Acceptor | D–H distance | H⋯A distance | D–A distance | A–H⋯D angle |
|---|---|---|---|---|---|---|
| Na2Mn(SO4)2·4H2O | ||||||
| O5 | H5b | O3 | 0.960(13) | 1.817(12) | 2.7634(13) | 168.3(15) |
| O5 | H5a | O4 | 0.960(17) | 1.774(16) | 2.7074(14) | 163.3(14) |
| O6 | H6a | O3 | 0.960(17) | 2.067(16) | 2.9400(14) | 150.4(16) |
| O6 | H6b | O4 | 0.960(10) | 1.901(10) | 2.8483(12) | 168.8(13) |
|
||||||
| Na2Ni(SO4)2·10H2O | ||||||
| O5 | H5a | O1 | 0.949(8) | 1.785(8) | 2.7263(11) | 171.2(10) |
| O5 | H5b | O4 | 0.955(9) | 1.800(10) | 2.7480(11) | 171.0(13) |
| O6 | H6a | O9 | 0.952(6) | 2.088(7) | 3.0022(11) | 160.5(12) |
| O6 | H6b | O2 | 0.939(10) | 1.817(10) | 2.7512(11) | 172.9(11) |
| O7 | H7a | O3 | 0.955(13) | 1.888(14) | 2.8137(12) | 162.4(12) |
| O7 | H7b | O2 | 0.952(12) | 1.988(15) | 2.9045(14) | 160.9(13) |
| O8 | H8a | O4 | 0.955(17) | 1.861(17) | 2.8111(14) | 172.9(15) |
| O8 | H8b | O1 | 0.952(11) | 2.236(13) | 3.1189(11) | 153.7(13) |
| O9 | H9a | O1 | 0.950(15) | 1.795(15) | 2.7389(12) | 172.3(11) |
| O9 | H9b | O2 | 0.951(12) | 1.889(13) | 2.8384(10) | 175.4(17) |
 | ||
| Fig. 6 Perspective view along the a axis of the crystal structure of Na2Mn(SO4)2·4H2O (a). The dashed blue lines correspond to the O–H⋯O hydrogen bonds interconnecting the [Mn(SO4)2(H2O)4]2− building blocks involving the water molecules H6a–O6–H6b (b) and H5a–O5–H5b (c). The sodium atoms are located in the tunnels (a). | ||
It is very interesting to notice that the manganese phases Na2Mn(SO4)2·4H2O [this work], Na2Mn(SO4)2·2H2O,20 Na2+γMn1−γ/2(SO4)3,20 Na12Mn7(SO4)12[S2O7]·12H2O19 and Na2Mn(SO4)219 that crystallize with the blödite-, kröhnkite-, alluaudite-, löweite- and glauberite-type of structures, respectively were prepared via a supersaturation method at different temperatures and using the same precursors (Na2SO4 + MnSO4·H2O + H2O). At 25 and 70 °C, pure kröhnkite- and löweite-phases were formed, respectively whereas at 22 °C a mixture of kröhnkite-, blödite- and löweite-phases was observed. Furthermore, the thermal treatment of the kröhnkite-phase at 227 °C, led to the formation of an alluaudite-phase with the composition Na2+γMn1−γ/2(SO4)3.20 A similar phase could also be prepared via a solid state synthesis route when a mixture of Na2SO4 and MnSO4 was ball milled then annealed at 350 °C for few hours.42 Moreover, at 227 °C the löweite-phase decomposed into the glauberite-phase Na2Mn(SO4)2 besides an impurity that could be MnS2O7.19 Although, the five phases were prepared from the same precursors, their crystal structures are different (Fig. 7). Indeed, the thermal treatments affected significantly the coordination sphere of the Mn atoms. In the blödite-Na2Mn(SO4)2·4H2O, the Mn atom is coordinated to two oxygen atoms and four water molecules forming the [MnO2(H2O)4]2− octahedron. This octahedron share two corners with the SO4 tetrahedra to form the [Mn(SO4)2(H2O)4]2− building block (Fig. 7a). In the kröhnkite-Na2Mn(SO4)2·2H2O, the Mn atom is coordinated to four oxygen atoms and two water molecules. The MnO6 octahedra share corners with four SO4 tetrahedra to form infinite chains along the c axis (Fig. 7b). These chains are condensed at 227 °C to form the 3d-framework of the alluaudite-Na2+γMn1−γ/2(SO4)3. In this structure the MnO6 octahedra share edges and form dimer units that are interconnected through SO4 tetrahedra (Fig. 7c). In the löweite-Na12Mn7(SO4)12[S2O7]·12H2O, two Mn atoms exist. Mn1 is coordinated to two water molecules and four oxygen atoms, whereas Mn2 is coordinated to six oxygen atoms. The Mn1O6 and Mn2O6 octahedra are bridged by the SO4 tetrahedra to form a 3d-framework (Fig. 7d). At 227 °C, the water molecules of the löweite evaporated and a structural transition to the glauberite-Na2Mn(SO4)2 was observed. In this structure the MnO6 octahedra share corners with six SO4 tetrahedra to form a 3d-framework (Fig. 7e).
 | ||
| Fig. 7 Synthesis methods and crystal structure views for Na2Mn(SO4)2·4H2O (a), Na2Mn(SO4)2·2H2O (b), Na2+γMn1−γ/2(SO4)3 (c), Na12Mn7(SO4)12[S2O7]·12H2O (d) and Na2Mn(SO4)2 (e). The coordination spheres of the Mn atoms are also provided for comparison. | ||
3.3. Crystal structure of Na2Ni(SO4)2·10H2O
The Na2Ni(SO4)2·10H2O compound is isostructural with Na2Mg(SO4)2·10H2O.12 The structure is built up of [Ni(H2O)6]2+ and [Na2(SO4)2(H2O)4]2− layers parallel to the (100) plane. These layers are interconnected through hydrogen bonds forming the structure of Na2Ni(SO4)2·10H2O. The interatomic distances and the hydrogen bonds are listed in Tables 3 and 4, respectively.The nickel cations are coordinated to six water molecules forming isolated distorted [Ni(H2O)6]2+ octahedra. These octahedra are interlinked by the O6–H6a⋯O9 hydrogen bonds forming the layer 2 parallel to the (100) plane (Fig. 8c). The d(Ni1–O) distances range from 2.0365 to 2.0892 Å with an average distance of 2.0555 Å which is slightly shorter than the sum of the effective ionic radii of the six-coordinated Ni2+ and O2− {IR(Ni2+) + IR(O2−) = 0.69 + 1.4 Å}.38 The BVS value of 2.03 is in good agreement with the oxidation state 2+ expected for the Ni atoms.39
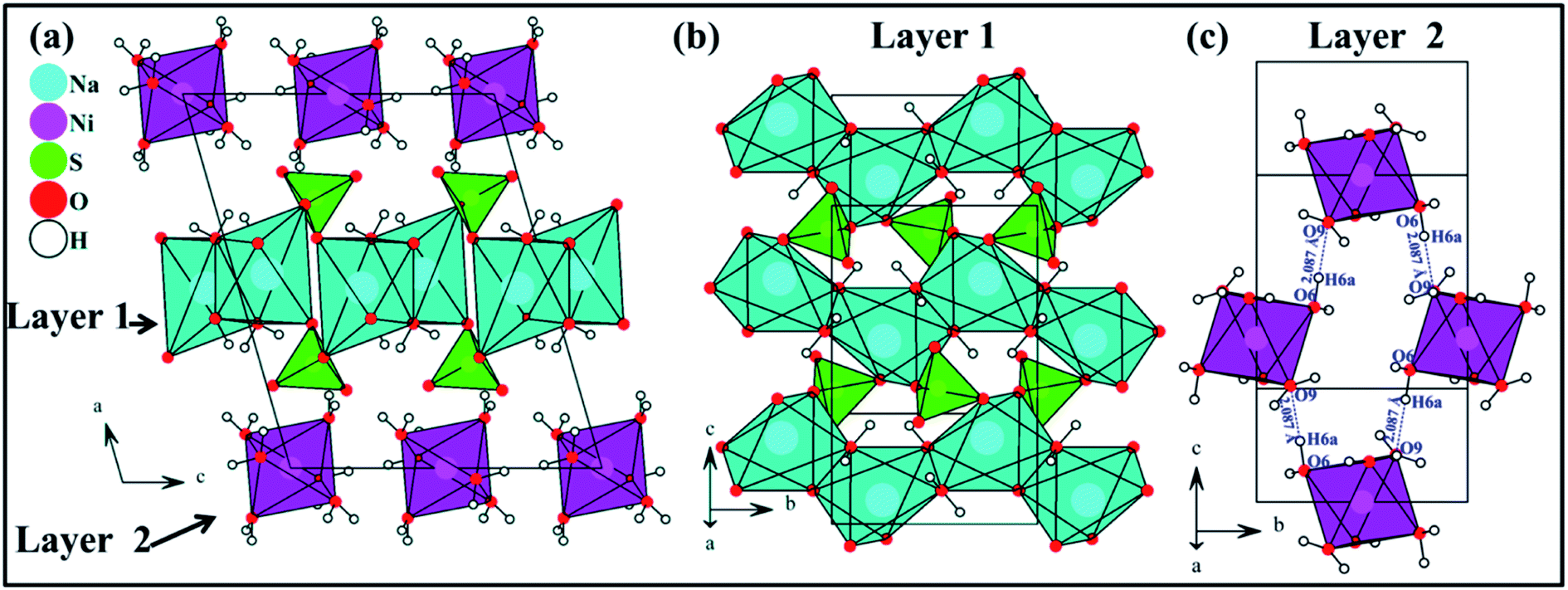 | ||
| Fig. 8 View along the b axis of the crystal structure of Na2Ni(SO4)2·10H2O (a), view of [Na2(SO4)2(H2O)4]2− layer 1 on the (100) plane (b) and view of [Ni(H2O)6]2+ layer 2 on the (100) plane (c). | ||
The SO4 tetrahedra are regular. The distances range from 1.4607 to 1.4842 Å with the average value of 1.4740 Å. This value is shorter than the sum of the effective ionic radii of the four-coordinated S6+ and O2−{IR(S6+) + IR(O2−) = 0.12 + 1.38 Å}. The BVS value of 6.00 is in good agreement with the oxidation state 6+ expected for sulfur.
The Na1+ cations are surrounded by six oxygen atoms forming distorted octahedra. These octahedra share edges and form infinite chains running along the b axis (Fig. 8b). These chains share corners with the SO4 tetrahedra forming [Na2(SO4)2(H2O)4]2− layers parallel to the (100) plane (see layer 1 in Fig. 8b). The average Na1–O distance of 2.4537 Å is consistent with the sum of the effective ionic radii of the six-coordinated Na+ and O2− {IR(Na+) + IR(O2−) = 1.02 + 1.40 Å}. The BVS value of 1.04 is in good agreement with the oxidation state 1+ expected for the Na atoms.
In Na2Ni(SO4)2·10H2O the water molecules are the hydrogen-bond donors, whereas the oxygen atoms O1, O3, O3, O4 and O9 play the role of hydrogen bond acceptors (Table 4 and Fig. 9). The hydrogen bonds can be divided into two categories; intra- and inter-layers bonds as depicted on Fig. 9a, c and b, respectively. Based on the classification of Jeffrey all the O–H⋯O hydrogen bonds are moderate (Table 4 and Fig. 9).40,41
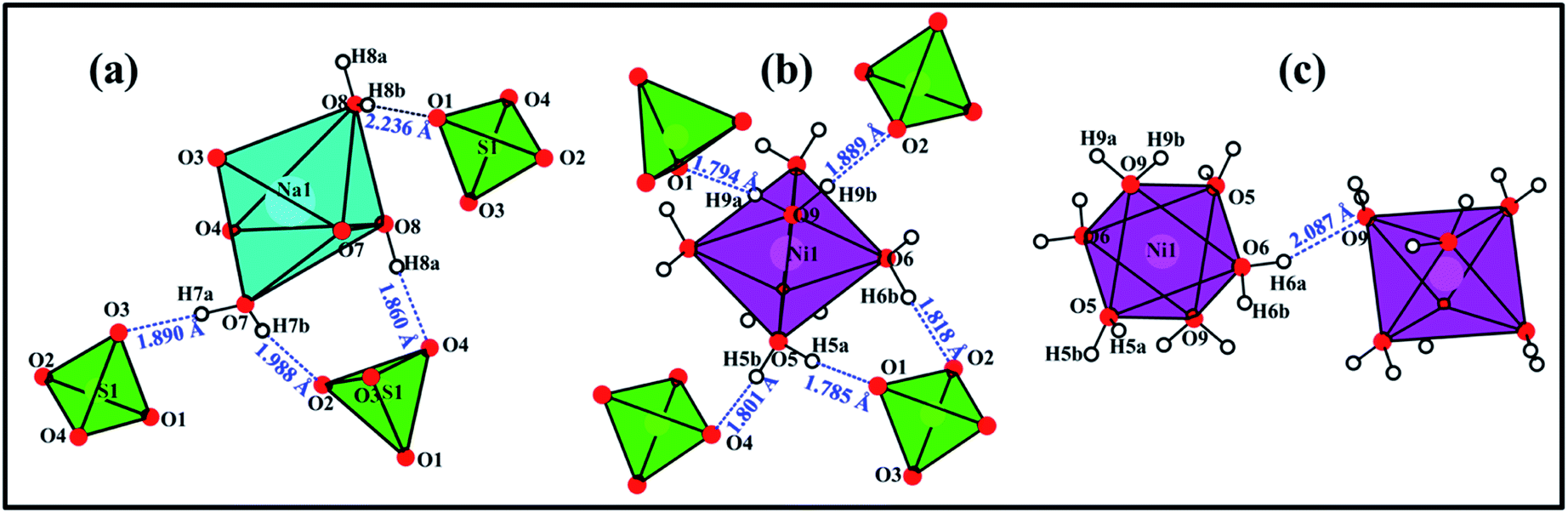 | ||
| Fig. 9 View of the hydrogen bonds within the layer 1 (a), between layer 1 and layer 2 (b), and within the layer 2 (c). | ||
It is worth to mention that in the nickel system Na2Ni(SO4)2·nH2O four phases have been reported (n = 0, 2, 4 and 6), however only the crystal structures of Na2Ni(SO4)2,24 Na2Ni(SO4)2·4H2O25 and Na2Ni(SO4)2·6H2O26 were solved (Fig. 10). Theses phases were prepared via solid state-, room temperature- and hydrothermal-synthesis routes, respectively and using different precursors and solvents. These experimental conditions are at the origin of the variation in the degree of hydration n of the Na2Ni(SO4)2·nH2O phases which induced several structural changes. Our careful analyses indicate that the presence of water molecules affects in first place the coordination sphere of the transition metal (nickel) due to its large electrical charge z. Indeed, for n = 0, the NiO6 octahedra share one edge and four corners with five SO4 tetrahedra leading to [Ni(SO4)2]2− 3d-framework (Fig. 10d). Whereas, for n = 4, the Ni atoms are coordinated to four water molecules and two oxygen atoms, from two adjacent SO4 tetrahedra, forming the isolated [Ni(SO4)2(H2O)4]2− building block of the blödite-type of structure (Fig. 10a). For n = 6 and 10, six water molecules are coordinated to the Ni atoms forming the isolated [Ni(H2O)6]2+ octahedra (Fig. 10b and c) which are connected to the SO4 tetrahedra only through hydrogen bonds. For n = 10, the (n − 6) extra water molecules are coordinated to the sodium atoms. Since n ≤ 10, in the four phases the water molecules are coordinated. One would expect to observe interstitial water only for n > 12. Indeed in the case of Na2Mg(SO4)2·16H2O, six water molecules are coordinated to the magnesium atoms, six water molecules are coordinated to the sodium atoms and four water molecules are interstitial.14 Therefore the chemical formula can be written as {[Na2(H2O)6][Mg(H2O)6][(SO4)2]}·4H2O.
 | ||
| Fig. 10 Synthesis methods and crystal structure views for Na2Ni(SO4)2·4H2O (a), Na2Ni(SO4)2·6H2O (b), Na2Ni(SO4)2·10H2O (c) and Na2Ni(SO4)2 (d). The coordination spheres of the Ni atoms are also provided for comparison. | ||
3.4. Comparison of the crystal structures
The quantitative comparison of the isotypic crystal structures within the Na2M(SO4)2·4H2O series (M = Mg, V, Mn, Fe, Co, Ni, Zn, Cd) and between the two isotypic structures of Na2M(SO4)2·10H2O (M = Mg, Ni), respectively was performed using the program compstru.43–46 The comparisons did not include the hydrogen atoms since in the various structural refinements different constrains/restrains on the water molecules were applied. Origin shifts of (0 ½ ½) and (0 ½ 0) were applied to the crystal structures of Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O, respectively. All the crystal structures in the Na2M(SO4)2·4H2O series were compared to Na2Ni(SO4)2·4H2O which was chosen as a reference. The numerical details of the comparisons are given in Table 5. The crystal structures of Na2Ni(SO4)2·4H2O and its Mg, Fe, Co, and Zn analogues show a very high similarity (Δ < 0.02) due to similar ionic radii of the five metal cations. Larger differences were observed with Mn and Cd (Δ = 0.044 and 0.079, respectively) due to their greater ionic radii. It should be also noted that the cell volume increases almost linearly with the ionic radii of the metal cations (Fig. 11).| Na2Ni(SO4)2·4H2O versus Na2M(SO4)2·4H2O | ||||||||
| M | Mg | Zn | Co | Fe | V | Mn | Cd | |
| Atom | |u|/Å | |u|/Å | |u|/Å | |u|/Å | |u|/Å | |u|/Å | |u|/Å | |
| Ni1/M | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | |
| Na1 | 0.0153 | 0.0088 | 0.0070 | 0.0091 | 0.0157 | 0.0285 | 0.0467 | |
| S1 | 0.0361 | 0.0197 | 0.0142 | 0.0241 | 0.0369 | 0.0523 | 0.0692 | |
| O1 | 0.0310 | 0.0216 | 0.0243 | 0.0505 | 0.0298 | 0.0626 | 0.0905 | |
| O2 | 0.0602 | 0.0172 | 0.0085 | 0.0226 | 0.0623 | 0.0325 | 0.0628 | |
| O3 | 0.0787 | 0.0320 | 0.0212 | 0.0343 | 0.0799 | 0.0797 | 0.1563 | |
| O4 | 0.0147 | 0.0183 | 0.0113 | 0.0115 | 0.0122 | 0.0456 | 0.1058 | |
| O5 | 0.0174 | 0.0228 | 0.0292 | 0.0580 | 0.0174 | 0.0965 | 0.1814 | |
| O6 | 0.0328 | 0.0337 | 0.0400 | 0.0719 | 0.0311 | 0.0971 | 0.1742 | |
| Degree of lattice distortion (S) | 0.0034 | 0.0030 | 0.0030 | 0.0046 | 0.0057 | 0.0082 | 0.0138 | |
| The maximum distance (dmax)/Å | 0.0787 | 0.0337 | 0.0400 | 0.0719 | 0.0799 | 0.0971 | 0.1814 | |
| Arithmetic mean (dav)/Å | 0.0337 | 0.0205 | 0.0183 | 0.0332 | 0.0336 | 0.0582 | 0.1043 | |
| Measure of similarity (Δ) | 0.019 | 0.014 | 0.014 | 0.018 | 0.027 | 0.044 | 0.079 | |
| Ref. | 7 | 7 | 7 | 34 | 33 | This work | 1 | |
|
||||||||
| Na2Mg(SO4)2·10H2O versus Na2Ni(SO4)2·10H2O | ||||||||
| Atom | |u|/Å | |||||||
| Mg1/Ni1 | 0.0000 | |||||||
| Na1 | 0.0117 | |||||||
| S1 | 0.0217 | |||||||
| O1 | 0.0444 | |||||||
| O2 | 0.0220 | |||||||
| O3 | 0.0430 | |||||||
| O4 | 0.0210 | |||||||
| O5 | 0.0132 | |||||||
| O6 | 0.0242 | |||||||
| O7 | 0.0181 | |||||||
| O8 | 0.0094 | |||||||
| O9 | 0.0377 | |||||||
| Degree of lattice distortion (S) | 0.0017 | |||||||
| The maximum distance (dmax)/Å | 0.0444 | |||||||
| Arithmetic mean (dav)/Å | 0.0232 | |||||||
| Measure of similarity (Δ) | 0.005 | |||||||
 | ||
| Fig. 11 Unit-cell volumes as a function of the ionic radius of the M cation in the blödite-type of compounds Na2M(SO4)2·4H2O (M = Mg, V, Mn, Fe, Co, Ni, Zn, Cd). | ||
The crystal structure of Na2Ni(SO4)2·10H2O was also compared to the Mg analogue using the compstru program. As indicated in Table 5, the two isotypic compounds are essentially coincident (Δ = 0.005). The largest deviations of 0.0444 and 0.0430 were observed for the atom pairs O1 and O3 of the MgO6 octahedron.
4. Conclusion
After the recent discovery of the new polymorphic modification of Na2Mn3(SO4)4 and the solid solution Na2Mn3−xMgx(SO4),47 two other sulfate phases were prepared for the first time by a wet chemistry route and their crystal structures were solved using single crystal XRD data. Na2Mn(SO4)2·4H2O with the blödite-type structure is the missing link in the series of Na2M(SO4)2·4H2O sulfates (M = Mg, V, Mn, Fe, Co, Ni, Zn, Cd), whereas Na2Ni(SO4)2·10H2O is the first compound isostructural with the aristotype Na2Mg(SO4)2·10H2O. The powder XRD data revealed that only the nickel phase was almost pure, whereas the manganese phase was a mixture of at least three phases. The TGA data provided the optimal conditions to fully dehydrate the nickel phase. When Na2Ni(SO4)2·10H2O is heated at temperatures between 300 and 600 °C, the anhydrous phase WC-Na2Ni(SO4)2 could be obtained. Interestingly, it is isostructural with SS-Na2Ni(SO4)2 which was prepared by solid state synthesis route and it is thermally more stable than Na2Fe(SO4)2 which is a suitable cathode material for NIBs. Therefore, the dehydration of hydrous sulfates and phosphates should be considered as a promising step toward further realization of novel cathode materials for NIBs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Authors gratefully acknowledge financial support from National Priorities Research Program (NPRP9-263-2-122) funded by Qatar National Research Fund (QNRF). Authors also would like to thank Dr Said Mansour for giving us access to the characterization tools in the core lab. The publication of this article was funded by the Qatar National Library.References
- D. K. Saha, G. Madras and T. N. Guru Row, Cryst. Growth Des., 2011, 11, 3213 CrossRef CAS.
- M. Wildner and D. Stoilova, Z. Kristallogr., 2003, 218, 201 CAS.
- L. M. Dikareva, Y. V. Zefirov, A. N. Zhilyaev, I. B. Baranovskii and M. A. Porai Koshits, Russ. J. Inorg. Chem., 1987, 32, 64 Search PubMed.
- V. I. Bukin and Y. Z. Nozik, J. Struct. Chem., 1974, 15, 616 CrossRef.
- M. Giglio, Acta Crystallogr., 1958, 11, 789 CrossRef CAS.
- M. E. Diaz Vivar, S. Baggio, A. Ibanez and R. F. Baggio, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, i30 CrossRef PubMed.
- D. Stoilova and M. Wildner, J. Mol. Struct., 2004, 706, 57 CrossRef CAS.
- P. Comodi, S. Nazzareni, T. Balic Zunic, A. Zucchini and M. Hanfland, Am. Mineral., 2014, 99, 511 CrossRef.
- S. J. Mills, S. A. Wilson, G. M. Dipple and M. Raudsepp, Mineral. Mag., 2010, 74, 903 CrossRef CAS.
- E. M. S. Leduc, R. C. Peterson and R. Wang, Am. Mineral., 2009, 94, 1005 CrossRef CAS.
- W. Wu, J. M. Xie, D. P. Xie and Y. W. Xuan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, i7 CrossRef CAS PubMed.
- A. V. Kasatkin, F. Nestola, J. Plášil, J. Marty, D. I. Belakovskiy, A. A. Agakhanov, S. J. Mills, D. Pedron, A. Lanza, M. Favaro, S. Bianchin, I. S. Lykova, V. Goliáš and W. D. Birch, Mineral. Mag., 2013, 77, 367 CrossRef CAS.
- E. M. S. Leduc, R. C. Peterson and R. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, i81 CrossRef CAS PubMed.
- K. Leftwich, D. L. Bish and C. H. Chen, Am. Mineral., 2013, 98, 1772 CrossRef CAS.
- I. A. Trussov, L. L. Male, M. L. Sanjuan, A. Orera and P. R. Slater, J. Solid State Chem., 2019, 272, 157 CrossRef CAS.
- M. Reynaud, G. Rousse, A. M. Abakumov, M. T. Sougrati, G. Van Tendeloo, J. N. Chotard and J. M. Tarascon, J. Mater. Chem. A, 2014, 2, 2671 RSC.
- P. Singh, K. Shiva, H. Celio and J. B. Goodenough, Energy Environ. Sci., 2015, 8, 3000 RSC.
- P. Barpanda, G. Oyama, C. D. Ling and A. Yamada, Chem. Mater., 2014, 26(3), 1297 CrossRef CAS.
- D. Swain and T. N. Guru Row, Inorg. Chem., 2009, 48, 7048 CrossRef CAS PubMed.
- D. Marinova, V. Kostov, R. Nikolova, R. Kukeva, E. Zhecheva, M. Sendova-Vasileva and R. Stoyanova, J. Mater. Chem. A, 2015, 3, 22287 RSC.
- P. Barpanda, G. Oyama, S. Nishimura, S.-C. Chung and A. Yamada, Nat. Commun., 2014, 5, 4358 CrossRef CAS PubMed.
- G. Oyama, S. Nishimura, Y. Suzuki, M. Okubo and A. Yamada, ChemElectroChem, 2015, 2, 1019 CrossRef CAS.
- G. Oyama, O. Pecher, K. J. Griffith, S. Nishimura, R. Pigliapochi, C. P. Grey and A. Yamada, Chem. Mater., 2016, 28, 5321 CrossRef CAS.
- A. M. Fry, O. T. Sweeney, W. A. Phelan, N. Drichko, M. A. Siegler and T. M. McQueen, J. Solid State Chem., 2015, 222, 129 CrossRef CAS.
- M. E. D. De Vivar, S. Baggio, M. T. Garland and R. F. Baggio, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i196 CrossRef.
- P. S. Zhao, F. F. Jian, Z. S. Bai, J. Zheng and R. R. Zhuang, Struct. Chem., 2006, 17, 519 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Crystallographic Computing System JANA2006: General features, Z. Kristallogr., 2006, 229, 345 Search PubMed.
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- E. Matzat, Neues Jahrb. Mineral., Abh., 1970, 113, 1 CAS.
- A. V. Kasatkin, M. Favaro, S. Bianchin, I. S. Lykova, V. Golias, W. D. Birch, F. Nestola, J. Plasil, J. Marty, D. I. Belakovskiy, A. A. Agakhanov, S. J. Mills, D. Pedron and A. Lanza, Mineral. Mag., 2013, 77, 367 CrossRef CAS.
- I. M. Rumanova, Dokl. Akad. Nauk SSSR, 1958, 118, 84 CAS.
- S. Peytavin and L. Cot, C. R. Seances Acad. Sci., Ser. C, 1969, 269, 1206 CAS.
- M. Hudak, J. G. Diaz and J. Kozisek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, i10 CrossRef CAS PubMed.
- E. H. Nickel and P. J. Bridge, Mineral. Mag., 1977, 4137, 41 Search PubMed.
- J. Schlueter, K.-H. Klaska and G. Gebhard, Neues Jahrb. Mineral., Monatsh., 1999, 3, 97 Search PubMed.
- D. Marinova, M. Wildner, T. Bancheva, R. Stoyanova, M. Georgiev and D. G. Stoilova, Phys. Chem. Miner., 2018, 45, 801 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- D. Dwibedi, R. B. Araujo, S. Chakraborty, P. P. Shanbogh, N. G. Sundaram, R. Ahuja and P. Barpanda, J. Mater. Chem. A, 2015, 3, 18564 RSC.
- G. de la Flor, D. Orobengoa, E. Tasci, J. M. Perez-Mato and M. I. Aroyo, J. Appl. Crystallogr., 2016, 49, 653 CrossRef CAS.
- C. Capillas, J. M. Perez-Mato and M. I. Aroyo, J. Phys.: Condens. Matter, 2007, 19, 275203 CrossRef.
- D. Orobengoa, C. Capillas, M. I. Aroyo and J. M. Perez-Mato, J. Appl. Crystallogr., 2009, 42, 820 CrossRef CAS.
- G. Bergerhoff, M. Berndt, K. Brandenburg and T. Degen, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 147 CrossRef PubMed.
- H. Ben Yahia, Z. Kristallogr.–Cryst. Mater., 2019, 234, 697 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The theoretical powder X-ray diffraction patterns (PXRD) of Na12Mn7(SO4)13·15H2O, Na2Mn(SO4)2·2H2O and Na2Mn(SO4)2·4H2O, the experimental PXRD and the TGA of the sample containing manganese and the anisotropic displacement parameters (in Å2) for Na2Mn(SO4)2·4H2O and Na2Ni(SO4)2·10H2O are given in supplementary information. CCDC 1962424 and 1962425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00301h |
| This journal is © The Royal Society of Chemistry 2020 |