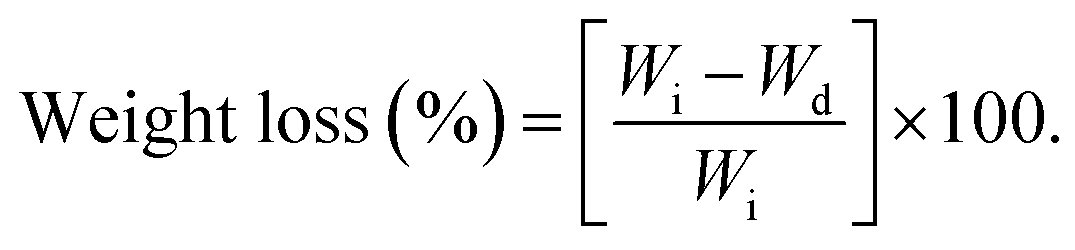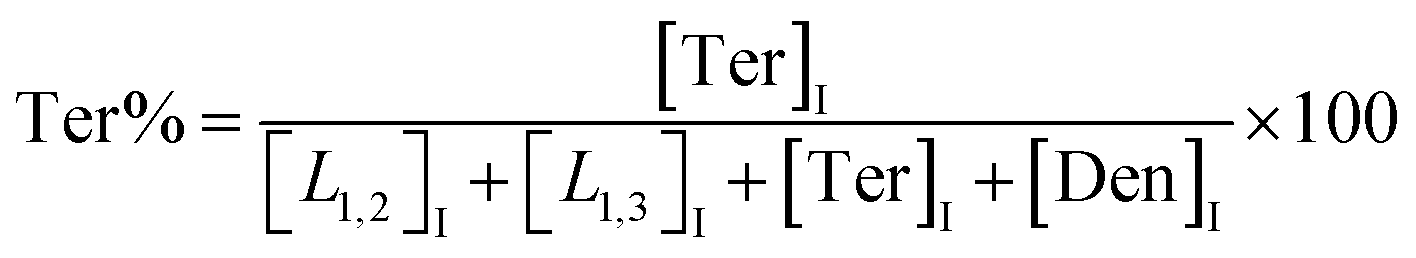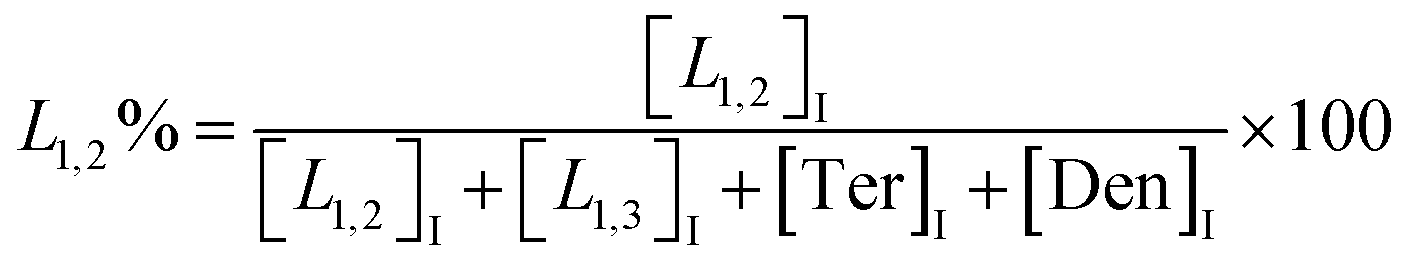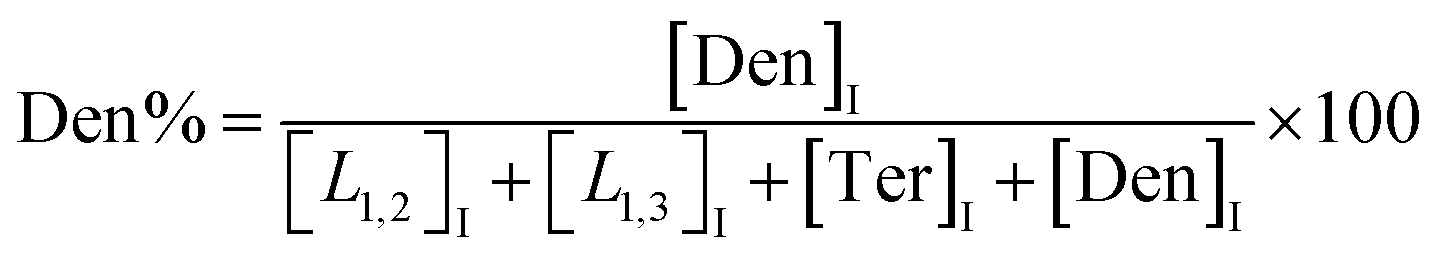DOI:
10.1039/D0RA00120A
(Paper)
RSC Adv., 2020,
10, 6414-6422
Syntheses of high molecular weight hydroxy functional copolymers by green and selective polycondensation methods†
Received
6th January 2020
, Accepted 3rd February 2020
First published on 11th February 2020
Abstract
Synthesizing hydroxy-functional linear copolymers with high molecular weights (Mn) and low branching degree (Den%) remains challenging, although there has been much headway in the area of functional copolymers. Here, we studied the effect of polymerization methods (one-step or two-step) and solvents (organic solvent: diphenyl ether or ionic liquids: [Cnmim]TF2N/BF4/PF6, n = 2, 4, 6, 8, or 10) on Mn and Den% of copolymers P(OA–GA) (1,8-octanediol adipate (O-A)/glycerol adipate (G-A)). The Mn of P(OA–GA) reached up to 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 937 g mol−1 in two-step in diphenyl ether, and the Den% of glycerol can be controlled within 30%. The physical properties of these copolymers were investigated by contact angles, differential scanning calorimetry (DSC), and in vitro biodegradation. With increasing glycerol content in the polyesters, both hydrophilic properties and degradation properties increased. This system not only facilitates the synthesis of functional polyesters with high molecular weight and low branching, but also expands the possibility of using bio-based monomers to synthesize functional polymers.
937 g mol−1 in two-step in diphenyl ether, and the Den% of glycerol can be controlled within 30%. The physical properties of these copolymers were investigated by contact angles, differential scanning calorimetry (DSC), and in vitro biodegradation. With increasing glycerol content in the polyesters, both hydrophilic properties and degradation properties increased. This system not only facilitates the synthesis of functional polyesters with high molecular weight and low branching, but also expands the possibility of using bio-based monomers to synthesize functional polymers.
Introduction
Aliphatic polyester is a member of the polyester family, and widely used in packaging, healthcare, pharmacies, and electronics because of its biodegradability, biocompatibility, and good physical and chemical properties versus traditional polymers.1,2 However, the lack of modifiable pendent functional groups on the backbone limits the large-scale application of these polyesters. Thus, much work has been focused on synthesizing polyesters derived from polyols. These are expected to find an efficient method to obtain polyesters with free functional hydroxyl groups which can be further functionalized.3–5
Glycerol is the simplest polyol with three hydroxyl groups. It was naturally synthesized from animals, plants, and microorganisms by diversified avenues,6 and the US Food and Drug Administration (FDA) has confirmed that it is safe for medical applications.7,8 It can be used directly in polyester synthesis via esterification or transesterification, but the synthesis of highly functional, linear polyesters by glycerol remains a major challenge due to its multiple hydroxyl groups. Special reaction conditions and an accurate control method are needed to form selective ester bonds. These steps play critical roles in the maintenance of the integrity and the linearity of the obtained polyester.
Methods using polyol for the synthesis of polymers with functional extension are under intense study. Shibata et al.4 used low temperature polycondensation technology to synthesize polyesters (Mn = ca. 1.0 × 104 g mol−1) with dangling hydroxyl groups by using Lewis acids catalysed in solvent-free systems. The reactivity of the pendent hydroxyl group was demonstrated through further glycosylation. Gustini et al.9 synthesized a renewable linear polyester derived from sorbitol through solvent-free polycondensation and then studied the efficiency and selectivity of different catalytic systems by incorporating sorbitol. Enzyme catalysis offers several advantages over chemical catalysis, including high enantio- and regio-selectivity, mild reaction conditions, and low energy consumption.10 Despite the many advantages of enzymatic catalysts, solvent-free systems are not an optimal approach to preparing functional polyesters. They require a substantial amount of energy, and polyesters synthesized via this method often have a lower molecular weight.
Solution systems may improve the rate of interchain transesterification when N-435 is the catalyst. Taresco V et al.11 used Novozyme-435 (N-435) as a catalyst to synthesize a series of poly(glycerol adipates) (PGA) in tetrahydrofuran (THF) and adjusted the structure (trisubstitution from ∼5% up to ∼30%) by reaction temperature, Mn was up to 1.3 × 104 g mol−1. Mahapatro et al.12 studied the influence of substrates and solvents on polycondensations catalysed by N-435. For poly-esterification compared other three solvents (xylene, tetra-ethylene glycol dimethyl ether, and 2-methoxyethyl ether), diphenyl ether was a more desirable solvent. The solvent should have a sufficiently high boiling point to remove the by-product from the reaction. Moreover, the catalyst activity in solvent is also a key factor in synthesizing polyesters with high molecular weight. Ionic liquids (ILs), compounds with low vapor pressures and high chemical, thermal stability, and solubility for many organic or inorganic substances13 and have tunable solubility and are recoverable,14 have been widely used in polycondensation reactions such as polyamides, polyimides,15,16 and other good performance polymers17 to obtain polymers in milder reaction conditions. Heise and others18–21 synthesized poly(ε-caprolactone) (Mn ≤ 10000 g mol−1) by N-435 catalysed ring opening polymerization, and polyesters (Mn ≤ 5400 g mol−1) by N-435 catalysed synthesized from transesterification. Fradet et al.22 synthesized poly (glycolic acid) from glycolic acid or oligomers in imidazolium-based ionic liquids. The number average of oligomers could be up to 45 via polymerization in 1-butyl-3-methylimidazolium bis[(trifluoromethyl)-sulfonyl]-imide. Poor miscibility of poly(glycolic acid) with solvent and inferior activity of metal catalysts in ILs results in low molecular weight. Studies by Lv et al.23 indicated that N-435 has higher activity in [PF6]−- and [NTf2]−-based ILs than [BF4]−- and [N(CN)2]−-based ILs. However, the relationship between the structure of hydroxy-functional polyesters and solvent properties remains largely unexplored.
The aim of this work is to explore a benign, eco-friendly method of polymerization for hydroxy-functional polyesters with high molecular weight. The first goal was to construct a model reaction of P(OA–GA) via solution polymerization. Adipic acid and 1,8-octanediol (OD) act as co-monomers for glycerol. These monomers are available from renewable sources.24,25 Diphenyl ether and [C6mim] Tf2N were selected as reaction solvents due to their sufficiently high boiling point and activities for N-435. Polymers were first synthesized via a one-step or two-step method. Relevant factors in this comparative study included pre-condensation and condensation conditions. Specifically, they include the influence of the feed ratio, the amount of catalyst used, the reaction time, and the reaction temperature on properties of the products. Further study evaluated different ionic liquids including [Cnmim]Tf2N/BF4/PF6 (n = 2, 4, 6, 8, or 10) to discover the influence of ILs characteristic on structure and molecular weight of hydroxy-functional polyesters. This is a new approach to synthesizing co-polyesters with high molecular weights and low degrees of branching.
The details of the copolyesters were studied in detail via 1H NMR, 13C NMR, and GPC. The hydrophilic, thermal properties and degradation properties of the hydroxyl-functional polyesters were tested via contact angles, DSC and vitro biodegradation, respectively.
Experimental section
Materials
Glycerol (99% pure) was purchased from Aldrich; 1,8-octanediol (98%, 1,8-OD) and adipic acid (98%) were purchased from Macklin. Diphenyl ether was purchased from Adamas. All ionic liquids were purchased from Shanghai Chengjie Chemical Co., Ltd. Tetrahydrofuran (THF) and methanol were purchased from Tianjin Fuyu Fine Chemical Co., Ltd. All reagents were used without further purification. Novozyme-435 (specified activity 7000 PLU g−1) was provided by Novozymes.
Syntheses of oligo-esters
Prepolymer P(OA–GA) (1,8-octanediol adipate (O-A)/glycerol adipate (G-A)) was synthesized by solvent-free polymerization. In this process, 2.92 g (0.02 mol) adipic acid, 2.34 g (0.016 mol) 1,8-OD, and 0.37 g (0.004 mol) glycerol were added in a 25 ml two-necked round bottom flask. The flask was placed into an oil bath with a magnetic stirrer at 500 rpm and heated at 130 °C, 140 °C, and 150 °C for 1 h and 160 °C for 2 h. Nitrogen is used to remove the byproduct water. The prepolymer was collected after cooling and directly applied to the polycondensation.
Lipase-catalysed post-polycondensations in different solvents
The prepolymer and catalyst (N-435) (10 wt% of monomer) were placed in a 10 ml round-bottom flask and then reacted in diphenyl ether (2:1 v/w of prepolymer) or [Cnmim]Tf2N/BF4/PF6 (n = 2, 4, 6, 8, 10) (35 wt% prepolymer, 65 wt% ionic liquid). The flask with the reactants was vacuumed and filled with dry nitrogen three times. The flask was then heated at 60 °C in an oil bath at 500 rpm for 24 h under vacuum (1.5–3 mmHg). The product was cooled and dissolved in tetrahydrofuran (THF); the N-435 was removed by filtration. Excess THF of the polymer solution was removed by rotary evaporation and precipitated in methanol. The precipitate was dried in a vacuum oven for 24 h.
Nuclear magnetic resonance
1H NMR and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer using chloroform-d6 as solvent. Chemical shifts (ppm) are expressed relative to the internal reference tetramethylsilane (TMS, 0.00 ppm).
Molecular weight
The number-average molecular weight and polydispersity of copolymers were determined by gel permeation chromatography (GPC) equipped with a Waters HPLC system. The Waters HPLC system consists of a 510 pump, a 717 autosampler, and a 410 refractive index detector. Separations were achieved using a Waters Stryragel HT3, HT4, and HT5 columns in series. Tetrahydrofuran (THF) was the solvent, and the flow rate was 0.8 ml min−1. The system was calibrated with polystyrene molecular weight standards from 2830 K to 2640 K (from Polymer Laboratory). The sample concentration was 3–4 mg ml−1, the injection volume was 10 μL, and then the data were analysed by Trisec GPC software version 3.
Contact angles
Contact angles of the copolymers were measured using a KSV Cam200 (KSV Instruments Ltd., Helsinki, Finland) at 25 °C. The resulting copolymers were adhered to the slides and compressed into flakes. The shape and the contact angle were analysed by dedicated software (CAM200). Each sample was measured at five different sites and then averaged.
Differential scanning calorimetry (DSC)
Differential scanning was documented by an analyser (Q2000, TA Instruments, Leatherhead, UK). Two heating/cooling cycles from −70 °C to 120 °C were used to analyse the sample at a heating rate of 10 °C min−1. Nitrogen gas at 50 ml min−1 purged the DSC cell. Data were reported and analysed with software (Version 4.5.05A).
Vitro biodegradation
The in vitro biodegradation rates of the P(OA–GA) sample were measured by changes in dry weight. The sample (20 mg) was weighed and melted at 60 °C in a 1.5 ml small vial for 6 h in a vacuumed drying oven. The sample was cooled to room temperature and treated with phosphate-buffered saline (1.5 ml) and then incubated on a shaking table. The temperature of the shaking table is 37 °C. It was rotated at 160 rpm. After 24 h, the sample was removed and washed with deionized water and then vacuum dried at 30 °C until the mass of the sample remained constant. The weight loss was calculated with eqn (1) (Wi: initial weight, and Wd: dried weight). Three replicates were averaged:| |
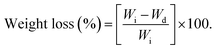 | (1) |
Results and discussion
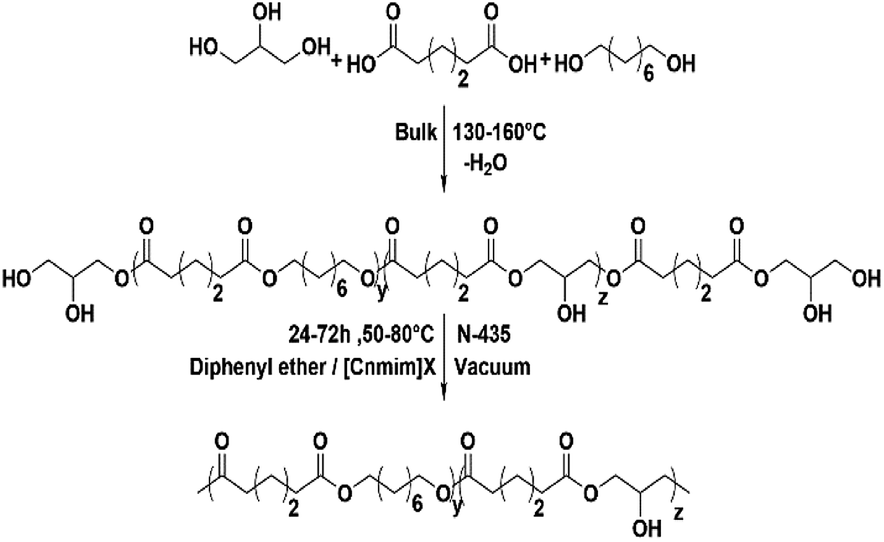
This work concentrated on the synthesis of functional polyester P(OA–GA) (Scheme 1) with a high molecular weight by N-435 catalysed in solvent polycondensations. Of particular interest is the construction of the P(OA–GA) model. Diphenyl ether and [C6mim]Tf2N were used as solvents to compare the effects of polycondensation method, pre-polycondensation, and polycondensation conditions on the structure and molecular weight of P(OA–GA). These included the feed ratio, species of solvent, catalyst amount, reaction time, and temperature. Further studies were conducted in different ionic liquids to understand the characteristics of ionic liquids as a polymerization solvent.
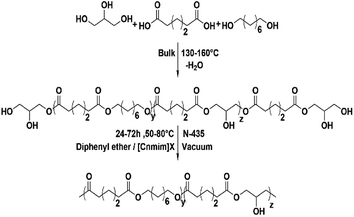 |
| | Scheme 1 Procedure for two step polycondensation. | |
Model reaction of P(OA–GA)
Glycerol has multiple hydroxyl groups, and esterification of carboxylic acid and their derivatives can be performed at both primary and secondary hydroxyls of glycerol. These systems can lead to the branching of the chain and even chain–chain crosslinking. The main challenge faced by many experiments is the preparation of highly functional linear polyesters.26 The P(OA–GA) copolymer can be dissolved in tetrahydrofuran, and no chain–chain crosslinking was seen.
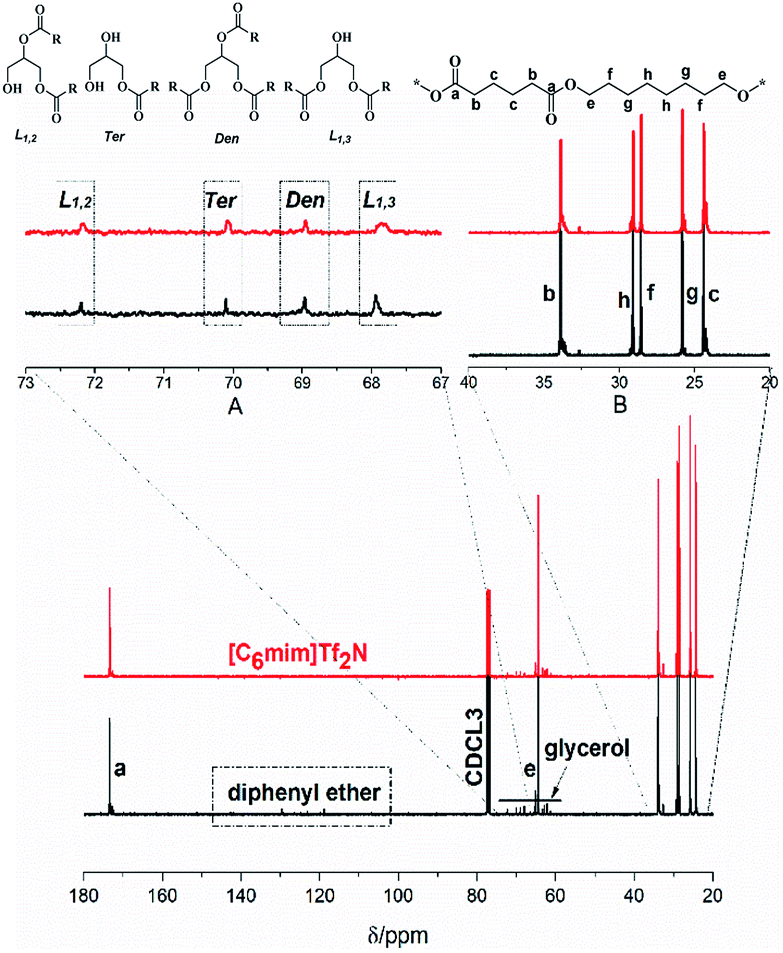
The P(OA–GA) structure was determined by 1H NMR (Fig. S1†). The main chain of the methylene protons of adipic acid and 1,8-octanediol in P(OA–GA) is clearly observed at 1.3–2.4 ppm and 4.05 ppm followed by the –OH proton peak at 3.7 ppm. All protons of glyceride with repeating units were found between 3.5 and 5.3 ppm. Peaks among 5.1–5.3 ppm were believed to be the secondary ester signal, which indicated that a part of secondary hydroxyl of glycerol participated in the reaction. The peak at 5.10 and 5.26 ppm (h′ and h′′ in Fig. S1 and Fig. S1† inset) corresponds to the methyl proton of a disubstituted glycerol unit (L1,2) by esterification of adipic acid with primary-secondary and a methine proton of trisubstituted glycerol unit (Den), respectively. Interestingly, the peak was not noticeable when [C6mim]Tf2N was the solvent (Fig. S1,† inset), which indicated that glycerol with lower branching degree in [C6mim]Tf2N than diphenyl ether. A sharp singlet was observed at 7.26 ppm due to CDCl3. The multiple peak groups next to the solvent peak were attributed to the diphenyl ether. The diphenyl ether has a high boiling point (259 °C) and melting point (27 °C); thus, it is difficult to separate from the product.
The macromolecular architecture of P(OA–GA) was further accurately defined by 13C NMR (Fig. 1). The 13C NMR qualitatively analysed the type of methine carbon (which determined the existence of different glyceride groups). It also quantitatively determined the proportion of each glyceride group. Peak distribution is consistent with Kulshrestha et al.27 Four methine (–CH–) signals of glycerol units from different substitution patterns were found in the 67–73 ppm (Fig. 1). The observation of a single peak at 70.3 ppm is due to the terminal glycerol units (Ter) that were formed via esterification of the adipic acid with a glycerol primary hydroxyl. The peak at 68.9 ppm is due to trisubstituted or dendritic glycerol units (Den). The signal of disubstituted glycerol units was seen at 72.1 and 68.2 ppm including L1,2 and L1,3 formed by esterification of adipic acid with primary–secondary or primary–primary hydroxyls of glycerol, respectively. The relative percentages of Ter, L1,2, L1,3, and Den were calculated by eqn (2)–(5) and are summarized in Table 1.
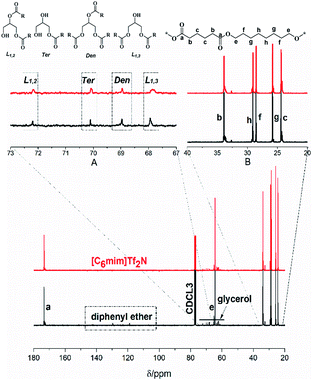 |
| | Fig. 1 13C-NMR spectrum of P(OA–GA), showing the signals corresponding to different methine groups of the synthesized P(OA–GA) polymers. | |
Table 1 The proportion of glycerol repeat unit substitution of copolymer P(OA–GA)a
| Entry |
Molar ratio A:O:Gb |
Pre-polymerization temperaturec (°C) |
Solvent |
Time (h) |
Terd (%) |
L1,2e (%) |
L1,3f (%) |
Deng (%) |
| The products were synthesized through two-step method (entries M2–M5, M7–M9) or directly synthesized from monomers (M1 and M6); all post-polymerization were conducted at 60 °C, N-435 (10 wt% of monomers), in vacuum 1.5–3 mmHg. A = adipic acid; O = 1,8-octanediol; G = glycerol. Pre-polymerization were conducted under N2 atmosphere. Data were analysed by 13C NMR and calculated based on. Eqn (2). Eqn (3). Eqn (4). Eqn (5). |
| M1 |
1:0.8:0.2 |
— |
Diphenyl ether |
12 |
11.76 |
14.12 |
58.82 |
15.29 |
| M2 |
1:0.8:0.2 |
100–130 |
Diphenyl ether |
24 |
18.66 |
10.07 |
39.93 |
31.34 |
| M3 |
1:0.8:0.2 |
110–140 |
Diphenyl ether |
24 |
14.98 |
10.58 |
39.96 |
34.49 |
| M4 |
1:0.8:0.2 |
130–160 |
Diphenyl ether |
24 |
12.80 |
12.40 |
40.00 |
34.80 |
| M5 |
1:0.84:0.21 |
130–160 |
Diphenyl ether |
24 |
12.89 |
15.11 |
44.44 |
27.56 |
| M6 |
1:0.8:0.2 |
— |
[C6mim]Tf2N |
24 |
13.74 |
17.53 |
47.39 |
21.33 |
| M7 |
1:0.8:0.2 |
130–160 |
[C6mim]Tf2N |
24 |
13.78 |
15.11 |
44.44 |
26.67 |
| M8 |
1:0.84:0.21 |
130–160 |
[C6mim]Tf2N |
24 |
20.42 |
19.58 |
40.83 |
19.20 |
| M9 |
1:0.88:0.22 |
130–160 |
[C6mim]Tf2N |
24 |
n.d. |
n.d. |
n.d. |
n.d. |
The effects of the polymerization method on the relative percentages were explored in diphenyl ether (entries M1 and M2, Table 1) and in [C6mim]Tf2N (entries M6 and M7, Table 1). The degree branching (Den) of products using the two-step method was higher than that using the one-step due to the higher pre-polymerization temperature. Entries M2–M4 (Table 1) showed the effect of pre-polymerization temperature on the polymer structure. The amount of Den, L1,2, and L1,3 increased with temperature; the content of Ter decreased slightly, which indicates that more glycerol is incorporated into the polymer backbone. The results of entries M4 and M5 and M7–M9 in Table 1 showed that the diol/diacid ratio was over unity is necessary to formulate low-branched polymers. In addition, more low-branched polymers were discovered in [C6mim]Tf2N (entry M8, Table 1) than in diphenyl ether (entry M5, Table 1). It's worth noting that the gel content in the reaction increased based on visual observation when using a one-step method in diphenyl ether after 24 h. The solvent test found that the product was even difficult to dissolve in a strong polar solvent such as dimethyl sulfoxide (DMSO). Gel formation is attributed to cross-linking reactions. No gelation was observed when the reaction time was shortened to 12 h. The copolymer can be completely dissolved in THF.
| |
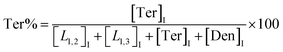 | (2) |
| |
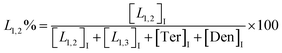 | (3) |
| |
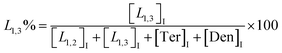 | (4) |
| |
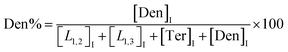 | (5) |
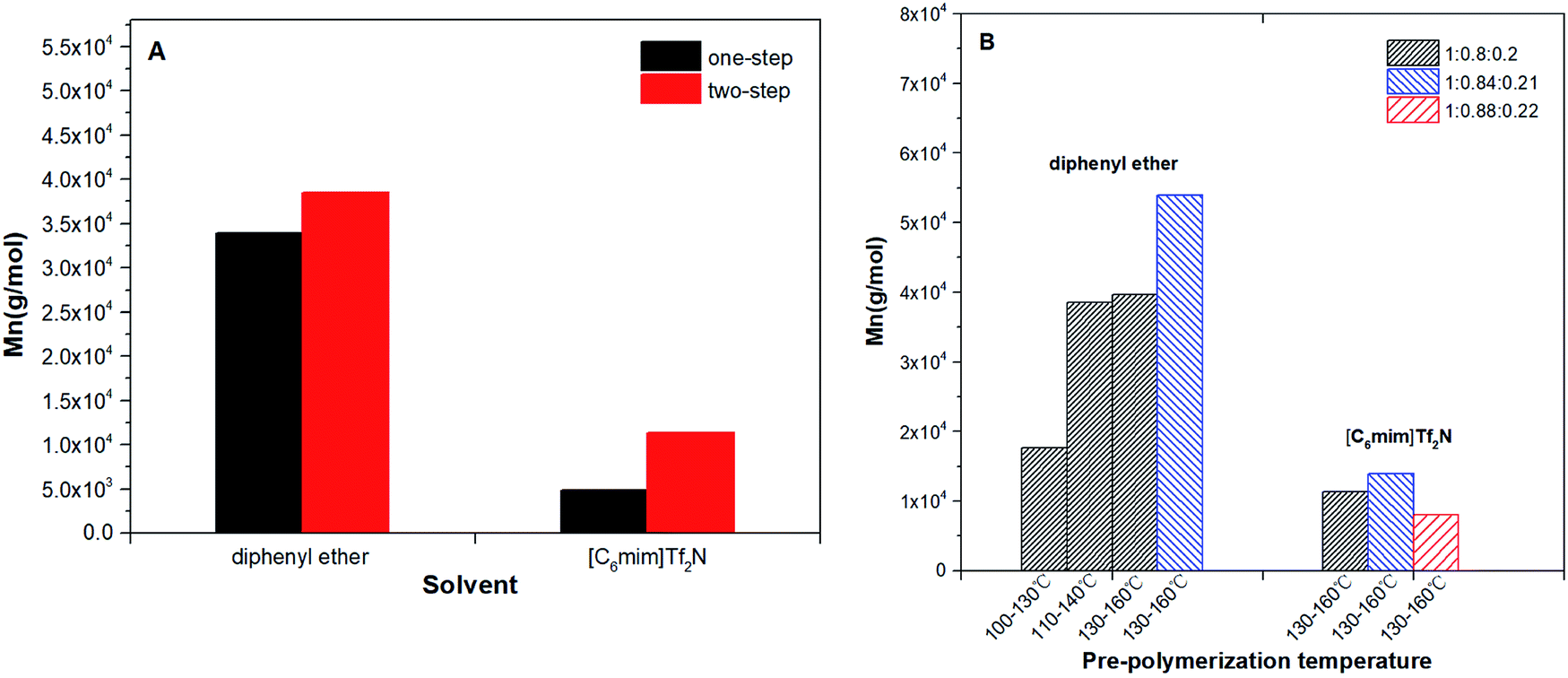
The results of number-average molecular weight (Mn) and polydispersity (PDI) were determined by GPC and summarized in Table 2. The highest Mn of the co-polyester was up to 53937 g mol−1 (diphenyl ether): This is far higher than that obtained in bulk (Mn < 2.0 × 104 g mol−1). The comparisons have been provided in Table S2.†4,8,11,28 This proves that when N-435 is the catalyst, the solvent system is an ideal reaction system for obtaining high molecular weight copolyesters. The detailed trend of the Mn of the products are shown in Fig. 2.
Table 2 Molecular weight and polydispersity of P(OA–GA) from model reactiona
| Entry |
Molar ratio A:O:Gb |
Pre-polymerization temperaturec (°C) |
Solvent |
Time (h) |
Mnd (g mol−1) |
PDId (Mw/Mn) |
| The products were synthesized through two-step method (entries M2–M5, M7–M9) or directly synthesized from monomers (M1 and M6); all post-polymerization were conducted at 60 °C, N-435 (10 wt% of monomers), in vacuum 1.5–3 mmHg. A = adipic acid; O = 1,8-octanediol; G = glycerol. Pre-polymerization were conducted at 130, 140, 150 °C for 1 h and 160 °C for 2 h under N2 atmosphere. Determined by GPC measurement in THF. |
| M1 |
1:0.8:0.2 |
— |
Diphenyl ether |
12 |
33921 |
1.49 |
| M2 |
1:0.8:0.2 |
100–130 |
Diphenyl ether |
24 |
17623 |
3.34 |
| M3 |
1:0.8:0.2 |
110–140 |
Diphenyl ether |
24 |
38516 |
1.28 |
| M4 |
1:0.8:0.2 |
130–160 |
Diphenyl ether |
24 |
39687 |
1.36 |
| M5 |
1:0.84:0.21 |
130–160 |
Diphenyl ether |
24 |
53937 |
1.79 |
| M6 |
1:0.8:0.2 |
— |
[C6mim]Tf2N |
24 |
4905 |
1.28 |
| M7 |
1:0.8:0.2 |
130–160 |
[C6mim]Tf2N |
24 |
11322 |
1.71 |
| M8 |
1:0.84:0.21 |
130–160 |
[C6mim]Tf2N |
24 |
13913 |
1.73 |
| M9 |
1:0.88:0.22 |
130–160 |
[C6mim]Tf2N |
24 |
8039 |
1.55 |
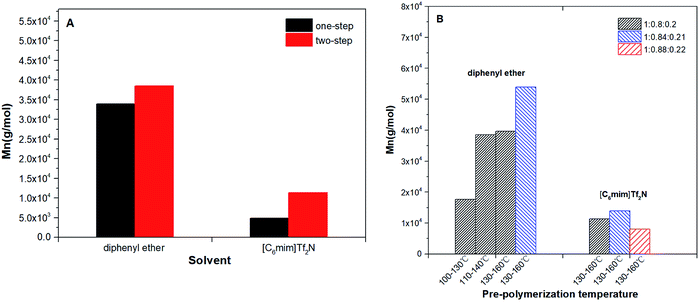 |
| | Fig. 2 P(OA–GA), synthesized by N-435 catalysis: (A) effect of solvent and polycondensation method on Mn and PDI (Mw/Mn); (B) effect of solvent and pre-condensation temperature and molar ratio on Mn and PDI (Mw/Mn). | |
Fig. 2A summarizes the influence of a condensation method on Mn of P(OA–GA). The two-step method has obvious advantages for the synthesis of high molecular weight copolyesters. The two-step method can reduce the acidity of the polycondensation system, thereby reducing the damage to the activity of the N-435. Furthermore, oligomers replaced the diacid and diol that can remove the water produced in the incipient phase of the formation of oligomers. Water affects the stability of fluorinated inorganic anions especially at elevated temperatures.29 Fig. 2B displays the influence of pre-condensation temperature and acid/alcohol ratio for the products' Mn. The Mn of products increased with pre-temperature and higher molecular weight products could be obtained when the diol/diacid ratio was slightly over unity. This because diol in excess is easier to get an oligomer with a hydroxyl end-group. This is conducive to the progress of postcondensation. The unreacted alcohol monomer can function as a chain extender in the post-polycondensation to obtain higher molecular weight products. Although it is a different reaction system, Fu et al.30 proved that the diol/diacid ratio of the oligomers is a key factor affecting the molecular weight of polyesters. The synthesis of P(OA–GA) by two-step method in diphenyl ether and [C6mim]Tf2N was repeated several times in order to evaluate the reproducibility of that procedure. It was found that the amount of trisubstituted units ranged between 15 and 30% while molecular weights in diphenyl ether up to 50000 g mol−1 and in [C6mim]Tf2N it stays around 13000 g mol−1 (Table S3†).
Post-condensation conditions of P(OA–GA) in [C6mim]Tf2N
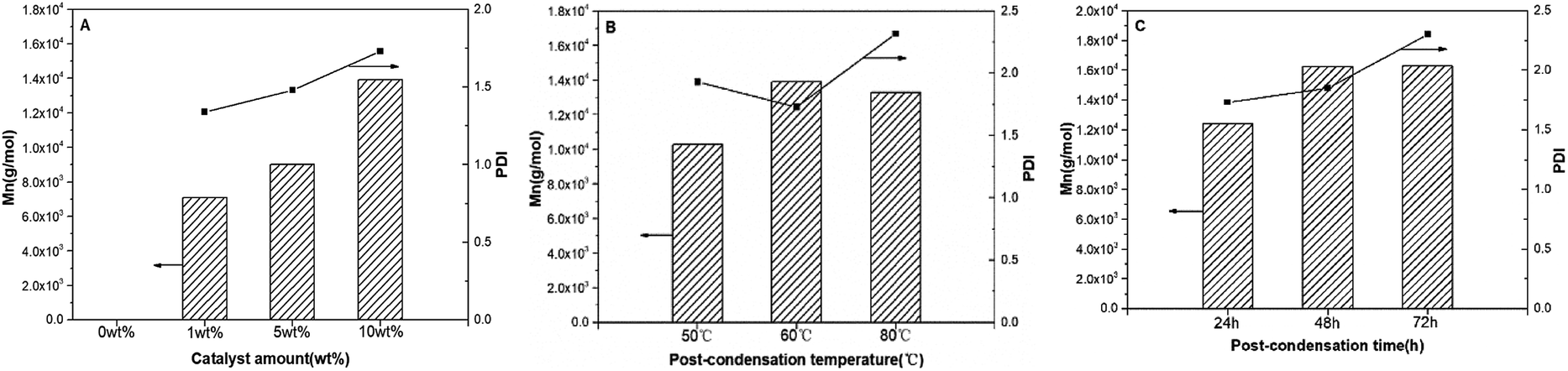
Copolyester synthesized in [C6mim]Tf2N has lower-branched and lower molecular weight, so the polycondensation of P(OA–GA) catalysed by N-435 was further explored at different postcondensation conditions (amount of catalyst, reaction temperature, and reaction time) to further increase the molecular weight of the copolyester. The results are listed in Table 3 and Fig. 3.
Table 3 Polycondensations of P(OA–GA) in different conditionsa
| Entry |
Tem. (°C) |
Time (h) |
Catalyst (wt%) |
Terb (%) |
L1,2c (%) |
L1,3d (%) |
Dene (%) |
Mnf (g mol−1) |
PDIf (Mw/Mn) |
Yield (%) |
| All products were synthesized by two-step method. Pre-condensation conditions: adipic acid:1,8-octanediol:glycerol = 1:0.84:0.21; 130, 140, 150 °C for 1 h and 160 °C for 2 h. Post-polycondensations of all entries were in vacuum 1.5–3 mmHg, N-435 catalysed. Data were analysed by 13C NMR and calculated based on. Eqn (2). Eqn (3). Eqn (4). Eqn (5). Determined by GPC measurement in THF n.d. = not determined. |
| C1 |
60 |
24 |
0 |
n.d. |
n.d. |
n.d. |
n.d. |
n.d. |
n.d. |
n.d. |
| C2 |
60 |
24 |
1 |
28.36 |
16.83 |
48.08 |
6.07 |
7087 |
1.34 |
54 |
| C3 |
60 |
24 |
5 |
25.46 |
17.13 |
46.76 |
10.65 |
9015 |
1.48 |
77 |
| C4 |
60 |
24 |
10 |
18.30 |
21.90 |
42.86 |
16.94 |
13913 |
1.73 |
56 |
| C5 |
50 |
24 |
10 |
20.82 |
19.18 |
43.83 |
16.17 |
10298 |
2.03 |
57 |
| C6 |
80 |
24 |
10 |
15.81 |
20.94 |
42.74 |
20.51 |
13303 |
2.32 |
43 |
| C7 |
60 |
48 |
10 |
19.97 |
17.49 |
42.84 |
19.70 |
16240 |
1.85 |
66 |
| C8 |
60 |
72 |
10 |
20.59 |
17.23 |
42.01 |
20.17 |
16282 |
2.30 |
73 |
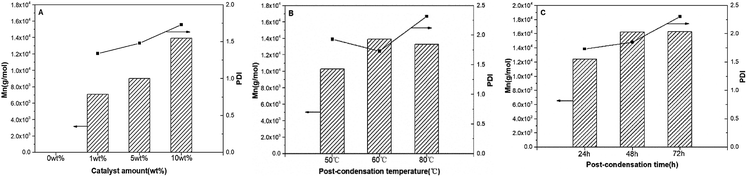 |
| | Fig. 3 P(OA–GA), synthesized by N-435 catalysis: plots of Mn and PDI (Mw/Mn) as a function of catalyst amount (A); post-condensation temperature (B); post-condensation time (C). | |
The amount of catalyst affects the structure of P(OA–GA) (entries C1–C4). L1,2 and Den increased, L1,3 and Ter decreased slightly with increasing catalyst, which suggesting that more glycerol units are incorporated into the polymer backbone with increasing catalyst dose. There was no significant glycerol signal in 1H NMR or 13C NMR without catalyst. In addition, the presence of glycerol repeat unit substitution patterns changed only slightly when the temperature increased from 50 °C to 60 °C; however, the percent of Den increased from 16.17% to 20.51% when the temperature increased to 80 °C. Concurrently, the percent of Ter decreased from 20.82% to 15.81%, and the percent of L1,2 and L1,3 showed only minor variation (<1%). This demonstrated that the degree of branching of the product increased with polycondensation temperature. This trend is consistent with the findings of Taresco et al.11 High temperatures lead to lower enzyme regioselectivity and high polymer branching. This is due to higher miscibility between the oligomer and the ionic liquid as the temperature increased. The degree of branching of the product also increased with reaction time (entries C4, C7, and C8). These results indicated that the quantity of catalyst is the determinants of the P(OA–GA) structure. Furthermore, a distinct correlation could be discovered on the Mn and PDI of P(OA–GA) with the amount of catalyst (Fig. 3A). There Mn increased from 7087 to 9015 and 13913 g mol−1 as the catalyst increased from 1 wt% to 5 wt% and 10 wt% suggesting that N-435 is also critical in the synthesis of high molecular weight P(OA–GA). The PDI of products remained under 2.0. Fig. 3B shows the variation of Mn and PDI of P(OA–GA) with increasing reaction temperature. Reactions at 50 °C, 60 °C, and 80 °C gave products with Mn values of 10298, 13913, and 13303 g mol−1, respectively. The PDI reached a maximum of 2.32 at 80 °C. Fig. 3C shows that Mn and PDI continued to increase as the reaction time became longer.
Development of the ionic liquid species
The type of ILs will affect the activity of the catalyst and the solubility of the oligomer/ILs and thus the structure and molecular weight of the copolyester. N-435 has higher activity and stability in ILs with [Tf2N]−, [PF6]−, and [BF4]− anions than ILs with anion containing stronger hydrogen bond ability such as Cl− or [OctSO4]−.31 Therefore, the design of this section is mainly to explore the effect of the types of ILs on Mn and Den of polyesters. The results are shown in Table 4. Higher Mn (Mn > 13725 g mol−1) was obtained in [Tf2N]−-based ILs (entries L1–L4) than [PF6]− (entries L5–L7) or [BF4]−-based (entries L8–L10) ILs. This is because ILs containing anions [PF6]− or [BF4]− have more nucleophilic properties than those containing [Tf2N]−, which reduces the activity of N-435.32
Table 4 Polycondensations of P(OA–GA) in different ionic liquidsa
| Entry |
Molar ratio A:O:Gb |
Ionic liquid |
Vis.c |
Terd (%) |
L1,2e (%) |
L1,3f (%) |
Deng (%) |
Mnh (g mol−1) |
PDIh |
| All products were synthesized through two-step method. Pre-condensation conditions: 130, 140, 150 °C for 1 h and 160 °C for 2 h. Post-polycondensations conditions: 60 °C, N-435 (10 wt% of monomers), 24 h, in vacuum 1.5–3 mmHg. A = adipic acid; O = 1,8-octanediol; G = glycerol. Viscosity (mPa s, 25 °C) were provide by Shanghai Chengjie Chemical Co., Ltd. Data were analyzed by 13C NMR and calculated based on. Eqn (2). Eqn (3). Eqn (4). Eqn (5). Determined by GPC measurement in THF n.d. = not determined. |
| L1 |
1:0.84:0.21 |
[C2mim]TF2N |
33 |
21.28 |
19.15 |
42.55 |
17.02 |
13885 |
2.35 |
| L2 |
1:0.84:0.21 |
[C4mim]TF2N |
52 |
20.23 |
19.84 |
41.25 |
18.68 |
17509 |
1.56 |
| L3 |
1:0.84:0.21 |
[C6mim]TF2N |
87.3 |
18.30 |
21.90 |
42.86 |
16.94 |
13913 |
1.73 |
| L4 |
1:0.84:0.21 |
[C10mim]TF2N |
142 |
21.04 |
20.25 |
42.67 |
16.04 |
13725 |
2.02 |
| L5 |
1:0.84:0.21 |
[C4mim]PF6 |
204 |
21.25 |
18.33 |
41.67 |
18.75 |
3280 |
1.04 |
| L6 |
1:0.84:0.21 |
[C6mim]PF6 |
585 |
19.05 |
19.44 |
42.86 |
18.65 |
11461 |
2.30 |
| L7 |
1:0.84:0.21 |
[C8mim]PF6 |
682 |
26.85 |
18.52 |
46.30 |
8.33 |
5640 |
1.32 |
| L8 |
1:0.84:0.21 |
[C2mim]BF4 |
38 |
n.d. |
n.d. |
n.d. |
n.d. |
10945 |
2.06 |
| L9 |
1:0.84:0.21 |
[C6mim]BF4 |
220 |
n.d. |
n.d. |
n.d. |
n.d. |
12310 |
1.69 |
| L10 |
1:0.84:0.21 |
[C10mim]BF4 |
930 |
n.d. |
n.d. |
n.d. |
n.d. |
11223 |
2.15 |
| L11 |
1:0.53:0.53 |
[C6mim]TF2N |
87.3 |
17.21 |
17.49 |
39.86 |
25.44 |
10795 |
1.32 |
| L12 |
1:0.53:0.53 |
[C4mim]PF6 |
204 |
16.87 |
19.28 |
40.16 |
23.69 |
10605 |
1.24 |
| L13 |
1:0.53:0.53 |
[C6mim]PF6 |
585 |
14.8 |
18.4 |
40.00 |
26.80 |
15158 |
1.37 |
| L14 |
1:0.53:0.53 |
[C2mim]BF4 |
38 |
15.98 |
18.26 |
45.66 |
20.09 |
12599 |
1.73 |
| L15 |
1:0.53:0.53 |
[C6mim]BF4 |
220 |
31.02 |
16.67 |
46.30 |
6.02 |
5371 |
1.27 |
| L16 |
1:0.53:0.53 |
[C10mim]BF4 |
930 |
33.93 |
16.07 |
44.64 |
5.36 |
4349 |
1.15 |
The Mn of copolymer increased at first and then decreased with viscosity of the ILs, which suggests that an increase in the viscosity of the ILs will limit further diffusion of the polymer chain. However, the viscosity of the ILs has little effect on the molecular structure of the copolymer. The proportion of Ter, L1,2, L1,3 and Den changes within 3%. Oligomers were stratified with BF4−-based ILs in post-polycondensation (entries L8–L10). The reaction is a heterogeneous reaction. Poor miscibility resulted in a lower glycerol incorporation rate, and no significant glycerol peak was observed in the 13C NMR.
The addition of glycerol was increased (entries L11–L16) to further understand the influence of the ILs on the synthesis efficiency and structure of P(OA–GA). The results indicated that oligomer can be miscible with [BF4]−-based ILs (entries L14–L16), and a distinct glycerol signal could be observed in 13C NMR. This is attributed to increases in the glycerin content causing an increase of the oligomer polarity. This facilitates good miscibility between oligomer and ILs with higher polarity such as BF4−. Nevertheless, the percent Ter in the polymer obtained in the BF4−-based (entries L15 and L16) ILs is still higher than that of PF6−-based (entries L12 and L13) and Tf2N−-based (entry L11) except [C2mim]BF4 (entry L14). Meanwhile, the degree of branching was reduced and terminated with glycerol, this led to lower molecular weights. [C2mim]BF4 (entry L14) differs from this situation with high Mn because of the low-viscosity, which indicated that an ideal viscosity of the reaction system will lead to a higher molecular weight.
Characterization
Hydrophilic test
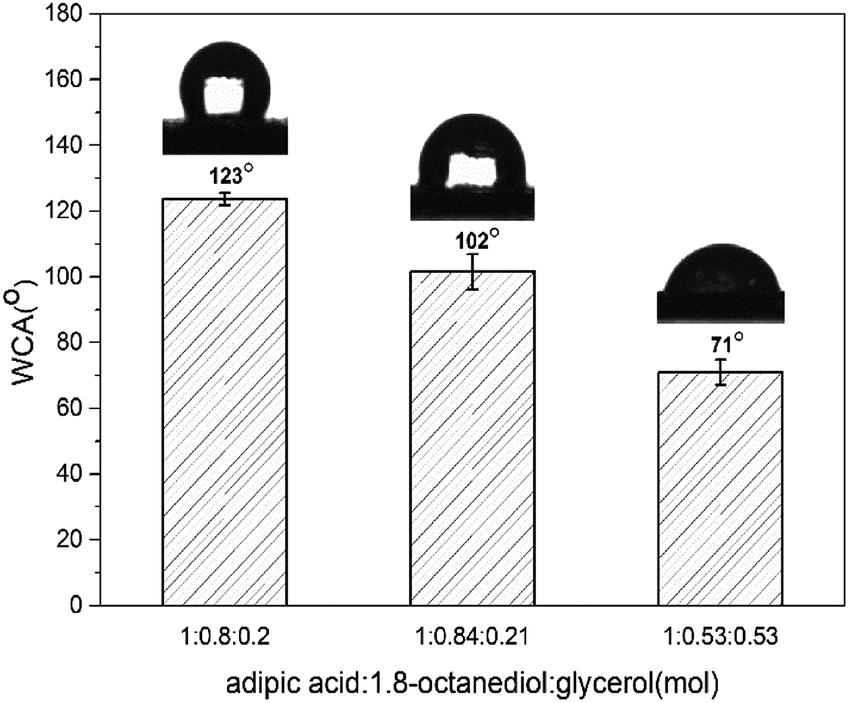
The wettability properties were measured on polymer-coated glass by contact angle (θ) to understand the hydrophilicity/hydrophobicity characteristics of copolymer (Fig. 4). Contact angle tests indicated that the value decreased from 120° to 70° as the amount of glycerol added. P(OA–GA) prepared at feed ratio (adipic acid:1,8-octanediol:glycerol = 1:0.8:0.2) had contact angles between 123.70° ± 1.86°. This value kept falling with the increase of the glycerol addition. The contact angles of the copolymers were θ = 101.54 ± 5.29 (adipic acid:1,8-octanediol:glycerol = 1:0.84:0.21) and 71.05 ± 3.85 (adipic acid:1,8-octanediol:glycerol = 1:0.53:0.53). These observations were due to the increased glycerol content and pendant hydroxyl groups of P(OA–GA). By increasing the glycerin content, more pendant hydroxyl groups are available. More hydroxyl groups pendent on the copolymer chain enhances the hydrophilicity of the material. The hydroxyl content of the products was quantitatively calculated by 31P NMR (Fig. S2†) as previously reported by Gustini et al.33 The data are summarized in Table S1.† Here, the 2-chloro-4,4,5,5-tetramethyl-dioxaphospholane compound reacts with the primary and secondary hydroxyl groups present on the polymer backbone. Cyclohexanol is an internal standard that quantifies the number of hydroxyl groups. Data afforded by these experiments are expressed in mgKOH gsample−1 (Table S1†). This confirmed that the hydrophilicity increased with hydroxyl content.
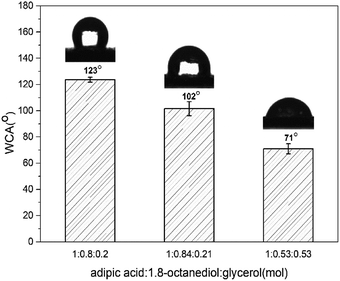 |
| | Fig. 4 Contact angle of P(OA-GA) synthesized in different feed ratio. | |
Thermal performance test
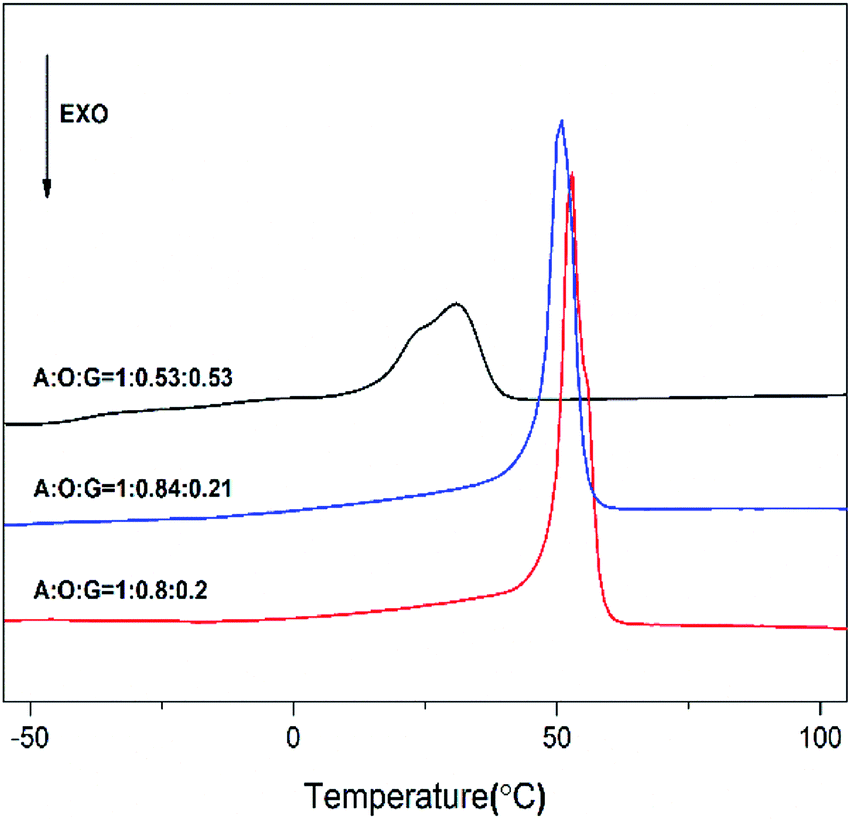
Fig. 5 displays the first scan of DSC curves of these products P(OA–GA). From Fig. 5, the melting endotherms peaks were observed at 30–53 °C. Obviously, the melting temperature decreases with increasing glycerol content. This is because the crystallinity of P(OA–GA) was disrupted by glycerol units along the polyester chain. In addition, thermodynamic measurements showed that we can adjust the melting temperature of the copolymer by fine-tuning the glycerol content.
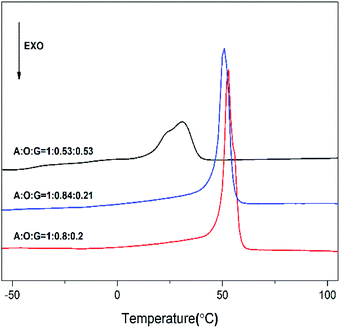 |
| | Fig. 5 DSC curves (first scan) of P(OA–GA) (content in material ratio on curves). | |
In vitro degradability test
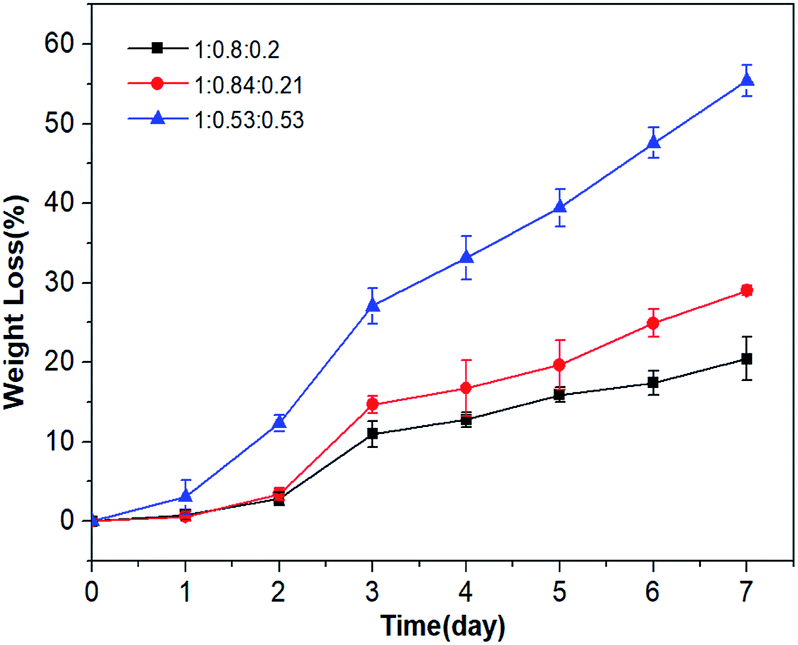
P(OA–GA) was immersed in phosphate buffer (37 °C) for a predetermined time, and the degradation was studied via weight loss. Fig. 6 shows that no samples had significant weight loss on the first day. Two days later, P(OA–GA) (adipate:1,8-octanoic acid:glycerol = 1:0.53:0.53) had a higher degradation than the other glycerin-doped samples. After 7 days, P(OA–GA) (adipic acid:1,8-octanediol:glycerol = 1:0.8:0.2) lost the least amount of weight 20.4 ± 2.7%. The weight loss of P(OA–GA) (adipic acid:1,8-octanediol:glycerol = 1:0.84:0.21) is 29 ± 0.6%. As expected, P(OA–GA) (adipic acid:1,8-octanediol:glycerol = 1:0.53:0.53) have the highest degradation rate, and the weight loss is 55.4 ± 2%. Which proved that the composition and hydrophobicity of the polymer can affect its degradation rate under the same environmental conditions34,35 and the amount of added glycerol can largely adjust the degradation rate of P(OA–GA).
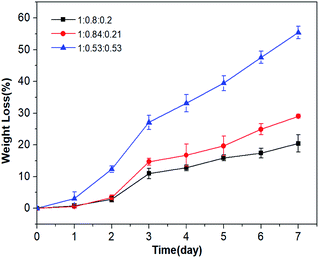 |
| | Fig. 6 P(OA–GA) co-polyester weight loss as a function of soaking time. | |
Conclusions
In this work, we described the polycondensation of adipic acid, 1,8-OD, and glycerol catalysed by N-435 to produce a hydroxyl functionalized polyester with high molecular weight and lower degree of branching via a benign and eco-friendly technique. Diphenyl ether was found to be the preferred solvent to prepare P(OA–GA) co-polyester with a high Mn (Mn = 53937 g mol−1 in 24 h). Although ILs gave functional polyesters with relatively low molecular weight, this is a more environmentally-friendly route. The molecular weight of the polyesters depends on the activity of the catalyst in the ILs and the miscibility of aliphatic polyester/ionic liquid. The N-435 remained active in Tf2N− and PF6−-based ionic liquids. This was substantially suppressed in BF4− systems. The miscibility of aliphatic polyester/ionic was determined by their viscosity and polarity. The contact angle, DSC, and in vitro biodegradation data confirmed that the character of products can be adjusted via the extent of glycerin addition. This will further expand the range of applications for hydroxyl functional polyesters. Examples include biomedical applications such as drug delivery applications and gene therapy as well as improved inks, paints, and adhesive materials. Moreover, ILs can extend the possibility of using polyol monomers to synthesize a higher molecular weight functional polymer. Our lab is extending this approach to other complex polyols, carbohydrate derivatives and polymer classes. We expect to generate polyesters of structurally unprecedented types.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 21661027, No. 21968028), and Program for Young Innovative Talents of Shihezi University (No. CXRC201704, No. CXPY201903).
References
- S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079–9084 CrossRef CAS PubMed
 .
. - A. Celli, P. Marchese, S. Sullalti, C. Berti, G. Barbiroli, S. Commereuc and V. Verney, Green Chem., 2012, 14, 182–187 RSC .
- H. Ghamgui, N. Miled, A. Rebaï, M. Karra-chaâbouni and Y. Gargouri, Enzyme Microb. Technol., 2006, 39, 717–723 CrossRef CAS .
- Y. Shibata and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5747–5759 CrossRef CAS .
- J. Liu, F.-F. Zeng, Z.-M. Liu, C.-X. Zhang, W.-h. Ling and Y.-M. Chen, Lipids Health Dis., 2013, 12, 159 CrossRef .
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, Worth Publishers, 6th edn, 2013 Search PubMed .
- M. Agach, S. Delbaere, S. Marinkovic, B. Estrine and V. Nardello-Rataj, Colloids Surf., A, 2013, 419, 263–273 CrossRef CAS .
- Y. Yang, W. Lu, J. Cai, Y. Hou, S. Ouyang, W. Xie and R. A. Gross, Macromolecules, 2011, 44, 1977–1985 CrossRef CAS .
- L. Gustini, C. Lavilla, W. W. Janssen, A. Martinez de Ilarduya, S. Munoz-Guerra and C. E. Koning, ChemSusChem, 2016, 9, 2250–2260 CrossRef CAS PubMed .
- I. K. Varma, A.-C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949–981 CrossRef CAS .
- V. Taresco, R. Creasey, J. Kennon, G. Mantovani, C. Alexander, J. C. Burley and M. C. Garnett, Polymer, 2016, 89, 41–49 CrossRef CAS .
- A. Mahapatro, B. Kalra, A. Kumar and R. A. Gross, Biomacromolecules, 2003, 4, 544–551 CrossRef CAS PubMed .
- S. Sun and X. Chen, Biochem. Eng. J., 2015, 97, 25–31 CrossRef CAS .
- A. A. Elgharbawy, F. A. Riyadi, M. Z. Alam and M. Moniruzzaman, J. Mol. Liq., 2018, 251, 150–166 CrossRef CAS .
- E. Lozinskaya, A. Shaplov and Y. S. Vygodskii, Eur. Polym. J., 2004, 40, 2065–2075 CrossRef CAS .
- C. R. Langrick, Macromol. Rapid Commun., 2002, 23, 676–680 CrossRef .
- E. I. Lozinskaya, A. S. Shaplov, M. V. Kotseruba, L. I. Komarova, K. A. Lyssenko, M. Y. Antipin, D. G. Golovanov and Y. S. Vygodskii, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 380–394 CrossRef CAS .
- H. Uyama, T. Takamoto and S. Kobayashi, Polym. J., 2002, 34, 94 CrossRef CAS .
- J. L. Kaar, A. M. Jesionowski, J. A. Berberich, R. Moulton and A. J. Russell, J. Am. Chem. Soc., 2003, 125, 4125–4131 CrossRef CAS PubMed .
- S. J. Nara, J. R. Harjani, M. M. Salunkhe, A. T. Mane and P. P. Wadgaonkar, Tetrahedron Lett., 2003, 44, 1371–1373 CrossRef CAS .
- R. Marcilla, M. De Geus, D. Mecerreyes, C. J. Duxbury, C. E. Koning and A. Heise, Eur. Polym. J., 2006, 42, 1215–1221 CrossRef CAS .
- S. Dali, H. Lefebvre, R. E. Gharbi and A. Fradet, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3025–3035 CrossRef CAS .
- S. Lv, X. Zou, H. Qian, J. Qin, Q. Jin and X. Wang, RSC Adv., 2016, 6, 108697–108707 RSC .
- S. Liu, K. Huang, H. Yu and F. Wu, J. Appl. Polym. Sci., 2018, 135, 46516 CrossRef .
- E. Karlsson, J. H. Shin, V. Saez-Jimenez, V. Mapelli and L. Olsson, Biotechnol. Adv., 2018, 36(8), 2248–2263 CrossRef PubMed .
- V. T. Wyatt and G. D. Strahan, Polymers, 2012, 4, 396–407 CrossRef .
- A. S. Kulshrestha, W. Gao and R. A. Gross, Macromolecules, 2005, 38, 3193–3204 CrossRef CAS .
- A. Kumar, A. S. Kulshrestha, W. Gao and R. A. Gross, Macromolecules, 2003, 36, 8219–8221 CrossRef CAS .
- J. Davis Jr, C. Gordon, C. Hilgers and P. Wasserscheid, Synthesis and Purification of Ionic Liquids, Wiley-VCH Verlag GmbH & Co, New York, 2003 Search PubMed .
- C. Fu and Z. Liu, Polymer, 2008, 49, 461–466 CrossRef CAS .
- B.-M. Lue, Z. Guo and X. Xu, Process Biochem., 2010, 45, 1375–1382 CrossRef CAS .
- Q. Pan, L. Yang and X. Meng, J. Am. Oil Chem. Soc., 2013, 90, 501–509 CrossRef CAS .
- L. Gustini, C. Lavilla, W. W. Janssen, A. Martinez de Ilarduya, S. Muñoz-Guerra and C. E. Koning, ChemSusChem, 2016, 9, 2250–2260 CrossRef CAS PubMed .
- J. Yang, A. R. Webb and G. A. Ameer, Adv. Mater., 2004, 16, 511–516 CrossRef CAS .
- Y.-Y. Linko, Z.-L. Wang and J. Seppälä, Enzyme Microb. Technol., 1995, 17, 506–511 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary P(OA–GA) 1H NMR, 31P NMR, hydroxyl content, the comparisons of different polymerization methods and the repeatability of synthetic methods. See DOI: 10.1039/d0ra00120a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab