
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHectogram-scale green synthesis of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures with Superb Congo red adsorption performance†
Leitao Zhang‡
ab,
Lilan Huang‡a,
Lei Zhang a,
Binzhong Lub,
Junbo Lib,
Yingfang Xiea,
Qiang Maa,
Qingping Xina,
Hui Yea,
Lizhi Zhaoa,
Yuzhong Zhang*a and
Hong Lia
a,
Binzhong Lub,
Junbo Lib,
Yingfang Xiea,
Qiang Maa,
Qingping Xina,
Hui Yea,
Lizhi Zhaoa,
Yuzhong Zhang*a and
Hong Lia
aState Key Laboratory of Separation Membranes and Membrane Processes, School of Materials Science and Engineering, Tianjin Polytechnic University, Tianjin 300387, P. R. China. E-mail: zhangyz2004cn@vip.163.com
bSchool of Chemical Engineering and Pharmaceutics, Henan University of Science and Technology, Luoyang 47100, Henan, P. R. China
First published on 11th February 2020
Abstract
Delicate design of hierarchical nanoarchitectures has become a highly effective strategy to develop novel adsorbents with improved adsorption capacity. Herein, hectogram-scale green fabrication of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures (4A-Cu-T, T was the calcination temperature) with terrific Congo red (CR) dye adsorption performance was achieved through a simple, template-free and surfactant-free hydrothermal approach. A series of characterization techniques, including scanning electron microscopy, transmission electron microscopy, X-ray diffraction and photoelectron spectroscopy demonstrated that all resultant adsorbents featured a core–shell structure with 4A zeolite as core ingredients and CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies as shell components. The adsorption experimental results pointed out that 4A-Cu-300 with a maximum adsorption capacity of 512.987 mg g−1 showed the best adsorption performance amongst all as-prepared adsorbents, and the adsorption capacity of shell component-CuOxCu(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies was calculated up to 3685.500 mg g−1. The shell thickness and phase ratio of CuO and Cu(OH)2 in CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies played key roles in improving the adsorption capacity. The successive tests suggested that the “carbon deposition” resulted in the decreased adsorption capacity of first-regenerated adsorbents, but little variance in adsorption performance among regenerated samples demonstrated the good stability of such adsorbents. This work unlocks a method for the rational design of high-performance adsorbents via delicate decoration of poor-performance materials with nanosheet assemblies, which will endow the non-active materials with enhanced adsorption properties.
Introduction
Water pollution, especially for dyes, has become a universal problem afflicting people all over the world owing to its negative impact on ecosystems and humans.1–3 Among the abatement processes for dye-contaminated effluents, adsorption has received tremendous attention due to its simplicity, high-efficiency and low-energy requirements.4,5 However, most state-of-the-art adsorbents often suffer from limited adsorption capacities and low adsorption rates.5–7 Thus, how to rationally design and develop high-performance adsorbents for dye wastewater remediation is of great urgency.As interest in adsorbent-specific nanomaterials with enhanced adsorption properties is increasing, core–shell hierarchical nanostructures with well-defined internal core components and functional shell micro/nanoarchitectures are capturing substantial attention because of their conducive and versatile compositions and structures to serve such purposes.8,9 As well, delicate design of three-dimensional (3D) nanosheet assemblies can also render improved adsorption capacity and faster adsorption rate.4,10,11 Hence, it is coveted to decorate core components with nanosheet assemblies to construct the core–shell hierarchical nanomaterials. In this regard, several methodologies have been unearthed for the fabrication of such hierarchical nanoarchitectures. Recently, Yu and his co-authors12,13 prepared various such core–shell nanostructures, in which few-layer CoFe or NiFe layered double hydroxide (LDH) nanosheets were grown on Cu nanowire for effective overall water splitting by means of a facile and scalable electrochemical approach. O'Hare's group14–16 reported an in situ precipitation method for the synthesis of core–shell hybrid materials with zeolite or silica as core components and organic-LDH nanosheet assemblies as shell structure. Based on a similar synthesis strategy with slight modification, Wang et al.17 fabricated a variety of core–shell structures by in situ coating a vertically and randomly aligned LDH layers on the surface of diverse zeolites and found that the adsorption capacity of such composites in wet conditions can be largely enhanced. Unfortunately, the preparation processes usually involve tedious procedures, the use of surfactants/templates and hazardous chemicals, which limit scalable production and do not noticeably meet the criteria of “Twelve Principles of Green Chemistry” in nanomaterials synthesis.18,19 As such, it is imperative to propose a simple, green and scalable material design framework towards the fabrication of adsorbent-specific core–shell hierarchical nanomaterials.
Herein, a simple, template-free and surfactant-free methodology was proposed and used in large scale preparation of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures, which were used as adsorbents in CR dye adsorption study. Firstly, a series of techniques, including scanning electron microscopy, transmission electron microscopy, X-ray powder diffraction and photoelectron spectroscopy so on, were applied to investigate the morphology, structure and component of as-obtained materials. Moreover, a plausible formation mechanism of precursor was put forward based on the literature analysis and experimental results. Secondly, the emphasis was focused on the CR adsorption study of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures. Lastly, the stability of adsorbents was investigated through successive adsorption experiments and multiple analysis characterizations.
Material and methods
Preparation of copper chloride aqueous solution (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 000 mg L−1)
000 mg L−1)
53.4322 g copper chloride powders (Shanghai Aladdin Biochemical Technology Co., Ltd., China) and proper amount of ultrapure water (18.2 MΩ cm−1) was added into 100 ml beaker. After the dissolution of the copper chloride, the aqueous solution was transferred into 1000 ml volumetric flask.
Preparation of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures
Typically, 10 ml 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1 CuCl2 aqueous solution was diluted 100 ml 2000 mg L−1 and the pH is 1.60 through the adjustment of hydrochloric acid or sulfuric acid (1.0 mol L−1) and sodium hydroxide (1.0 mol L−1). Then, 2 g 4A zeolite (Shanghai Jiuzhou Chemicals Co. Ltd., China) was added to 100 ml 2000 mg L−1 (pH ≈ 1.60) CuCl2 aqueous solution. After shaking for 3.5 h in the air-shaker (200 rpm), light blue 4A-Cu could be obtained through vacuum filtration and lyophilization. Finally, 4A-Cu was annealed for 5 h in air at different temperature and a series of hierarchical 4A zeolite@CuOx(OH)2−2x (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures with different ratio of CuO/Cu(OH)2 could be obtained and they were named as 4A-Cu-T (where T refers to temperature). For example, 4A-Cu-300 refers to hierarchical 4A zeolite@CuOx(OH)2−2x (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures obtained after the high-temperature treatment of 4A-Cu for 5 h at 300 °C.
000 mg L−1 CuCl2 aqueous solution was diluted 100 ml 2000 mg L−1 and the pH is 1.60 through the adjustment of hydrochloric acid or sulfuric acid (1.0 mol L−1) and sodium hydroxide (1.0 mol L−1). Then, 2 g 4A zeolite (Shanghai Jiuzhou Chemicals Co. Ltd., China) was added to 100 ml 2000 mg L−1 (pH ≈ 1.60) CuCl2 aqueous solution. After shaking for 3.5 h in the air-shaker (200 rpm), light blue 4A-Cu could be obtained through vacuum filtration and lyophilization. Finally, 4A-Cu was annealed for 5 h in air at different temperature and a series of hierarchical 4A zeolite@CuOx(OH)2−2x (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures with different ratio of CuO/Cu(OH)2 could be obtained and they were named as 4A-Cu-T (where T refers to temperature). For example, 4A-Cu-300 refers to hierarchical 4A zeolite@CuOx(OH)2−2x (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures obtained after the high-temperature treatment of 4A-Cu for 5 h at 300 °C.
The reference adsorbent was prepared based on the same synthesis procedure above without cupric aqueous. To be exact, 2 g 4A zeolite (Shanghai Jiuzhou Chemicals Co. Ltd., China) was added to 100 ml pure water with pH = 1.60. After shaking for 3.5 h in the air-shaker (200 rpm), the reference precursor could be obtained through vacuum filtration and lyophilization. Finally, reference precursor was annealed for 5 h in air at 300 °C.
Adsorption experiments for Congo red
In usual adsorption experiments, the desired amounts of as-obtained adsorbents were mixed with 100 ml 300 mg L−1 Congo red (CR) aqueous solution in 250 ml conical flask to initiate the adsorption reaction. After stirring at 250 rpm for 30 min, 2 ml of the solution was pipetted and the adsorbents were filtrated and separated through a 0.45 μm membrane filter. The adsorption capacity was calculated according to the residual CR concentration in the filtrates.In the adsorption kinetics experiments, 0.2 g of as-obtained adsorbents were mixed with 100 ml CR aqueous solution with a concentration of 1000 mg L−1 in 250 ml conical flask to initiate the adsorption reaction. At different time intervals, 2 ml of the solution was pipetted and the adsorbents was filtrated and separated through a 0.45 μm membrane filter. The adsorption capacity was calculated according to the residual CR concentration in the filtrates.
In the successive adsorption recycling experiments, 0.2 g of as-obtained adsorbents (4A-Cu-300) were mixed with 100 ml CR aqueous solution with a concentration of 1000 mg L−1 in 250 ml conical flask to initiate the adsorption reaction. After stirring at 250 rpm for 50 min, the adsorption-saturated adsorbents were separated through a centrifuge (10000 rpm, 10 min) and washed three times with ultrapure water. Then adsorbent-regeneration experiments were carried out at 300 °C for 5 h. After each run, the CR concentration in supernatant solutions was determined to calculate the adsorption capacity.
Among all experiments, Congo red (CR) aqueous solution with different concentrations was prepared through the dilution of high concentration CR aqueous solution (10000 mg L−1). The CR concentration was analysed using a ultraviolet-visible (UV-Vis) spectrophotometer (Purkinje General TU-1901) and a linear calibration curve over 1–100 mg L−1 based on the adsorbance value at 497 nm. Of note, all the experiments were performed at neutral pH and room temperature.
Characterization methods
XRD (X-ray diffraction) patterns were recorded using a BRUKER D8-DISCOVER X-ray diffractometer (Cu Kα radiation, λ = 1.540598 Å).The morphology and structure were characterized using a field emission scanning electron microscope (ZEISS, GeminiSEM 500, Germany) and a high resolution transmission electron microscope (HRTEM, JEM-2100F, JEOL; HiTACHI H7650). The TEM samples were prepared by dispersing in ethanol and then dropping onto the copper grid coated with carbon film. The XPS (X-ray photoelectron spectroscopy) studies were carried out with K-Alpha (Thermo Fisher Scientific, America), using an aluminum anode (Al Kα = 1486.6 eV). The binding energy was referenced to the C1s peak at 284.8 eV of the surface adventitious carbon. The IR (infrared radiation) spectra were recorded through a TENSOR37 spectrophotometer (Bruker AXS, USA). The UV-Vis spectra measurements were performed on a Purkinje General TU-1901 spectrophotometer. N2 adsorption–desorption isotherm was determined by Quantachrome NOVA 1000 Gas Sorption Analyzer and Micrometritics ASAP 2020 Physisorption Analyzer.
Results and discussion
The fabrication process for hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures is illustrated in Fig. 1. The adsorbent precursors (labeled as 4A-Cu) were gained through the vacuum filtration and lyophilization (−55 °C, 48 h) of the mixture of 2 g 4A zeolite and 100 ml 1000 mg L−1 CuCl2 solution at pH 1.6. After the calcination treatment of the adsorbent precursor in air, hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures could be gained and was noted as 4A-Cu-T (T is the calcination temperature). Of particular note, no template or surfactant was used in whole preparation process, indicating that this synthetic methodology was an environment-friendly approach and it conformed to the “Twelve Principles of Green Chemistry”.18,19 Additionally, hectogram-scale production of such nanomaterials were also achieved via the addition of 100 g 4A zeolite into 5000 ml 1000 mg L−1 aqueous solution containing bivalent copper ions. | ||
| Fig. 1 The schematic illustration of the synthesis procedures of hierarchical 4A zeolite@CuOx(OH)2−2x (0 ≤ x < 1) nanosheet assemblies core–shell nanomaterials. | ||
The morphologies and microstructures of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures were investigated by SEM and TEM in Fig. 2. Compared to the smooth surface of 4A zeolite with cubic configuration (Fig. S1†), the SEM images (Fig. 2a and b) presented that 4A-Cu and 4A-Cu-300 had an open, highly porous surface morphology constructed by thousands of interconnected nanosheets. The typical TEM images of 4A-Cu (Fig. 2c and d) and 4A-Cu-300 (Fig. 2f and g) highlighted a core–shell feature with the 4A zeolite surrounded by interconnected nanosheet assemblies. The SADE pattern in Fig. 2e revealed the growth of an amorphous nanosheet-assembly on the surface of 4A zeolite in 4A-Cu, while Fig. 1h demonstrated that surface nanosheet-assemblies in 4A-Cu-300 contained ultrasmall crystal domains with the domain-size of less than 5 nm (inset in Fig. 2h), which were surrounded by the vast majority of amorphous regions. The average shell thickness of copper oxides/hydroxides nanosheet assemblies was measured based on at least 20 TEM images (Fig. S2†). The shell thickness was ca.150 nm for 4A-Cu, 125 nm for 4A-Cu-200, 100 nm for 4A-Cu-300, 75 nm for 4A-Cu-400 and 55 nm for 4A-Cu-500, and the cause for shell structure shrinkage (decreased thickness) might result from the high-temperature dehydration reaction of Cu(OH)2 followed by CuO formation in calcination treatment.
 | ||
| Fig. 2 The morphology and structure characterization. The SEM images of (a) 4A-Cu and (b) 4A-Cu-300. The insets in the top right corner of (a) and (b) panels were the corresponding zoomed-in images. The (c) and (d) low-magnification TEM images of 4A-Cu, and (e) was a SADE pattern that corresponded the square area in (d). The (f) and (g) low-magnification TEM images of 4A-Cu-300. (h) HRTEM image of the square part in panel (g). The inset in (h) was an exploded view of the circular region in (h). | ||
To determine the phase purity and crystallinity of as-obtained adsorbents, the powder X-ray diffraction patterns of 4A-Cu and 4A-Cu-300 were recorded in Fig. 3a. As was seen in this figure, the diffraction peaks positions of 4A-Cu were consistent with the counterparts of the original 4A zeolite but 4A-Cu presented poor signal-noise ratio, demonstrating that partial structure of 4A zeolite was destroyed during the growth of copper oxide/hydroxides shell structure. With a close observation of diffraction peak angles, as shown by the red arrow in Fig. 3b, the diffraction peaks positions of 4A-Cu shifted to the higher angle, which might result from the unit-cell shrinkage of core 4A zeolite due to the ion exchange reaction between the sodium ions (ionic radius 0.102 nm) in the zeolite structure and bivalent copper ions (ionic radius 0.073 nm) in aqueous solution.20 After calcination at 500 °C for 5 h, 4A-Cu-500 presented poorer crystallization degree compared to 4A zeolite, but two peaks located at 35.50° and 38.73° appeared and they could be well indexed to monoclinic CuO (a = 4.6853 Å, b = 3.4257 Å, c = 5.1303 Å, α = γ = 90°, β = 99.549°), which undoubtedly demonstrated the presence of CuO phase in 4A-Cu-500 sample. Nevertheless, it was rather difficulty to distinguish phase structure of nanosheet-assemblies shells from the XRD pattern for 4A-Cu and 4A-Cu-300 samples. Given the strong dependence of adsorption property on the phase structure,21 other techniques were employed to further discriminate phase structure. The infrared spectrum is extremely sensitive to valence-band vibration mode and can provide specific information about material structure.22 The IR spectra presented in Fig. S3† disclosed that there were three weak vibration peaks at 460 cm−1, 610 cm−1 and 690 cm−1, proving that the envelope phase of 4A-Cu was copper hydroxide, while copper hydroxides and copper oxides coexisted in 4A-Cu-T samples due to the occurrence of three weak vibration bands centered at 460 cm−1, 610 cm−1 and 710 cm−1.23,24 Based on the analysis of Cu2p XPS spectrum in Fig. 3c, there was a wider shake-up satellite at about 8.4 eV higher than the Cu2p3/2 main peak located at 943.4 eV. The fact ruled out the possibility of the presence of mono-valence copper ion and provided a solid evidence that CuO and Cu(OH)2 phase co-existed in as-obtained materials.25,26 In addition, a subtle decrease in binding energy was observed in main Cu2p3/2 and Cu2p1/2 peaks within the range of 4A-Cu to 4A-Cu-500. This results suggested a gradual increase of CuO content from 4A-Cu to 4A-Cu-500 due to the lower coordination capability of O2− ligand than OH− ligand.27
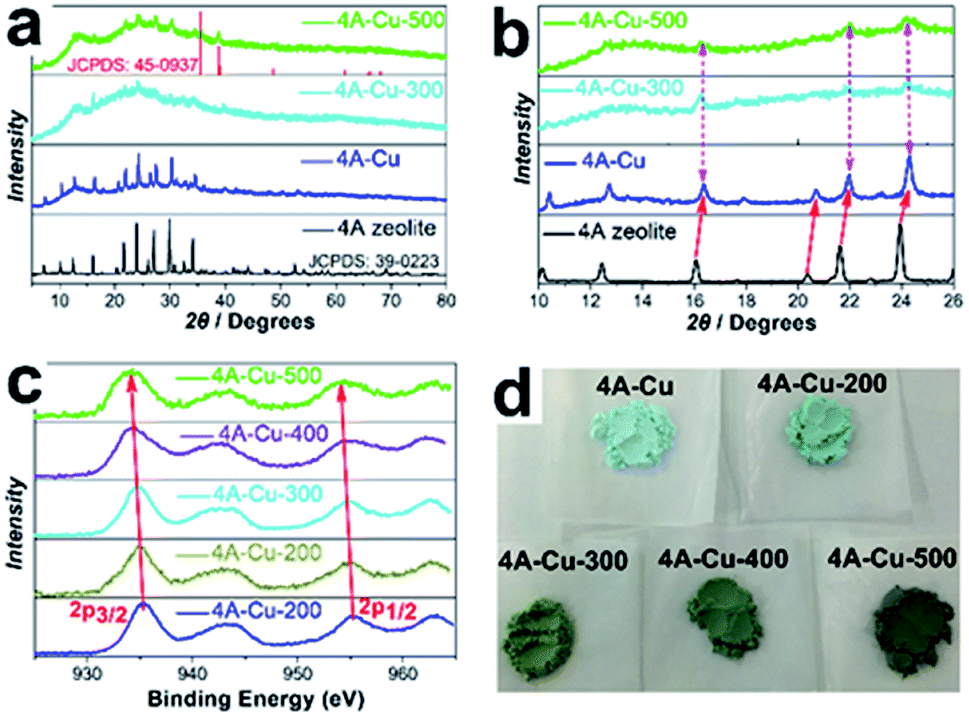 | ||
| Fig. 3 (a) The XRD pattern of 4A-Cu-500, 4A-Cu-300, 4A-Cu and 4A zeolite samples; (b) the enlarged view ranged from 10° to 26° in figure (a) confirmed the shift-to-high-angle after the growth of 3D nanosheet-assemblies. (c) The high-resolution Cu2p XPS spectra of 4A-Cu-500, 4A-Cu-300 and 4A-Cu. (d) The digital pictures of all as-prepared samples. | ||
Additionally, a series of snapshots from 4A-Cu to 4A-Cu-500 were displayed in Fig. 3d. The color turned gradually from light blue of 4A-Cu to black brown of 4A-Cu-500, which inferred that the higher the incineration temperature was, the more CuO phase appeared. This result agreed well with the XPS analysis. In a word, based on the above analysis, it seemed safe to conclude that only amorphous Cu(OH)2 appears in 4A-Cu precursor, while amorphous Cu(OH)2 and partially-crystalline CuO co-existed in 4A-Cu-T materials and the molecular formula of shell components could be expressed as CuOx(OH)2−2x (0 ≤ x < 1).
To examine the surface and pore characteristics of 4A-Cu and 4A-Cu-300, N2 adsorption–desorption measurements were carried out. According to the International Union of Pure and Applied Chemistry (IUPAC) definition, the adsorption–desorption isotherms (Fig. S4†) exhibited a hybrid characteristics of type III and type IV isotherm with a hysteresis loop, indicating the presence of a wide pore distribution from micropore to macropore. The calculation results of Non Local Density Functional Theory (NLDFT) method (the insets in Fig. S4†) indicated the pore size range from 2 nm to 80 nm. All the samples showed tiny difference in the specific surface area (Table S1†). Compared to 4A zeolite, the surface area of as-prepared materials reduced dramatically (Table S1†) and this might be caused by the structure destruction of 4A zeolite and coverage of copper oxides/hydroxides nanosheet-assemblies.
The formation mechanism of 4A-Cu precursor was investigated based on the hydrothermal growth process from two hands – the seeds and the driving force. As well known, the zeolite dealuminum reaction will happen in acidic conditions and this can lead to irreversible structural damage (loss in crystallinity) and the generation of micro/meso-pore defect (Fig. 4).28,29 The formed defect might be refilled by the residual silicon species, thus resulting in the generation of new Al–O–Si chain segments.29 The Al–O–Si segments are metastable and therefore can serve as seeds for the growth of Cu(OH)2 nanosheets.14,29 As for the driving force, there are several papers to assist to understand this hand. Dermot O'Hare et al.14 indicated the optimum pH 10 was the driving force for the growth of three-dimensional assembly composed of layered double hydroxide nanosheets on the surface of silica and Y zeolite. Xu and co-authors30 studied the formation process of porous Pd nanosheets constructed by Pd nanowires, and they concluded that pH played a pivotal role in determining the morphology of end-product. In fact, we also found that pH 1.6 was the optimal condition for the controllable synthesis of 4A-Cu (Fig. S5†). To this end, we carried out a twin experiment where 4A zeolite was added into the aqueous solution with pH = 1.60 and found that 4A zeolite could release a large amount of OH−1 in a short time followed by a rapid pH increase to 5.44 in 2.5 seconds. So the released OH−1 by 4A zeolite in acid aqueous solution can be considered as the driving force for Cu(OH)2 shell formation. Besides, we found other metal ions but copper ion was unfavorable for the growth of corresponding metal hydroxide nanosheet-shell on the surface of 4A zeolite. In our previous study, we had demonstrated that other metal ions could be used to prepare the materials containing isolated metal species, such as iron.28 As for the detailed mechanism, we will perform more experiments to answer. The plausible formation mechanism of 4A-Cu precursor was presented in Fig. 4.
 | ||
| Fig. 4 The plausible formation mechanism of 4A-Cu precursor. | ||
Here, we selected the Congo red (CR) as probe molecule to investigate the adsorption performance of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies nanoarchitectures. CR is a benzidine-based anionic diazo dye containing NH2 and SO32− group and shows strong resistance to biodegradation and photodegradation.10,31 Adsorption is usually used as an appealing approach for CR removal.32–34 Fig. 5a manifested a pre-experiment result of 0.2 g 4A-Cu with 100 ml 300 mg L−1 CR aqueous solution. In this experiment, 81.50% and 91.50% CR could be removed by 4A-Cu adsorbents in 2 min and 4 min, respectively. The adsorption capacity at 30 min was 142.50 mg g−1. Likewise, in an identical-twin experiment, it could be found that 4A-Cu-300 functioned better than 4A-Cu in CR adsorption experiments, with removal efficiency of 95.01% in 1 min (Fig. 5b and ESI Video 1†). However, only 12.46% and 12.10% CR (100 ml 100 mg L−1) can be decolored by 0.2 g 4A zeolite and reference adsorbents with prolonged adsorption time up to 100 h and the adsorption capacity is 6.23 mg g−1 and 6.05 mg g−1, respectively (Fig. 5c and d). These results proved that the adsorption performance could be improved sharply upon the growth of the components-CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies on the surface of 4A zeolite.
 | ||
| Fig. 5 The CR adsorption study of (a) 4A-Cu, (b) 4A-Cu-300, (c) 4A-zeolite and (d) reference adsorbents. Other conditions: CR = 100 ml, 300 mg L−1 (CR concentration in 4A zeolite and reference adsorbents study is 100 mg L−1); adsorbent = 0.2 g; pH = 6.30; T = 25 °C. | ||
The CR adsorption performance of as-obtained adsorbents was furtherly studied in detail and the results were exhibited in Fig. S6.† The pseudo-first-order and pseudo-second-order functions were used to fit these adsorption kinetic processes, as shown in S3 part.† Based on the analysis of fitting parameters in Table S2,† one could conclude that pseudo-second-order adsorption kinetic equation can have a better description for CR adsorption experiments than pseudo-first-order model, suggesting that the rate-limiting step in adsorption process might be chemical sorption or chemisorption involving valence forces through sharing or exchange of electrons between sorbents and sorbates. The dependence of adsorption capacity at equilibrium on the incineration temperature was presented in Fig. 6a and it showed that, with the increase of incineration temperature, the equilibrium adsorption capacity behaved like a roller coaster with the maxima occurring at between 300 °C and 400 °C. Additionally, in order to probe how the solutes interact with the adsorbents, the isothermal adsorption experiments of 4A-Cu-300 were also carried out and the adsorption isotherm was recorded in Fig. 6b. According to the fitting results (Table S3†), Langmuir isotherm model (R2 = 0.957) had a better description than Freundlich isotherm model (R2 = 0.911), providing the evidence that monolayer adsorption reaction occured on the surface of 4A-Cu-300 samples. According to the Langmuir equation, the maximum adsorption capacity of 4A-Cu-300 is 512.987 mg g−1, which was not the highest reported value in up-to-date references. However, in consideration of the fact that only the shell components worked in CR adsorption study, the adsorption capacity of the shell components-CuOxCu(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies could be calculated up to 3685.500 mg g−1 (detailed method in S4 part†), which would be among the highest reported values in state-of-the-art literatures (Table 1).
 | ||
| Fig. 6 (a) The dependence of adsorption capacity at equilibrium on incineration temperature. (b) The experimental adsorption isotherm curves and its fitting models of CR adsorption on 4A-Cu-300. Note: the temperature of 4A-Cu precursor was labeled as −55 °C because the lyophilization preparation reaction occurred at −55 °C. | ||
| Type of adsorbent | Qm (mg g−1) | Reference |
|---|---|---|
| MgO–SiO2 composites | 4000 | 41 |
| CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies | 3685.500 | Present study |
| 4A-Cu-300 | 512.987 | Present study |
| Porous rod-like MgO | 3236 | 32 |
| Hierarchical MgO | 2409 | 4 |
| Ultrathin CeO2 NWs | 2382.75 | 42 |
| MgO nanoparticles | 2375 | 43 |
| Gemini surfactant/GO composites | 2325 | 44 |
| Leather shavings biochar | 1916 | 45 |
| Magnetic chitosan “fluid” | 1724 | 46 |
| Ni/Mg/Al layered double oxides | 1250 | 10 |
| CNT/Mg(Al)O nanocomposites | 1250 | 47 |
| MIL-68(In) nanorods | 1204 | 48 |
| Histidine/layered double hydroxide hybrid nanosheets | 1112 | 49 |
| CeO2 nanowire arrays | 987 | 50 |
| Hierarchical CeO2 nanospheres | 942.7 | 51 |
| Pentiptycene-based microporous organic polymer | 932 | 33 |
| Ni/Co-LDH hollow dodecahedron | 909.2 | 52 |
| NiO nanospheres | 440 | 53 |
| Magnesium borate microspheres | 202.84 | 54 |
To explore the possible adsorption-enhanced mechanism of shell components-CuOxCu(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies, multiple factors were investigated. Firstly, the zeta potential of as-prepared adsorbents was investigated, which usually rules the adsorption performance in some previous reports.35–37 The measurement results showed that all the adsorbents in neutral pH were positively charged, evidencing the favorable adsorption for the negatively charged CR molecules. However, the little difference amongst all adsorbents (zeta potential was in the range of +15 to +20 mV) ruled out the possibility that zeta potential made a big contribution to the improved adsorption capacity. Secondly, the specific surface area, which is of key significance in improving the adsorption capacity,38,39 was calculated according to N2 adsorption–desorption isotherms. The obtained results showed that there was only a tiny difference among all adsorbents (Table S1†), indicating that specific surface area exerted a negligible influence on the adsorption capacity. Thirdly, the dependence of adsorption capacity on the shell thickness and the phase ratio of CuO and Cu(OH)2 in CuOxCu(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies was investigated. As discussed above, the shell components acted a crucial role in improving CR adsorption capacity. The fact meant that a larger thickness was favorable for the improved adsorption capacity, consistent with the results reported by Du's group.40 As incineration temperature increased, the shell thickness decreased gradually as revealed by TEM images (Fig. S2†) and the phase ratio of CuO and Cu(OH)2 increased as demonstrated by XPS analysis (Fig. 3c) and color change (Fig. 3d). These results justified that a critical shell thickness and phase ratio of CuO and Cu(OH)2 resulted in the improved adsorption capacity. A series of further experiments are carrying out to give a detailed interpretation on the adsorption-enhanced mechanism of the shell thickness and the phase ratio of CuO and Cu(OH)2 in CR removal process.
The stability is a critical indicator to evaluate the availability of adsorbents in dye wastewater treatment. Here, the stability of adsorbents was studied through successive recycling experiments of 4A-Cu-300 adsorbents. After first adsorption experiment finished (fresh adsorbents), the adsorbents were regenerated by annealing in air at 300 °C for another 5 h, followed by the three consecutive adsorption experiments in 100 ml 1000 mg L−1 CR solution and the results were shown in Fig. 7. The regenerated adsorbents showed little difference in adsorption capacity (412.830 mg g−1 at 50 min), but had a poorer CR removal than that of the fresh adsorbents (476.357 mg g−1 at 50 min), proving the good stability of regenerated adsorbents. It was very fascinating to explore the reason why a decreased adsorption performance of regenerated absorbents occur, and hence we performed various characterizations to discriminate the difference between fresh materials and regenerated ones. The SEM and TEM images of third (3rd) regenerated adsorbents showed they still remained a core–shell structure with interconnected nanosheet assemblies on the surface of 4A zeolite (Fig. 8a and b). The Cu 2p XPS spectrum in Fig. 8c gave the evidence that only cupric ions were in 3rd regenerated adsorbents, consistent with that of fresh 4A-Cu-300.23,26 Fig. 8d displayed the digital photos of fresh adsorbents and 3rd regenerated adsorbents, where the regenerated adsorbents appeared darker green than the fresh ones. The C1s XPS spectra (Fig. 8e) showed that carbon atom ratio in the regenerated adsorbent was slightly higher than that in fresh samples (20.57% at% vs. 17.76% at%), while the content of C–C species increased obviously (43.99 at% vs. 30.09 at%). This fact demonstrated that, during the regeneration of adsorbent, partial carbon elements in CR dye molecules turned into “carbon deposition”55 or coke carbon56 and thus existed on the surface of regenerated adsorbents. Nowadays, many studies56–59 have stated that coke carbon could occupy the active sites and led to the decrease of catalysis or adsorption activity. Therefore, the coke carbon species residual on the surface of the regenerative adsorbent presumably decreased the adsorption property of the regenerative adsorbents.
 | ||
| Fig. 7 The successive recycling experiments of 4A-Cu-300. Other conditions: CR = 100 ml, 1000 mg L; adsorbent = 0.2 g; pH = 6.30; T = 25 °C. | ||
 | ||
| Fig. 8 The characterization of 4A-Cu-300 (fresh adsorbents) and regenerated adsorbents. (a) SEM image and (b) TEM image of 3rd regenerated adsorbents. (c) The Cu2p1/2 XPS spectra of 4A-Cu-300 and 3rd regenerated adsorbents. (d) The digital pictures of 4A-Cu-300 and 3rd regenerated adsorbents. (e) The C1s XPS spectra of 4A-Cu-300, 3rd regenerated adsorbents. | ||
In addition, the residual concentration of copper(II) ions in remaining solution after adsorption reaction finished (4A-Cu-300) was measured due to its high toxicity to aquatic lives and human beings.60,61 The measurement results of inductively coupled plasma-mass spectrometry (ICP-MS) exhibited that the concentration of total copper in aqueous solution was about 0.13 mg L−1, which was far below the regulatory limit of the People' Republic of China drinking water standard (GB5749-2006) and integrated wastewater discharge standard (GB8978-2002). At the same time, only ca. 0.072% copper element loses from 4A-Cu-300 adsorbents, which would not compromise its adsorption performance. Based on the discussion above, hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures are expected to be promising candidates for dye wastewater treatment due to their easy-to-prepare, high adsorption capacity and excellent stability.
Conclusions
To sum up, a simple, green and scale-up method was developed to achieve hectogram-scale synthesis of hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures, which exhibited excellent CR adsorption performance. Various characterization techniques, including scanning electron microscopy, transmission electron microscopy, X-ray diffraction and photoelectron spectroscopy so on, identified undoubtedly that shell components-CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies grew uniformly on the surface of 4A zeolite to form core–shell-type adsorbents. The adsorption experiments demonstrated that 4A-Cu-300 showed best adsorption performance amongst all adsorbents, with maximum adsorption capacity of 512.987 mg g−1. The shell components-CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies played a pivotal role in enhancing the adsorption performance of as-prepared adsorbents and the adsorption capacity of shell components could reached up to 3685.500 mg g−1. Adsorption dynamics as studied by time variation and thermodynamics by concentration variation indicated that the adsorption data followed the pseudo second order kinetics and the Langmuir isotherm model, respectively. The adsorption capacity exhibited a strong dependence on the shell thickness and phase ratio of CuO and Cu(OH)2 in CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies. The successive adsorption experiments demonstrated the good stability of the regenerated adsorbents, but the coke carbon led to their poorer adsorption performance than fresh ones. It is expected that hierarchical 4A zeolite@CuOx(OH)(2−2x) (0 ≤ x < 1) nanosheet assemblies core–shell nanoarchitectures can be promising candidates in dye wastewater remediation. Meanwhile, this discovery highlights the importance of rational decoration of an inactive material with hierarchical nanoarchitectures in transforming it into high-performance adsorbent.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21676201, 21706189, 51503146, 21878277), National Key Research and Development Plan (2017YFC0404001), Tianjin Municipal Education Commission Scientific Research Project (2017KJ074), Science and Technology Plans of Tianjin (18JCQNJC06800, 17PTSYJC00050, 18PTSYJC00190), Technology Research Funds Projects of Ocean (201305004-5), Program for Innovative Research Team in University of Tianjin (No. TD13-5044), and Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education of China (Grant no. IRT13084).Notes and references
- J. Liu, S. Cheng, N. Cao, C. Geng, C. He, Q. Shi, C. Xu, J. Ni, R. M. DuChanois, M. Elimelech and H. Zhao, Nat. Nanotechnol., 2019, 14, 64–71 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS.
- Y. Pi, X. Li, Q. Xia, J. Wu, Y. Li, J. Xiao and Z. Li, Chem. Eng. J., 2018, 337, 351–371 CrossRef CAS.
- P. Tian, X.-y. Han, G.-l. Ning, H.-x. Fang, J.-w. Ye, W.-t. Gong and Y. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 12411–12418 CrossRef CAS.
- S. De Gisi, G. Lofrano, M. Grassi and M. Notarnicola, Sustainable Mater. Technol., 2016, 9, 10–40 CrossRef CAS.
- N. Huang, L. Zhai, H. Xu and D. Jiang, J. Am. Chem. Soc., 2017, 139, 2428–2434 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS.
- Q. Al-Naddaf, H. Thakkar and F. Rezaei, ACS Appl. Mater. Interfaces, 2018, 10, 29656–29666 CrossRef CAS.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D.-L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- C. Lei, X. Zhu, B. Zhu, C. Jiang, Y. Le and J. Yu, J. Hazard. Mater., 2017, 321, 801–811 CrossRef CAS PubMed.
- K. Peng and H. Yang, Chem. Commun., 2017, 53, 6085–6088 RSC.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- L. Yu, H. Zhou, J. Sun, F. Qin, D. Luo, L. Xie, F. Yu, J. Bao, Y. Li, Y. Yu, S. Chen and Z. Ren, Nano Energy, 2017, 41, 327–336 CrossRef CAS.
- C. Chen, C. F. H. Byles, J.-C. Buffet, N. H. Rees, Y. Wu and D. O'Hare, Chem. Sci., 2016, 7, 1457–1461 RSC.
- W. L. J. Kwok, D.-G. Crivoi, C. Chen, J.-C. Buffet and D. O'Hare, Dalton Trans., 2018, 47, 143–149 RSC.
- J.-C. Buffet, C. F. H. Byles, R. Felton, C. Chen and D. O'Hare, Chem. Commun., 2016, 52, 4076–4079 RSC.
- R. Li, T. Xue, R. Bingre, Y. Gao, B. Louis and Q. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 34834–34839 CrossRef CAS PubMed.
- L. M. Gilbertson, J. B. Zimmerman, D. L. Plata, J. E. Hutchison and P. T. Anastas, Chem. Soc. Rev., 2015, 44, 5758–5777 RSC.
- H. Duan, D. Wang and Y. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- A. Mundstock, N. Wang, S. Friebe and J. Caro, Microporous Mesoporous Mater., 2015, 215, 20–28 CrossRef CAS.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- J. F. Edwards and G. L. Schrader, J. Phys. Chem., 1984, 88, 5620–5624 CrossRef CAS.
- G. Papadimitropoulos, N. Vourdas, V. E. Vamvakas and D. Davazoglou, Thin Solid Films, 2006, 515, 2428–2432 CrossRef CAS.
- G. Papadimitropoulos, N. Vourdas, V. E. Vamvakas and D. Davazoglou, J. Phys.: Conf. Ser., 2005, 10, 182 CrossRef CAS.
- S. Gong, X. Wu, J. Zhang, N. Han and Y. Chen, CrystEngComm, 2018, 20, 3096–3104 RSC.
- Z. Hong, Y. Cao and J. Deng, Mater. Lett., 2002, 52, 34–38 CrossRef CAS.
- X.-Q. Xin and L.-M. Zheng, J. Solid State Chem., 1993, 106, 451–460 CrossRef CAS.
- L. Zhang, H. Ye, L. Zhao, L. Zhang, L. Yao, Y. Zhang and H. Li, Chem. Commun., 2015, 51, 16936–16939 RSC.
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS.
- X. Qiu, H. Zhang, P. Wu, F. Zhang, S. Wei, D. Sun, L. Xu and Y. Tang, Adv. Funct. Mater., 2017, 27, 1603852–1603860 CrossRef.
- B. Huang, Y. Liu, B. Li, H. Wang and G. Zeng, RSC Adv., 2019, 9, 32462–32471 RSC.
- Z. Bai, Y. Zheng and Z. Zhang, J. Mater. Chem. A, 2017, 5, 6630–6637 RSC.
- S. Luo, Q. Zhang, Y. Zhang, K. P. Weaver, W. A. Phillip and R. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 15174–15182 CrossRef CAS.
- S. Zhao, D. Kang, Z. Yang and Y. Huang, Appl. Surf. Sci., 2019, 488, 522–530 CrossRef CAS.
- M. Dai, J. Colloid Interface Sci., 1994, 164, 223–228 CrossRef CAS.
- X.-D. Du, C.-C. Wang, J.-G. Liu, X.-D. Zhao, J. Zhong, Y.-X. Li, J. Li and P. Wang, J. Colloid Interface Sci., 2017, 506, 437–441 CrossRef CAS PubMed.
- X.-Q. Qiao, F.-C. Hu, F.-Y. Tian, D.-F. Hou and D.-S. Li, RSC Adv., 2016, 6, 11631–11636 RSC.
- N. Hegyesi, R. T. Vad and B. Pukánszky, Appl. Clay Sci., 2017, 146, 50–55 CrossRef CAS.
- A. W. Lounsbury, J. S. Yamani, C. P. Johnston, P. Larese-Casanova and J. B. Zimmerman, J. Hazard. Mater., 2016, 310, 117–124 CrossRef CAS PubMed.
- Y. Du, W. Liu, R. Qiang, Y. Wang, X. Han, J. Ma and P. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 12997–13006 CrossRef CAS PubMed.
- M. Hu, X. Yan, X. Hu, J. Zhang, R. Feng and M. Zhou, J. Colloid Interface Sci., 2018, 510, 111–117 CrossRef CAS PubMed.
- X.-F. Yu, J.-W. Liu, H.-P. Cong, L. Xue, M. N. Arshad, H. A. Albar, T. R. Sobahi, Q. Gao and S.-H. Yu, Chem. Sci., 2015, 6, 2511–2515 RSC.
- H. Zhang, J. Hu, J. Xie, S. Wang and Y. Cao, RSC Adv., 2019, 9, 2011–2017 RSC.
- S. He, X. Liu, P. Yan, A. Wang, J. Su and X. Su, RSC Adv., 2019, 9, 4908–4916 RSC.
- X. Huang, F. Yu, Q. Peng and Y. Huang, RSC Adv., 2018, 8, 29781–29788 RSC.
- M. Hui, P. Shengyan, H. Yaqi, Z. Rongxin, Z. Anatoly and C. Wei, Chem. Eng. J., 2018, 345, 556–565 CrossRef CAS.
- S. Yang, L. Wang, X. Zhang, W. Yang and G. Song, Chem. Eng. J., 2015, 275, 315–321 CrossRef CAS.
- L.-N. Jin, X.-Y. Qian, J.-G. Wang, H. Aslan and M. Dong, J. Colloid Interface Sci., 2015, 453, 270–275 CrossRef CAS PubMed.
- M. Shamsayei, Y. Yamini and H. Asiabi, J. Colloid Interface Sci., 2018, 529, 255–264 CrossRef CAS PubMed.
- X.-h. Lu, D.-z. Zheng, J.-y. Gan, Z.-q. Liu, C.-l. Liang, P. Liu and Y.-x. Tong, J. Mater. Chem., 2010, 20, 7118–7122 RSC.
- X. Ouyang, W. Li, S. Xie, T. Zhai, M. Yu, J. Gan and X. Lu, New J. Chem., 2013, 37, 585–588 RSC.
- H. Hu, J. Liu, Z. Xu, L. Zhang, B. Cheng and W. Ho, Appl. Surf. Sci., 2019, 478, 981–990 CrossRef CAS.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2012, 116, 6873–6878 CrossRef CAS.
- R.-F. Guo, Y.-Q. Ma and Z.-H. Liu, RSC Adv., 2019, 9, 20009–20018 RSC.
- H. Park, X. Li, S. Y. Lai, D. Chen, K. S. Blinn, M. Liu, S. Choi, M. Liu, S. Park and L. A. Bottomley, Nano Lett., 2015, 15, 6047–6050 CrossRef CAS PubMed.
- Q. Yang, F. Chai, C. Ma, C. Sun, S. Shi and L. Chen, J. Mater. Chem. A, 2016, 4, 18031–18036 RSC.
- Q. Liu, M. Gong, B. Cook, D. Ewing, M. Casper, A. Stramel and J. Wu, Adv. Mater. Interfaces, 2017, 4, 1601064 CrossRef.
- S. Gomez Sanz, L. McMillan, J. McGregor, J. A. Zeitler, N. Al-Yassir, S. Al-Khattaf and L. F. Gladden, Catal. Sci. Technol., 2016, 6, 1120–1133 RSC.
- D. Li, M. Lu, K. Aragaki, M. Koike, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2016, 192, 171–181 CrossRef CAS.
- Y. Wang, J. Qin, S. Zhou, X. Lin, L. Ye, C. Song and Y. Yan, Water Res., 2015, 73, 252–264 CrossRef CAS PubMed.
- M. K. Uddin, Chem. Eng. J., 2017, 308, 438–462 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09899b |
| ‡ These authors contributed equally to this work and were the co-first authors. |
| This journal is © The Royal Society of Chemistry 2020 |