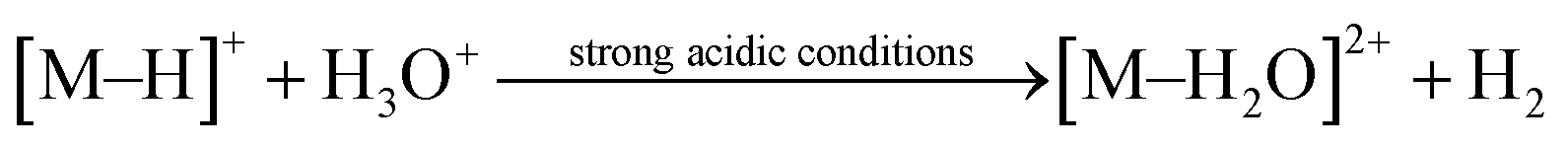DOI:
10.1039/C9RA10651K
(Paper)
RSC Adv., 2020,
10, 10411-10419
pH-Dependent transfer hydrogenation or dihydrogen release catalyzed by a [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ complex: a DFT mechanistic understanding†
Received
18th December 2019
, Accepted 15th February 2020
First published on 11th March 2020
Abstract
The reaction mechanism of the pH-dependent transfer hydrogenation of a ketone or the dehydrogenation of formic acid catalyzed by a [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ complex in aqueous media has been investigated using the density functional theory (DFT) method. The TM-catalyzed TH of ketones with formic acid as the hydrogen source proceeds via two steps: the formation of a metal hydride and the transfer of the hydride to the substrate ketone. The calculated results show that ruthenium hydride formation is the rate-determining step. This proceeds via an ion-pair mechanism with an energy barrier of 14.1 kcal mol−1. Interestingly, the dihydrogen release process of formic acid and the hydride transfer process that produces alcohols are competitive under different pH environments. The investigation explores the feasibility of the two pathways under different pH environments. Under acidic conditions (pH = 4), the free energy barrier of the dihydrogen release pathway is 4.5 kcal mol−1 that is higher than that of the hydride transfer pathway, suggesting that the hydride transfer pathway is more favorable than the dihydrogen release pathway. However, under strongly acidic conditions, the dihydrogen release pathway is more favorable compared to the hydride transfer pathway. In addition, the ruthenium hydride formation pathway is less favorable than the ruthenium hydroxo complex formation pathway under basic conditions.
Introduction
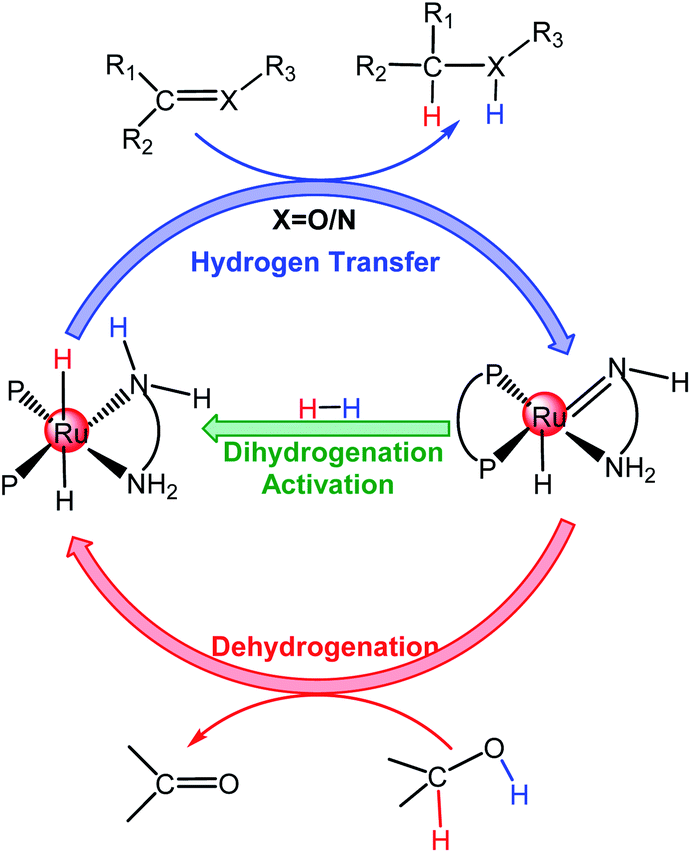
The hydrogenation of polar double bonds, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN, catalyzed by transition-metal (TM) complexes is a fundamentally important method to produce value-added alcohol- and amine-containing chemicals in organic syntheses and chemical industries.1,2 The transfer hydrogenation (TH) of ketones using organic hydrogen sources as well as H2 hydrogenation (HH) using molecular hydrogen resources have gained more attention.3 The TH of ketones can be performed either in water or in organic solvents.4–8 However, due to environmental and ecological advantages and reaction-specific pH selectivity, using water as the solvent for the TH of ketones is very convenient and attractive compared to traditional organic solvents.9–12 Two hydrogen sources have primarily been used in TM-catalyzed TH, namely isopropanol and formic acid. The conception of metal–ligand bifunctional cooperation is often used to explain the reaction mechanism of TH and the HH of polar double bonds catalyzed by TM complexes with different ligand environments such as diphosphine-diamine ruthenium catalyst A (see Scheme 1)13–19 as well as those of TH using formic acid hydrogen sources.20–23 For the mode of hydrogen transfer, inner-sphere and outer-sphere mechanisms are proposed based on the direct/indirect interactions between the metal center and the atoms of the ketones/imines, excluding the hydrogen atoms.
O and CN, catalyzed by transition-metal (TM) complexes is a fundamentally important method to produce value-added alcohol- and amine-containing chemicals in organic syntheses and chemical industries.1,2 The transfer hydrogenation (TH) of ketones using organic hydrogen sources as well as H2 hydrogenation (HH) using molecular hydrogen resources have gained more attention.3 The TH of ketones can be performed either in water or in organic solvents.4–8 However, due to environmental and ecological advantages and reaction-specific pH selectivity, using water as the solvent for the TH of ketones is very convenient and attractive compared to traditional organic solvents.9–12 Two hydrogen sources have primarily been used in TM-catalyzed TH, namely isopropanol and formic acid. The conception of metal–ligand bifunctional cooperation is often used to explain the reaction mechanism of TH and the HH of polar double bonds catalyzed by TM complexes with different ligand environments such as diphosphine-diamine ruthenium catalyst A (see Scheme 1)13–19 as well as those of TH using formic acid hydrogen sources.20–23 For the mode of hydrogen transfer, inner-sphere and outer-sphere mechanisms are proposed based on the direct/indirect interactions between the metal center and the atoms of the ketones/imines, excluding the hydrogen atoms.
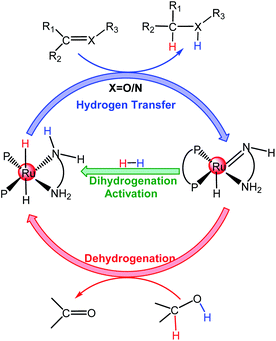 |
| | Scheme 1 The metal–ligand bifunctional mechanism for ketone/imine hydrogenation catalyzed by Ru complexes. | |
A number of TH reactions of polar double bonds catalyzed by cyclometallated TM complexes have been developed in the past decades.11,24–27 Most of the TM catalysts for ketone/imine hydrogenation have a Lewis acidic site in the TM center and a Lewis basic site in the ligand moiety of the structure (TM-LB catalyst) or a Lewis basic site in the TM center and a Lewis acidic site in the ligand moiety (TM-LA catalyst).2,28 In 1986, Shvo et al. reported a useful TH catalyst (B in Scheme 2) for ketones, which is an example of a ligand–metal bifunctional catalyst wherein the redox activity is distributed between the metal center and a cyclopentadienone ligand.29 In 1995, Noyori et al. synthesized η6-arene-Ru complexes bearing monotosylated 1,2-diamine moieties (C in Scheme 2), which are efficient TH catalysts for ketones.30 In 2006, Xiao et al. made a breakthrough by improving catalyst efficiency using iridium catalysts with N-sulfonyl ethylenediamine as the ligand (D in Scheme 2) in water with HCOONa as the hydrogen source.31 Some cyclometallated TM complexes are single-site catalysts. They can be modified in the ligand moiety to provide a Lewis basic or acidic site and then can become bifunctional catalysts like B, C, and D. In 2002, Ogo et al. reported the TH of ketones using HCOONa or HCOOH in water with cyclometalated single-site Ru complexes (E in Scheme 2).32 In 2012, Fukuzumi et al. reported a single-site cyclometalated Ir complex bearing a bpyO ligand (F in Scheme 2), which could catalyze aliphatic alcohol dehydrogenation at room temperature in a basic aqueous solution.33 In 2013, Xiao et al. reported another cyclometalated single-site Ir complex (G in Scheme 2), which was shown to be an excellent catalyst for the TH of carbonyl compounds in water using formate as the hydrogen source.27 As discussed above, it is obvious that the mechanism of TH reactions catalyzed by cyclometallated single-site TM complexes is different from that of bifunctional TM catalysts. However, reports concerning the mechanism of TH with formic acid as the hydrogen source and catalyzed by cyclometallated single-site TM complexes have been sporadic.22 The TM-catalyzed TH of ketones with formic acid as the hydrogen source proceeds via two sequential steps. First, hydride transfer from the formate moiety to the metal center (formation of the metal hydride) occurs and then a hydride transfer step from the metal to the ketone substrate follows. There are two modes for metal hydride formation: the β-hydrogen elimination mechanism and the ion-pair mechanism (see Scheme 3a).22
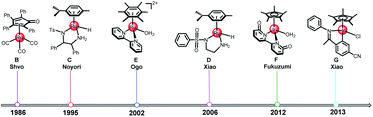 |
| | Scheme 2 The development of cyclometallated TM complexes. | |
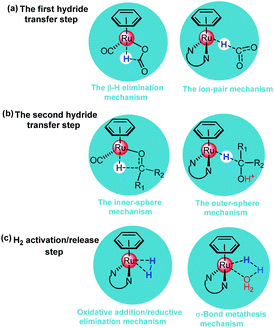 |
| | Scheme 3 (a) Representative reaction modes of formate decarboxylation to produce the metal hydride (b) Representative reaction modes of hydride transfer from the metal to the ketone substrate. (c) Representative reaction modes of H2 activation/release. | |
For the β-hydrogen elimination mechanism, it has been stated that the formate anion must occupy two coordination sites before cleavage of the C–H bond of the formate can occur.34 In 2013, Yang et al.35 investigated the mechanism of formic acid dehydrogenation catalyzed by an iron complex via the DFT method and their study indicated that β-hydrogen elimination was involved in the transition state (TS) of metal hydride formation. In the ion-pair mechanism, the metal hydride is formed by the direct hydride transfer from the formate moiety to the metal center in the form of an ion pair and the Ru–O bond is broken before metal hydride formation.36 In 2015, Xiao et al.22 investigated the mechanism of imine reduction with formic acid catalyzed by a single-site cyclometallated Ir catalyst, the calculated results showed that metal hydride formation proceeds via an ion-pair mechanism.37 If the TM center of the metal hydride species is coordinatively unsaturated, the substrates prefer to coordinate with the TM center according to the β-hydrogen elimination mechanism along an inner-sphere pathway.38 If it is coordinatively saturated, the substrate will coordinate with the TM center according to the ion-pair mechanism along an outer-sphere pathway, which is supported by experimental observations.22,27,32,39 The reaction pathways of the second hydride transfer step from the metal to the ketone substrate are similar to the reversible processes of the first hydride transfer step mentioned above (see Scheme 3b).
The non-cooperation mechanism of H2 activation/release for single-site TM catalysts can be divided into the classical oxidative addition/reductive elimination mechanism and the σ-bond metathesis (hydrogenolysis) mechanism (see Scheme 3c).28 In the classical oxidative addition/reductive elimination mechanism, the coordination of H2 affords a typical dihydrogen complex [M–H2], which quickly undergoes oxidative addition to form a dihydride intermediate [H–M–H] along a homolytic splitting pathway. The oxidation state of the metal center increases as the dihydride complex is formed in the oxidative addition of H2. In the σ-bond metathesis mechanism, the metal hydride [M–H] generally prefers to activate/release H2 via a heterolytic splitting pathway. During the dehydrogenation, solvents, including water or formate, can assist H2 release.
Previous studies on the TH of ketones in aqueous media have revealed that water can accelerate ketone reduction and that pH has a dramatic effect on the catalytic activities of cyclometallated Ru, Rh, and Ir complexes.12,24,25,31,32,40–42 This effect was also observed in the dehydrogenation reactions of formic acid.43–48 In 2003, Ogo et al. reported pH-dependent TH of a variety of carbonyl compounds catalyzed using a cyclometallated Ir catalyst in water.25 They proposed that the dehydrogenation of formic acid is achieved under strongly acidic conditions, which was confirmed via a GC analysis. The TH of ketones and formic acid dehydrogenation catalyzed by the same cyclometallated catalyst in water are two competitive pathways at different pH values.
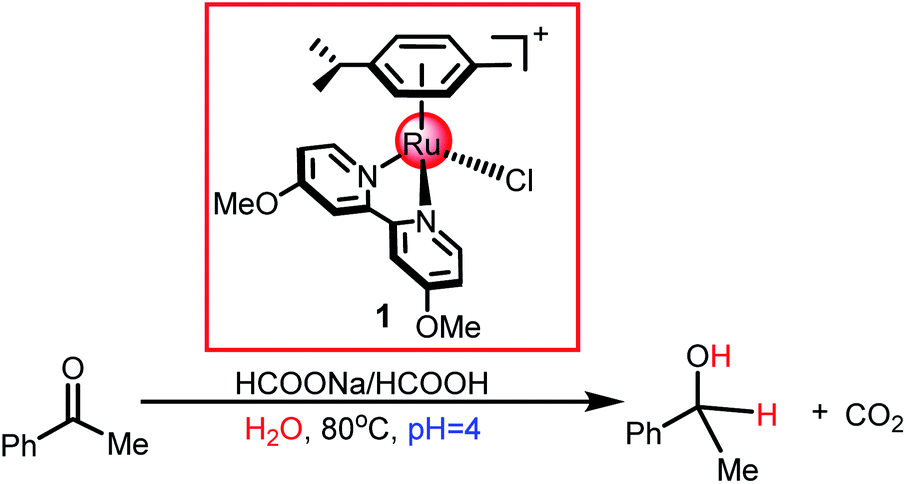
Recently, Espino et al.49 developed a pH-dependent catalytic system for the TH of acetophenone using functional cyclometallated [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ as the catalyst precursor, which showed only one open coordination site upon chloride dissociation (see Scheme 4). They proposed a tentative reaction mechanism for cyclometallated [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ (1) catalyzed acetophenone hydrogenation, which included two steps: hydride transfer from the formate to the metal center via the β-hydrogen elimination mechanism and hydride transfer to substrates via the inner-sphere mechanism. In the past decades, a lot of experimental and theoretical studies have been performed to unveil the nature of the TH and HH of ketones catalyzed by TM complexes and to understand the preference for different hydrogen resources in different catalytic systems. However, the reaction mechanism of pH-dependent transfer hydrogenation or dihydrogen release catalyzed by a [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ complex is still unclear. Herein, a DFT study was performed to investigate the nature of this system, which may provide insights to develop an understanding on the TH and HH of polar double bonds catalyzed by cyclometallated single-site TM complexes.
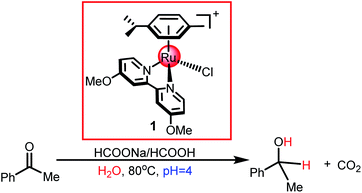 |
| | Scheme 4 Reduction of ketones catalyzed by the cyclometallated ruthenium single-site complex. | |
Computational methods
In accordance with our previous computational studies,3,19,50–55 all calculations in this study were carried out using the DFT method with ωB97X-D56 using the Gaussian 09 program.57 The SMD polarizable continuum model in water as the solvent was employed in the calculations.58 The effective core potential (ECP) of Ru with a double-ζ valence basis set (LANL2DZ) was chosen to describe Ru and the 6-31G(d,p) basis set for other atoms.59,60 All the transition states were confirmed via vibrational analysis and characterized by only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed to confirm all transition states connecting two desired minima. All relative energies of the stationary points along the reaction pathway are relative to complex 2. Unless otherwise stated, the energy values in the following parts are free energies. Gibbs free energies were calculated at 298.15 K.
Results and discussion
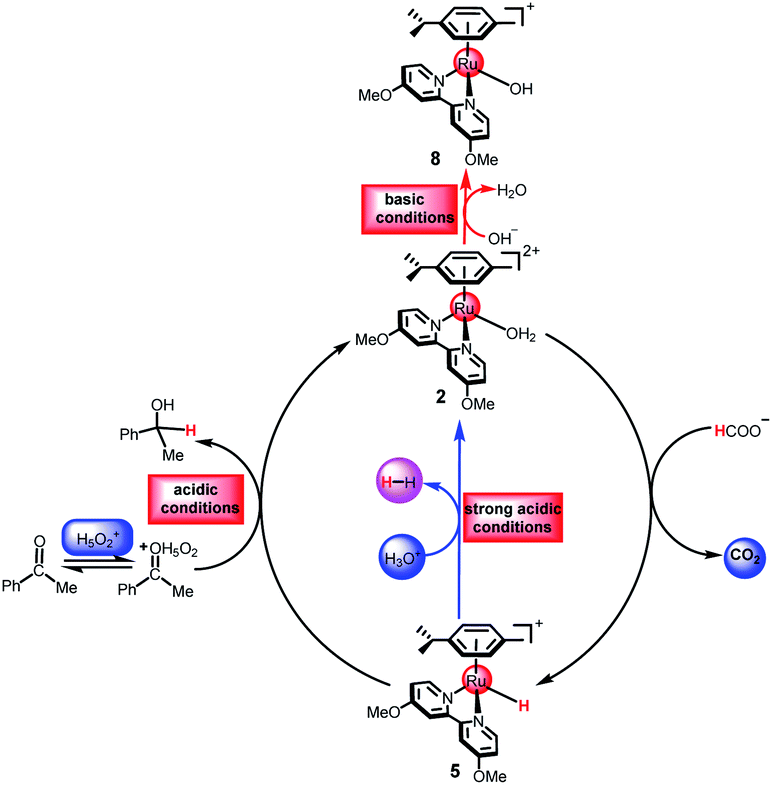
The reaction mechanism of ketone transfer hydrogenation or dehydrogenation of formic acid catalyzed by the [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ complex is shown in Fig. 1. Two steps exist in this process: the first step is the formation of a metal hydride and the second step is hydride transfer from the metal to the ketone substrate to complete TH or the dihydrogen release. The mechanisms of TH using ketones and the dehydrogenation of formic acid have the same first step. Initially, the catalytic precursor 1 undergoes an aquation step to give complex 2, which is the active catalytic species in the catalytic cycle.49 Then, the H2O of complex 2 is substituted with the formate anion to form 3. The next step is the metal hydride formation from 3 to 5. Subsequently, the dihydrogen release pathway and the hydride transfer pathway compete to release dihydrogen or complete the TH of the ketone. The hydride of 5 coordinates to the protonated ketone (in acidic aqueous media) to form intermediate 6 or reacts with hydrated protons to release dihydrogen. The reverse process of dihydrogen release, the dihydrogen activation, is also considered. Finally, the active catalytic species 2 is regenerated.
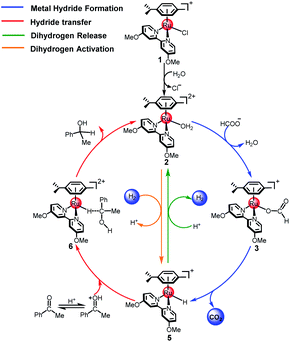 |
| | Fig. 1 The catalytic cycle for the TH of ketones catalyzed by cyclometallated ruthenium single-site complexes. | |
Metal hydride formation
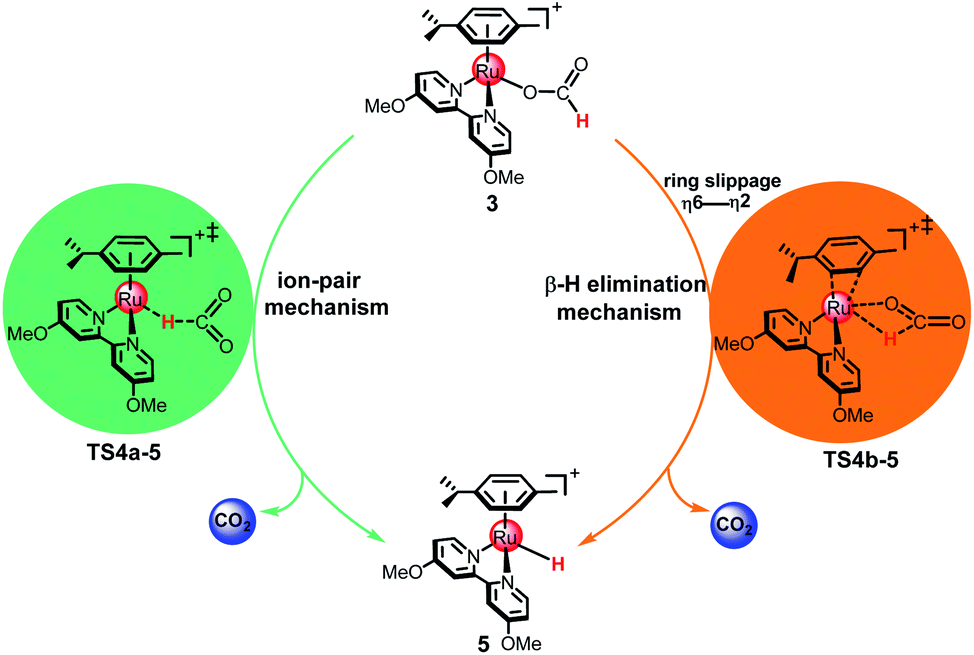
As shown in Fig. 2, two possible mechanisms for the formation of metal hydride 5 from 3 exist: the ion-pair mechanism and the β-hydrogen elimination mechanism. In the ion-pair mechanism, the intermediate 5 is formed via TS4a-5, which forms an ion pair between the dissociated formate and the cationic 16e Ru(II) complex. The β-hydrogen elimination mechanism involves a change in the arene ring from η6- to η2-coordination by ring slippage from 3 to TS4b-5. Then, the metal hydride 5 is formed via TS4b-5 with a four-membered ring structure.
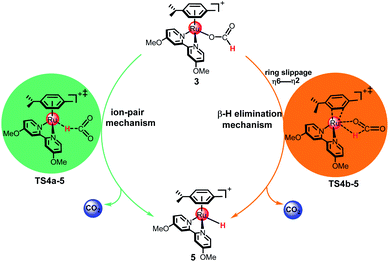 |
| | Fig. 2 The ion-pair mechanism and β-hydrogen elimination mechanism for the formation of metal hydride 5. | |
The ion-pair mechanism. In the ion-pair mechanism, the H2O ligand in the catalytic species 2 is substituted with HCOO− to form intermediate 3 in the presence of the formate anion. This process is exergonic by 12.1 kcal mol−1 (see Fig. 3). Then, 3 undergoes conformational conversion to produce intermediate 4a. This process is endergonic by 12.7 kcal mol−1. Subsequently, the metal hydride intermediate 5 is formed by a hydride transfer step from the formate to the Ru center via TS4a-5, with an energy barrier of 1.4 kcal mol−1. Finally, the C–H bond cleavage affords the metal hydride 5 and releases one molecule CO2. The Ru–H distance is 2.953 Å in 3, indicating that there is little interaction between the hydrogen atom of the formate and the Ru(II) center. From 4a to 5, the Ru–H distance decreases from 1.840 Å to 1.598 Å, showing that a covalent Ru–H bond is formed in 5. Notably, the Ru–O bond cleavage is accompanied by charge separation and is more difficult than the C–H bond activation of the formate moiety in 3. The energy barrier for this ion-pair pathway is 14.1 kcal mol−1 from 3 to TS4a-5.
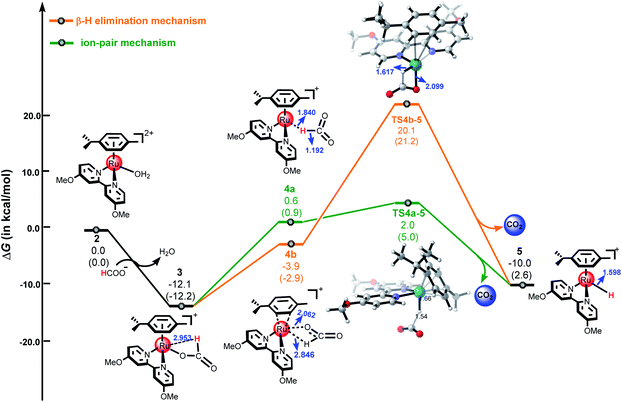 |
| | Fig. 3 The free energy profiles for the formation of metal hydride 5. All energies are denoted in kcal mol−1 and interatomic distances are shown in Å, the values in parentheses are electronic energies. | |
The β-hydrogen elimination mechanism. A feature of the β-hydrogen elimination mechanism is the ring slippage of the arene ring from η6-coordination to η2-coordination, i.e., from intermediate 3 to intermediate 4b. This process is endergonic by 8.2 kcal mol−1. Then, 5 is formed after hydride transfer from the formate to the Ru center via TS4b-5, which has a four-member ring structure with an energy barrier of 24.0 kcal mol−1. In comparison to TS4a-5 from the ion-pair mechanism, the most notable feature here is that the oxygen of the formate moiety coordinates with the Ru center while the Ru–O bond distance changes from 2.062 Å in 4b to 2.099 Å in TS4b-5. The energy barrier for this β-hydrogen elimination pathway is 32.2 kcal mol−1 from 3 to TS4b-5, which is 18.1 kcal mol−1 higher than that of the ion-pair pathway (14.1 kcal mol−1), indicating that the β-hydrogen elimination mechanism is much less favorable than the ion-pair mechanism.
Ketone hydrogenation or dihydrogen release
Fig. 4 shows four possible pathways for the second hydride transfer to complete the TH of ketones. There are two outer-sphere pathways and two inner-sphere pathways for hydride transfer from the metal center to the carbon atom of the ketone substrate, which belong to the ion-pair mechanism and β-hydrogen elimination mechanism, respectively.
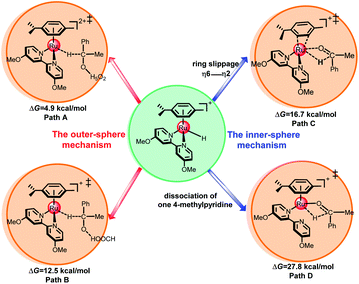 |
| | Fig. 4 Four possible pathways of hydride transfer from the metal to the ketone substrate. The values of ΔG (in kcal mol−1) indicate the free energy barriers of the four possible pathways. | |
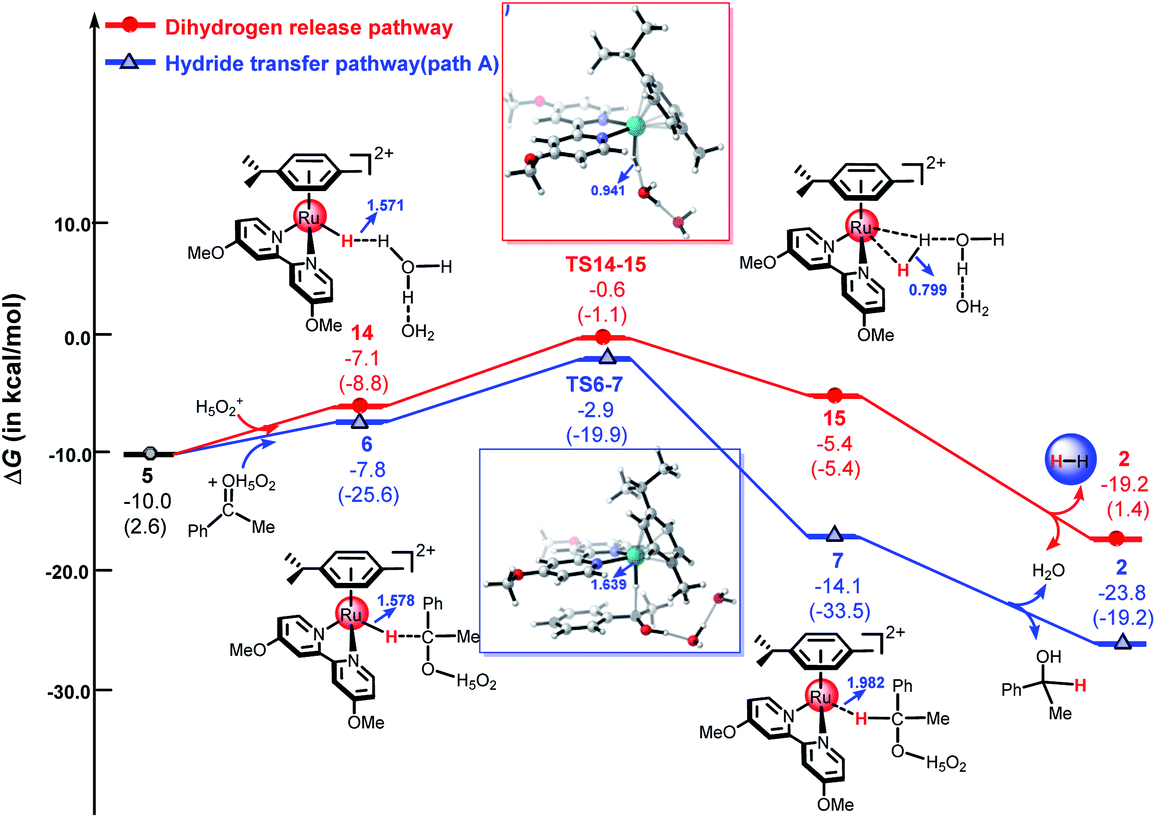
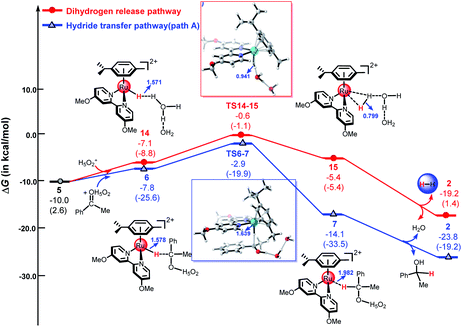
As shown in Fig. 5, the ketone substrate can be stabilized with a hydrated proton (H5O2+) through hydrogen bonding under acidic conditions in path A, this process is endergonic by 3.6 kcal mol−1 (see Fig. S8†). The use of the hydrated proton (H5O2+) here corresponds to an acidic aqueous medium.12 First, the protonated ketone coordinates with metal hydride 5 to form intermediate 6. Then, the hydride is transferred from the Ru center to the carbon atom of the protonated ketone via TS6-7 with an energy barrier of 4.9 kcal mol−1. Subsequently, intermediate 7 is formed, the free energy of complex 7 is −14.1 kcal mol−1. From 6 to TS6-7, the Ru–H bond length increases from 1.578 Å to 1.639 Å and the C–H distance decreases from 2.431 Å to 1.697 Å. The calculated energy barriers of path B, path C, and path D are 12.5 kcal mol−1, 16.7 kcal mol−1, and 27.8 kcal mol−1, respectively (see Fig. 4 above and Fig. S1–S3 in ESI†). The calculated results demonstrate that hydride transfer from the metal to the ketone substrate via the ion-pair mechanism (path A) is more favorable than the three other possible pathways.
 |
| | Fig. 5 The free energy profiles of the hydride transfer pathway (path A) and dihydrogen release pathway. All energies are denoted in kcal mol−1 and interatomic distances are shown in Å, the values in brackets are electronic energies. | |
Recently, several studies have shown that metal hydride species ([M–H]+) can react with hydrated protons to release H2 in an acidic aqueous medium.43,44,47,48,61 As mentioned above, the dihydrogen release pathway and the second hydride transfer pathway (path A) could be two competitive processes driven by the cyclometallated ruthenium single-site species 5. As shown in Fig. 5, the energy barrier of the dihydrogen release pathway is 9.4 kcal mol−1, which is 4.5 kcal mol−1 higher than that of path A (4.9 kcal mol−1). Obviously, the hydride transfer pathway via an outer-sphere ion-pair mechanism (path A) is more favorable than the dihydrogen release pathway, which is in agreement with experimental results.49
The influence of pH on reactivity
Espino et al. found that this [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ system is pH-dependent in experiments.49 A pH value of 4 was found to be the most favorable and the reduction becomes slow or stagnant outside of a reasonable pH value. Furthermore, the experimental results have shown that the reversible formation of unreactive [Ru–OH]+ at high pH levels (basic condition) leads to inactivity. Several cases of pH-dependent selective TH of ketones have been reported by several groups.24,32,62 For those active cyclometallated TM single-site complexes [M–H2O]2+ and metal hydrides [M–H]+, Ogo et al. proposed that (1) under strongly acidic conditions, the protonation of the [M–H]+ leads to the release of H2, which was confirmed by GC analysis (see eqn (1)); (2) under basic conditions, [M–H2O]2+ is predominantly deprotonated to form a hydroxo complex [M–OH]+, which easily leads to the termination of the reaction (see eqn (2)). In 2018, Xu et al.11 pointed out that even though a metal hydride is generated, reduction did not occur under neutral or basic conditions. Thus, the pH-dependence is also related to the proton-mediated activation of the ketones.| |
 | (1) |
| |
 | (2) |
That is to say, different pH values may result in different reaction reactivities. As shown in Fig. 6, the active species 5 can react with H3O+ to release H2 under strongly acidic conditions or with the protonated ketone to undergo the second hydride transfer pathway. The active intermediate 2 can form the hydroxo intermediate 8 leading to the termination of the reaction under basic conditions or the formation of the metal hydride 5 under acidic conditions. The metal hydride formation pathway and the metal hydroxo formation pathway are the two processes open to the catalytic species 2 and the chosen pathway is dependent on the pH values.
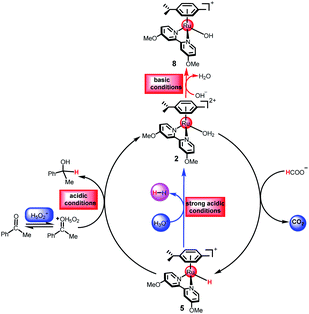 |
| | Fig. 6 Proposed mechanism for TH of ketones catalyzed by cyclometallated ruthenium single-site complex at different pH values. | |
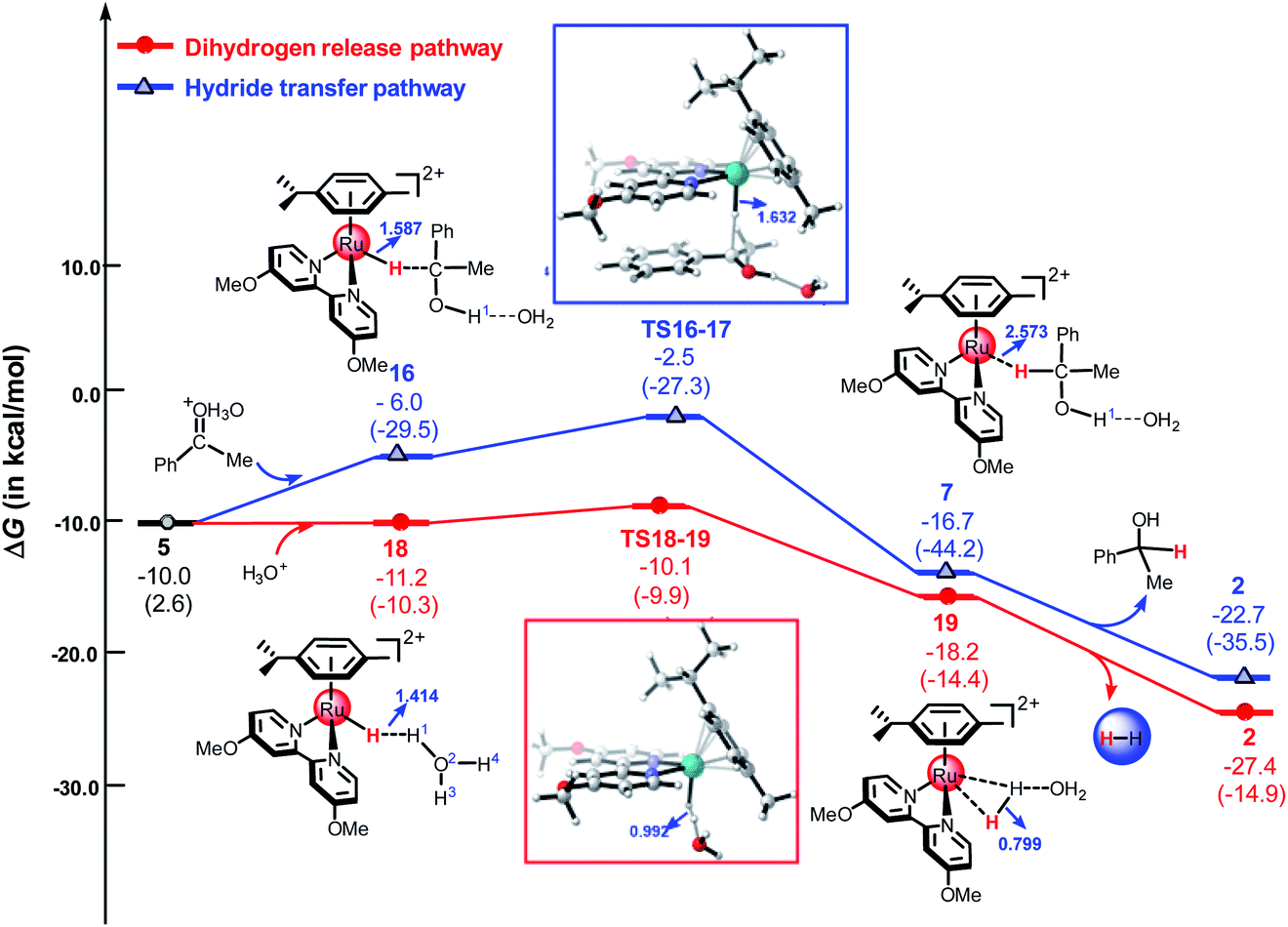
Herein, we used the DFT method to investigate the reaction mechanism for the TH of ketones catalyzed by a cyclometallated ruthenium single-site complex at different pH values. We used different hydrated protons including H3O+, H5O2+, and H7O3+ to indicate the different pH values of the solution. Fig. 7 shows the free energy profiles of the dihydrogen release pathway and the hydride transfer pathway mediated by H3O+. The energy barrier of the dihydrogen release pathway is 1.1 kcal mol−1. Interestingly, the reaction energy barrier along the hydride transfer pathway (Path A) is 3.5 kcal mol−1. In general, the dihydrogen release pathway, driven by the cyclometallated TM single-site system, is more favorable than the hydride transfer pathway under strongly acidic conditions, which is in agreement with the experimental results.11,25 Compared with that of the H1 atom of H5O2+ and H7O3+, the H1 atom in H3O+ carries a more positive charge (see Table S1 in ESI†). This means that H3O+ is more acidity. The calculated results indicated that the dihydrogen release pathway is less favorable than the hydride transfer pathway (Path A) for the reaction mediated by H5O2+ or H7O3+ (see Fig. 5 and S4 in ESI†).
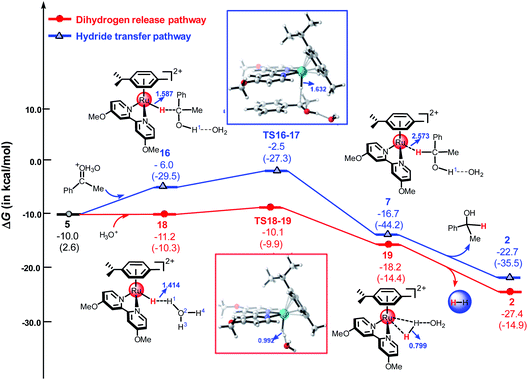 |
| | Fig. 7 The free energy profiles of the hydride transfer pathway and the dihydrogen release pathway mediated by H3O+. All energies are denoted in kcal mol−1 and interatomic distances are shown in Å, the values in parentheses are electronic energies. | |
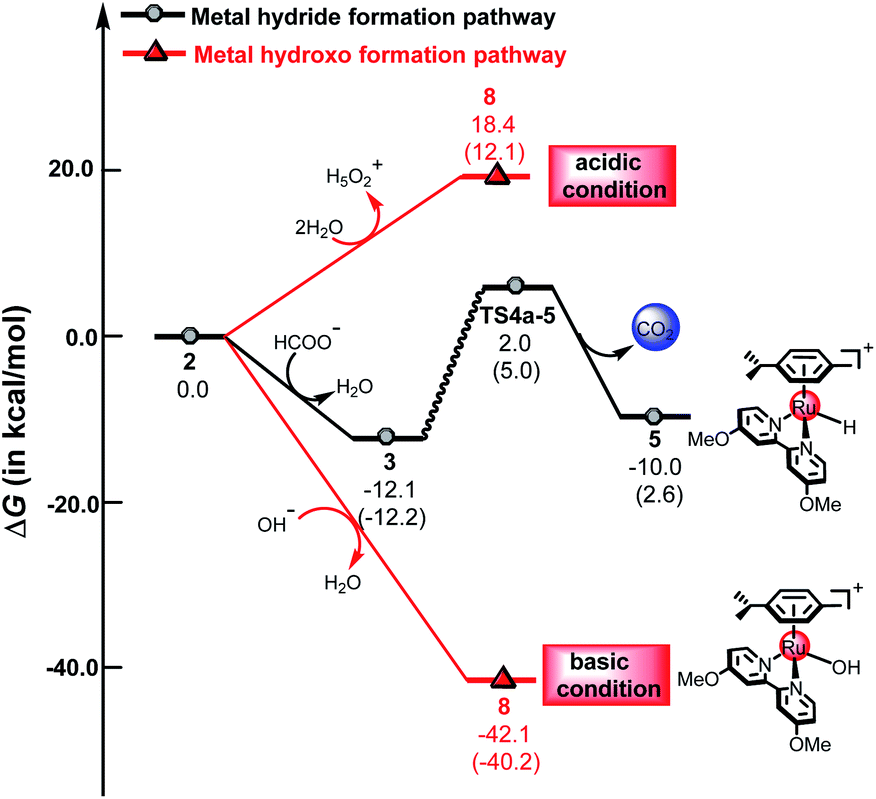
Fig. 8 demonstrates the energy profiles of the metal hydride formation pathway and the metal hydroxo formation pathway under acidic and basic conditions. The energy barrier of the metal hydride formation is 14.1 kcal mol−1, which may also be formed under acidic, neutral or basic conditions. However, for the metal hydroxo formation pathway, the free energy changes dramatically in solutions of different pH values. Under acidic conditions, the metal hydride formation pathway is more favorable than the metal hydroxo formation pathway, which is endergonic by 18.4 kcal mol−1. In contrast, the metal hydride formation pathway is less favorable than the metal hydroxo formation pathway under basic conditions. The metal hydroxo formation pathway is exergonic by 42.1 kcal mol−1 under basic conditions, indicating that 8 is very easily formed under basic conditions. The calculated results indicate that different pH values can change the reactivity of the reaction, which may switch the preference for ketone hydrogenation or dihydrogen release in the mechanism.
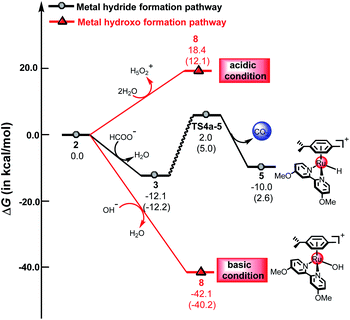 |
| | Fig. 8 The free energy profiles of the metal hydride formation pathway and the metal hydroxo formation pathway, the values in parentheses are electronic energies. | |
The modes of dihydrogen activation
Fig. 9 shows three typical HH or TH catalysts and their modes of dihydrogen activation. While catalyst A30 is an efficient HH catalyst, catalysts C30,63 and 2 (ref. 49) show good TH activities. Meanwhile, catalyst C was reported to be able to drive HH reactions under acidic conditions.17,64,65 Our previous theoretical works investigated the preference for the TH or HH of ketones and concluded that H2 coordination is essential for dihydrogen activation.3 Under neutral or basic conditions, the 16e species RuNC, with strong delocalized π-bonds, is hard to break to provide a vacant d-orbital for H2 coordination, thus the H2 activation barrier is high. On the contrary, RuNA is a 16e species, which could provide a vacant d-orbital for H2 coordination, thus H2 could be activated. Whereas under acidic conditions, dihydrogen can easily coordinate with RuNC to form a stable η2-H2 intermediate with the assistance of TfOH, which indicates that protonic acid interrupts the hyperconjugative effect of the Ru–N double bonds, resulting in a significant reduction in the energy barrier for dihydrogen activation. The single-site TM catalysts usually operate via non-cooperation mechanisms including the classical oxidative addition/reductive elimination mechanism and the σ-bond metathesis mechanism.28 Interestingly, the catalytic species 2 does not have a M–H/N–H bifunctional framework and the mode of dihydrogen activation catalyzed by 2 is different from those catalyzed by A and C (metal–ligand bifunctional cooperation). It is obvious that dihydrogen heterolytic splitting, activated by 2, belongs to the ion-pair mechanism, which requires metal-solvent cooperation (see TS18-19 in Fig. 9). The H2 with assistance from the solvent, H2O, prefer to coordinate with the TM center similar to an end-on coordination mode. The complex 2 shows only one open coordination site upon H2O ligand dissociation. The dihydrogen activation mode of the single-site complex 2 does not change the oxidation state of the Ru center of complex 2. On the contrary, the energy barrier of H2 activation is only 17.4 kcal mol−1 (see Fig. 7), which is the reverse reaction of H2 release. This implies that complex 2 is not only an efficient catalyst for TH but also an efficient catalyst for H2-hydrogenation in acidic aqueous media based on this calculation.
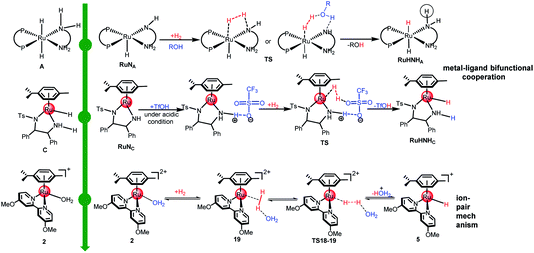 |
| | Fig. 9 Three typical TM catalysts for HH or TH of ketones and the modes of dihydrogen activation. | |
Conclusions
In summary, we investigated the transfer hydrogenation (TH) of ketones and dihydrogen release catalyzed by [(η6-arene)RuCl(κ2-N,N-dmobpy)]+ complexes in different pH environments using the DFT method. TH of ketones proceeds via two sequential steps: the formation of a metal hydride and the second hydride transfer via an ion-pair mechanism. The calculated results show that metal hydride formation is the rate-determining step with a free energy barrier of 14.1 kcal mol−1. In addition, we analyzed the origin of pH-dependent transfer hydrogenation and dihydrogen release catalyzed by this single-site cyclometallated ruthenium complex. Under weakly acidic conditions, the favorable pathway is the metal hydride formation pathway and the hydride transfer pathway via an ion-pair mechanism to complete the TH of ketones. Under strongly acidic conditions, the dehydrogenation of formic acid can be achieved to release dihydrogen following the metal hydride formation pathway and the dihydrogen release pathway via an ion-pair mechanism. This is in agreement with experimental results. The dihydrogen activation mode of this reaction adopts an ion-pair mechanism, which is metal-solvent cooperative in nature. Such single-site cyclometallated TM catalysts are predicted to be able to drive H2-hydrogenation under acidic conditions. Meanwhile, under basic conditions, the active species [(η6-arene)Ru(H2O)(κ2-N,N-dmobpy)]2+ is easily deprotonated to form the hydroxo complex [(η6-arene)Ru(OH)(κ2-N,N-dmobpy)]+, which leads to the termination of the reaction.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21672018), the State Key Laboratory of Physical Chemistry of Solid Surfaces (Xiamen University) (No. 201811), and the Fundamental Research Funds for the Central Universities (XK1802-6). We thank the National Supercomputing Center in Tianjin (TianHe-1) for providing part of the computational sources.
References
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- Y. Liu, X. Yue, C. Luo, L. Zhang and M. Lei, Energy Environ. Mater., 2019, 1–21 Search PubMed.
- M. Lei, W. Zhang, Y. Chen and Y. Tang, Organometallics, 2010, 29, 543–548 CrossRef CAS.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051–1069 CrossRef CAS.
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Huskens, Synthesis, 1994, 1994, 1007–1017 CrossRef.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750–1770 CrossRef CAS PubMed.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- F. Pena-Pereira, A. Kloskowski and J. Namieśnik, Green Chem., 2015, 17, 3687–3705 RSC.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- J.-t. Liu, S. Yang, W. Tang, Z. Yang and J. Xu, Green Chem., 2018, 20, 2118–2124 RSC.
- X. Wu, X. Li, F. King and J. Xiao, Angew. Chem., Int. Ed., 2005, 44, 3407–3411 CrossRef CAS PubMed.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed.
- P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS PubMed.
- O. Pàmies and J.-E. Bäckvall, Chem.–Eur. J., 2001, 7, 5052–5058 CrossRef.
- Y. Chen, Y. Tang, S. Liu, M. Lei and W. Fang, Organometallics, 2009, 28, 2078–2084 CrossRef CAS.
- X. Zhang, X. Guo, Y. Chen, Y. Tang, M. Lei and W. Fang, Phys. Chem. Chem. Phys., 2012, 14, 6003–6012 RSC.
- R. Feng, A. Xiao, X. Zhang, Y. Tang and M. Lei, Dalton Trans., 2013, 42, 2130–2145 RSC.
- T. Koike and T. Ikariya, Adv. Synth. Catal., 2004, 346, 37–41 CrossRef CAS.
- D. G. Blackmond, M. Ropic and M. Stefinovic, Org. Process Res. Dev., 2006, 10, 457–463 CrossRef CAS.
- H. Y. Chen, C. Wang, X. Wu, X. Jiang, C. R. Catlow and J. Xiao, Chem.–Eur. J., 2015, 21, 16564–16577 CrossRef CAS PubMed.
- Z. Yang, Z. Zhu, R. Luo, X. Qiu, J.-t. Liu, J.-K. Yang and W. Tang, Green Chem., 2017, 19, 3296–3301 RSC.
- S. Ogo, N. Makihara and Y. Watanabe, Organometallics, 1999, 18, 5470–5474 CrossRef CAS.
- T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 4149–4154 CrossRef CAS.
- J. Canivet, L. Karmazin-Brelot and G. Süss-Fink, J. Organomet. Chem., 2005, 690, 3202–3211 CrossRef CAS.
- Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629–634 RSC.
- Z. Ke, Y. Li, C. Hou and Y. Liu, Phys. Sci. Rev., 2018, 3, 20170038 Search PubMed.
- Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675–2676 CrossRef CAS.
- X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718–6722 CrossRef CAS PubMed.
- S. Ogo, T. Abura and Y. Watanabe, Organometallics, 2002, 21, 2964–2969 CrossRef CAS.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 9417–9427 CrossRef CAS PubMed.
- E. N. Yurtchenko and N. P. Anikeenko, React. Kinet. Catal. Lett., 1975, 2, 65–72 CrossRef.
- X. Yang, Dalton Trans., 2013, 42, 11987–11991 RSC.
- J. H. Merrifield and J. A. Gladysz, Organometallics, 1983, 2, 782–784 CrossRef CAS.
- C. P. Casey, S. W. Singer and D. R. Powell, Can. J. Chem., 2001, 79, 1002–1011 CrossRef CAS.
- J. S. Samec, A. H. Ell, J. B. Aberg, T. Privalov, L. Eriksson and J. E. Backvall, J. Am. Chem. Soc., 2006, 128, 14293–14305 CrossRef CAS PubMed.
- J. E. Martins, G. J. Clarkson and M. Wills, Org. Lett., 2009, 11, 847–850 CrossRef CAS PubMed.
- X. Wu, D. Vinci, T. Ikariya and J. Xiao, Chem. Commun., 2005, 4447–4449, 10.1039/b507276j.
- J. Xiao, X. Li, J. Blacker, I. Houson and X. Wu, Synlett, 2006, 2006, 1155–1160 CrossRef.
- X. Wu and J. Xiao, Chem. Commun., 2007, 2449–2466 RSC.
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Wurtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS PubMed.
- C. Guan, D. D. Zhang, Y. Pan, M. Iguchi, M. J. Ajitha, J. Hu, H. Li, C. Yao, M. H. Huang, S. Min, J. Zheng, Y. Himeda, H. Kawanami and K. W. Huang, Inorg. Chem., 2017, 56, 438–445 CrossRef CAS PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- W. H. Wang, S. Xu, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ChemSusChem, 2014, 7, 1976–1983 CrossRef CAS PubMed.
- C. Aliende, M. Pérez-Manrique, F. A. Jalón, B. R. Manzano, A. M. Rodríguez and G. Espino, Organometallics, 2012, 31, 6106–6123 CrossRef CAS.
- H. Li, X. Ma, B. Zhang and M. Lei, Organometallics, 2016, 35, 3301–3310 CrossRef CAS.
- L. Li, M. Lei and S. Sakaki, Organometallics, 2017, 36, 3530–3538 CrossRef CAS.
- L. Li, M. Lei, Y. Xie, H. F. Schaefer, 3rd, B. Chen and R. Hoffmann, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9803–9808 CrossRef CAS PubMed.
- L. Li, H. Zhu, L. Liu, D. Song and M. Lei, Inorg. Chem., 2018, 57, 3054–3060 CrossRef CAS PubMed.
- M. Xiao, X. Yue, R. Xu, W. Tang, D. Xue, C. Li, M. Lei, J. Xiao and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 10528–10536 CrossRef CAS PubMed.
- X. Yue, L. Li, P. Li, C. Luo, M. Pu, Z. Yang and M. Lei, Chin. J. Chem., 2019, 37, 883–886 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- Z. Wang, S. M. Lu, J. Li, J. Wang and C. Li, Chem.–Eur. J., 2015, 21, 12592–12595 CrossRef CAS PubMed.
- C. A. Mebi, R. P. Nair and B. J. Frost, Organometallics, 2007, 26, 429–438 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- C. A. Sandoval, T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata and R. Noyori, Chem.–Asian J., 2006, 1, 102–110 CrossRef CAS PubMed.
- T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255–257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10651k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zexing Cao
a,
Zexing Cao