
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUse of flow field-flow fractionation and single particle inductively coupled plasma mass spectrometry for size determination of selenium nanoparticles in a mixture†
Luluil Maknuna,
Jitapa Sumranjitb and
Atitaya Siripinyanond *a
*a
aDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Mahidol University, Rama VI Road, Bangkok 10400, Thailand. E-mail: atitaya.sir@mahidol.ac.th; Fax: +66-2-354-7151; Tel: +66-2-201-5195
bNational Nanotechnology Center, National Science and Technology Development Agency, 111 Phahonyothin Rd., Klongluang, Pathumthani 12120, Thailand
First published on 11th February 2020
Abstract
Various analytical techniques have been used for size analysis of selenium nanoparticles (SeNPs). These include flow field-flow fractionation (FlFFF), single particle inductively coupled plasma mass spectrometry (SP-ICP-MS), dynamic light scattering (DLS) and transmission electron microscopy (TEM). For hydrodynamic diameter estimation, the FlFFF technique was used and the results were compared with those analyzed by DLS. For core diameter estimation, the results obtained from SP-ICP-MS were compared with those from TEM. Two types of FlFFF channel were employed, i.e., symmetrical FlFFF (Sy-FlFFF) and asymmetrical FlFFF (Asy-FlFFF). Considering the use of FlFFF, optimization was performed on a Sy-FlFFF channel to select the most appropriate carrier liquid and membrane in order to minimize problems due to particle membrane interaction. The use of FL-70 and 10 kDa RC provided an acceptable compromise peak quality and size accuracy for all samples of SeNPs which were coated by proteins (positively charged SeNPs) and sodium dodecyl sulfate (negatively charged SeNPs). FlFFF always yielded the lower estimate of the hydrodynamic size than DLS as a reference method. The results obtained by SP-ICP-MS were consistent with the TEM method for the core diameter estimation. The results from FlFFF and the DLS reference method were significantly different as confirmed by paired t-test analysis, while the results provided by SP-ICP-MS and the TEM reference method were not significantly different. Furthermore, consecutive size analysis by SP-ICP-MS for the fractions collected from FlFFF was proposed for sizing of SeNP mixtures. The combined technique helps to improve the size analysis in the complex samples and shows more advantages than using only SP-ICP-MS.
Introduction
Nanoparticles have been used in wide applications owing to their unique chemical and physical properties such as large surface area and nanoscale size.1 Selenium nanoparticles (SeNPs) have gained attention due to their functions of having anticancer,2 and antimicrobial properties,3 as dietary supplements,4 and in food packaging.5 The synthesis and characterization of various functional inorganic nanoparticles by different techniques to provide chemical composition, morphology, and size have been reported in the literature.6–10 According to the European Commission, not only the size of particles but also the size distribution should be considered when dealing with the hazard or risk of nanomaterials to human health and environment.11 Therefore, various instruments have been used to provide the particle size and size distribution of NPs such as dynamic light scattering (DLS),12 transmission electron microscopy (TEM),13,14 and field-flow fractionation (FFF) coupled with inductively coupled plasma mass spectrometry (ICP-MS).15,16Each characterization technique provides a different meaning of “diameter”. For example, DLS is used to determine the average intensity of brownian nanoparticles in colloidal suspension and recorded as “hydrodynamic diameter (dh)”.17 This technique is generally used for engineered nanoparticles characterization including synthesized SeNPs characterization to control the growth of NPs from agglomeration.18 Transmission electron microscopy (TEM) is known as a powerful imaging method of NPs presented in the sample. By using computer imaging analysis software, the particles can be counted for size characterization. The core diameter information is obtained. However, the correctness of size analysis by TEM depends on the contrast of the TEM image.19 The area-equivalent size obtained by TEM is also limited by the number of measured particles. By using this technique, the aggregation of particles contributes to the erroneous size analysis.16 Therefore, an alternative technique for size based separation and quantification such as FlFFF or single particle ICP-MS is required.
Flow field-flow fractionation (FlFFF), a separation-based method, has been reported for hydrodynamic size determination of nanoparticles or colloidal particles, both as synthesized and as in samples containing complex matrices.4,20,21 Two different types of FlFFF are available including symmetrical flow field-flow fractionation (Sy-FlFFF) and asymmetrical flow field-flow fractionation (Asy-FlFFF). FlFFF deserves special attentions for nanoparticles characterization due to their high separation performance.22,23 In order to provide robust protocol and high reliability for inter-laboratory comparison of NPs size measurement, size characterization of SeNPs was tested by using two different techniques of FlFFF including Sy-FlFFF and Asy-FlFFF.
With FlFFF, the retention time is used to calculate the particle size (hydrodynamic diameter, “dh”) of nanoparticles according to the Stoke's law theory. Nonetheless, the retention time of nanoparticles might be shifted due to the occurrence of particle membrane-interactions which is being a challenge in FlFFF, resulting in invalid size calculation of nanoparticles. This phenomenon has been investigated depending on some factors such as type of membrane material, carrier liquid, stabilizing agent of particles and operating condition, for example cross flow rate.24–30 Saenmuangchin and Siripinyanond26 estimated the hydrodynamic diameter of AuNPs with different types of surface coating agent by using Sy-FlFFF. The effect of various types of surface coating agents: i.e., electrostatically stabilizing agent, e.g., tannic acid (TA) and citrate (CT); and sterically stabilizing agents, e.g., polyethylene glycol (PEG), polyvinylpyrrolidone (PVP), and branched polyethylene imine (BPEI) in different membrane materials and carrier liquids to the retention behavior of AuNPs was observed because of the strong attractive interactions between particles and membrane as discussed by Meisterjahn et al.24 The polarity and hydrophobicity of coating agents have a critical influence on the variability of retention time and relative recovery.31 Hence, two types of coating agents (protein and SDS), which resulted in SeNPs of different charges, were used to examine the performance of FlFFF as to expand the function of FlFFF in wide range of nanoparticles analysis.
Single particle inductively coupled plasma mass spectrometry (SP-ICP-MS) is known as an emerging technique for characterization, quantification and identification of nanoparticles.32,33 SP-ICP-MS offers the information on the number of particles detected per time and their mass concentration.34 The diameter information from this technique is recorded as “dmass”. This technique has been applied for SeNPs at low concentrations in commercial Se-rich yeast.35 For complex samples, particle size analysis is still a challenge due to the presence of unaccounted forms of metal. Therefore, SP-ICP-MS has been used in combination with another analytical approach such as Asy-FlFFF as reported by Loeschner et al.36 for analysis of AgNPs in chicken meat. As the analysis of the particle size in the complex sample such as synthesized NPs is still a challenge, an alternative technique is required.
Overall aims of this work are focused on the application of Sy-FlFFF under optimum condition to determine the hydrodynamic diameter of various types of SeNPs compared with Asy-FlFFF and SP-ICP-MS to determine the core diameter of SeNPs; and SP-ICP-MS analysis after size fractionation by Sy-FlFFF for core diameter determination of SeNPs in the mixtures. TEM and DLS are selected as reference methods because both methods are known as routine technique for nanoparticle size analysis. Use of combined techniques of Sy-FlFFF and SP-ICP-MS was demonstrated for size analysis of synthesized SeNPs size mixtures as a model of complex sample.
Experimental
Reagents and materials
The deionized water used in this study was obtained from a water purification system (18.2 MΩ cm−1) (EASY pure®II model D7031, Barnstead Thermolyne Corporation, Dubuque, Iowa, USA). Two protocols were used for SeNPs synthesis. In the first protocol, the following chemicals were used: 0.01 mol L−1 of sodium dodecyl sulfate (SDS) (Fluka Steinheim, Germany) as stabilizing agent; 520 mmol L−1 solution of sodium thiosulfate (Sigma-Aldrich, St.Louis, Missouri, USA) as reducing agent; and 5.2 mmol L−1 of selenium(IV) dioxide (Alfa Aesar, Massachusetts, USA) as precursor. In the second protocol, the reagents for preparation of SeNPs (protein as stabilizing agent) was 30 mmol L−1 of sodium selenite as precursor, 2.5% (w/w) of β-lactoglobulin from bovine milk and 2.5% (w/w) albumin from bovine serum as stabilizing agents. All chemicals used in this second protocol were purchased from Sigma-Aldrich, St.Louis, Missouri, USA. 300 mmol L−1 of L-ascorbic acid (Fisher Scientific UK Limited, Leicestershire, UK) was used as reducing agent. FL-70 was purchased from Fisher Scientific UK Limited, Leicestershire, UK (composition: 1.4% tetrasodium ethylenediaminetetraacetate, 0.5% sodium oleate, 0.1% sodium bicarbonate, 2.7% sodium carbonate, 3.8% triethanolamine oleate 3.8%, 88.8% water and others). Sodium azide (NaN3) was purchased from Merck, Darmstadt, Germany. Commercial gold nanoparticle standards (AuNPs) 20 nm, 60 nm, and 80 nm (Nanocomposix, Inc., San Diego, California, USA) were used to check the sensitivity of SP-ICP-MS and test the performance of Sy-FlFFF system. Commercial polystyrene (PS) nanoparticle standards 20 nm, 40 nm, 60 nm and 100 nm (Sigma-Aldrich, St.Louis, Missouri, USA) were used for checking the performance of Asy-FlFFF system. 0.1% HNO3 (Merck, Darmstadt, Germany) was used for preparation of selenium standard solution in solution mode ICP-MS measurement. The pure 99% methane gas (S.I.Technology Co.Ltd., Bangkok, Thailand) was used as a reaction gas.SeNPs synthesis
SeNPs were prepared by using chemical reduction method with two different protocols, as shown in Fig. 1. For the first protocol which is SDS stabilized, sodium thiosulfate was used as the reducing agent. Both reducing agent (520 mmol L−1 sodium thiosulfate) and precursor solution (5.2 mmol L−1 selenium dioxide) were prepared in the 0.01 mol L−1 SDS. SDS was used not only as stabilizing agent but also as diluent. Various amounts of sodium thiosulfate were added in selenium(IV) dioxide, as summarized in Fig. 1. At room temperature, the selenium solutions converted from colorless to various colors. Stirring was carried out up to 5 hours. Then, the resulting SeNPs was stored at room temperature. All detailed reactions are mentioned elsewhere.14 | ||
| Fig. 1 Schematic diagram showing the synthesis protocol of SeNPs stabilized by (a) SDS and (b) proteins. | ||
SeNPs by protein stabilized (second protocol) was prepared by adding ascorbic acid into protein solution, as illustrated in Fig. 1. The solutions were allowed to react for 15 minutes to ensure the high yield of selenium nanoparticles. To prevent SeNPs from aggregation, SeNPs were stored at 4 °C.
Instrumentation
Another FlFFF called Asy-FlFFF (AF2000, Postnova Analytics, Salt Lake City, USA) with the dimension of 33.5 cm long × 4.0 cm wide × 250 μm thick was employed. A trapezoidal shape of 350 μm spacer with 10 kDa RC membrane (Postnova Analytics) was used. The Asy-FlFFF system used in this study was coupled with UV detector SPD-20A (Postnova Analytics). The detection wavelength at 410 nm was used for small particles (sample A–E; sample H and sample I) and 550 nm for large particles (sample F and sample G). Carrier liquid (0.02% FL-70 + 0.02% NaN3) was delivered by two HPLC pumps (Postnova Analytics). To determine the channel thickness, 100 nm PS standard particle was introduced into the FlFFF. Then, the hydrodynamic diameters of synthesized SeNPs were determined. PS standards of 20 nm, 40 nm, and 60 nm were injected into the Asy-FlFFF for checking the sensitivity of system. The details of running condition are shown in Table S2.†
| A = πr2 | (1) |
| d = 2 × r | (2) |
| f = CSTD × ηSTD × Qsample | (3) |
| W = CSTD × ηSTD × Qsample | (4) |
Pulse intensity was converted into the mass flux, which is further converted into diameter. The calibration curve was constructed by using 1, 3, and 5 μg L−1 of ionic selenium standard solution. All individual samples were diluted at: 500×; 50×; and followed by 25× in deionized water before introduction into the SP-ICP-MS. The dilution processes of the samples were supposedly optimized to avoid coincidence events. The Syngistix™ nano application software was used for data evaluation. The instrumental operating conditions are listed in Table S3.†
Data evaluation of hydrodynamic diameter and core diameter from FlFFF and SP-ICP-MS
In Sy-FlFFF and Asy-FlFFF system, peak area of each sample was analyzed by using OriginPro 8.6 software (OriginLab Corporation, MA, USA). The retention time (tr) of peak elution for all samples was obtained to determine the hydrodynamic diameter (dh), then was calculated using eqn (5) below:
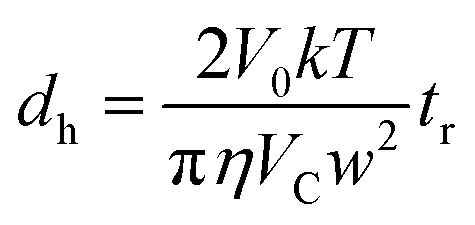 | (5) |
Statistical analysis (paired t-test method) for comparison of the results by each technique
In order to compare results obtained by two different techniques, paired t-test analysis was used for checking if there is any significant difference between two techniques. The t calculation (tcal) was compared with the t critical or theory (from t-table). The tcal was calculated from the data of experiment as shown in eqn (6):
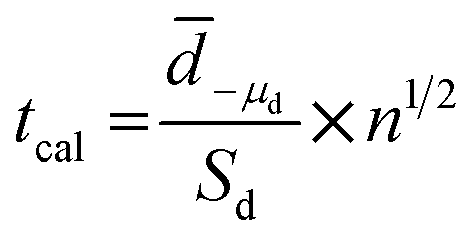 | (6) |
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) is the sample mean difference, Sd is the standard deviation of differences, μd is the true mean difference, and n is number of paired sample data of the test. In our experiment, nine samples were tested resulting in 9 paired data (n). If the tcal is smaller than tcrit, there is no significant difference between the two methods.40
is the sample mean difference, Sd is the standard deviation of differences, μd is the true mean difference, and n is number of paired sample data of the test. In our experiment, nine samples were tested resulting in 9 paired data (n). If the tcal is smaller than tcrit, there is no significant difference between the two methods.40
Result and discussion
DLS for hydrodynamic diameter determination of SeNPs
SeNPs with different types of stabilizing agents were successfully synthesized (Fig. 2a). These include sodium dodecyl sulfate (SDS) stabilized SeNPs (sample A–G) (Fig. 2a from A to G) and proteins stabilized SeNPs including β-lactoglobulin (Fig. 2a, H) and albumin (Fig. 2a, I). These coating agents were used to prevent SeNPs from aggregation.41 SeNPs were prepared by ascorbic acid (C6H8O6) reduction of selenite (SeO32−) to selenium with zero oxidation state.4| SeO32− + 2C6H8O6 + 2H+ → Se + 2C6H6O6 + 3H2O |
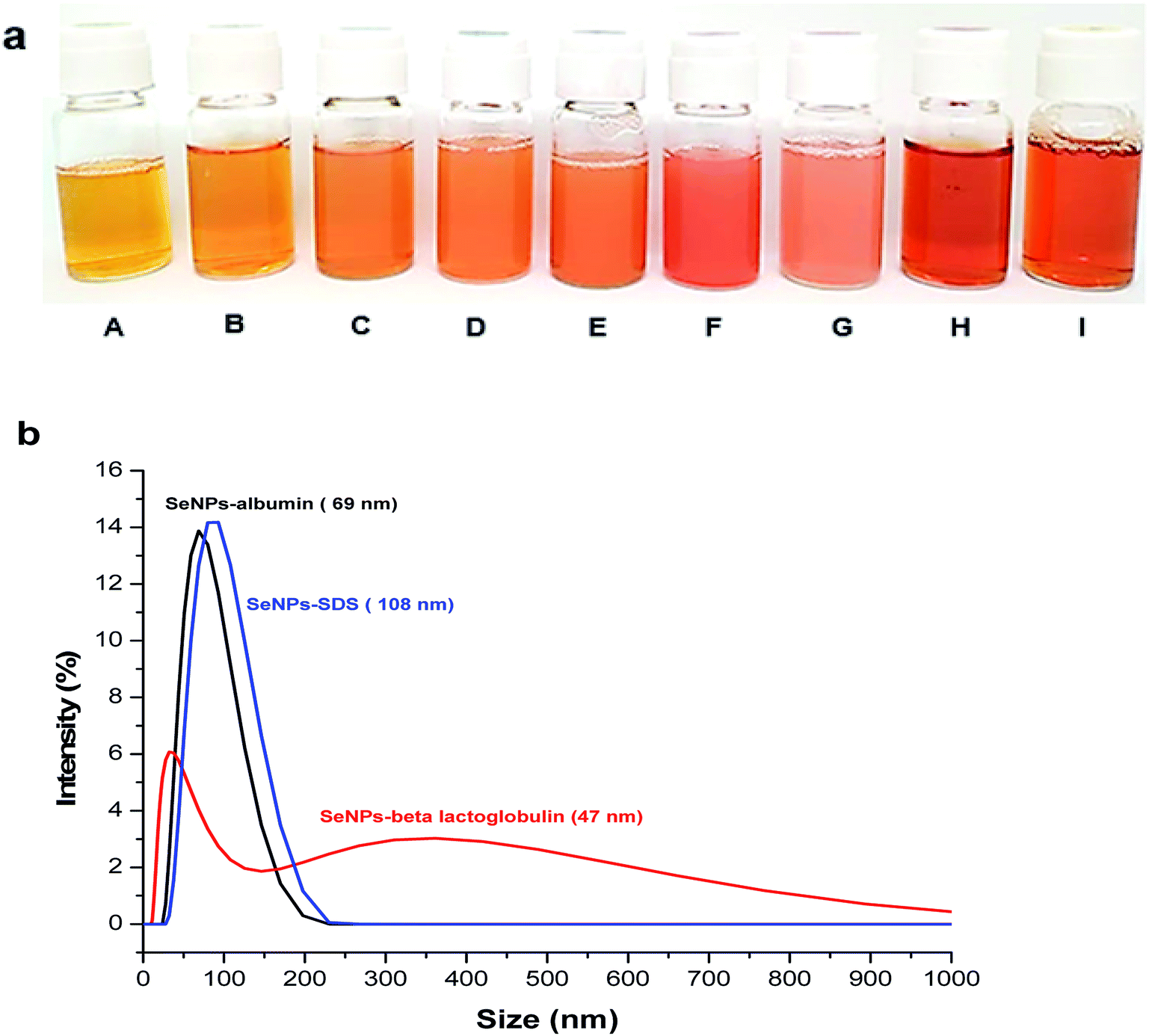 | ||
| Fig. 2 (a) Image of SeNPs synthesized by using SDS as stabilizing agent with various amounts of sodium thiosulfate (sample A–G) and protein as stabilizing agents (sample H and I) (b) size distribution observed from DLS for SeNPs stabilized by SDS (sample D); SeNPs stabilized by β-lactoglobulin; and SeNPs stabilized by albumin (n = 3). | ||
For the synthesis of SeNPs coated by SDS, various amounts of sodium thiosulfate as reducing agent were used. The formation of SeNPs occured when sodium thiosulfate as reducing agents was added into the solution containing selenium dioxide as precursor and SDS as stabilizing agents. According to Lin et al. (2002),14 selenite in the second step was rapidly reduced to selenium with zero valent in the presence of sodium thiosulfate.
| SeO2 (s) + H2O (l) → H2SeO3 (aq) (1st step) |
| H2SeO3 + 4H+ + 4e− → Se + 3H2O (2nd step) |
| Se2O32− + 5H2O → 2SO42− + 10H+ + 8e− (3rd step) |
As the amount of sodium thiosulfate decreased, the orange color changed from light to dark (from A to F) except SeNPs with the lowest amount of thiosulfate (G). The lowest amount of thiosulfate resulted in the largest particle size and the highest possibility to undergo aggregation as it happened in the SeNPs (G). According to Lin et al. (2002),14 at higher concentrations of thiosulfate reducing agent the nucleation process was faster than the growth of the particle, resulting in smaller particles. Considering the absorption spectra of SeNPs in Fig. S4,† decreasing the amounts of thiosulfate resulted in the red-shift of SeNPs absorption, indicating the larger particle size.
Different types of stabilizing agents provided different zeta potential values. The zeta potential values of SeNPs coated by proteins are positive (23.3 ± 0.5 mV) for SeNPs stabilized by β-lactoglobulin (sample H) and (35.8 ± 1.5 mV) for SeNPs stabilized by albumin (sample I) and that of SDS is negative (see Table 1) which is due to the different functional groups of each stabilizer. The zeta potential values of the sample should be known before FlFFF experiment to ensure a good selection of carrier liquid and membrane materials.38
| SeNPs synthesized | Hydrodynamic diameter | Core diameter | Zeta potential (mV) | |||
|---|---|---|---|---|---|---|
| Sy-FlFFF | Asy-FlFFF | DLS | TEM | SP-ICP-MS | ||
| a Note: N is the total numbers of particles detected. | ||||||
| SeNPs coated by SDS with various amounts of thiosulfate | ||||||
| Sample A | 33.6 ± 0.5 | 28.1 ± 0.3 | 36.1 ± 1.0 | 26.9 ± 3.5 (N = 172) | 27.6 ± 1.8 | −67.8 ± 1.5 |
| Sample B | 52.3 ± 1.2 | 61.7 ± 1.1 | 58.7 ± 0.2 | 32.0 ± 3.1 (N = 212) | 35.1 ± 2.2 | −60.2 ± 2.7 |
| Sample C | 82.7 ± 1.8 | 78.3 ± 0.9 | 88.3 ± 0.2 | 70.1 ± 3.2 (N = 157) | 67.9 ± 1.4 | −31.5 ± 1.6 |
| Sample D | 100.9 ± 1.8 | 107.5 ± 0.6 | 108.9 ± 0.8 | 93.0 ± 1.6 (N = 78) | 90.3 ± 0.2 | −31.2 ± 1.1 |
| Sample E | 127.7 ± 1.2 | 123.7 ± 0.3 | 134.9 ± 0.9 | 107.9 ± 2.6 (N = 51) | 110.9 ± 3.3 | −32.5 ± 1.1 |
| Sample F | 164.6 ± 2.4 | 156.1 ± 0.2 | 175.7 ± 0.7 | 149.4 ± 3.1 (N = 22) | 143.9 ± 1.9 | −37.2 ± 3.6 |
| Sample G | 188.4 ± 1.5 | 189.1 ± 0.4 | 210.6 ± 1.9 | 180.9 ± 2.4 (N = 40) | 180.8 ± 3.2 | −37.5 ± 2.4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| SeNPs coated by proteins | ||||||
| Sample H | 44.8 ± 1.4 | 50.2 ± 0.6 | 47.3 ± 1.0 | 31.8 ± 2.5 (N = 101) | 33.0 ± 1.4 | 23.3 ± 0.5 |
| Sample I | 75.2 ± 2.3 | 56.6 ± 0.9 | 68.7 ± 0.4 | 63.6 ± 4.6 (N = 105) | 61.8 ± 2.2 | 35.8 ± 1.5 |
The information of dh is also helpful prior to FlFFF experiment for monitoring of the behavior of NPs.38 Hence, DLS was used for hydrodynamic diameter determination of SeNPs. Table 1 summarizes the hydrodynamic diameters of all SeNPs by using DLS. Considering the hydrodynamic diameters of SeNPs stabilized by proteins and SDS with the smallest size, SeNPs coated by proteins (sample H and sample I) showed larger size than SeNPs coated by SDS (sample A). This is due to the differences between molecular weight of each stabilizing agent which are 288.4 g mol−1 (∼0.28 kDa) for SDS, 18.4 kDa for β-lactoglobulin and 66.5 kDa for albumin. However, the size of the NPs does not depend only on the molecular weight of coating agent but also the concentration of stabilizing agent and reducing agent.14
In order to better evaluate the stability of SeNPs coated by protein before introduction into FlFFF, the size distribution of those SeNPs was monitored. Focusing on the results from DLS for size distribution of SeNPs, SeNPs coated by β-lactoglobulin does not show a monomodal peak such as for those SeNPs coated by albumin and SDS, as illustrated in Fig. 2b. The hydrodynamic diameter at the larger value around 350 nm was observed for SeNPs stabilized by β-lactoglobulin and contributed more than 50% of intensity. The zeta potential value of SeNPs coated by β-lactoglobulin is less than 30 mV, implying that agglomeration from SeNPs coated by β-lactoglobulin is more likely as compared to other NPs mentioned. The bimodal peak of β-lactoglobulin observed from DLS is due to that at pH = 2, β-lactoglobulin is presented as dimers and other larger aggregates. Above pH 7, the dimers of β-lactoglobulin can be dissociated into monomers.42 Adjusting the pH above 7, the zeta potential of SeNPs coated by β-lactoglobulin changed from positive to negative charge.4,15 In this case, the stabilizing agents or ligands used for NPs stabilization may affect the diffusion behavior of nanoparticles such as the shifting of peak intensity towards the larger value.38,43 It should be noted that challenge still remains for analysis by DLS, particularly for nanoparticles coated by proteins due to their possibility for aggregation as it was also previously mentioned in another literature for β-lactoglobulin characterization.44 To reduce the aggregate formation of SeNPs coated by β-lactoglobulin by using DLS, adjusting the pH and setting the right temperature is highly recommended.
FlFFF for hydrodynamic diameter determination of SeNPs
For accurate hydrodynamic diameter estimation of SeNPs by Sy-FlFFF, the optimization of SyFlFFF system is necessary. In order to minimize particle membrane interaction, types of membrane materials and carrier liquids were carefully chosen to obtain good peak quality. All details of the condition optimization were performed and the optimum condition was selected as explained in ESI (see Sections 1–3 in ESI; Fig. S1–S3†). All SeNPs samples were introduced into the Sy-FlFFF by using FL-70 and sodium azide as carrier liquid (approximately pH 8–9) and 10 kDa RC as membrane material. In order to evaluate the efficiency of Sy-FlFFF system, we also introduced all SeNPs-SDS samples into Asy-FlFFF system under the same conditions. The fractograms of SeNPs obtained from two different systems of FlFFF are illustrated in Fig. 3.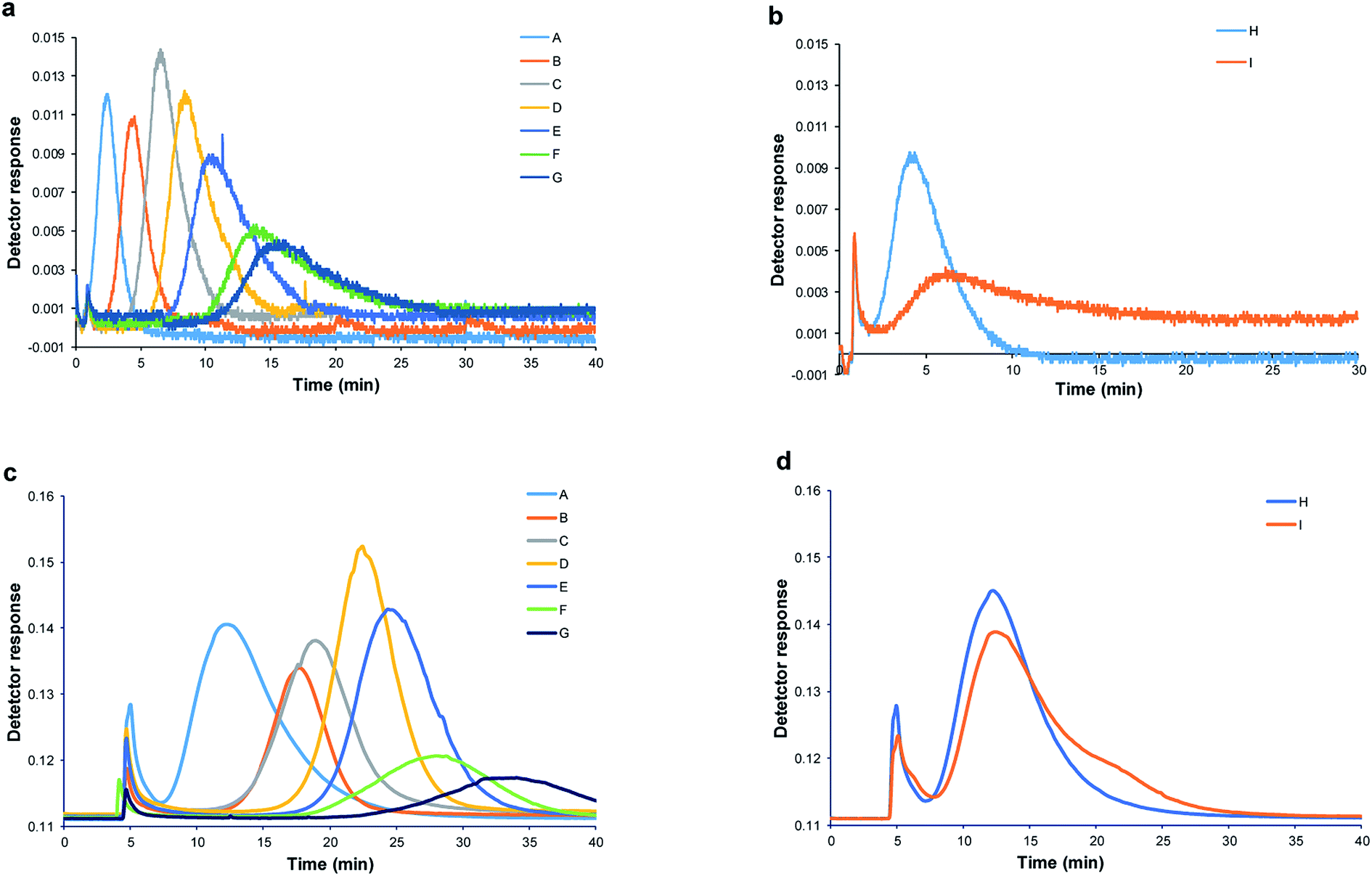 | ||
| Fig. 3 Overlaid fractograms of SeNPs synthesized with various types of stabilizing agents from Sy-FlFFF (a and b) and Asy-FlFFF (c and d). (a) and (c) SeNPs stabilized by SDS with various amounts (mL) of thiosulfate as reducing agent [sample A–G represents the shifting retention time of SeNPs synthesized by reduced the amount of thiosulfate]. (b) and (d) SeNPs stabilized by protein [β-lactoglobulin (sample H) and albumin (sample I)] with the use of 0.02% FL-70 and 0.02% NaN3 and 10 kDa RC membrane (n = 3). | ||
The fractograms of SDS stabilized SeNPs from Sy-FlFFF are shown in Fig. 3a, and those of protein stabilized are illustrated in Fig. 3b. The fractograms of SeNPs obtained from Asy-FlFFF are displayed in Fig. 3c and d. It is interesting to have a closer look at the elution peak of SeNPs stabilized by β-lactoglobulin. As mentioned earlier by using DLS method, bimodal peaks were observed. With Sy-FlFFF and Asy-FlFFF, however, only one peak was observed for SeNPs coated by β-lactoglobulin. By converting the fractograms into particle size distribution shown in Fig. S5(a and b),† the size detectable was in the range of 50–100 nm under this operating condition.
The hydrodynamic diameter (dh) estimation from Sy-FlFFF and Asy-FlFFF was calculated based on the retention time observed from the fractogram and converted into the diameter. Following method by Giddings et al. and Dou et al. (2015),39,45 we calculated the hydrodynamic diameter of SeNPs based on the channel thickness measurements from the known diameter of standard PS particle which is a suitable procedure for all types of FlFFF. But this method is also challenging due to the uncertainties from membrane compression. The type of standard such as PS nanoparticles are suitable to use for channel thickness determination. PS standard particles are recommended to use because it is inert avoiding particle membrane interaction and it is rather temperature independence.45 According to the FlFFF retention theory (see eqn (5)) following the method by Saenmuangchin and Siripinyanond in 2018 for AuNPs26 by using Sy-FlFFF, the size calculation of NPs can be directly determined based on the retention time if the value of the channel thickness is known. The channel thickness (w) is important to be measured by using standard nanoparticle at the same condition as for sample measurement.45 Hence, 100 nm standard polystyrene particle was selected for channel thickness determination.
According to the results from statistical analysis (see eqn (6)), the hydrodynamic diameters obtained from Sy-FlFFF and Asy-FlFFF were not significantly different as tcal (0.72) was less than tcrit (2.31) as shown in Table 1. It shows that the different channel construction of FlFFF does not affect on the size measurements of NPs but affects on the peak widths. As Asy-FlFFF channel has only one bottom frit and lower surface area than that of Sy-FlFFF due to its trapezoidal shape, it can minimize the particle membrane interaction and decrease peak widths.46 As shown in Fig. 3b and d for SeNPs stabilized by protein, the peak elutions of SeNPs (sample I) are different. For sample I, SeNPs coated by albumin showed two peaks due to the aggregation of particles (Fig. 3d). By using this technique, the determination of the hydrodynamic diameter based on retention time is very dependent on the particles type and coating. In summary, under optimum conditions Sy-FlFFF has ability to estimate the hydrodynamic diameters of SeNPs as the results were similarly to those from Asy-FlFFF.
Concerning the size comparison of SeNPs from both techniques with DLS by using paired t-test method, the t calculation (tcal) from the experiment is 3.29 for Asy-FlFFF and 3.08 for Sy-FlFFF, whereas the t critical (tcrit) is 2.31 (at P = 0.05). Therefore, the hydrodynamic diameters from both techniques (FlFFF and DLS) are significantly different as tcal was higher than tcrit. The difference of both FlFFF techniques from the DLS was mainly due to the possibility of DLS to provide larger particle size than FlFFF. According to the light diffusion theory, a particle having an “n” times larger diameter diffuses “n2” times more light. The larger particles show more scattering than the smaller particles, which can affect to the average mean diameter of NPs. As observed in the earlier study by other investigators for SiO2 NPs,47 DLS measured the z-averaged diameter whereas FlFFF with UV detection offered the information on the volume-averaged diameter, causing the DLS to show high sensitivity for larger size ranges and low sensitivity for smaller size ranges. Another plausible explanation for larger size observed from DLS especially for SeNPs coated by SDS was the extension of the electrical double layer of SDS micelles48 around SeNPs, and thereby reducing the diffusion speed of particles which resulted in the larger experimentally observed hydrodynamic diameter.
TEM for core size determination of SeNPs
The information on core diameter is obtained by using TEM. With TEM, size distribution of NPs could be analyzed by using NIH-image/ImageJ program. This program can be used for counting the number of representative particles in the image. The interpretation of results by using this technique was reported by Woehrle et al.19According to the TEM image (see Fig. 4), the size of SeNPs increased as a result of decreasing the amount of thiosulfate. Particle size distributions from TEM are shown in the Fig. 5A (sample A–C) and ESI (see Fig. S6† (sample D–I)). The particle size distribution as shown in those figures was obtained by data conversion from the area to diameter (see eqn (1) and (2)) of each image of SeNPs (A–I), and was converted to the particle size distribution by using OriginPro 8.6 software. It is clearly seen from those figures that the population of nanoparticles decreases as the size increases. For the large particle, agglomeration is a main cause for decreasing numbers of particles, as shown in Fig. S6(f)† for sample F. For SeNPs coated by protein (sample H and I), the agglomeration of particles accurred (Fig. 3(H) and (I)). As shown in Fig. S6(h) and (i),† the particle size distribution detected in this sample was until around 180 nm which indicated the aggregation of samples. By using only this technique without confirmation by another technique for all SeNPs samples, the largest particle detected as shown in the particle size distribution (Fig. S6†) can not be distinguished due to the agglomeration of SeNPs presented in the suspension or during the preparation on the TEM grid, as the sample was prepared by drying for few minutes before introducing into the TEM as also mentioned by Loeschner et al. (2013).36 But, this technique allowed the detection of particle size < 10 nm in the sample as shown in Fig. 5A(a–c) for small particles of SeNPs coated by SDS. In contrast, the actual number concentration of nanoparticle population cannot be provided by TEM (quantification analysis). Therefore, SP-ICP-MS is used for providing specific information on number of particles and their mass concentration.
 | ||
| Fig. 4 TEM images of SeNPs. Image (A)–(G) represent the SeNPs stabilized by SDS with decreasing amount of thiosulfate with the mean diameter of 26.9 ± 2.5 (sample A), 32.0 ± 2.1 (sample B), 68.4 ± 0.2 (sample C), 93.0 ± 2.6 (sample D), 114.5 ± 1.6 (sample E), 147.2 ± 3.1 (sample F), and 179.9 ± 2.4 nm (sample G). Image (H) and (I) represent the sample by using protein as stabilizing agent with the mean diameter of 32.3 ± 1.6 (sample H) and 62.5 ± 2.5 (sample I). | ||
 | ||
| Fig. 5 Particle size distribution of SeNPs-SDS (sample A; sample B; sample C) from (A) TEM (n = 5) and (B) SP-ICP-MS (n = 2). The number of particle selected for area-equivalent diameter measurements from TEM was 20 to over 100 particles in 5 TEM image for one sample. | ||
SP-ICP-MS for core size determination of SeNPs
The differences between SP-ICP-MS and TEM is the sample concentration needed for the analysis. SP-ICP-MS allows the concentration in ppb–ppt level to be analyzed, meaning less sample consumption. In contrast, TEM can detect the smallest particle size down to few nanometers (see Fig. 5). The slight differences in the particle size for SP-ICP-MS are shown in the Fig. 5B of (a–c) sample (represents sample A–C), the particle size below 10 nm was not detected as compared with TEM technique (see Fig. 5A). The smallest particle size detectable by SP-ICP-MS as shown for sample A–C (Fig. 5B(a–c)) is between 20 to 25 nm. Based on this result, TEM can be used for sample with size lower than 10 nm but not for sample with low concentration and small amount of samples. In this case, SP-ICP-MS is a well-suited method to perform the particle size of NPs at low number concentration of sample, but high sample dilution is required which could cause particle instability and more attention should be paid. Although the number of classes in SP-ICP-MS size distribution was not the same for all samples, we diluted all samples to contain between 1.0 to 4.0 × 104 particles per mL to ensure reliable particle size analysis.Table 1 shows the particle size of SeNPs by SP-ICP-MS and TEM. The results obtained from SP-ICP-MS are in good agreement with those from TEM which ranged from 30 nm to 180 nm. There was no significant difference between the two techniques, as checked by paired t-test method. The tcal (0.5) is less than tcrit (2.31) at P = 0.05. Eventhough TEM and SP-ICP-MS techniques provide similar results, the apparent difference is the number population of nanoparticles present when plotted as size distribution which is shown in Fig. S6 and S7.†
In the following experiment, the mixed size of SeNPs-SDS was also introduced into SP-ICP-MS to test the feasibility of system for more complex sample such as synthesized NPs. The mixed size sample B and sample C; and sample B and sample E were observed as shown in Fig. 6a and b. It is not clear how many size presents in the suspension as shown by the red arrow. For further investigation, SP-ICP-MS size analysis was carried out for the fractions collected from Sy-FlFFF since the system showed the best performance for SeNPs separation based on their different sizes under optimum condition of membrane material and carrier liquid.
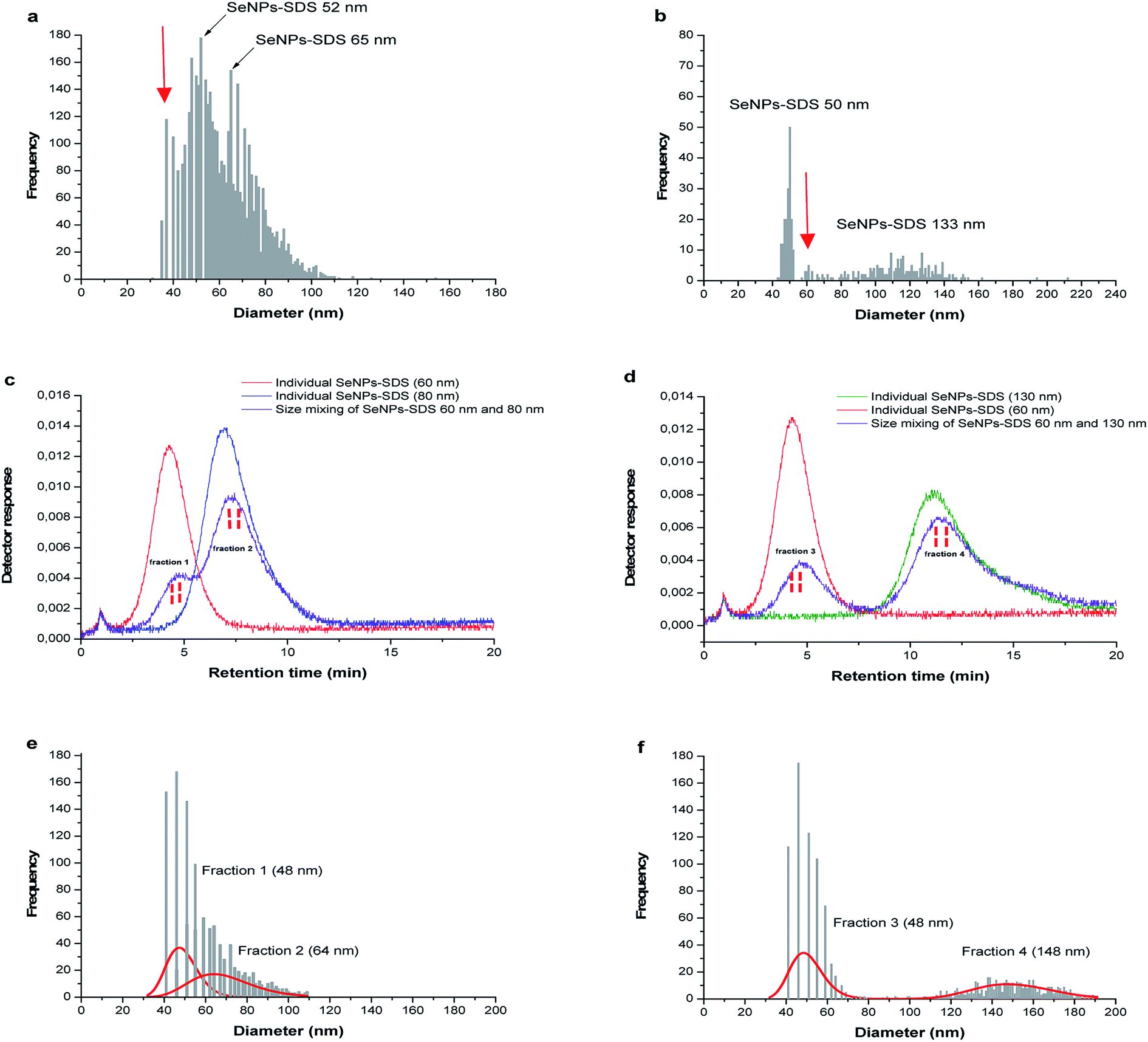 | ||
| Fig. 6 (a) and (b) Direct measurement of size mixture of 60 nm (sample B) and 80 nm (sample C); and 60 nm (sample B) and 130 nm (sample E) (DLS analysis) into SP-ICP-MS. (c) and (d) Sy-FlFFF fractograms of individual SeNPs of 60 nm, 80 nm and 130 nm and fractions of SeNPs 60 nm and 80 nm; and 60 nm and 130 nm collected for size analysis by SP-ICPMS. (e) and (f) Number size distribution of SeNPs fractions collected from Sy-FlFFF (n = 3). | ||
Comparison of the diameter information obtained from various techniques
As described earlier, the difference in particle size values obtained from various techniques is due to the different principles of each technique. According to the data in Table 1, DLS and FlFFF yielded larger sizes than those from TEM and SP-ICP-MS. In theory, DLS and FlFFF measure the hydrodynamic diameter, which refers not only to the core diameter of particle but also everything around the particle core including stabilizing agents. DLS detection is based on the translational diffusion of particles which depends not only on the size of the particle but also the surface structure including type of ions in the medium. Changes in the surroundings of the particle affect on the diffusion speed of NPs, and thereby changing in the hydrodynamic diameter.49 According to the results in Table 1, different medium and surface structure of stabilizing agents between SeNPs coated by protein (dissolved in water) and SeNPs coated by SDS (dissolved in SDS) affected on the stability of NPs which was shown by the zeta potential values. All SeNPs coated by SDS exhibited monomodal peak but not with SeNPs coated by protein (β-lactoglobulin, Fig. 2b) which had the zeta potential lower than ±30 mV.Considering the results from TEM and SP-ICP-MS, both techniques gave similar results according to the paired t-test analysis. From Table 1, TEM showed the largest values of standard deviation as compared to the other techniques. This might be due to the aggregation of NPs during the sample preparation process as the samples need to be dried under ultrahigh vacuum before TEM analysis. As shown in Fig. 4 for β-lactoglobulin and albumin stabilized SeNPs, the overlap of larger particle with smaller particle was observed. Without careful adjustment of the TEM image contrast in ImageJ software, the apparent reading is not assigned to a single particle. As the agglomeration was observed as shown in Fig. 4, the size distribution in Fig. S6† broadened and shifted to larger size, which was mentioned by Zook et al. in 2011 for AuNPs agglomeration.50 The ability of TEM to observe the agglomeration of NPs is considered as an advantage of TEM over SP-ICP-MS. Size distributions obtained by SP-ICP-MS were shown to be narrow with the size limit detection of approximately 20 nm (Fig. 5 and S6†). However, the average size values obtained by SP-ICP-MS are in acceptable agreement with TEM analysis.
Consecutive size analysis by SP-ICP-MS after Sy-FlFFF for SeNPs mixtures
As mentioned in the previous part, two mixtures of different size SeNPs stabilized by SDS (sample B) and (sample C); ((sample B) and (sample E)) were introduced into Sy-FlFFF. Then, the fractions were collected from Sy-FlFFF with different interval times for further size observation by SP-ICP-MS. Fractions were collected from Sy-FlFFF in the certain area of elution peak as shown in the dashed line in Fig. 6c (sample B and sample C) and Fig. 6d (sample C and sample E). The retention times observed for the particles in the mixture were slightly different from the retention times when individual size was introduced into the system. This is due to the different stability of SeNPs as individual and in the mixture.All the fractions collected from Sy-FlFFF were introduced into SP-ICP-MS. Fig. 6e and f shows the size distributions by SP-ICP-MS of fractions collected from Sy-FlFFF. Fraction 1, 2, and 4, which corresponds to the individual particle of sample B, sample C, and sample E, shows a different size value from individual particle (see Fig. S8 in ESI†), especially fraction 4 (see Table 2). This might be due to the fact that larger particles are easier to agglomerate due to the high dilution by using SP-ICP-MS as reported by Loeschner et al.36 for AgNPs fraction. The amount of particles was reduced after fractionation by FlFFF which is correlated with the number pulse counts of particle. This might be due to some particle loss during fractionation in Sy-FlFFF.
| Mixtures of SeNPs | Mean size of SeNPs in mixtures (nm) | ||
|---|---|---|---|
| Obtained from FlFFF (n = 3) | Obtained from SP-ICP-MS (n = 2) | Obtained from SP-ICP-MS after fraction collection from FlFFF (n = 2) | |
| a a = fraction 1; b = fraction 2; c = fraction 3; d = fraction 4. | |||
| Sample B + sample C (1:2 volume ratio) |
56.7 ± 1.2a/79.1 ± 1.8b | 52.4 ± 4.8/64.6 ± 3.2 | 48.1 ± 1.0a/64.0 ± 2.9b |
| Sample C + sample E (1:2 volume ratio) |
58.2 ± 1.7c/130.2 ± 1.2d | 52.4 ± 4.8/133.9 ± 2.4 | 48.7 ± 1.1c/148.5 ± 1.9d |
Conclusion
Different techniques were exploited to determine the hydrodynamic diameter and core diameter of synthesized SeNPs. For implementing FlFFF with the goal to provide hydrodynamic size information of nanoparticles, the selection of carrier liquid and membrane are fundamental to obtain accurate particle size results. For complex samples, the different sizes obtained by FlFFF and DLS depend on the properties of stabilizing agents and the possibility of particle agglomeration that affect to the reliability of size information. TEM yields a good visualization of sample, but high particle concentration is required. Furthermore, SP-ICP-MS is a well-suited method to provide number-based size distribution with the use of very low concentration and short time analysis. For the analysis of mixtures containing more than one size, fractionation by FlFFF was carried out prior to size analysis by SP-ICP-MS. This approach is highly recommended for samples with mixed size. Finally, it is clearly presented that no single technique provides high size accuracy and high reliability of sizing analysis for complex samples. SP-ICP-MS can not be applied in a stand-alone manner when the suspension composed more than one size. The capability of the combined technique of FlFFF and SP-ICP-MS should be further explored for characterization of NPs in the complex sample. For future work, the developed method can be applied to detect NPs in complex matrices such as in the environment. Attention will be paid on careful optimization of sample dilutions to provide accurate size results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the National Science and Technology Development Agency (NSTDA) according to the Thailand Graduate Institute of Science and Technology (TGIST) scholarship agreement TG-55-14-61-078M for financial support. We also sincerely acknowledge the Thailand Research Fund (TRF) and Mahidol University for providing the research grant under grant number BRG 6180006. The support from Center of Excellence for Innovation in Chemistry (PERCH-CIC) is gratefully acknowledged.References
- I. Khan, K. Saeed and I. Khan, Arabian J. Chem., 2017, 12, 908–931 CrossRef.
- T. Chen, Y.-S. Wong, W. Zheng, Y. Bai and L. Huang, Colloids Surf. B Biointerfaces, 2008, 67, 26–31 CrossRef CAS PubMed.
- P. A. Tran and T. J. Webster, Int. J. Nanomed., 2011, 6, 1553–1558 CAS.
- M. M. Pornwilard, W. Somchue, J. Shiowatana and A. Siripinyanond, Food Res. Int., 2014, 57, 203–209 CrossRef CAS.
- M. Palomo-Siguero, P. Vera, Y. Echegoyen, C. Nerin, C. Cámara and Y. Madrid, Spectrochim. Acta Part B At. Spectrosc., 2017, 132, 19–25 CrossRef CAS.
- A. Elsaesser and C. V. Howard, Adv. Drug Deliv. Rev., 2012, 64, 129–137 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- C. V. Reddy, I. N. Reddy, B. Akkinepally, K. R. Reddy and J. Shim, J. Alloys Compd., 2020, 814, 152349 CrossRef.
- C. V. Reddy, I. N. Reddy, K. R. Reddy, S. Jaesool and K. Yoo, Electrochim. Acta, 2019, 317, 416–426 CrossRef CAS.
- R. Cai, D. Yang, K. Te Lin, T. S. Cao, Y. Lyv, K. Chen, Y. Yang, J. Ge, L. Xia, G. Christou, Y. Zhao, Z. Chen and W. Tan, Nanoscale, 2019, 11, 20968–20976 RSC.
- E. Union, Commision Recommendation of 18 October on the definition of nanomaterial, 2011, 275, p. 38 Search PubMed.
- P. Verma and S. Maheshwari, Journal of Microscopy and Ultrastructure, 2018, 6, 182 Search PubMed.
- J. Zhang, Z. Teng, Y. Yuan, Q.-Z. Zeng, Z. Lou, S.-H. Lee and Q. Wang, J. Ultrastruct. Mol. Struct. Res., 2018, 107, 1406–1413 CAS.
- Z.-H. Lin and C. C. Wang, Mater. Chem. Phys., 2005, 92, 591–594 CrossRef CAS.
- M.-M. Pornwilard and A. Siripinyanond, J. Anal. At. Spectrom., 2014, 29, 1739–1752 RSC.
- T. K. Mudalige, H. Qu and S. W. Linder, J. Chromatogr. A, 2015, 1420, 92–97 CrossRef CAS.
- H. Kato, M. Suzuki, K. Fujita, M. Horie, S. Endoh, Y. Yoshida, H. Iwahashi, K. Takahashi, A. Nakamura and S. Kinugasa, Toxicol. In Vitro, 2009, 23, 927–934 CrossRef CAS.
- K. Bai, B. Hong, J. He, Z. Hong and R. Tan, Int. J. Nanomed., 2017, 12, 4527 CrossRef CAS PubMed.
- G. H. Woehrle, J. E. Hutchison, S. Özkar and R. G. Finke, Turk. J. Chem., 2006, 30, 1–13 CAS.
- J. Bae, W. Kim, K. Rah, E. C. Jung and S. Lee, Microchem. J., 2012, 104, 44–48 CrossRef CAS.
- A. F. Thünemann, P. Knappe, R. Bienert and S. Weidner, Anal. Methods, 2009, 1, 177–182 RSC.
- J. C. Giddings, Science, 1993, 260, 1456–1465 CrossRef CAS PubMed.
- M. Amde, Z. Q. Tan and J. Liu, Talanta, 2019, 200, 357–365 CrossRef CAS PubMed.
- B. Meisterjahn, S. Wagner, F. von der Kammer, D. Hennecke and T. Hofmann, J. Chromatogr. A, 2016, 1440, 150–159 CrossRef CAS PubMed.
- N. Bendixen, S. Losert, C. Adlhart, M. Lattuada and A. Ulrich, J. Chromatogr. A, 2014, 1334, 92–100 CrossRef CAS.
- R. Saenmuangchin and A. Siripinyanond, Anal. Bioanal. Chem., 2018, 410, 6845–6859 CrossRef CAS.
- A. Ulrich, S. Losert, N. Bendixen, A. Al-Kattan, H. Hagendorfer, B. Nowack, C. Adlhart, J. Ebert, M. Lattuada and K. Hungerbühler, J. Anal. At. Spectrom., 2012, 27, 1120–1130 RSC.
- H. Hagendorfer, R. Kaegi, J. Traber, S. F. Mertens, R. Scherrers, C. Ludwig and A. Ulrich, Anal. Chim. Acta, 2011, 706, 367–378 CrossRef CAS.
- E. Bolea, J. Jiménez-Lamana, F. Laborda and J. R. Castillo, Anal. Bioanal. Chem., 2011, 401, 2723 CrossRef CAS PubMed.
- S. Dubascoux, F. Von Der Kammer, I. Le Hécho, M. P. Gautier and G. Lespes, J. Chromatogr. A, 2008, 1206, 160–165 CrossRef CAS PubMed.
- H. Qu, I. R. Quevedo, S. W. Linder, A. Fong and T. K. Mudalige, J. Nanopart. Res., 2016, 18, 292 CrossRef.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Anal. Chem., 2013, 85, 2270–2279 Search PubMed.
- R. Aznar, F. Barahona, O. Geiss, J. Ponti, T. J. Luis and J. Barrero-Moreno, Talanta, 2017, 175, 200–208 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jimenez-Lamana, Trends Environ. Anal. Chem., 2016, 9, 15–23 CrossRef CAS.
- J. Jiménez-Lamana, I. Abad-Álvaro, K. Bierla, F. Laborda, J. Szpunar and R. J. Lobinski, J. Anal. At. Spectrom., 2018, 33, 452–460 RSC.
- K. Loeschner, J. Navratilova, C. Købler, K. Mølhave, S. Wagner, F. von der Kammer and E. H. Larsen, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- S. Motellier, N. Pelissier and J. G. Mattei, J. Anal. At. Spectrom., 2017, 32, 1348–1358 RSC.
- J. C. Giddings, P. S. Williams and M. A. J. Benincasa, J. Chromatogr. A, 1992, 627, 23–35 CrossRef CAS.
- J. N. Miller and J. C. Miller, Statistic and Chemometrics for Analytical Chemistry, Science, 5th edn, 2005, p. 268 Search PubMed.
- I. Donati, A. Travan, C. Pelillo, T. Scarpa, A. Coslovi, A. Bonifacio, V. Sergo and S. Paoletti, Biomacromolecules, 2009, 10, 210–213 CrossRef CAS PubMed.
- K. Sakurai, M. Oobatake and Y. Goto, Protein Sci., 2001, 10, 2325–2335 CrossRef CAS.
- I. Montes-Burgos, D. Walczyk, P. Hole, J. Smith, I. Lynch and K. J. Dawson, Int. J. Mol. Sci., 2010, 12, 47–53 Search PubMed.
- S. Blake, S. Amin, W. Qi, M. Majumdar and E. N. Lewis, Int. J. Mol. Sci., 2015, 16, 17719–17733 CrossRef CAS PubMed.
- H. Dou, E. C. Jung and S. Lee, J. Chromatogr. A, 2015, 1393, 115–121 CrossRef CAS PubMed.
- F. A. Messaud, R. D. Sanderson, J. R. Runyon, T. Otte, H. Pasch and S. K. R. Williams, Prog. Polym. Sci., 2009, 34, 351–368 CrossRef CAS.
- H. Kato, A. Nakamura and N. Noda, Mater. Express, 2014, 4, 144–152 CrossRef CAS.
- H. Kato, A. Nakamura, K. Takahashi and S. Kinugasa, Nanomaterials, 2012, 2, 15–30 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- J. M. Zook, R. I. MacCuspie, L. E. Locascio, M. D. Halter and J. T. Elliott, Nanotoxicology, 2011, 5, 517–530 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07120b |
| This journal is © The Royal Society of Chemistry 2020 |