
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly responsive methanol sensor based on graphene oxide/polyindole composites†
Katesara Phasuksomab,
Walaiporn Prissanaroon-Ouajaic and
Anuvat Sirivat *ab
*ab
aConductivity and Electroactive Polymers Research Unit, The Petroleum and Petrochemical College, Chulalongkorn University, Bangkok, 10330, Thailand. E-mail: anuvat.s@chula.ac.th; Fax: +66 2 611 7221; Tel: +66 2 218 4131
bCenter of Excellence on Petrochemical and Materials Technology, Bangkok, 10330, Thailand
cDepartment of Industrial Chemistry, King Mongkut's University of Technology North Bangkok, Bangkok, 10800, Thailand
First published on 17th April 2020
Abstract
Graphene-based materials, namely commercial graphene (cm-G), commercial graphene oxide (cm-GO), reduced graphene oxide (rGO), and synthesized graphene oxide (OIHM-GO), and their composites with polyindole (PIn) were used as sensing materials for methanol vapor. The synthesized graphene oxide was prepared by the optimized improved Hummers' method. rGO was prepared from cm-GO by two different methods: thermally mild reduction at 120 °C to yield T-rGO and chemical reduction by ascorbic acid to yield C-rGO. Graphene-based material/polyindole composites were prepared by in situ polymerization. In this report, the sensing responses were evaluated from the responsive electrical currents at room temperature. cm-GO showed the highest methanol response because it possessed the highest number of oxygen species, which act as the active sites. The relative electrical conductivity response of the in situ cm-GO/dPIn composite to methanol was the highest amongst the composites. The in situ OIHM-GO/dPIn composite possessed the high relative conductivity response of 81.89 ± 2.12 at 11.36 ppm, a sensitivity of 7.37 ppm−1 with R2 of 0.9967 in the methanol concentration range of 1.14–11.36 ppm, a theoretical LOD of 0.015 ppm, and repeatability of at least 4 cycles with good selectivity. This work represents the first report of the preparation and testing of graphene-based materials/polyindole composites as methanol sensors.
1. Introduction
Presently, graphene is a highly interesting and utilized material because of its extraordinary 2D crystal structure consisting of sp2-hybridized carbon atoms; it provides a larger surface area compared to other nano-scaled particles,1 high flexibility, good mechanical strength (Young modulus ∼ 0.05 TPa), high thermal conductivity (5000 W m−1 K−1 at 27 °C) and extremely high electron mobility (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1) under ambient conditions, and high electrical conductivity owing to its near zero band gap (resistivity 10−6 Ω cm).2 Due to these unique properties of graphene, it can be used in various applications, such as batteries,3,4 anti bacterial activities,5 transistors,6 drug delivery,7 photovoltaics,8 capacitors,9 solar cells,10 and sensors.11
000 cm2 V−1 s−1) under ambient conditions, and high electrical conductivity owing to its near zero band gap (resistivity 10−6 Ω cm).2 Due to these unique properties of graphene, it can be used in various applications, such as batteries,3,4 anti bacterial activities,5 transistors,6 drug delivery,7 photovoltaics,8 capacitors,9 solar cells,10 and sensors.11
However, graphene is poorly reactive because it has no reactive species; thus, it is insensitive to other chemicals.12,13
Graphene oxide (GO) is covalently bonded and functionalized with oxygen functional groups, such as hydroxyl, carbonyl, carboxyl, and epoxide groups, attached to its structure, leading to some partial sp3 hybridized carbon atoms14 and a decrease in its electrical conductivity. However, the electrical conductivity of GO depends on the number of oxygens in its structure and can be controlled by its band gap.15 GO has received much interest in gas sensor applications because its structure contains various oxygen species, which provide a variety of possible active sites. These oxygen species, namely hydroxyl, carbonyl, carboxyl, and peroxide groups, provide the active sites for interaction with the methanol vapor and increase the methanol response relative to the pristine conductive polymer. Generally, GO can be prepared by the oxidation of graphite using various procedures, such as Brodie's method using KClO3 and HNO3,16 Hofmann's method using KClO3, HNO3, and H2SO4,17 Staudenmaier's method18 using KClO3, HNO3, and H2SO4, and the Hummers' method using NaNO3, H2SO4, and KMnO4.19 However, the Hummers' method is more popular because of its safe synthesis procedure compared to the other methods, lack of toxic reactants (KClO3), easily controlled synthesis temperature, and high degree of oxidation.20 Although the Hummers' method is popular, NaNO3 used in oxidation can emit harmful gases.20 To remedy this problem, the Hummers' method was improved by replacing NaNO3 with H2SO4, H3PO4, and a doubled amount of KMnO4; this technique is sometimes called the “improved Hummers' method” or “Tour method”.21 In 2016, the optimized improved Hummers' method was reported as a novel synthetic method for modified GO by Lavin-Lopez et al.;22 its advantages are low production cost, short reaction time and lack of toxicity. The reaction time decreases from 12 hours to 3 hours because the coagulation step is omitted; the wash step is reduced, the use of H3PO4 is not necessary, the production per batch of GO is increased, and the method is easily scalable.
Reduced graphene oxide (rGO) is a graphene-based material which has received much attention because it not only possesses high electrical conductivity but also exhibits adaptable surface properties. Two major reduction strategies can be performed to prepare rGO, namely chemical reduction and thermal reduction. Chemical reduction can be divided into four techniques: (i) chemical reagent reduction; (ii) photocatalyst reduction; (iii) solvothermal reduction; and (iv) electrochemical reduction.14 Commonly, reduction with a chemical reagent is popular because of its large scale synthesis in which GO can be reduced with strong reducing reagents such as sodium hydride, sodium borohydride, lithium aluminium hydride, and hydrazine. Although these reducing agents provide very powerful reduction ability, they are highly toxic.23 Recently, green reducing reagents such as sugars, amino acids, plat extracts, and, especially, ascorbic acid or vitamin C have emerged as interesting alternative reducing agents because of their non-toxicity, powerful reduction ability, low cost, production of more stable rGO, and lack of heteroatom creation.14,24,25 Thermal reduction is classified in two methods, (i) thermal annealing and (ii) microwave and photo reduction; thermal annealing provides a higher degree of reduction and lower cost.14,23
Gas sensing materials fabricated from graphene-based materials exhibit good sensing properties. They show high surface areas and provide low limits of detection; facilitate gas interaction owing to the oxygen species functionalized on their structures, which promote energy and gas adsorption;26 and can achieve high sensitivity and selectivity by hybridization or compositing with other materials.27 Sensing materials fabricated from combinations of conductive polymers and graphene-based materials have recently attracted interest. For example, Al-Mashat et al. (2010) prepared a graphene/polyaniline (PANI) nanocomposite via chemical polymerization as a H2 sensor; the sensor provided a higher H2 resistive response and sensitivity compared to sensors based on pristine PANI and graphene alone because the graphene enhances the overall surface area of the composite nanostructure.28 Konwer et al. (2013) synthesized GO/PANI composites by in situ polymerization for use as a methanol sensing material; the composite sensor provided a higher sensitive response and reproducibility and a shorter response time relative to pure PANI because the larger surface area and the more reactive oxygen species on GO promoted the methanol interaction.29 Tiwari et al. (2015) fabricated a rGO/PPy composite film for ammonia detection by drop-cast in situ oxidative polymerization. The thin film sensor showed homogeneous dispersion of graphene in the polymer matrix; this enhanced the mechanical and electrical properties, including the surface-to-volume ratio. The sensitivity of the rGO/PPy composite was higher than that of pure PPy, and the response time slightly decreased; however, the recovery was not fully complete because of the higher energy binding sites of rGO.30 Graphene/conductive polymer composites can be prepared by three main methods: (i) in situ polymerization; (ii) mechanical mixing; and (iii) melt mixing. However, in situ polymerization is extensively used because it facilitates homogeneous dispersion of loaded graphene in the polymer matrix, resulting in a strong interaction between the graphene filler and the polymer.12
Conductive polymers are organic polymers that possess extraordinary properties compared to conventional polymers because they can induce charge and electron transfer through their chemical structures, called conjugated structures.31 Thus, they have been suitably used in electronic applications such as light emitting diodes, sensors, solar cells, capacitors, and transistors. Generally, the electrical conductivity of a conductive polymer is controlled by a dopant. Mostly, the dopants are anions; thus, most conductive polymers are typically p-type semiconductors.32 Conductive polymers have been used in sensor applications because they behave as either transducers or sensing materials as chemiresistive sensors; also, they are inexpensive and portable, can be used at room temperature, and enable real time monitoring.32 The conductive polymers that are ubiquitously investigated in sensor applications are polyaniline, polypyrrole, and polythiophene owing to their high electrical conductivity, facile preparation, and ease of surface modification;31,33 however, there have been a few reports on polyindole in sensor applications.
Polyindole (PIn) is a conductive polymer with a conjugated structure of alternative single bonds and double bonds along the polymer chain. The PIn structure consists of a benzene ring linked with a pyrolytic ring. Although PIn possesses low electrical conductivity, it has other advantages, such as good thermal stability, high redox activity, and stable electrical conductivity in air. Our previous work reported that the doping of PIn with perchloric acid (HClO4) could improve its electrical conductivity by many orders of magnitude to support various electronic applications.34 Recently, PIn was applied as an antifungal material for the fungi Penicillium chrysogenum35 and as a capacitor,36 humidity sensor,37 methanol sensor,38 etc. Furthermore, PIn-composited graphene based materials can be applied in various electronic applications; for example, GO/PIn composite was utilized as a supercapacitor,36 GO/PIn-incorporated Ag was applied as an electrode material,39 and rGO/PIn nanocomposite was used as a capacitor.40 However, graphene-based material/PIn composites have not been previously reported in sensor applications.
Methanol is a volatile organic compound; it easily evaporates into the surrounding air at room temperature because of its high vapor pressure. Methanol is widely used as a solvent or cleaning agent in both laboratories and industry. Inhalation of much methanol vapor can cause a variety of health effects, such as headache, drowsiness, nausea, blurred vision, convulsion, and even blindness, tumour, and cancer. Moreover, it is highly flammable and toxic; thus, it is harmful to humans and animals. The American Conference of Governmental Industrial Hygienists (ACGIH) allows a threshold concentration of methanol vapor released during a working day (8 hours) for repeated exposure without producing adverse health effects of 200 ppm.41,42 Thus, it is necessary to detect methanol at a very low concentration.
The aim of this work was to prepare composites of polyindole and graphene-based materials (commercial graphene (cm-G), commercial GO (cm-GO), synthesized GO (OIHM-GO), T-rGO, and C-rGO) by in situ polymerization for application as methanol sensors. rGO was prepared by both thermally mild reduction and chemical reduction with ascorbic acid or vitamin C, where cm-GO was used as a starting raw material. The synthesized GO was prepared by the optimized improved Hummers' method (OIHM) in order to compare it with cm-GO. The graphene-based materials and their composites were characterized by various techniques: FT-IR, XRD, XPS, FE-SEM, and 2-point probe. The graphene-based materials and their composites with polyindoles were used as sensing materials to detect methanol vapor. The sensor response was recorded in terms of the electrical current signal change at room temperature using nitrogen as a base gas. The most suitable composite material was chosen to determine the effects of the filler content (% v/v) on the methanol response. Moreover, the sensitivity, limit of detection, reversibility, repeatability, and selectivity were investigated and are reported here.
2. Experimental
2.1 Materials
Indole powder was used as a monomer in the synthesis, and anhydrous ferric chloride (FeCl3, analytical grade) as an oxidizing agent was purchased from Merck. 37% wt/wt hydrochloric acid (HCl, analytical grade) and 98% wt/wt sulfuric acid (H2SO4, analytical grade) were obtained from RCI Labscan. Ethanol (EtOH, analytical grade) for monomer dissolution was purchased from HAYMAN. 30% wt/wt ammonium hydroxide (NH4OH, analytical grade) and 70% wt/wt perchloric acid (HClO4, analytical grade) were purchased from KEMAUS and were used as a de-doping agent and a doping agent, respectively. Potassium permanganate (KMnO4, analytical grade) as an oxidant for the graphite oxidation was acquired from Ajax Finechem Pty Ltd. 30% hydrogen peroxide (H2O2, analytical grade) was purchased from Chem-supply. Graphene (grade M) was purchased from XG Science Company, and commercial GO was purchased from Xiamen Tob New Energy Technology Co., Ltd. Synthetic graphite powder with a particle size lower than 20 μm purchased from Aldrich Chemistry was used as a raw material in the GO synthesis. L-Ascorbic acid and vitamin C (analytical grade) were used as reducing agents and were acquired from Carlo Erba Reagents. The methanol (CH3OH, analytical grade) used in the sensor testing was purchased from Honeywell. Acetone (analytical grade) was purchased from Scharlau. Hexane and tetrahydrofuran (THF, analytical grade) were purchased from RCI Labscan. Ultra-high purity nitrogen (99.998% purity) from Praxair Company was used as a carrier gas. Double distilled water was used as a solvent in the synthesis.2.2 Reduction of graphene oxide
In the thermal reduction, the cm-GO was treated by thermal heating at 120 °C under atmospheric air in an oven for 24 hours to obtain the thermally reduced graphene oxide (T-rGO). In the chemical reduction, cm-GO was sonicated in double distilled water for 30 min using an ultrasonic sonicator (Transsonic 570H, Elma). To cool the heat produced during the sonication, the sonicated cm-GO solution was stirred at room temperature for 1 hour. Then, ascorbic acid powder was gradually added to the cm-GO solution at room temperature and then stirred for 24 hours. Next, the chemically reduced graphene oxide (C-rGO) was filtered, washed with double distilled water, and dried at 80 °C for 24 hours, respectively.43 The suitable GO to ascorbic acid weight ratio was fixed at 1:3.3 according to the work of Z. Sui et al. in 2011.44 The analytical results of the rGOs are reported in Fig. S1 and S2 in the ESI.†
2.3 Graphene oxide preparation by the optimized improved Hummers' method
To prepare the synthesized GO, the oxidation step in the 2016 work of M. del P. Lavin-Lopez et al. was followed; however, the amount of graphite was reduced to 3 g and the graphite:KMnO4 mass ratio was maintained at 1:3 in order to obtain the same oxidation degree.22 3 g of graphite powder was mixed with 80 mL of 98% wt/wt H2SO4 and then continuously stirred for 1 h. Next, a strong oxidizing agent (KMnO4, 9 g) was gradually dropped into the mixed solution, where the temperature was slowly raised to 45 °C in this step because of an exothermic reaction. Then, the mixed solution was heated to 50 °C under stirring. The reaction was continuously allowed to proceed for 3 hours by controlling the reaction temperature under 55 °C to prevent overheating; the black color of the graphite slowly changed to dark brown and the solution became viscous due to the oxidation with KMnO4. After that, the mixture was poured over 400 g of ice flakes. Next, 3 mL of 30% wt/wt H2O2 was slowly added to the mixture with the ice flakes to terminate the oxidation, and the mixture was simultaneously stirred by hand until all gas bubbles disappeared. Next, the mixture was separated by filtration with a vacuum pump to obtain a GO slurry. Finally, the GO slurry was washed with double distilled water (200 mL), 5% v/v HCl (200 mL), and ethanol (200 mL), respectively. The GO slurry was dried at room temperature for 24 hours and at 50 °C in the oven for 24 hours to obtain the solid GO. Finally, the solid GO was ground with a coffee grinder to obtain GO powder.45 Herein, the synthesized GO was named OIHM-GO. The OIHM-GO synthesis procedure is shown in Scheme 1. The analytical results of OIHM-GO are reported in Fig. S3 of the ESI.†
 | ||
| Scheme 1 GO synthesis by the optimized improved Hummers' method (OIHM). | ||
2.4 Composite preparation by in situ chemical oxidative polymerization
Each graphene-based material (cm-G, cm-GO, OIHM-GO, T-rGO, and C-rGO) was separately sonicated by a sonicator (Elma, Transsonic 570H) for 30 min in 0.1 M HCl (100 mL), which was used as the solvent in the PIn synthesis. 10.66 g of FeCl3 as the oxidant in the PIn was separately dissolved in 0.1 M HCl (80 mL). The sonicated solution was cooled to room temperature by stirring at room temperature in order to reduce the heat that arose from continuous sonication before mixing with the oxidant solution. Next, an indole monomer solution (3 g of monomer dissolved in 10 mL of ethanol) was gradually dropped into the mixture solution and then constantly stirred for 24 hours. Then, the composite was filtered and washed with double distilled water until the yellow mixture solution became clear. Finally, the precipitate was dried at 70 °C for 24 hours and gently milled with a mortar for 2–3 min to obtain a composite powder. The composites of PIn with various graphene-based materials synthesized by in situ polymerization, cm-G, cm-GO, OIHM-GO, T-rGO, and C-rGO, were named in situ cm-G/PIn, in situ cm-GO/PIn, in situ OIHM-GO/PIn, in situ T-rGO/PIn, and in situ C-rGO/PIn, respectively, in which the graphene-based material content was fixed at 10% v/v. PIn was synthesized by the same procedure without the graphene-based materials in order to compare it with the composites. The in situ polymerization steps are shown in Scheme 2.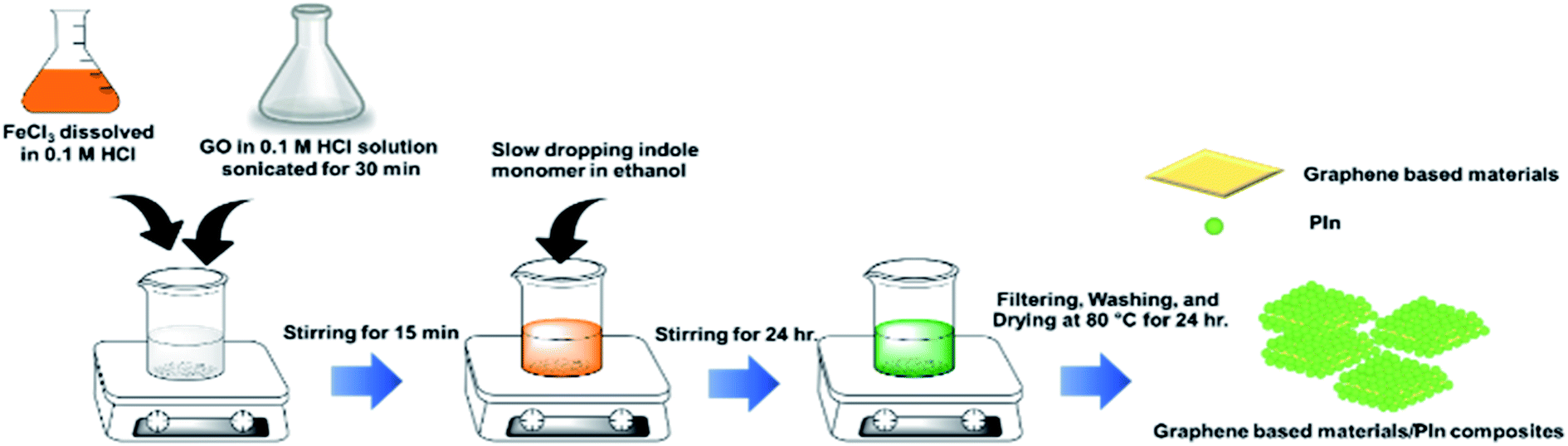 | ||
| Scheme 2 In situ polymerization of the graphene-based materials/PIn composites. | ||
2.5 Doping–dedoping process
The obtained PIn powder was dedoped in 5 M NH4OH solution for 24 hours at the dedoping NH4OH/PIn mole ratio of 10:1. The dedoped-PIn was filtered, rinsed with double distilled water, and dried at 70 °C for 24 hours. The dedoped-PIn powder was then doped with 2.5 M HClO4 for 24 hours at a doping mole ratio of HClO4/PIn of 10:1 (the doping mole ratio is defined as (acid mole)/(PIn mole), where PIn denotes the average indole repeating units).38 The doped-PIn (dPIn) was filtered by a cellulose acetate membrane (pore size of 0.45 μm, Whatman) and dried at 70 °C for 24 hours. The graphene-based materials/PIn composites prepared by in situ polymerization were dedoped and doped under the same method. Also, the cm-GO/dPIn composite was fabricated by simple physical mixing; the dPIn was mixed with cm-GO in order to compare its response with the composites prepared by the in situ polymerization. Herein, the cm-GO/dPIn composite fabricated by simple physical mixing was named SM cm-GO/dPIn.
2.6 Methanol vapor detection
The sample powder was compressed in a pellet with a thickness of 1.5 ± 0.3 mm and a diameter of 1.3 cm. The constructed gas chamber system consisted of two stainless steel chambers with volumes of 8 inch3 connected in series. The first chamber was used to prepare a gas at a certain concentration before releasing it into the second chamber. The sample pellet was loaded into a custom-built 2-point probe and placed into the second chamber, where the electrical signal was real-time recorded in this chamber using an electrometer (6517A, Keithley) and the 6517 Hi-R Test software under applied voltage in the linear ohmic regime. First, air and humidity inside the chambers were evacuated by a vacuum pump; the electrical signal tended to decrease with decreasing pressure inside the chamber owing to air and humidity removal until the electrical signal became relatively constant. Next, N2 gas was injected into the chambers; the electrical conductivity (σ) of the sample under the initial N2 atmosphere in the equilibrium state was defined as σinitial,N2. After that, N2 was removed by the vacuum pump. Methanol vapor was prepared by flowing N2 gas at a constant flow rate of 5.59 L min−1 controlled by a mass flow controller (AALBORG, GFC171S) into the methanol solvent without dilution; this vapor was injected into the first chamber and maintained for 5 min to obtain a homogeneous vapor before releasing it into the second chamber. The relative humidity was around 20 ± 3% as recorded by a Bluetooth wireless temperature and humidity data logger (EL-BT-2, Lascar Electronic). The electrical conductivity of the sample under the methanol vapor in the equilibrium state is defined as σmethanol. The electrical conductivity of the sample under final N2 in the equilibrium state is defined as σfinal,N2. The methanol vapor detection was tested at 1.1 atm pressure to prevent interfering gas or humidity from the environment from penetrating through the tested chamber, and the temperature was controlled at 26 ± 1 °C. N2 gas with 99.998% purity obtained from Praxair was used as a base gas. The sensor testing system was set up as in a previous report.38The relative conductivity response was utilized to identify the sensor response, which was defined as the normalized electrical conductivity under methanol vapor minus that under initial N2 and divided by that under initial N2, as in eqn (1):
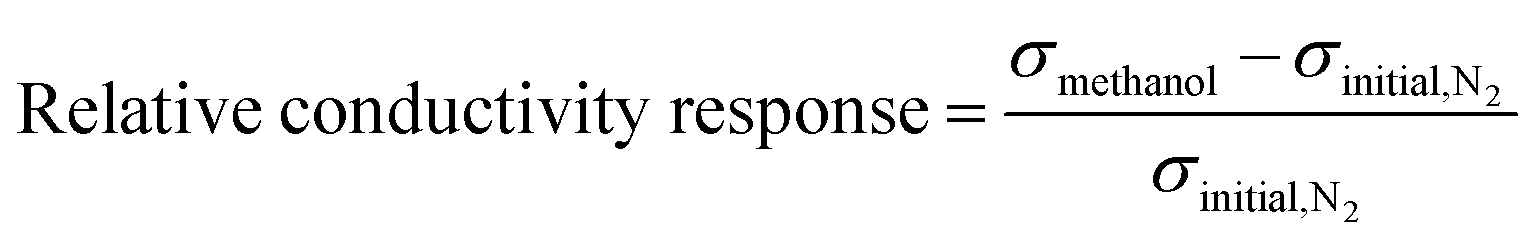 | (1) |
The electrical conductivity during the methanol vapor detection was measured and calculated as in eqn (2):
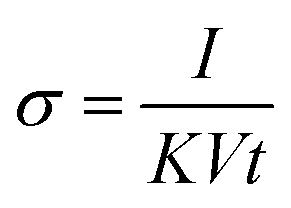 | (2) |
The sensitivity of the sensor is defined as the slope of the calibration curve acquired from the plot of the relative conductivity response (y-axis) and the methanol vapor concentration in ppm (x-axis); thus, the sensitivity unit is ppm−1. The methanol concentration in ppm was obtained by altering the ratio between the % methanol vapor volume and % total vapor volume. The theoretical limit of detection (LOD) is defined as three times the standard deviation of the electrical conductivity signal in the N2 gas (without methanol vapor) divided by the sensitivity, as in eqn (3):46,47
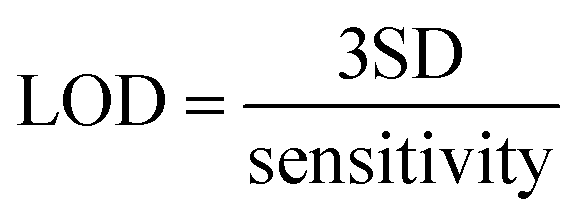 | (3) |
The response and recovery times are defined as the time required for electrical signal of a sensor to reach 90% of the equilibrium value under vapor exposure and the time required for the electrical signal of sensor to return to 10% of its original electrical signal under N2 after gas desorption, respectively.
2.7 Characterization
The functional groups of the graphene-based materials and their composites with polyindole were characterized by a Fourier transform infrared spectrometer in transmittance mode (Nicolet iS5, Thermo Scientific). The FT-IR spectra were recorded in the 4000–600 cm−1 wave number range with a resolution of 4 cm−1 and a scan number of 64, and potassium bromide (KBr) (spectroscopy grade, Carlo Erba) was used as a reference. The sample powder was mixed with KBr by milling in a mortar and was then compressed into a pellet before testing. The crystal structures of the samples were analyzed by a wide-angle X-ray spectrometer (Smartlab XRD, Rigaku), using a scan step of 0.01° at 2θ from 5° to 80° with a scan speed of 10° min−1, where the Cu-Kα X-ray source was generated at 40 kV/30 mA. The morphological properties of the materials were investigated by high magnification field emission scanning electron microscopy (FE-SEM, HITACHI, S-4800), with an accelerating voltage of 5 kV and an emission current of 10 μA. The sample powders were dried at 80 °C for 24 hours and then distributed on a carbon tape mounted on the stub; then, they were sputtered with pure platinum for 150 s before testing. Element and chemical bonding analysis was carried out by an X-ray photoelectron spectrometer, XPS (Kratos Analytical Shimadzu Group Company, Axis Ultra DLD), operated using a monochromated Al Kα X-ray radiation source. The wide scan XPS spectra were obtained at a pass energy of 160 eV and the high-resolution XPS spectra were analyzed at a pass energy of 40 eV. The binding energy was calibrated by using C 1s as a reference (binding energy of 284.8 eV). The XPS spectra were obtained using Casa-XPS software. The true densities of graphene-based materials and PIn were characterized by a gas pycnometer (Quantachrome, Ultrapycnometer 1000) operated by determining the pressure changes resulting from displacement of highly pure helium gas by the sample powder. The true density values of cm-G, cm-GO, OIHM-GO, C-rGO, T-rGO, and PIn were 1.6218 g cm−3, 1.8541 g cm−3, 1.8305 g cm−3, 1.7426 g cm−3, 1.7331 g cm−3, and 1.3321 g cm−3, respectively.The electrical conductivity was measured using a custom-built 2-point probe with two gold electrodes with a diameter of 1 mm and a probe spacing of 1.05 cm. Each sample pellet with a diameter of 1.3 cm and a thickness of 1.5 ± 0.3 mm was placed on the sample holder, and the two point probes were connected to the sample surface. The 2-point probe was linked with the electrometer (6517A, Keithley), and the voltage was applied in a linear ohmic regime to record the resulting current. The current–voltage slope (I–V slope) was obtained by the plot between the applied voltage (x-axis) and the recorded current (y-axis). The electrical conductivity (S cm−1) of the material in air at 26° ± 1° was calculated by eqn (4):
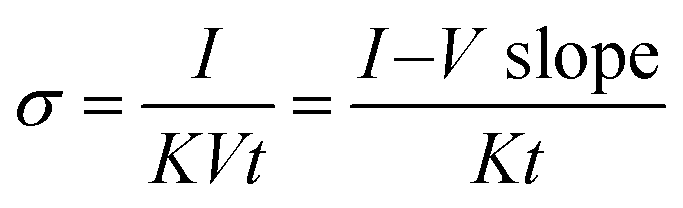 | (4) |
3. Results and discussion
3.1 Characterization of graphene-based material/PIn composites and doping
Graphene-based materials/PIn composites were prepared by in situ polymerization, where the graphene-based material was fixed at 10% v/v. The functional groups of the composites and pristine PIn were characterized by FT-IR. For PIn, the main absorption characteristics of the N–H stretching appeared at 3211 cm−1, the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1617 cm−1, the N–H deformation at 1567 cm−1, the C–N stretching at 1456 cm−1, the CN stretching at 1378 cm−1, the out-of-plane deformation of C–H at 742 cm−1, and the CO stretching at 1730 cm−1 resulting from the over-oxidation of PIn.34 The characteristic IR absorptions of the composites and PIn are very similar, except that the N–H stretching and the out of plane deformation of C–H are slightly shifted to lower wavenumbers for the composites. The shifts of the IR peaks to lower wavenumbers may correspond to the restricted growth and the confined modes of vibration of PIn in the presence of the graphene-based materials owing to the pi–pi interactions generated by the overlapping pi-orbitals of the non-planar aromatic rings of graphene and PIn.36 After doping PIn with HClO4, the FT-IR spectrum of dPIn showed strong characteristic IR absorptions of Cl–O at 1141 and 1082 cm−1, and the N–H stretching shifted to a lower wavenumber; this confirms that the doping process was successful.34 In the case of the composites doped with HClO4, the FTIR spectra of the composites are slightly different and difficult to discern. The FTIR spectra of the in situ cm-GO/dPIn and in situ OIHM-GO/dPIn show OH-stretching at 3372 and 3353 cm−1, respectively. Moreover, the N–H stretching of PIn composited with GO or OIHM-GO shows higher peak wavenumbers relative to the other samples. This is because of the higher interaction between the N–H sites of PIn and the oxygen species of GO. The in situ cm-G/dPIn spectrum apparently shows the C–H stretching of graphene at 2961 and 2923 cm−1. However, the FTIR spectra of in situ C-rGO/dPIn and in situ T-rGO/dPIn are quite similar. A summary of the peak assignments of the FTIR spectra of the composites is shown in Table S1 in the ESI.† All composites show the characteristic peaks of the dopant ClO4− at 1141 and 1082 cm−1.
C stretching at 1617 cm−1, the N–H deformation at 1567 cm−1, the C–N stretching at 1456 cm−1, the CN stretching at 1378 cm−1, the out-of-plane deformation of C–H at 742 cm−1, and the CO stretching at 1730 cm−1 resulting from the over-oxidation of PIn.34 The characteristic IR absorptions of the composites and PIn are very similar, except that the N–H stretching and the out of plane deformation of C–H are slightly shifted to lower wavenumbers for the composites. The shifts of the IR peaks to lower wavenumbers may correspond to the restricted growth and the confined modes of vibration of PIn in the presence of the graphene-based materials owing to the pi–pi interactions generated by the overlapping pi-orbitals of the non-planar aromatic rings of graphene and PIn.36 After doping PIn with HClO4, the FT-IR spectrum of dPIn showed strong characteristic IR absorptions of Cl–O at 1141 and 1082 cm−1, and the N–H stretching shifted to a lower wavenumber; this confirms that the doping process was successful.34 In the case of the composites doped with HClO4, the FTIR spectra of the composites are slightly different and difficult to discern. The FTIR spectra of the in situ cm-GO/dPIn and in situ OIHM-GO/dPIn show OH-stretching at 3372 and 3353 cm−1, respectively. Moreover, the N–H stretching of PIn composited with GO or OIHM-GO shows higher peak wavenumbers relative to the other samples. This is because of the higher interaction between the N–H sites of PIn and the oxygen species of GO. The in situ cm-G/dPIn spectrum apparently shows the C–H stretching of graphene at 2961 and 2923 cm−1. However, the FTIR spectra of in situ C-rGO/dPIn and in situ T-rGO/dPIn are quite similar. A summary of the peak assignments of the FTIR spectra of the composites is shown in Table S1 in the ESI.† All composites show the characteristic peaks of the dopant ClO4− at 1141 and 1082 cm−1.
Fig. 1(b) shows the X-ray diffraction spectra of the composites and dPIn. The main diffraction peaks of dPIn are broad and are visible at 2θ = 7.4°, 18.7°, and 25.8°, indicating a relatively amorphous structure.48 The smallest diffraction angle at 2θ = 7.4° is related to the spacing between the dopant and the positively charged nitrogen atom in the pyrolytic ring, and the diffraction peaks at 2θ = 18.7° and 25.8° can be referred to as the reflection of the polymer backbone.49 The interchain spacing of PIn is 4.31 Å as evaluated from the diffraction peak at 2θ = 25.8°.36 For an amorphous polymer, the interchain spacing is attributed to the distance between the polymer chains as induced by the specific arrangement of chemical bonds; the larger interchain spacing, the higher the disorder between polymer chains.50 For in situ cm-G/dPIn, the spectrum shows three diffraction peaks at 2θ = 6.2°, 17.6°, and 25.7°. The intense diffraction peak at 2θ = 25.7° indicates uniform and tight graphene layers and poor exfoliation by PIn.51 In addition, the weak and broader diffraction peaks of PIn are slightly shifted to lower diffraction angles of 2θ = 6.2° and 17.6°. The other composites prepared by in situ polymerization exhibit three diffraction peaks, similar to dPIn; however, all the peaks became broader and are slightly shifted to lower diffraction angles, corresponding to the exfoliation of their graphene layers by PIn and the irregular stacking of graphene-based materials with the PIn matrix.51 The XRD spectrum of SM cm-GO/dPIn, which was prepared by simple physical mixing, can be compared to that of in situ cm-GO/dPIn; the XRD spectra are quite different, as shown in Fig. 1(b). The SM cm-GO/dPIn retains the (001) diffraction peak at 2θ = 10.9° belonging to GO; however, it does not appear in the case of the in situ cm-GO/dPIn. This is because the in situ polymerization causes complete growth of PIn on the GO layers, whereas for SM cm-GO/dPIn, only physical interactions occur between GO and PIn; hence, the major diffraction peak of GO remains.
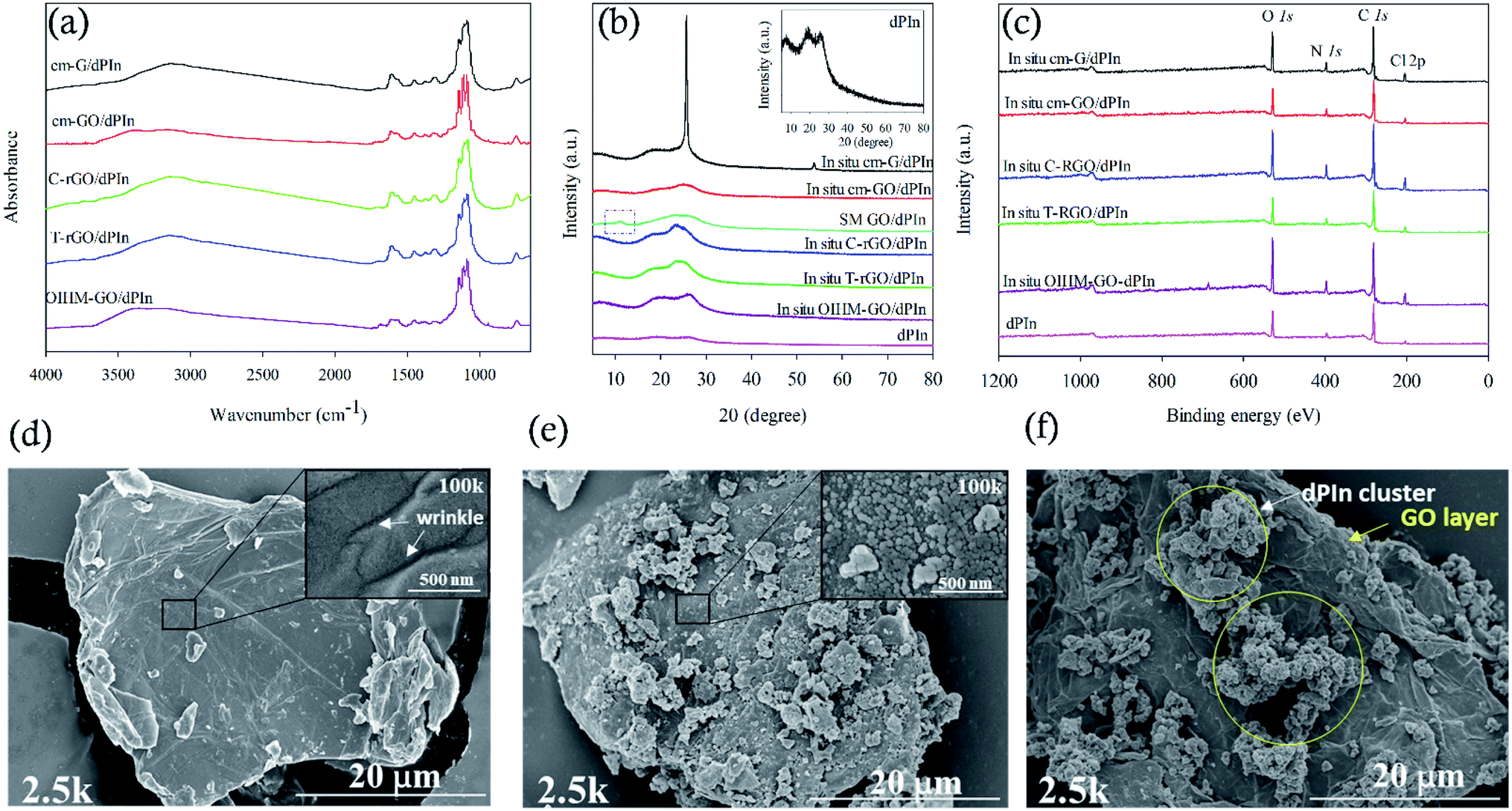 | ||
| Fig. 1 Characterization of the graphene-based materials/PIn composites: (a) FT-IR; (b) XRD; (c) wide scan XPS. The composites were prepared by in situ polymerization. (d–f) SEM images at 2.5k magnification of cm-GO, in situ 10% v/v cm-GO/dPIn, and SM 10% v/v cm-GO/dPIn, respectively. | ||
Wide scan XPS was used to identify the element compositions of the composites, as shown in Fig. 1(c), and the details are reported in Table 1. PIn consists of four elements: C, O, N, and Cl, in which the presence of oxygen is the result of overoxidation and the existence of chlorine is owing to self-doping of Cl− dopant from HCl and FeCl3 with the oxidized PIn chain by electrostatic interactions.34 After doping PIn with HClO4, the same elements were observed; however, the amounts of O and Cl increase because of the incorporation of ClO4− by doping, as identified by the characteristic IR absorption of ClO4− in the FT-IR spectrum. The high resolution scan of the N 1s XPS spectra was used to study the doping level related to the electrical conductivity of a material. The doping level is defined as (N˙+ + N++)/Ntotal,34 where N˙+ and N++ are the numbers of polarons and bipolarons and Ntotal is the amount of all nitrogen species on the PIn backbone, consisting of N˙+, N++, and N–H (amine nitrogen), observed at the binding energies of 399.6–400.3 eV, 400.7–400.9 eV, and 401.7–402.1 eV, respectively. Doping with HClO4 increases the doping level from 37.65 to 42.87 due to the increment of the number of charge carriers. In the cases of the composites, the wide scan XPS spectra are similar to that of PIn and consist of four elements: C, O, N, and Cl. However, the doping levels appear to be higher. The % doping levels of the composites are ordered as follows: in situ cm-G/PIn (48.16) > in situ C-rGO/PIn (45.10) > in situ T-rGO/PIn (41.70) > in situ OIHM-GO/PIn (40.25) > in situ cm-GO/PIn (39.27). For the composites after doping, the wide scan XPS shows the same elements; however, the numbers of oxygen and chlorine atoms are increased owing to the electrostatic interactions between ClO4− and the oxidized PIn. In addition, bipolarons can be distinctly observed after doping with HClO4 because ClO4− compensates for charges and generates a hole-like structure of PIn, inducing an increase in the bipolaron state. The % doping levels of the composites after the doping are ordered as follows: in situ cm-G/dPIn (59.79) > in situ C-rGO/dPIn (56.29) > in situ T-rGO/dPIn (54.16) > in situ OIHM-GO/dPIn (47.37) > in situ cm-GO/dPIn (44.85). For SM cm-GO/dPIn, the % doping level is 53.30.
| Material | Element (at%) | Doping level (%) | Electrical conductivity in air (S cm−1) | ||||
|---|---|---|---|---|---|---|---|
| C 1s | O 1s | N 1s | S 2p | Cl 2p | |||
| a Remarks: (1) sulfur atoms observed in graphene-based materials were obtained from residual H2SO4. (2) Doping of PIn and composites was fixed at a 10:1 doping mole ratio of HClO4/indole. (3) Doping level (%) of the conductive polymer is defined as [(N˙+ + N++)/Ntotal] × 100. |
|||||||
| PIn | 85.13 | 8.27 | 5.92 | — | 0.68 | 37.65 (ref. 34) | (1.15 ± 0.28) × 10−3 (ref. 34) |
| dPIn | 73.44 | 20.07 | 4.73 | — | 1.76 | 41.87 | 1.26 ± 0.08 (ref. 34) |
| cm-G | 93.49 | 6.51 | — | — | — | — | 31157.68 ± 6 |
| In situ 10% v/v cm-G/PIn | 86.60 | 2.45 | 9.34 | — | 1.61 | 48.95 | 0.51 ± 0.05 |
| In situ 10% v/v cm-G/dPIn | 70.63 | 19.11 | 6.92 | — | 3.34 | 59.79 | 19.17 ± 0.86 |
| cm-GO | 67.12 | 31.31 | — | 1.57 | — | — | 1.23 ± 0.38 |
| In situ 10% v/v cm-GO/PIn | 87.06 | 2.62 | 9.26 | — | 1.06 | 39.27 | (3.82 ± 0.95) × 10−3 |
| In situ 10% v/v cm-GO/dPIn | 71.28 | 20.39 | 5.59 | — | 2.73 | 44.85 | 2.11 ± 0.38 |
| SM 10% v/v cm-GO/dPIn | 67.91 | 20.56 | 9.04 | — | 2.48 | 53.30 | 4.05 ± 1.07 |
| C-rGO | 80.94 | 19.06 | — | — | — | — | 1143.11 ± 3.089 |
| In situ 10% v/v C-rGO/PIn | 87.86 | 0.79 | 9.99 | — | 1.36 | 45.10 | (3.65 ± 0.44) × 10−2 |
| In situ 10% v/v C-rGO/dPIn | 67.59 | 19.65 | 9.18 | — | 3.58 | 56.29 | 12.15 ± 2.90 |
| T-rGO | 77.05 | 21.34 | — | 1.61 | — | — | 684.57 ± 25.36 |
| In situ 10% v/v T-rGO/PIn | 86.57 | 2.08 | 10.10 | — | 1.25 | 43.73 | (1.86 ± 0.08) × 10−2 |
| In situ 10% v/v T-rGO/dPIn | 69.61 | 19.22 | 8.21 | — | 2.95 | 54.16 | 7.64 ± 0.35 |
| OIHM-GO (the synthesized GO) | 66.62 | 31.77 | — | 1.61 | — | — | 1.45 ± 0.02 |
| In situ 10% v/v OIHM-GO/PIn | 88.29 | 1.66 | 8.75 | — | 1.30 | 40.25 | (2.88 ± 0.65) × 10−3 |
| In situ 10% v/v OIHM-GO/dPIn | 64.96 | 21.82 | 9.75 | — | 3.47 | 47.37 | 2.22 ± 0.43 |
The electrical conductivities of the composites were measured by the 2-point probe and are listed in Table 1. The electrical conductivity of the materials is directly related to the doping level, as confirmed by XPS. The electrical conductivity of the composites is as follows: in situ cm-G/PIn (0.51 ± 0.05 S cm−1) > in situ C-rGO/PIn ((3.65 ± 0.44) × 10−2 S cm−1) > in situ T-rGO/PIn ((1.86 ± 0.08) × 10−2 S cm−1) > in situ cm-GO/PIn ((3.82 ± 0.95) × 10−3 S cm−1) > in situ OIHM-GO/PIn ((2.88 ± 0.65) × 10−3 S cm−1). For the pristine PIn, the electrical conductivity is (1.15 ± 0.28) × 10−3 S cm−1. Clearly, the electrical conductivity of the composites is higher than that of pure PIn because of the pi–pi interactions between PIn and the graphene-based materials, which induce the creation of charge carrier pathways.29,52 Another reason is that the order of the PIn molecules is enhanced because the PIn molecules are oriented on the layers of graphene-based materials with induced chain expansion; consequentially, the electrical conductivity of PIn is enhanced.52 For the composites doped with HClO4, the electrical conductivity sequence is as follows: in situ cm-G/dPIn (19.17 ± 0.86 S cm−1) > in situ C-rGO/dPIn (12.15 ± 2.90 S cm−1) > in situ T-rGO/dPIn (7.64 ± 0.35 S cm−1) > in situ cm-GO/dPIn (2.11 ± 0.38 S cm−1) > in situ OIHM-GO/dPIn (1.45 ± 0.02 S cm−1). This indicates that the doping improves the electrical conductivity by several orders of magnitude. When the electrical conductivity of SM cm-GO/dPIn is compared with that of in situ cm-GO/dPIn, the electrical conductivity of SM cm-GO/dPIn is higher than that of in situ cm-GO/dPIn. For SM cm-GO/dPIn, GO is not fully covered by the available PIn molecules, resulting in poor interfacial interaction and a non-homogeneous phase in the mixture. On the other hand, GO can easily interact with moisture in the environment, leading to an increase in electrical conductivity.
The surface morphologies of the composites were characterized by SEM. All the composites showed that the graphene-based materials were covered with PIn because indole monomers can adsorb and grow on the surfaces of graphene-based materials.40 For example, this phenomenon is illustrated in Fig. 1(d) and (e), which show the surface morphologies of cm-GO and the in situ cm-GO/dPIn composite, respectively. It can be observed that their morphologies are not alike. The pure GO shows a wrinkled surface, as clearly marked in the inset of Fig. 1(d), which shows the SEM image at 100k magnification. Meanwhile, for in situ cm-GO/dPIn, GO is fully covered by PIn molecules and the wrinkles disappear, as shown in the inset of Fig. 1(e), which shows the SEM image at 100k magnification. This suggests that PIn nucleation was generated on the GO surface. Fig. 1(f) shows the surface morphology of SM cm-GO/dPIn, which is clearly different from that of in situ cm-GO/dPIn because the cm-GO sheet is not covered by PIn, as observed from the wrinkled surface of the cm-GO.
The intermolecular interactions between the conductive polymer and the graphene-based materials can be based on various interactions, such as hydrophobic interactions; pi–pi interactions; electrostatic or coulombic interactions; hydrogen bonding; and van der Waals interactions.53 For in situ cm-G/PIn, the intermolecular interactions between graphene and PIn mainly arise from pi–pi interactions, hydrophobic interactions, and van der Waals interactions because the graphene structure does not contain heteroatoms. For the other composites, the interactions are not only pi–pi interactions, hydrophobic interactions, and van der Waals interactions, but also include electrostatic or coulombic interactions and hydrogen bonding because the oxygen functional groups in the GO and rGO layers can interact with the heterocyclic atoms of PIn and promote strong interfacial interactions.53
3.2 Graphene-based materials and methanol responses
cm-G, cm-GO, OIHM-GO, T-rGO, and C-rGO in pellet form were used to detect 11.36 ppm methanol vapor under N2 base gas at room temperature (26 ± 1 °C). The relative conductivity response is a factor used here to compare different material responses for methanol vapor detection, as shown in Fig. 2 and as detailed in Table 2. The methanol responses are ordered as follows: cm-G < C-rGO < T-rGO < OIHM-GO < cm-GO, whereas the corresponding relative conductivity responses toward the methanol vapor are (7.90 ± 3.58) × 10−3, −0.10 ± 0.03, −0.48 ± 0.12, 1.53 ± 0.02, and 3.79 ± 0.3, respectively. Interestingly, the relative conductivity responses of the graphene-based materials do not depend on the original electrical conductivity of the material but are related to the available active sites of the materials. Methanol is a highly polar vapor; its adsorption on a sensing material is induced by hydrogen bonding interactions. A material containing a large amount of oxygen functional groups with hydrophilic properties produces a high methanol response. Herein, the amount of oxygen atoms of GO is higher than those of the rGOs and graphene, as confirmed by the XPS data in Table 1. Thus, it can be concluded that GO provides the highest methanol response because it possesses the highest hydrophilicity and more active sites compared to the rGOs and graphene. The high response of GO can be attributed to the interpretation of S. Prezioso et al. (2013) that the response to a target vapor is strongly enhanced when graphene is functionalized as GO to produce a higher number of active sites.54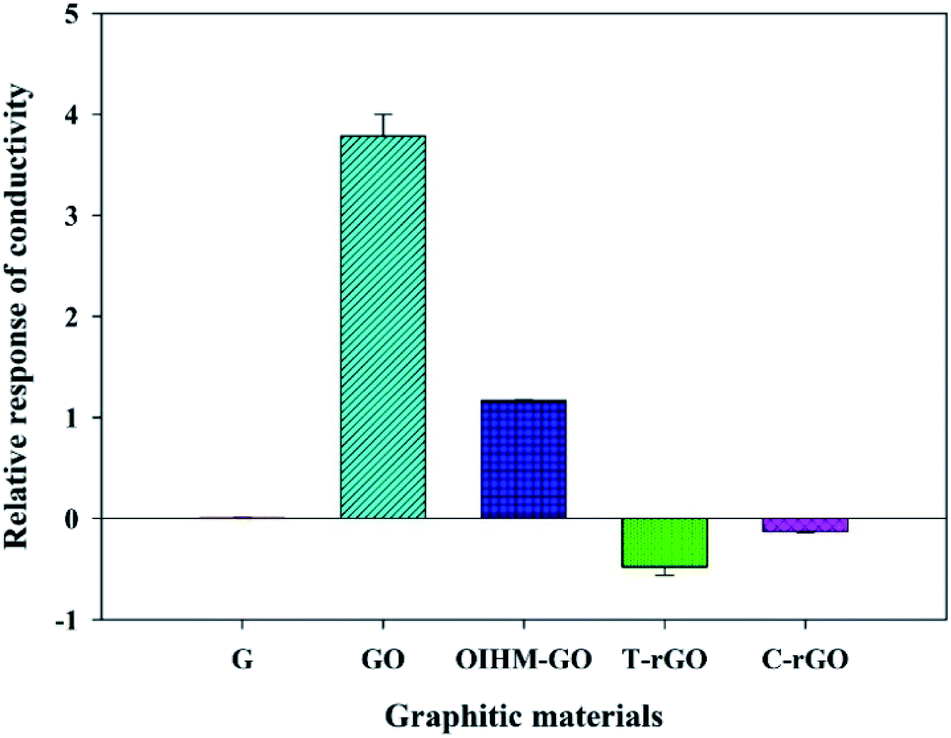 | ||
| Fig. 2 Relative responses of the conductivity of the graphitic materials under exposure to 11.36 ppm methanol vapor at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH. | ||
| Material | Relative conductivity response | Response time (min) | Recovery time (min) | Reversibility |
|---|---|---|---|---|
| a Remarks: (1) methanol concentration was constant at 11.36 ppm, 1.1 atm pressure, and 20 ± 3% RH. (2) Doping of PIn and composites was fixed at a 10:1 doping mole ratio of HClO4/indole. (3) In situ means the graphene-based materials/PIn composites were prepared by in situ polymerization. (4) SM means the graphene-based material/PIn composite was prepared by simple mixing. |
||||
| cm-G | (7.90 ± 3.58) × 10−3 | 11.67 ± 1.17 | — | Irrecoverable |
| cm-GO | 3.79 ± 0.3 | 16.15 ± 3.03 | 12.81 ± 3.43 | Fully recoverable |
| C-rGO | −0.10 ± 0.03 | 21.67 ± 4.50 | — | Partly recoverable |
| T-rGO | −0.48 ± 0.12 | 15.14 ± 0.31 | — | Partly recoverable |
| OIHM-GO | 1.53 ± 0.02 | 16.25 ± 3.78 | 1.29 ± 0.51 | Fully recoverable |
| dPIn38 | 57.83 ± 5.03 | 25.93 ± 1.44 | 18.88 ± 0.27 | Fully recoverable |
| In situ 10% v/v cm-G/dPIn | 0.71 ± 0.13 | 59.48 ± 7.26 | 4.52 ± 0.65 | Fully recoverable |
| In situ 10% v/v C-rGO/dPIn | 27.37 ± 2.22 | 54.84 ± 1.51 | 17.49 ± 1.01 | Fully recoverable |
| In situ 10% v/v T-rGO/dPIn | 20.03 ± 3.78 | 44.76 ± 2.31 | 19.83 ± 1.31 | Fully recoverable |
| In situ 10% v/v cm-GO/dPIn | 95.76 ± 3.77 | 54.03 ± 1.08 | 17.68 ± 0.82 | Fully recoverable |
| SM 10% v/v cm-GO/dPIn | 19.98 ± 1.63 | 78.23 ± 5.08 | 8.29 ± 1.78 | Fully recoverable |
| In situ 10% v/v OIHM-GO/dPIn | 81.89 ± 2.12 | 28.08 ± 1.93 | 19.91 ± 3.50 | Fully recoverable |
For OIHM-GO, although the number of oxygen atoms of OIHM-GO is slightly higher than that of cm-GO, the methanol response is lower. This may occur because of the low degree of oxidation of OIHM-GO, as confirmed by the low intensity (001) diffraction peak and the existing (002) diffraction peak.52 The low oxidation degree may be responsible for the low exfoliation, which provides a low surface area. Despite the large surface area and high electrical conductivity of graphene, it hardly responds to methanol vapor because its surface is hydrophobic, which is unreactive to the highly polar vapor. For the rGOs, the methanol responses are lower than those of GO; this is because the decrease in oxygen amount after the reduction induces low hydrophilicity, reducing the methanol response.
Negative responses were observed in both T-rGO and C-rGO, arising from the decreases in the electrical conductivity during methanol vapor exposure. A negative response of rGOs can originate from the p-type semiconducting behavior of rGO under ambient conditions.55 The negative responses of the rGOs toward methanol vapor are consistent with the results of H. Ahn et al., who reported in 2014 that rGO responded with a resistance increase (electrical conductivity decrease) during exposure to electron-donating methanol vapor.56 A positive response was clearly observed for GO, in which the electrical conductivity increased during methanol vapor exposure; this is due to the n-type semiconducting behavior of GO, as reported by H. Ahn et al. in 2014.56
For the sensor recoverability, complete recoveries were found only for cm-GO and OIHM-GO, whereas partial recoveries were observed for T-rGO and C-rGO and no recovery was observed for cm-G. The partial recoveries and non-recovery may correspond to the limited desorption of chemisorbed gas molecules by flushing with N2 gas,57 the pore sizes of the materials,58 and the high-binding energy sites.30
Pure graphene contains a low amount of oxygen, which provides fewer active sites. Thus, it requires a shorter response time to reach equilibrium compared to the other samples. The shorter time for graphene corresponds to its lower sensitivity due to its smaller amount of active sites.
3.3 Graphene-based material/dPIn composites and methanol responses
The graphene-based material/dPIn composites prepared by in situ polymerization were used to detect 11.36 ppm methanol vapor at room temperature (26 ± 1 °C), and N2 was used as a base gas. All the composites contained graphene-based materials at 10% v/v. The composites and PIn were doped with HClO4 at a doping mole ratio of 10:1 before the methanol vapor exposure testing.38 Herein, the role of PIn, as the main phase of the composite, is to interact with methanol by H-bonding; the interaction occurs between the N–H or +N–H site of the PIn chain and the O–H site of the methanol molecule. The methanol interaction results in expansion of the PIn chain, leading to higher electron mobility; consequentially, the electrical conductivity increases.
The relative conductivity responses of the composites are shown in Fig. 3 and detailed in Table 2. The methanol responses of the composites are arranged as follows: in situ cm-GO/dPIn > in situ OIHM-GO/dPIn > in situ C-rGO/dPIn > in situ T-rGO/dPIn > in situ cm-G/dPIn; the corresponding relative conductivity responses are equal to 95.76 ± 3.77, 81.88 ± 2.10, 27.37 ± 2.22, 20.03 ± 3.78, and 0.71 ± 0.13, respectively. All the composites manifested positive responses, indicating that the electrical conductivity of the composites increases under exposure to methanol vapor.
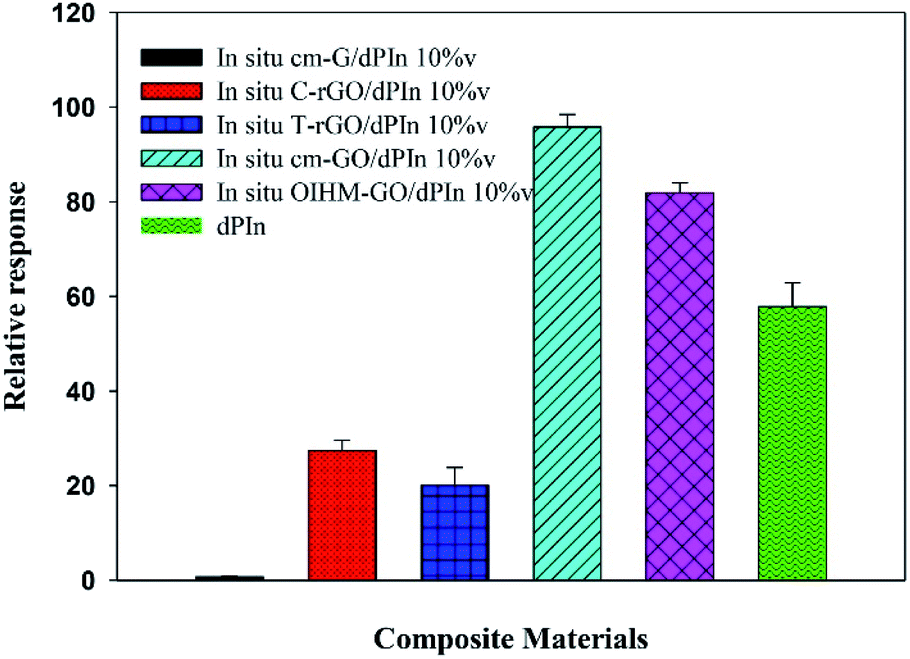 | ||
| Fig. 3 Relative responses of the conductivity of dPIn and various graphene-based materials/dPIn composites under exposure to 11.36 ppm methanol vapor at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH. | ||
The response behavior toward methanol vapor of the composites based on rGOs is different from that of the pristine materials, which gave negative responses. The key reason for this is the dPIn covered on the surface of the rGOs.28 For the composites based on graphene and rGOs, their methanol responses are lower than that of the pristine dPIn. This is because graphene and rGO have low hydrophilicity, resulting in low adsorption and diffusion of methanol into the inner layers of the composites. Thus, the gas diffusion and the methanol interaction are more important than the electrical conductivity. However, the methanol responses of the composites are higher than those of their pristine graphene-based materials alone. For instance, in situ cm-G/dPIn provides a higher relative response than cm-G alone, and the methanol responses of in situ T-rGO/dPIn and in situ C-rGO/dPIn are higher than those of T-rGO and C-rGO, respectively.
Herein, the two composites affording the highest responses to dPIn are the composites based on cm-GO and OIHM-GO; this is probably because (i) they contain more active sites for methanol molecules to adsorb to;54 (ii) the methanol diffusion into the inner layers is higher as a result of the hydrophilicity of GO, which induces intercalation or swelling when exposed to the polar vapor;70 and (iii) a synergistic effect is involved in the supplementary responses of both GO and dPIn.71 However, the relative conductivity responses of the composites based on OIHM-GO and GO are comparable. The response times of in situ cm-GO/dPIn and in situ OIHM-GO/dPIn are longer than that of dPIn due to the higher interaction between the oxygen species of cm-GO and OIHM-GO, which act as high energy active sites, defects, and vacancies; thus, the composites take more time to reach equilibrium.72 Moreover, on comparing the methanol responses of SM cm-GO/dPIn to in situ cm-GO/dPIn, the relative conductivity response of SM cm-GO/dPIn is equal to 19.98 ± 1.63; this is lower than that of in situ cm-GO/dPIn, which is 95.76 ± 3.77. This is probably because of the poor interfacial interaction between dPIn and the GO layers, which results in instability of the methanol interaction. Herein, the methanol sensors based on these composites are completely recoverable, as observed from the complete recoveries of the electrical signals to the original values after methanol removal.
The sensing mechanism of the graphene-based material/dPIn composites is significantly related to the methanol adsorption and diffusion into the inner layers of the composite and can be ordered as follows: in situ cm-GO/dPIn > in situ OIHM-GO/dPIn > in situ C-rGO/dPIn > in situ T-rGO/dPIn > in situ cm-G/dPIn, respectively. The methanol diffusion ability into the inner layers depends upon the surface properties, such as hydrophobic/hydrophilic properties.73 Generally, GO is well known as a highly hydrophilic material owing to the fact that it consists of many oxygen functional groups, inducing high diffusion of methanol molecules into the inner layers to interact with the active sites. For rGO and graphene, the hydrophilicity is low because oxygen species were eliminated; thus, the diffusion of methanol into the inner layers is lower. Thus, it can be suggested that methanol diffusion into the inner layers of the composites is obstructed by the rGO and graphene layers. For the graphene/dPIn composite, graphene as a hydrophobic material physically obstructs the interaction of methanol with polyindole as the main phase. Thus, this composite requires a longer response time to reach equilibrium relative to GO/dPIn and rGO/dPIn. The diffusion mechanism of methanol vapor into the inner layer of the composites is proposed in Fig. 4.
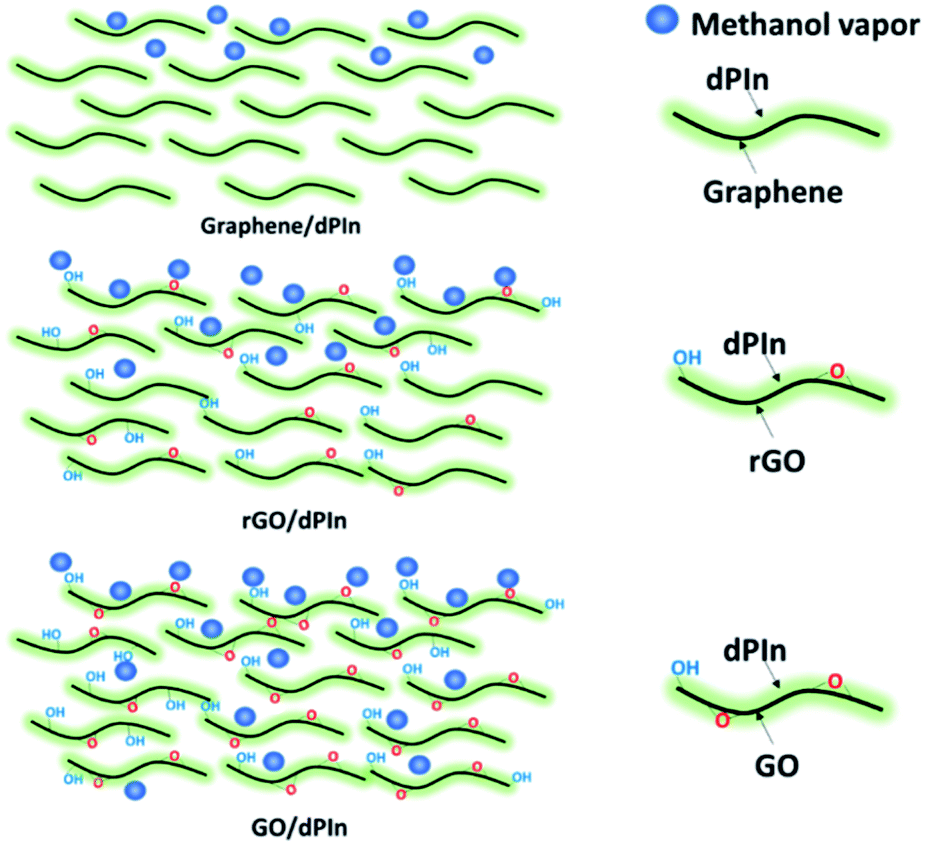 | ||
| Fig. 4 Proposed diffusion of methanol molecules into different graphene-based material/dPIn composites. | ||
The positive responses of the composites toward methanol vapor can be attributed to these mechanisms: (i) the increase in electrical conductivity of the composites is associated with the major interaction of the dPIn acting as the main phase of the composite; the methanol interaction induces expansion of the compact PIn chains to more stretched PIn chains and increases the charge carrier mobility resulting from the hydrogen bonding between methanol and the PIn chain;38 (ii) the partial composite swelling related to the molecular interaction between methanol and the composites by hydrogen bonding leads to volume expansion and a decrease of the interparticle distance; consequentially, the electrical conductivity increases. This last phenomenon is called the negative vapor coefficient effect (NVC).74 According to the swelling effect, the composite based on GO tends to greatly and easily swell because of its highly hydrophilic properties and good adsorption and diffusion of methanol molecules into the inner layers of the composite; thus, it demonstrates a higher response toward methanol compared to the composites based on rGO and graphene.
3.4 Performance of the OIHM-GO/dPIn composite in methanol sensing
In situ OIHM-GO/dPIn composites were prepared to investigate the effects of the OIHM-GO content. Although its response to methanol is lower than that of cm-GO, it is relatively effective and inexpensive to produce. Samples with OIHM-GO contents of 2.5, 5, 10, 15, and 20% v/v were prepared in order to determine the relative conductivity response under methanol vapor. Fig. 5 illustrates the relative conductivity response of the in situ OIHM-GO/dPIn samples with various OIHM-GO contents. The results show that the relative conductivity response tends to increase up to 10% v/v OIHM-GO. However, the relative conductivity response tends to decrease if the OIHM-GO content becomes excessive. Although OIHM-GO promotes the sensing response toward methanol vapor, the excessive OIHM-GO provides the composite with a denser physical barrier and thus diminishes the direct interaction between dPIn and methanol molecules; eventually, the sensing response is decreased.75 Therefore, the OIHM-GO content of 10% v/v is suitable because it provides the highest relative conductivity response toward methanol vapor. However, the sensors based on the in situ OIHM-GO/dPIn composites with all OIHM-GO contents were capable of full recovery because the electrical signal returned to its original value after methanol removal.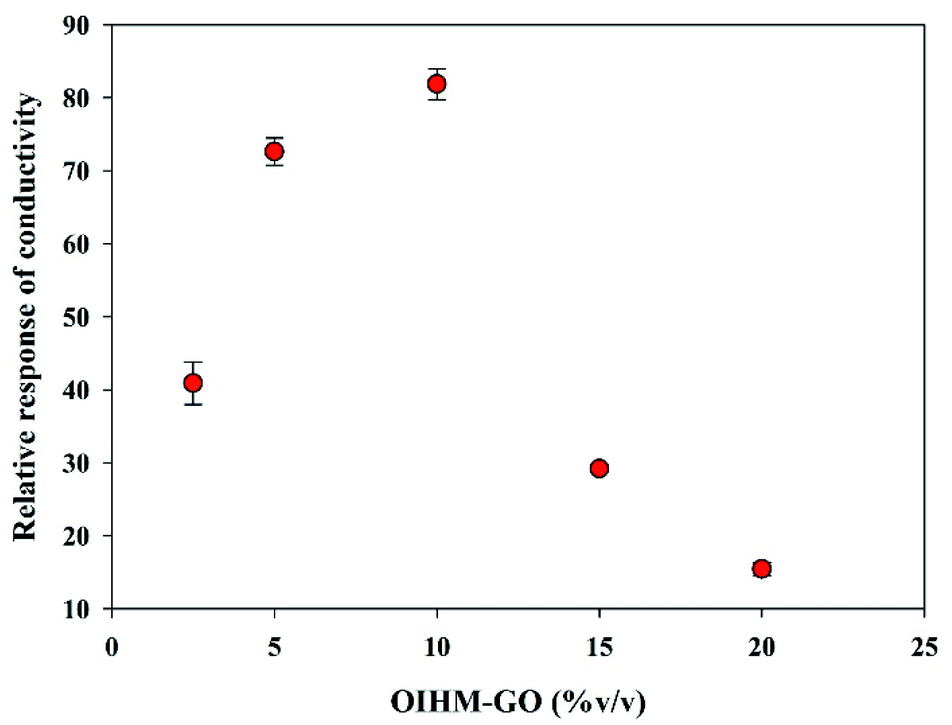 | ||
| Fig. 5 Relative responses of conductivity of in situ OIHM-GO/dPIn at various OIHM-GO contents under exposure to 11.36 ppm methanol vapor at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH. | ||
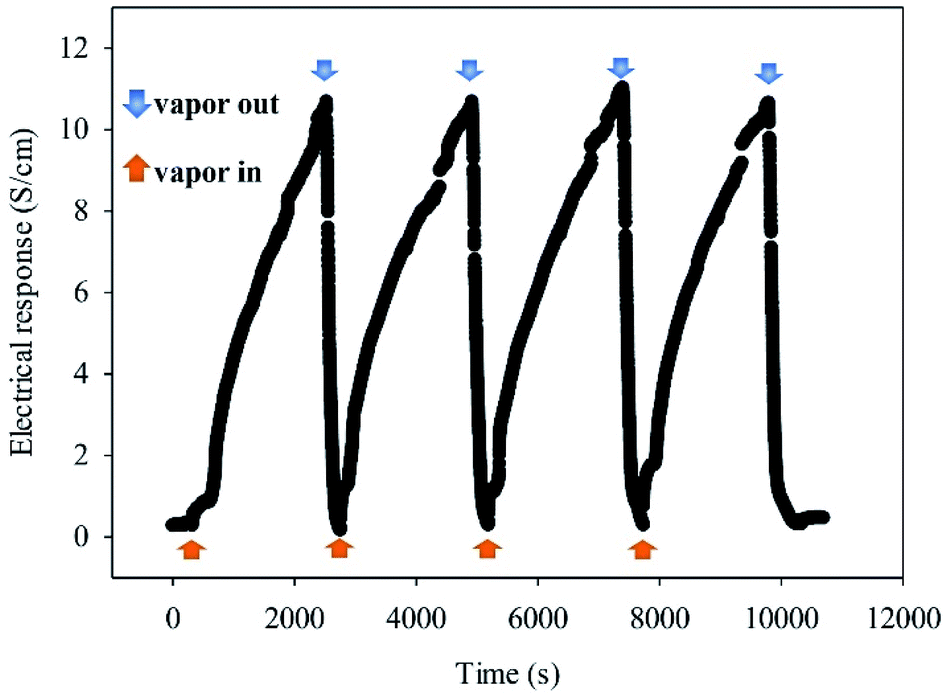 | ||
| Fig. 6 Cyclic response of a sensor based on in situ 10% v/v OIHM-GO/dPIn toward 3.41 ppm methanol vapor at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH. | ||
The reversibility of the methanol sensor based on in situ 10% v/v OIHM-GO/dPIn was confirmed by FT-IR spectroscopy. The in situ OIHM-GO/dPIn powder was homogeneously mixed with KBr (reference material) and then compressed to a thin pellet before being placed in a FT-IR gas cell. The FT-IR spectra of methanol vapor and the in situ 10% v/v OIHM-GO/dPIn composite before, during and after methanol exposure are shown in Fig. 7. Before being exposed to methanol, the in situ OIHM-GO/dPIn composite shows characteristic IR absorptions of O–H stretching at 3353 cm−1, N–H stretching at 3152 cm−1, CO stretching at 1714 cm−1, aromatic CC stretching at 1613 cm−1, C–N stretching at 1455 cm−1, CN stretching at 1374 cm−1, Cl–O stretching at 1144 and 1088 cm−1 and out-of-plane deformation of C–H at 747 cm−1. During methanol exposure, it can be observed that the IR absorption characteristic peaks slightly shift to higher wave numbers, such as the O–H and N–H stretching peaks, and some peaks slightly shift to lower wavenumbers, such as the CO and CN stretching peaks. These results may be associated with hydrogen bonding interactions, as proposed in Fig. 8. After methanol vapor removal, the IR absorption characteristics are similar to those of in situ OIHM-GO/dPIn before exposure to methanol. Therefore, it can be inferred that the interaction between in situ 10% v/v OIHM-GO/dPIn and methanol vapor is completely reversible.
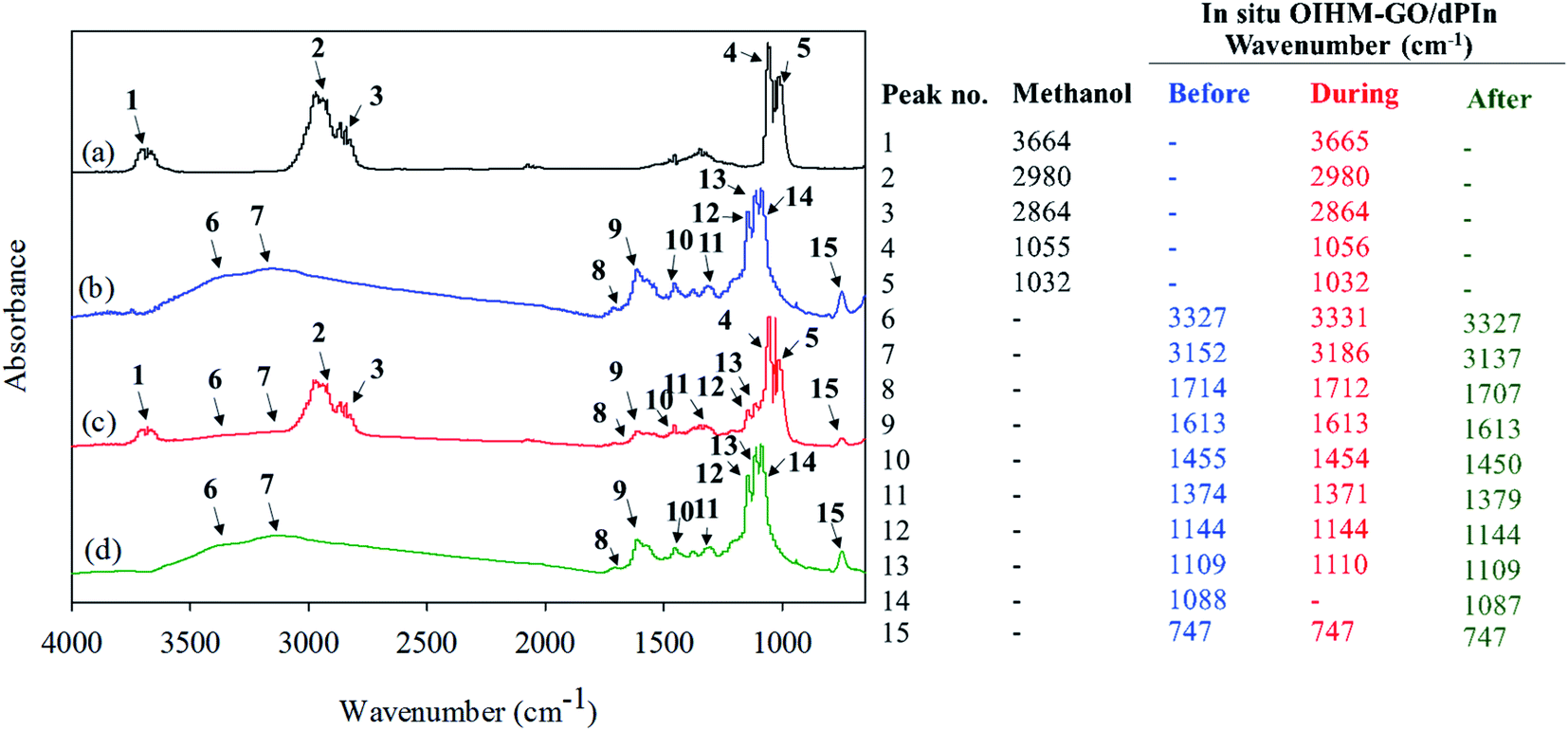 | ||
| Fig. 7 FTIR spectra of (a) methanol vapor; (b–d) in situ 10% v/v OIHM-GO/dPIn before, during, and after methanol exposure, respectively. | ||
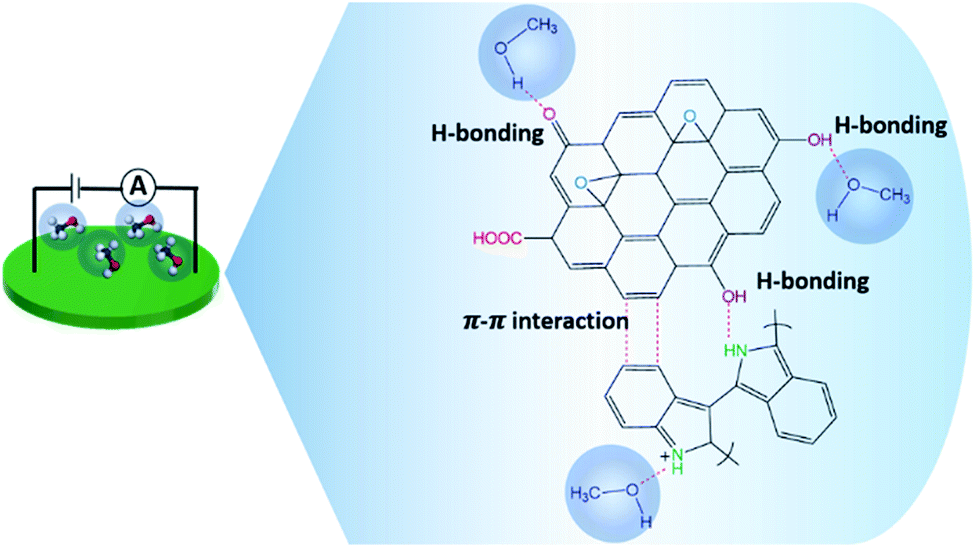 | ||
| Fig. 8 Proposed interactions between the in situ OIHM-GO/dPIn materials and methanol vapor. | ||
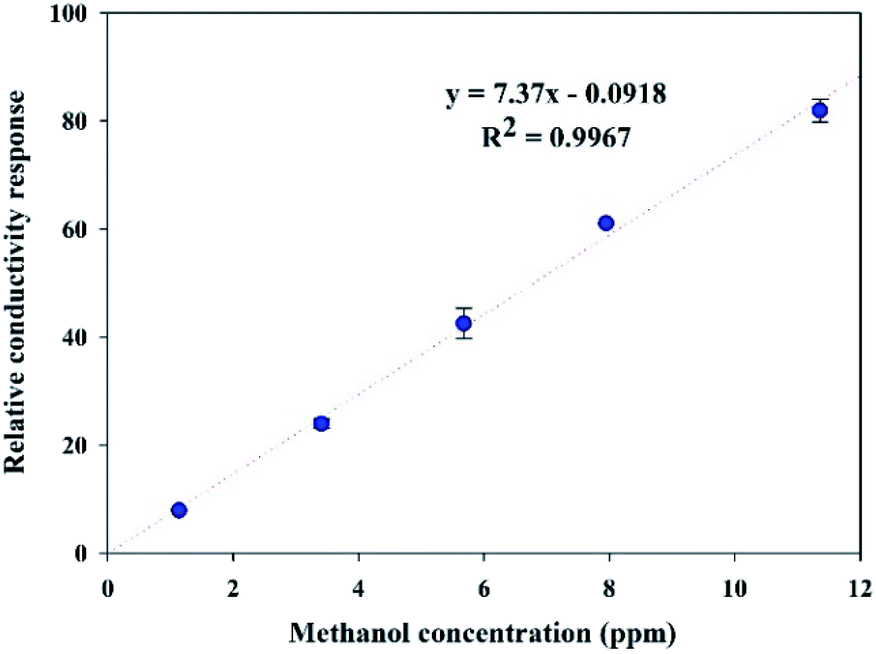 | ||
| Fig. 9 Calibration curve of the methanol sensor based on the in situ 10% v/v OIHM-GO/dPIn composite at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH. | ||
The sensitivity of dPIn reported in our previous work was 5.27 ppm−1 with R2 of 0.9965 in the same methanol concentration range.38 Thus, compositing dPIn with OIHM-GO by in situ polymerization can improve its methanol sensitivity. This result can be associated with the increase in the hydrogen bonding interactions between methanol and the active sites of dPIn and the oxygen species on the GO layer as proposed in Fig. 8; the hydrogen bonding interaction induces effective diffusion into the inner layer of the composite. The slope of the calibration curve not only indicates the sensitivity of the sensor but can also be used to calculate the theoretical LOD, as in eqn (3). For the in situ OIHM-GO/dPIn 10% v/v composite, the theoretical LOD is 0.015 ppm. The apparent LOD was also obtained from the extrapolated calibration curve to the x-axis; it is equal to 0.012 ppm. The LOD is lower than the threshold limit value (TLV) of methanol which is equal to 200 ppm.42 In the previous report, the theoretical LOD and apparent LOD of the methanol sensor based on dPIn were 0.048 ppm and 0.43 ppm, respectively.38 Therefore, the in situ OIHM-GO/dPIn composite can improve both the sensitivity and LOD toward methanol vapor, and it can be operated at room temperature. The performance of methanol sensors prepared from various sensing materials is compared to this work in Table 3. It can be observed that the methanol sensors based on hybrid materials from metals must be operated at high temperature (over 100 °C). However, their advantages are fast response and recovery times. The sensing materials based on conductive polymers and their carbon-based composites show relatively high responses and low LOD, short response and recovery times, and room temperature operation. The sensor based on the in situ 10% v/v OIHM-GO/dPIn composite shows higher methanol response than other graphene-based material/conductive polymer composites, such as PANI/GO, rGO–PIL/PEDOT, N-GQDs/PEDOT:PSS, and PEDOT–PSS/UL-GO, as shown in Table 3. However, the methanol sensors herein require long response and recovery times, probably because of the larger numbers of available active sites and defects as well as the sensing configuration. This may be also related to the preparation of the sensor as a thick pellet; as a result, methanol molecules take more time to diffuse into the inner layers of the composites and to reach an equilibrium state.
| Material | Relative response calculation | Sensitivity | Concentration range (ppm) | Response | Response/recovery times | LOD | Operating temperature | Base gas | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Remarks: Rg and R0 are the resistances of the sensing material to the target gas and base gas, respectively. RT is room temperature. σg and σN2 are the electrical conductivities of the sensing material in the presence of the target gas and N2 base gas, respectively. * Theoretical LOD, derived from (3σb)/sensitivity, where σb is the standard deviation of the electrical conductivity signal under N2. | |||||||||
| PPy/PVA | (Rg − R0)/R0 × 100 | — | 49–1059 | 2.6% (49 ppm) | 384 s/496 s | — | — | Air | 59 |
| PANI–Pd | (Rg − R0)/R0 | 8.90 × 105 Ω ppm−1 | 1–2000 | 104 (2000 ppm) | —/— | <1 ppm | RT | Air | 60 |
| Pd–Pt–In2O3/SnO2 | R0/Rg | — | 0.1–100 | 320.73 (100 ppm) | 32 s/47 s | 0.1 ppm | 160 °C | Air | 61 |
| ZnO/MoO3 | (R0 − Rg)/R0 × 100 | — | 5–500 | 56% (500 ppm) | 54 s/118 s | 34 ppm | 200 °C | Air | 62 |
| PANI/GO | (Rg − R0)/R0 | — | 100–500 | 37 (500 ppm) | 120 s/— | — | RT | Air | 29 |
| rGO–TiO2 nanotubes | (R0 − Rg)/R0 × 100 | — | 0.62–200 | 59% (1 ppm) | 41 s/46 s | 0.62 ppm | 110 °C | Air | 63 |
| Carboxylated MWCNT/PANI | (Rg − R0)/R0 | — | 50–500 | 0.897 (50 ppm) | — | — | RT | Air | 64 |
| s-PEI–LCP/rGO | (Rg − R0)/R0 × 100 | — | 300–1200 | 92% (1200 ppm) | 4 s/— | 4.6 ppm | RT | Air | 65 |
| 5% Co3O4–rGO | (Rg − R0)/R0 × 100 | — | 300–1000 | 4% (300 ppm) | ∼1–2 min/∼1–2 min | — | RT | O2/N2 | 66 |
| rGO–PIL/PEDOT | (Rg − R0)/R0 × 100 | — | 1–90 | ∼3% (1 ppm) | 20 s/149 s | 1 ppm | RT | N2 | 67 |
| N-GQDs/PEDOT:PSS | (Rg − R0)/R0 × 100 | — | 1–1000 | 154.4% (100 ppm) | 12 s/32 s | 5 ppm | RT | Air | 68 |
| PEDOT–PSS/UL-GO | (Rg − R0)/R0 × 100 | — | 35–1000 | 11% (35 ppm) | 3.2 s/16 s | — | RT | N2 | 69 |
| dPIn pellet | (σg − σN2)/σN2 | 5.27 ppm−1 | 1.14–11.36 | 57.83 (11.36 ppm) | 26 ± 1.44 min/19 ± 0.27 min | *48 ppb | 26 ± 1 °C | N2 | 38 |
| In situ 10% v/v OIHM-GO/dPIn pellet | (σg − σN2)/σN2 | 7.37 ppm−1 | 1.14–11.36 | 81.88 (11.36 ppm) | 28 ± 1.93 min/20 ± 3.50 min | *15 ppb | 26 ± 1 °C | N2 | Present work |
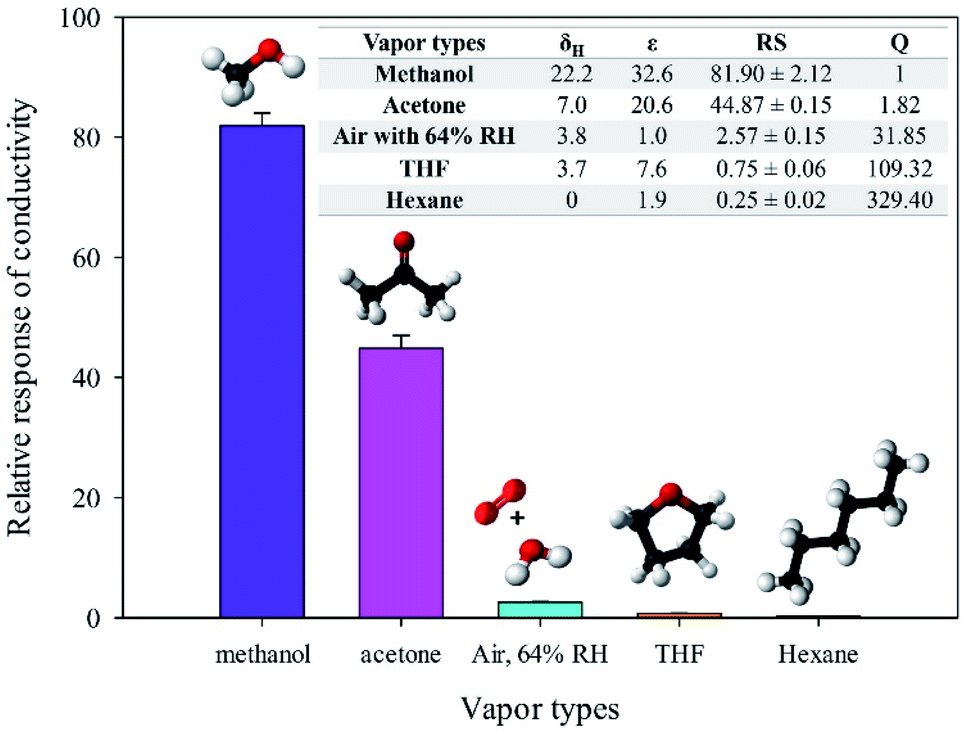 | ||
| Fig. 10 Relative conductivity response of in situ 10% v/v OIHM-GO/dPIn toward various vapor types at 26 ± 1 °C, 1.1 atm pressure, and 20 ± 3% RH under N2 base gas, including the selectivity coefficient and vapor properties. δH is the hydrogen interaction (MPa1/2), ε is the dielectric constant of the substance, RS is the relative response of conductivity, and Q is the selectivity coefficient. The δH and ε of air with 64% RH are based on oxygen. | ||
4. Conclusion
The methanol sensing abilities and the methanol conductivity responses of the carbon-based materials were arranged as follows: cm-GO > OIHM-GO > T-rGO > C-rGO > cm-G. The methanol response was related to the amounts of oxygen content present in the materials acting as the active sites. However, obtaining the positive or negative response depended on the n-type or p-type semiconducting behavior of the materials. Among the composites, the in situ 10% v/v cm-GO/dPIn was the most effective methanol sensing material because it provided the highest relative conductivity response of 95.76 ± 3.77 at a methanol concentration of 11.36 ppm. The in situ 10% v/v OIHM-GO/dPIn relative conductivity response was comparable, with a value of 81.90 ± 2.12. The sensitivity of the in situ 10% v/v OIHM-GO/dPIn composite was 7.37 ppm−1, with R2 of 0.9967 and a theoretical LOD of 0.015 ppm. In addition, the methanol sensor response based on the 10% v/v OIHM-GO/dPIn composite was reversible and quite selective; the response was nearly reproducible for at least 4 cycles. Although the methanol sensor herein exhibited good sensing performance, it still requires long response and recovery times. The short response time can be improved by preparing the composite as a thin film on a screen-printed electrode or an interdigitated electrode.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Royal Golden Jubilee (RGJ) Ph.D. Program (Grant No. PHD/0126/2557); the PETROMAT PhD Scholarship; the Conductive and Electroactive Polymers Research Unit of Chulalongkorn University; the Thailand Science Research and Innovation Fund (TSRI); and the National Research Council of Thailand (NRCT).References
- V. B. Mohan, K.-t. Lau, D. Hui and D. Bhattacharyya, Composites, Part B, 2018, 142, 200–220 CrossRef CAS.
- S. Basu and P. Bhattacharyya, Sens. Actuators, B, 2012, 173, 1–21 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- S. Szunerits and R. Boukherroub, J. Mater. Chem. B, 2016, 4, 6892–6912 RSC.
- B. K. Sharma and J.-H. Ahn, Solid-State Electron., 2013, 89, 177–188 CrossRef CAS.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS PubMed.
- Q. Ke and J. Wang, J. Materiomics, 2016, 2, 37–54 CrossRef.
- X.-F. Lin, Z.-Y. Zhang, Z.-K. Yuan, J. Li, X.-F. Xiao, W. Hong, X.-D. Chen and D.-S. Yu, Chin. Chem. Lett., 2016, 27, 1259–1270 CrossRef CAS.
- C. I. L. Justino, A. R. Gomes, A. C. Freitas, A. C. Duarte and T. A. P. Rocha-Santos, TrAC, Trends Anal. Chem., 2017, 91, 53–66 CrossRef CAS.
- J. Phiri, P. Gane and T. C. Maloney, Mater. Sci. Eng., B, 2017, 215, 9–28 CrossRef CAS.
- S. P. Lonkar, Y. S. Deshmukh and A. A. Abdala, Nano Res., 2014, 8, 1039–1074 CrossRef.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- V. Gupta, N. Sharma, U. Singh, M. Arif and A. Singh, Optik, 2017, 143, 115–124 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 14, 11 Search PubMed.
- U. Hofmann and E. König, Z. Anorg. Allg. Chem., 1937, 234, 311–336 CrossRef CAS.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W.-W. Liu and C. H. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- M. d. P. Lavin-Lopez, A. Romero, J. Garrido, L. Sanchez-Silva and J. L. Valverde, Ind. Eng. Chem. Res., 2016, 55, 12836–12847 CrossRef CAS.
- M. P. Lavin-Lopez, A. Paton-Carrero, L. Sanchez-Silva, J. L. Valverde and A. Romero, Adv. Powder Technol., 2017, 28, 3195–3203 CrossRef CAS.
- K. K. H. De Silva, H. H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef.
- Y. Peng and J. Li, Front. Environ. Sci. Eng., 2013, 7, 403–411 CrossRef CAS.
- K. Toda, R. Furue and S. Hayami, Anal. Chim. Acta, 2015, 878, 43–53 CrossRef CAS PubMed.
- L. Al-Mashat, K. Shin, K. Kalantar-zadeh, J. D. Plessis, S. H. Han, R. W. Kojima, R. B. Kaner, D. Li, X. Gou, S. J. Ippolito and W. Wlodarski, J. Phys. Chem. C, 2010, 114, 16168–16173 CrossRef CAS.
- S. Konwer, A. K. Guha and S. K. Dolui, J. Mater. Sci., 2012, 48, 1729–1739 CrossRef.
- D. C. Tiwari, P. Atri and R. Sharma, Synth. Met., 2015, 203, 228–234 CrossRef CAS.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- C. M. Hangarter, N. Chartuprayoon, S. C. Hernández, Y. Choa and N. V. Myung, Nano Today, 2013, 8, 39–55 CrossRef CAS.
- H. Bai and G. Shi, Sensors, 2007, 7, 267–307 CrossRef CAS.
- K. Phasuksom and A. Sirivat, Synth. Met., 2016, 219, 142–153 CrossRef CAS.
- D. Devadathan and R. Raveendran, Int. J. Chem. Eng. Appl., 2014, 5, 240–243 CAS.
- H. Mudila, S. Rana, M. G. H. Zaidi and S. Alam, Fullerenes, Nanotubes, Carbon Nanostruct., 2014, 23, 20–26 CrossRef.
- M. Tebyetekerwa, X. Wang, I. Marriam, P. Dan, S. Yang and M. Zhu, Mater. Lett., 2017, 209, 400–403 CrossRef CAS.
- K. Phasuksom, W. Prissanaroon-Ouajai and A. Sirivat, Sens. Actuators, B, 2018, 262, 1013–1023 CrossRef CAS.
- P. Dubey, A. Kumar and R. Prakash, Appl. Surf. Sci., 2015, 355, 262–267 CrossRef CAS.
- Q. Zhou, D. Zhu, X. Ma, J. Xu, W. Zhou and F. Zhao, RSC Adv., 2016, 6, 29840–29847 RSC.
- R. Paulraj, G. K. Mani, L. Nallathambi and J. B. B. Rayappan, J. Nanosci. Nanotechnol., 2016, 16, 8315–8321 CrossRef CAS.
- A. Mirzaei, S. G. Leonardi and G. Neri, Ceram. Int., 2016, 42, 15119–15141 CrossRef CAS.
- J.-L. Tian and H.-Y. Zhang, Fullerenes, Nanotubes, Carbon Nanostruct., 2016, 25, 17–22 CrossRef.
- Z. Sui, X. Zhang, Y. Lei and Y. Luo, Carbon, 2011, 49, 4314–4321 CrossRef CAS.
- A. A. Moosa and J. N. Jaafar, Nanosci. Nanotechnol., 2017, 7(2), 38–47 CAS.
- S. Pirsa and N. Alizadeh, Sens. Actuators, B, 2012, 168, 303–309 CrossRef CAS.
- M. Babaei and N. Alizadeh, Sens. Actuators, B, 2013, 183, 617–626 CrossRef CAS.
- G. Ürkmez, B. Sarı and H. İ. Ünal, J. Appl. Polym. Sci., 2011, 121, 1600–1609 CrossRef.
- B. Gupta, D. S. Chauhan and R. Prakash, Mater. Chem. Phys., 2010, 120, 625–630 CrossRef CAS.
- A. F. Halasa, G. D. Wathen, W. L. Hsu, A. Matrana and J. M. Massie, J. Appl. Polym. Sci., 1991, 43, 183–190 CrossRef CAS.
- W.-K. Jang, J. Yun, H.-I. Kim and Y.-S. Lee, Colloid Polym. Sci., 2012, 291, 1095–1103 CrossRef.
- L. Chen, W. Liu, X. Su, S. Xiao, H. Xie, C. Uher and X. Tang, Synth. Met., 2017, 229, 65–71 CrossRef CAS.
- K. Hu, D. D. Kulkarni, I. Choi and V. V. Tsukruk, Prog. Polym. Sci., 2014, 39, 1934–1972 CrossRef CAS.
- S. Prezioso, F. Perrozzi, L. Giancaterini, C. Cantalini, E. Treossi, V. Palermo, M. Nardone, S. Santucci and L. Ottaviano, J. Phys. Chem. C, 2013, 117, 10683–10690 CrossRef CAS.
- X. Huang, N. Hu, R. Gao, Y. Yu, Y. Wang, Z. Yang, E. Siu-Wai Kong, H. Wei and Y. Zhang, J. Mater. Chem., 2012, 22, 22488 RSC.
- H. S. Ahn, H. J. Park, J. H. Oh, J. C. Joo and D. J. Kim, Appl. Mech. Mater., 2013, 440, 64–68 Search PubMed.
- M. Parmar, C. Balamurugan and D. W. Lee, Sensors, 2013, 13, 16611–16624 CrossRef PubMed.
- Y. Yang, S. Li, W. Yang, W. Yuan, J. Xu and Y. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 13807–13814 CrossRef CAS PubMed.
- L. Jiang, H.-K. Jun, Y.-S. Hoh, J.-O. Lim, D.-D. Lee and J.-S. Huh, Sens. Actuators, B, 2005, 105, 132–137 CrossRef CAS.
- A. A. Athawale, S. V. Bhagwat and P. P. Katre, Sens. Actuators, B, 2006, 114, 263–267 CrossRef CAS.
- Y. Li, D. Deng, X. Xing, N. Chen, X. Liu, X. Xiao and Y. Wang, Sens. Actuators, B, 2016, 237, 133–141 CrossRef CAS.
- B. Mandal, Aaryashree, R. Singh and S. Mukherjee, IEEE Sens. J., 2018, 18, 2659–2666 CAS.
- D. Acharyya, S. Acharyya, K. Huang, P. Chung, M. Ho and P. Bhattacharyya, IEEE Trans. Nanotechnol., 2017, 16, 1122–1128 CAS.
- A. Bora, K. Mohan, D. Pegu, C. B. Gohain and S. K. Dolui, Sens. Actuators, B, 2017, 253, 977–986 CrossRef CAS.
- S. Bag, K. Rathi and K. Pal, Nanotechnology, 2017, 28, 205501 CrossRef PubMed.
- N. Chen, X. Li, X. Wang, J. Yu, J. Wang, Z. Tang and S. A. Akbar, Sens. Actuators, B, 2013, 188, 902–908 CrossRef CAS.
- T. T. Tung, M. Castro, J.-F. Feller, T. Y. Kim and K. S. Suh, Org. Electron., 2013, 14, 2789–2794 CrossRef CAS.
- J. N. Gavgani, H. S. Dehsari, A. Hasani, M. Mahyari, E. K. Shalamzari, A. Salehi and F. A. Taromi, RSC Adv., 2015, 5, 57559–57567 RSC.
- A. Hasani, H. S. Dehsari, J. N. Gavgani, E. K. Shalamzari, A. Salehi, F. Afshar Taromi and M. Mahyari, Microchim. Acta, 2015, 182, 1551–1559 CrossRef CAS.
- S. You, B. Sundqvist and A. V. Talyzin, ACS Nano, 2013, 7, 1395–1399 CrossRef CAS PubMed.
- F.-L. Meng, Z. Guo and X.-J. Huang, TrAC, Trends Anal. Chem., 2015, 68, 37–47 CrossRef CAS.
- N. Hu, Z. Yang, Y. Wang, L. Zhang, Y. Wang, X. Huang, H. Wei, L. Wei and Y. Zhang, Nanotechnology, 2014, 25, 025502 CrossRef PubMed.
- R. Kumar, D. K. Avasthi and A. Kaur, Sens. Actuators, B, 2017, 242, 461–468 CrossRef CAS.
- J. Lu, B. J. Park, B. Kumar, M. Castro, H. J. Choi and J.-F. Feller, Nanotechnology, 2010, 21, 255501 CrossRef PubMed.
- J. Sun, X. Shu, Y. Tian, Z. Tong, S. Bai, R. Luo, D. Li and C. C. Liu, Sens. Actuators, B, 2017, 241, 658–664 CrossRef CAS.
- A. T. Mane, S. T. Navale and V. B. Patil, Org. Electron., 2015, 19, 15–25 CrossRef CAS.
- S. T. Navale, G. D. Khuspe, M. A. Chougule and V. B. Patil, Ceram. Int., 2014, 40, 8013–8020 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The analytical results of reduced graphene oxide and the synthesized graphene oxide (OIHM-GO). See DOI: 10.1039/d0ra00158a |
| This journal is © The Royal Society of Chemistry 2020 |