DOI:
10.1039/D0RA00072H
(Paper)
RSC Adv., 2020,
10, 12492-12503
A green self-assembled organic supermolecule as an effective flame retardant for epoxy resin†
Received
3rd January 2020
, Accepted 10th February 2020
First published on 27th March 2020
Abstract
Toxicity and environmental issues have elicited research attention regarding the need to prepare a green flame retardant with high flame retardancy. Here, a supermolecular self-assembly technology was used to prepare nickel phytate as shell materials aggregated on aminated silica nanotemplates through electrostatic interactions as a green novel flame retardant (Ni@SiO2-PA). After incorporating the obtained core–shell structured Ni@SiO2-PA into epoxy resin (EP), the supermolecular shell effectively enhanced the adhesive property between the nanoparticles and the EP matrix. The thermal stability was improved, and the peak heat release rate decreased significantly after introducing the well-characterized Ni@SiO2-PA. The absorbance intensity of the toxic aromatic compounds also decreased. Moreover, the char yield of the EP composites was improved because of the synergetic coupled effects between the nickel phytate supermolecules and SiO2 nanotemplates. The possible fire-retardancy mechanism was hypothesized as follows. The crosslinking structure of the silica initially enabled the formation of a polymer network to prevent further decomposition. The N–P synergistic flame-retardancy system then generated a gas barrier and P-rich intumescent char. Besides, char-residue generation was catalyzed by introducing Ni2+, which isolated the heat and the exchange between oxygen and the matrix. Overall, this study proposes a novel green flame retardant that may enable significant improvements in preparing environmentally friendly organic–inorganic materials with applications in the fields of flame-retardant composites.
1. Introduction
Epoxy resin (EP) is a crucial thermosetting material that is extensively used in various industrial fields/devices, such as adhesive, electronic materials, and coating areas, because of its dimensional stability and excellent electrical and mechanical performances.1–3 However, similar to most polymer materials, the applications of EP in several areas are limited by its high flammability and release of a large amount toxic gases during combustion.4–7 Therefore, high-efficiency flame retardants should be developed to improve the flame retardancy of EP. Traditionally, halogenated compounds have been widely used to improve the flame retardancy of polymer materials. Unfortunately, halogenated compounds generate considerable amounts of corrosive and toxic gases during combustion, and the subsequent environmental pollution concerns restrain their use.8,9 In solving these problems, environmentally friendly flame retardants have become a topic of considerable research interest at present.10–12
Currently, several halogen-free flame retardants, such as graphene,13–15 layered double hydroxide (LDH),16,17 boron nitride (BN),18,19 MoS2,20 and carbon nanotubes,12,21 have been rapidly developed. A small amount of nanofiller added into the polymer matrix can result in excellent flame retardancy while simultaneously maintaining or enhancing the other properties of the composites. Zhang et al.22 built a crosslinked network on the basis of cyanate ester (CE) and graphene oxide with P and Si (FGO). Compared with pure CE, FGO/CE resins could obtain outstanding flame retardancy when the P content was as low as 0.18 wt%, which was much lower than those of available P flame retardants. Zhou et al.23 prepared by self-assembly exfoliated MoS2 and LDH through electrostatic force and then introduced these into EP to reduce their fire hazards. Yu et al.4 performed the thermal oxidation of BN to obtain hydroxylated functionalized (BNO) and then covalently incorporated it into modified EP. The addition of BNO into the modified EP enhanced the thermal stability and oxidative stability efficiently. The dispersion quality of the nanofillers mentioned above in the polymer matrix determines their effectiveness in improving the performance of a material. Considerable effort has been dedicated to nanofillers functionalization thus far. The surface chemical modification of nanofillers by materials containing flame-retardant elements, such as N and P, can efficiently improve the flame retardancy and its compatibility with most polymers.24 Flame-retardant compounds are commonly used to modify the nanofillers. However, most of the flame-retardant compounds, such as 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide and its derivatives, are obtained from nonrenewable resources, such as oil.25 Bio-based materials existing in nature as novel flame retardants are the inevitable requirement in developing a green strategy. Therefore, related research has become a topic of considerable concern.
Phytic acid (PA), which is an abundant natural product found in plant seeds and stems, has six phosphate groups that provide a variety of viable crosslinking sites.26,27 As an environmentally friendly and biocompatible organic acid, PA has a potential value in flame-retardant polymers because of its high P content. Wang et al.28 wrapped a PA-doped polypyrrole shell on bulky BN nanosheets as a flame retardant for thermoplastic polyurethane. The resulting system showed a significantly decreased peak heat release and dramatic suppression of CO and HCN releases. Shang et al.29 used melamine and bio-based PA to prepare 2D nanomaterials as flame retardants through self-assembly technology. The catalytic charring performance of the phytate structure generated P-rich intumescent char residue and volatile PO˙, which could rapture OH˙ and H˙ to terminate combustion reactions, respectively.30 Thus, PA, which can enhance the fire safety of polymers, can be used as an eco-friendly flame retardant. Cheng et al. prepared silica coating for improving the flame retardancy of silk fabric using naturally occurring phytic acid as a phosphorus precursor. The results showed that compared with the untreated silk, there was an obvious reduction in PHRR and THR as well as a significant increment in char residue.31 However, to the best of our knowledge, the combination of the aminated SiO2 and PA supermolecular technology and its application in polymer composites has never been reported.
In this work, we developed a facile procedure to prepare a new 3D organic–inorganic nanoparticle to form a core–shell flame retardant that could protect the EP matrix. Nickel phytate acted as the shell wrapped on aminated silica nanotemplates via electrostatic interaction as a green novel flame retardant (Ni@SiO2-PA). Supermolecular self-assembly technology was used and is easily available. The dispersion of Ni@SiO2-PA was observed. The thermal stability and flame retardancy were investigated. This work will trigger additional scientific interest in the development and application of bio-based materials to enhance thermal stability and fire safety during EP combustion.
2. Experimental
2.1 Materials
EP (DGETA) was purchased from Guangzhou Sui Xin Chemical Industrial Co., Ltd. (Guangdong, China). 3-Aminopropyltriethoxysilane (APTES) was provided by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). PA solution (70% in H2O), NiCl2·6H2O, and 4,4′-methylenedianiline (DDM) were obtained from Aladdin Reagent Co., Ltd. (Shanghai, China). Ethanol absolute and NH3 solution were supplied by Beijing Chemical Works (Beijing, China). Silicic acid tetraethyl ester was provided by Tianjin Yong Sheng Fine Chemical Co., Ltd. (Tianjin, China). All the chemical used in this work were of analytical grade and used without further treatment.
2.2 SiO2 synthesis
SiO2 nanospheres were prepared according to the previous method with a slight modification.32 In brief, first, 100 mL of NH3·H2O and 300 mL of absolute ethanol were mixed to obtain solution A, and 15 mL of TEOS was mixed with 30 mL of absolute ethanol to obtain solution B. Then, solution B was slowly added to solution A and stirred for 12 h at 30 °C. Finally, the suspension was centrifuged at 8500 rpm for 3 min. The obtained products were washed with absolute ethanol and deionized water several times, and then dried in an oven at 100 °C for 12 h.
2.3 Ni@SiO2-PA synthesis
The synthesis procedure of Ni@SiO2-PA is shown in Fig. 1. First, 0.5 g SiO2 was ultrasonically mixed in 150 mL of absolute ethanol and 40 mL of deionized water for 10 min. Then, 1 g APTES was slowly added to the solution above, and the mixture was stirred at 75 °C for 24 h. Finally, SiO2 was modified through the APTES (SiO2–NH2) and the suspension was centrifuged and repeatedly washed with absolute ethanol and deionized water, followed by drying in an oven at 100 °C for 12 h.
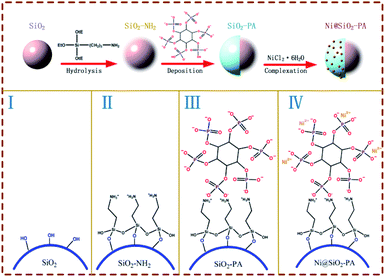 |
| | Fig. 1 Synthesis route of the nanostructure Ni@SiO2-PA. | |
Next, SiO2-PA was prepared via the strong interactions between the positive and negative charges. First, 1 g SiO2–NH2 was mixed with 100 mL of absolute ethanol under ultrasonication. Next, 5 g 70% PA solution was dissolved in 100 mL of absolute ethanol and 20 mL of deionized water and was then added into the solution above, followed by stirring for 12 h at 70 °C. The suspension was centrifuged and washed with absolute ethanol and deionized water. Then, the products were dried in an oven at 100 °C for 12 h.
The final step was the addition of Ni2+ to prepare the Ni@SiO2-PA. A total of 1 g SiO2-PA powder was dispersed in 100 mL of absolute ethanol, followed by ultrasonication for 10 min. Then, 2 g NiCl2·6H2O was dissolved in 40 mL of deionized water and added to the suspension above, and the resulting mixture was stirred for 3 h at 100 °C. Finally, the product was washed with absolute ethanol and deionized water and dried in an oven at 100 °C for 12 h.
2.4 Preparation of Ni@SiO2-PA/EP composites
The epoxy-based nanocomposites containing the Ni@SiO2-PA flame retardant were prepared as follows. First, pure EP was placed in a three-necked bottle and continuously stirred for 10 min at 80 °C. Then, Ni@SiO2-PA was added into the EP and stirred for 20 min, and DDM (25 wt% relative to EP) was added into the system above and further stirred for another 10 min. Finally, the mixture was slowly poured into a PTFE mold and cured at the programmed temperatures of 105 °C for 2 h, 155 °C for 2 h, and 199 °C for 2 h. The composites were marked as EP/Ni@SiO2-PAX, where X represents the Ni@SiO2-PA weight content relative to EP.
2.5 Characterization
Fourier transform infrared (FTIR) spectroscopy (Bruker Vertex 70, German) was performed in transmission mode with KBr pellets in the range of 4000–400 cm−1. X-Ray diffraction (XRD, Bruker D8 ADVANCE) was performed using a diffractometer equipped with Cu Kα radiation (λ = 0.154 nm). X-Ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250) was performed to characterize the elements in Ni@SiO2-PA and char. The excitation source was an Al Kα line at 1486.6 eV. Thermogravimetric analysis (TGA, PerkinElmer) was performed under a N2 atmosphere at a heating rate of 10 °C min−1 to investigate the pyrolysis behavior of the EP composites. Environment scanning electron microscopy (ESEM, FEI XL-30) with an acceleration voltage of 10 kV was utilized to observe the morphologies of SiO2 and its derivatives, as well as the internal structure of the residual char from the cone test. An energy-dispersive X-ray (EDX) spectroscopy (OXFORD) system connected to ESEM was used to investigate the elemental composition of the samples. The structure of SiO2 and its derivatives were observed by transmission electron microscopy (TEM; FEI Tecnai F30 G2, USA). Differential scanning calorimetry (TA Q20) measured the glass transition behavior of the EP composites at a heating rate of 10 °C min−1 under a N2 atmosphere. Limiting oxygen index (LOI) values were measured using an LOI meter (FESTEC JF-3, Korea) with sheet dimensions of 80 mm × 6.5 mm × 3.2 mm in accordance with ASTMD 2863-97. The UL-94 vertical burning properties of the EP composites were examined on a vertical burning tester (Motis Fire Technology UL94-X, China) with the sheet dimensions of 125 mm × 12.7 mm × 3.2 mm according to ASTMD 3801.
Cone calorimetry tests were performed using a calorimeter (iCone, FTT0242) in accordance with the ISO 5660-1 standard under a heat flux of 35 kW m2 and a sample size of 100 mm × 100 mm × 5 mm. TGA-infrared spectrometry (TG-IR) was performed using a TGA (iCone, 209F3) thermogravimetric analyzer linked to an FTIR (Brook, TENSOR27) spectrophotometer from 30 °C to 800 °C at 15 °C min−1 (N2 atmosphere, flow rate of 30 mL min−1). The graphitization degree of the samples was obtained using an inVia-Plus532 confocal Raman microprobe (Renishaw, UK). The detected wavenumber was between 800 and 2000 cm−1. The mechanical properties of the EP composites were investigated using an electronic universal testing machine (WSM-5KN, China) in accordance with GBT 2567.
3. Results and discussion
3.1 Ni@SiO2-PA characterizations
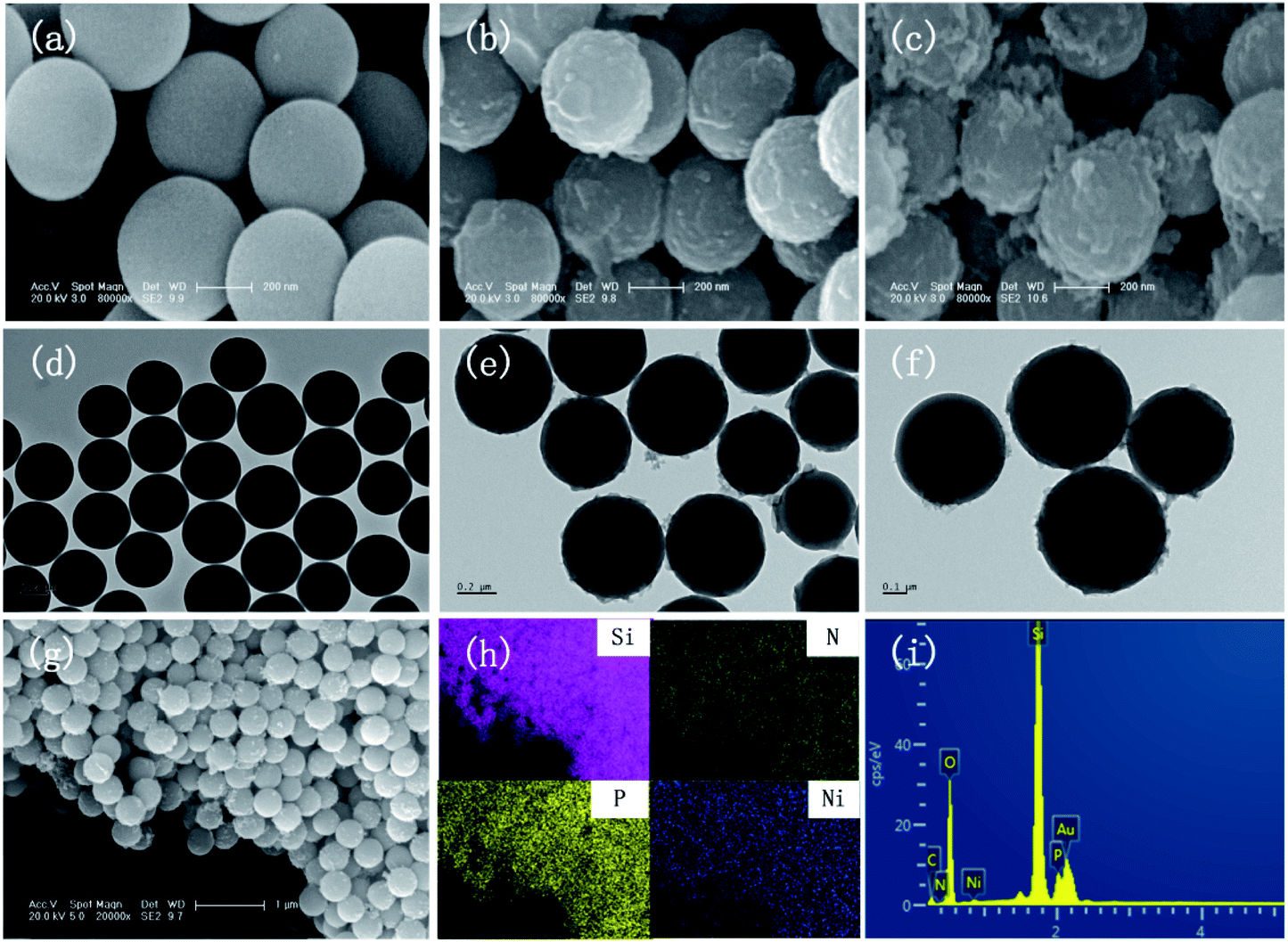
The SEM images of SiO2, SiO2-PA, and Ni@SiO2-PA are shown in Fig. 2. The smooth and regular SiO2 nanospheres with rich OH groups are presented in Fig. 2a. After processing by APTES, the surface of the SiO2 template with amino groups could provide attached sites for further treatment. When the PA wrapping was observed on SiO2–NH2 by electrostatic interaction, a viscous film appeared, as shown in Fig. 2b. The Ni@SiO2-PA particles showed a similar morphology to SiO2-PA. However, the surface was rough, and the PA wrapping on the surface of the aminated SiO2 formed a coordination compound with the added Ni2+, which made the surface substance of the nanospheres increasingly dense, as shown in Fig. 2c. The morphology of Ni@SiO2-PA was further examined by TEM (Fig. 2e and f). Compared with SiO2 (Fig. 2d), the core–shell structure was observed, and the surface of the SiO2 with a diameter of about 410 ± 20 nm was covered with the nickel phytate complex compound. After modification, the surface morphology became rough, and the complex wrapped around the microspheres completely. It was shown that the Ni@SiO2-PA nanospheres were monodispersed with a diameter of 490 ± 25 nm, a bit bigger than that of pure SiO2. Thus, it could be speculated that the thickness of the shell was about 40 nm. Besides, the presence of Si, N, P, and Ni was also found in the EDX spectrum, and all of these elements were homogeneously distributed at the surface (Fig. 2g–i). Thus, the three-step reaction from SiO2 to Ni@SiO2-PA occurred.
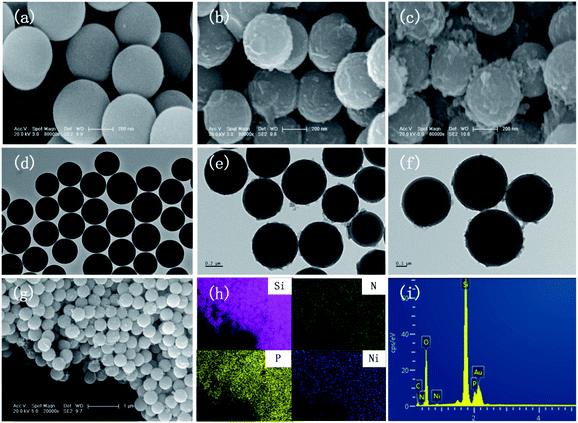 |
| | Fig. 2 SEM images of (a) SiO2, (b) SiO2-PA, and (c) Ni@SiO2-PA. TEM patterns of (d) SiO2 and (e and f) Ni@SiO2-PA. SEM image of (g) Ni@SiO2-PA with (h) elemental mapping of Si, N, P, and Ni and the EDX results of (i) Ni@SiO2-PA. | |
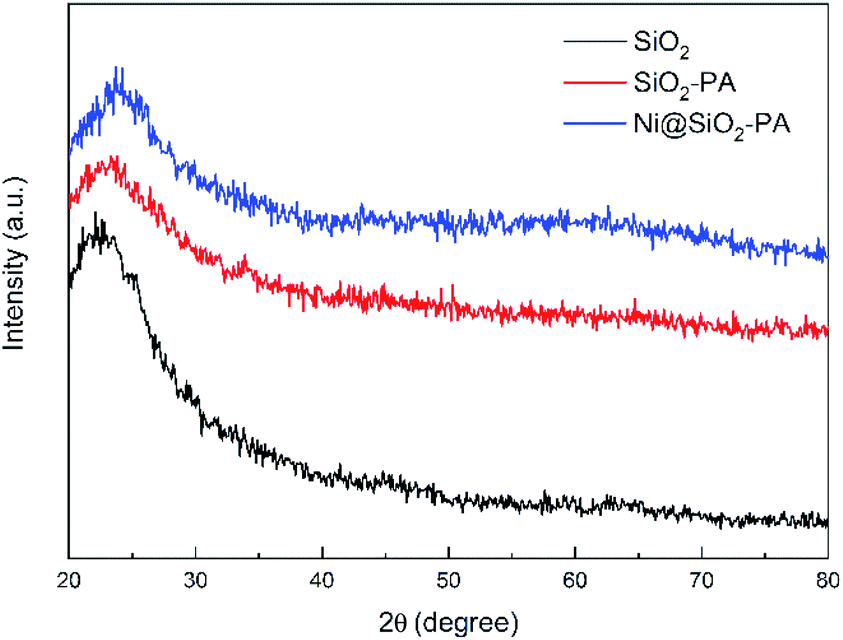
The XRD pattern for SiO2 and its derivatives are shown in Fig. 3. The diffraction pattern of SiO2 showed the reflection characteristics of amorphous silica.5 Compared with the pattern of SiO2, the peak positions of SiO2-PA and Ni@SiO2-PA were the same. However, the peak intensity significantly decreased in the 2θ range from 20° to 30°. This result may be due to the formation of a less ordered stacking structure. The results suggest that the presence of the flame-retardant particles wrapped on SiO2 surface changed its regularity and uniformity but still kept their amorphous features.
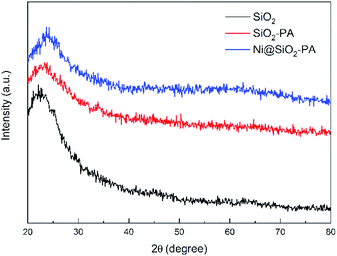 |
| | Fig. 3 XRD patterns of SiO2, SiO2-PA, and Ni@SiO2-PA. | |
The TGA curves of SiO2, SiO2-PA, and Ni@SiO2-PA under the N2 atmosphere are shown in Fig. 4 and its DTG curves are shown in Fig. S1.† SiO2-PA and Ni@SiO2-PA exhibited an evident weight loss between 50 °C and 800 °C, and the solid residue was decreased at 800 °C. This result was due to the presence of unstable N–P-containing components, which decomposed the product early compared with SiO2. Ni@SiO2-PA showed a higher solid residual yield than SiO2-PA at 800 °C, thereby suggesting an improved thermal stability after the decoration of Ni2+.
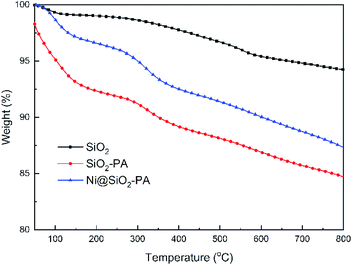 |
| | Fig. 4 TGA curves of SiO2, SiO2-PA, and Ni@SiO2-PA under a N2 atmosphere. | |
To confirm the structures of SiO2-PA further, we used XPS to clarify the elemental components on the SiO2 surface. As shown in Fig. 5, the heteroatom (N, P, and Ni) peaks at 398 (N 1s), 133 (P 2p), 190 (P 2s), 856 (Ni 2p3/2), and 645 eV (Ni LMM) in the XPS spectrum of Ni@SiO2-PA appeared compared with that of pure SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33,34 suggesting that N, P, and Ni atoms were doped into SiO2 successfully. Fig. 5b shows that the C 1s XPS spectrum of Ni@SiO2-PA was divided into four peaks. The characteristic peaks at 287.3, 286.5, 284.8, and 283.9 eV could be attributed to C–O, C–N, C–C, and C–Si, respectively.35,36 Three peaks located at 531.3, 532.4, and 533 eV were observed in the O 1s spectrum, which were attributed to P–O (Si–O), P–O–Ni, and absorbed water, respectively, as shown in Fig. 5c.2 Three peaks were detected in the N 1s XPS (Fig. 5d) at 401.1, 399.8, and 401.6 eV, corresponding to the positively charged N atoms in SiO2–NH2, N–C, and N–H bonds, respectively.37 As shown in Fig. 5e, the P 2p survey spectrum showed two main fitting peaks at 134.2 and 133.5 eV, thereby corresponding to a phosphate group and P–O bonds, respectively.28 Fig. 5f also shows peaks at 856 eV, corresponding to Ni 2P3/2, thereby indicating that Ni ions existed in Ni@SiO2-PA.34 These results demonstrated successful Ni@SiO2-PA synthesis.
33,34 suggesting that N, P, and Ni atoms were doped into SiO2 successfully. Fig. 5b shows that the C 1s XPS spectrum of Ni@SiO2-PA was divided into four peaks. The characteristic peaks at 287.3, 286.5, 284.8, and 283.9 eV could be attributed to C–O, C–N, C–C, and C–Si, respectively.35,36 Three peaks located at 531.3, 532.4, and 533 eV were observed in the O 1s spectrum, which were attributed to P–O (Si–O), P–O–Ni, and absorbed water, respectively, as shown in Fig. 5c.2 Three peaks were detected in the N 1s XPS (Fig. 5d) at 401.1, 399.8, and 401.6 eV, corresponding to the positively charged N atoms in SiO2–NH2, N–C, and N–H bonds, respectively.37 As shown in Fig. 5e, the P 2p survey spectrum showed two main fitting peaks at 134.2 and 133.5 eV, thereby corresponding to a phosphate group and P–O bonds, respectively.28 Fig. 5f also shows peaks at 856 eV, corresponding to Ni 2P3/2, thereby indicating that Ni ions existed in Ni@SiO2-PA.34 These results demonstrated successful Ni@SiO2-PA synthesis.
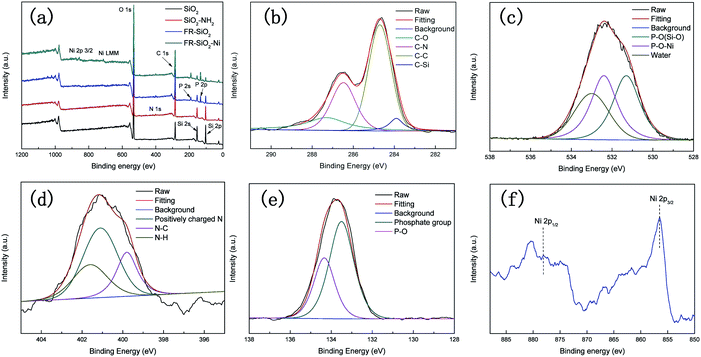 |
| | Fig. 5 XPS scans of (a) SiO2, SiO2-PA, and Ni@SiO2-PA. (b) C 1s, (c) O 1s, (d) N 1s, (e) P 2p, and (f) Ni 2p spectra of Ni@SiO2-PA. | |
3.2 Thermal properties of the EP composites
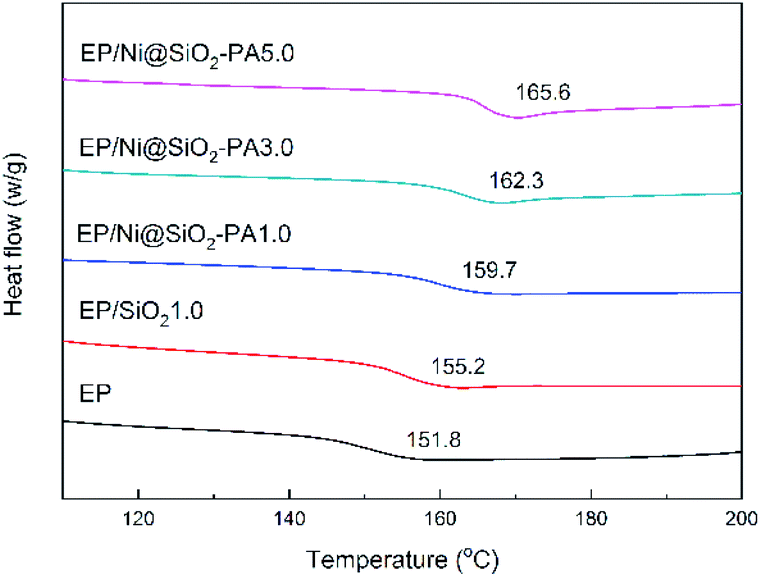
The glass-transition temperature (Tg) of the investigated EP nanocomposites are displayed in Fig. 6. Generally, Tg is a macroscopic indication of the relaxation behavior of nanocomposites.38 The incorporation of SiO2 and Ni@SiO2-PA had a remarkable influence on the Tg of the EP composites. Pure EP exhibited a relatively low Tg value at 151.8 °C. With the incorporation of 1 wt% SiO2, the Tg value of EP/SiO2 increased by 3.4 °C. This result was attributed to the addition of silica microspheres, which limited the movement of the molecular chain in the EP matrix because of steric hindrance. A 1.0 wt% loading of Ni@SiO2-PA in EP enhanced the Tg to 159.7 °C. As the Ni@SiO2-PA content increased to 5.0 wt%, the Tg value of the EP/Ni@SiO2-PA nanocomposites increased to 165.6 °C, which was 13.8 °C higher than that of pure EP. Therefore, the enhancement in Tg could be explained by the existence of steric hindrance by SiO2 and due to the rigid structure of the inositol structures in PA. The supermolecule adhesive formed strong interfacial adhesion between the nanoparticles and EP matrix. This effect would restrict the motions of the polymer chain segments. Besides, aminated SiO2 increased the crosslinking density and reduced the free volume of the EP nanocomposite system. Thus, the incorporation of Ni@SiO2-PA could improve the Tg values of the cured EP composites successfully.
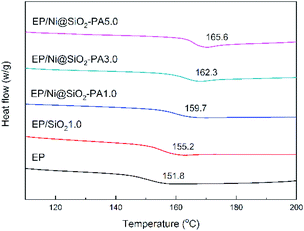 |
| | Fig. 6 DSC thermograms of EP and its composites. | |
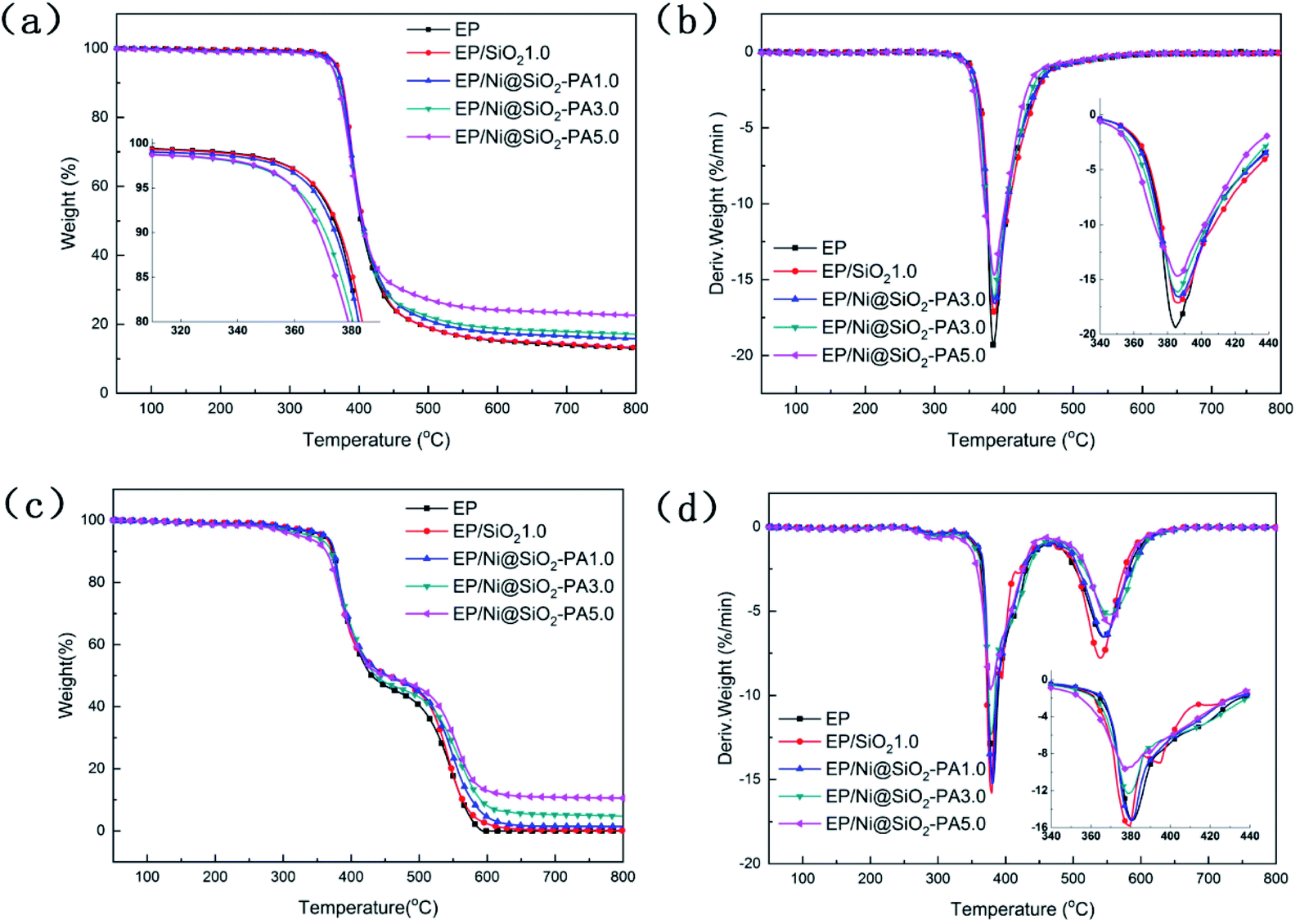
In general, the TG and DTG analyses of the EP composites were performed and the corresponding curves are shown in Fig. 7. In the inert atmosphere, the obtained thermal data are displayed in Table 1, and all the tested materials showed a similar one-stage degradation behavior from 300 °C to 450 °C, which could be attributed to the thermal decomposition of the epoxy chain network.39 As shown in Table 1, the T5 wt% values of the composites with a small amount of SiO2 and Ni@SiO2-PA were higher than those of pure EP, which could be attributed to the considerable thermal stability of SiO2 and its derivatives. When the incorporation of SiO2 and Ni@SiO2-PA reached 5 wt%, the initial thermal decomposition temperature of below 370 °C decreased significantly because of the early degradation of the nickel phytate shell. PA was thermally decomposed into pyrophosphate and polyphosphate, which can catalyze and cause composite decomposition.40 However, the composites incorporating SiO2 and the Ni@SiO2-PA nanospheres showed a relatively better thermal stability than pure EP due to the significant increase in residual char and the reduced mass loss rate (Fig. 7a and b). The obtained thermal data in air are displayed in Table 2. Two degradation stages were observed in the TGA curves for pure EP and its composites (Fig. 7c). The first degradation behavior for EP composites showed no difference compared with in the N2 atmosphere. The second degradation stage occurred from 500 °C to 600 °C, thereby corresponding to char degradation.33 The degradation temperature of char and its decomposition rate decreased as the amount of incorporated Ni@SiO2-PA increased (Fig. 7d). The char residues at 800 °C were also improved after adding SiO2 and Ni@SiO2-PA. When the amount of added Ni@SiO2-PA was 5 wt%, the highest char residue of approximately 10.52% was obtained, and the best thermal stability was exhibited. The evidence of the improving char yield and reducing mass loss rate can be attributed to two factors. First, the EP molecular chains connected by the amino-rich SiO2 as the crosslinking network acted as a physical barrier to protect the EP matrix from thermal degradation. Second, the PA was decomposed into polyphosphates, which were further degraded to form P-containing oxides. This process was beneficial for the formation of char residues, which can inhibit the release of the volatile products and heat exchange during combustion. Overall, adding Ni@SiO2-PA promoted catalytic char residue generation and inhibited EP matrix decomposition efficiently.
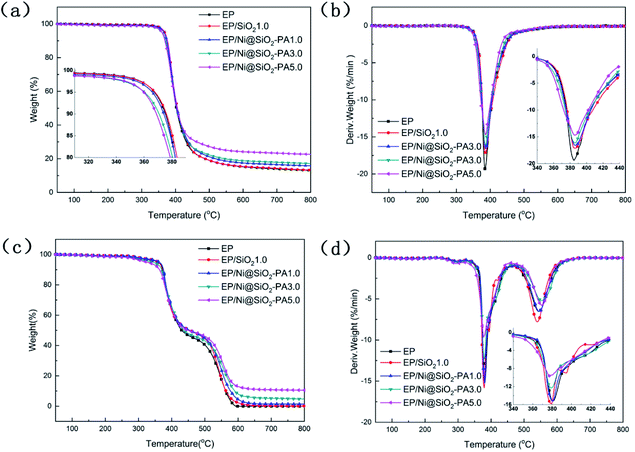 |
| | Fig. 7 TGA (a) and DTG (b) curves of EP and its composites under N2 atmosphere. TGA (c) and DTG (d) curves of EP and its composites in air. | |
Table 1 Thermal data of pure EP and its composites under a N2 atmosphere
| Samples |
T5 wt% (°C) |
T50 wt% (°C) |
Tmax (°C) |
Residue at Tmax °C (wt%) |
Residue at 800 °C (wt%) |
| EP |
367.3 |
402.1 |
384.7 |
75.78 |
13.06 |
| EP/SiO21.0 |
367.8 |
405.5 |
386.5 |
75.52 |
13.26 |
| EP/Ni@SiO2-PA1.0 |
365.8 |
404.5 |
387.0 |
73.07 |
15.83 |
| EP/Ni@SiO2-PA3.0 |
368.5 |
405.7 |
387.1 |
74.02 |
18.44 |
| EP/Ni@SiO2-PA5.0 |
359.6 |
403.8 |
385.6 |
71.28 |
22.60 |
Table 2 Thermal data of pure EP and its composites under an air atmosphere
| Samples |
T5 wt% (°C) |
T50 wt% (°C) |
Tmax1 (°C) |
Tmax2 (°C) |
Residue at Tmax1 °C (wt%) |
Residue at 800 °C (wt%) |
| EP |
358.1 |
430.4 |
380.8 |
544.6 |
82.16 |
0.00 |
| EP/SiO2 |
359.1 |
454.8 |
379.3 |
539.9 |
80.86 |
0.12 |
| EP/Ni@SiO2-PA1.0 |
361.4 |
453.6 |
380.3 |
548.3 |
83.18 |
1.40 |
| EP/Ni@SiO2-PA3.0 |
342.0 |
436.4 |
378.9 |
553.9 |
82.21 |
4.73 |
| EP/Ni@SiO2-PA5.0 |
324.8 |
450.6 |
377.5 |
553.9 |
80.81 |
10.52 |
3.3 Combustion properties
LOI and UL-94 tests are widely used to evaluate the flame-retardancy properties of materials and the related LOI and UL-94 data are summarized in Table 3. As shown in the table, the LOI of pure EP was only 25.7%. EP/Ni@SiO2-PA composites presented an increased tendency of LOI values as the Ni@SiO2-PA content increases. Besides, pure EP cannot pass the UL-94 flammability rating test and easily dripped after ignition. When the amount of Ni@SiO2-PA reached 3 wt%, its UL-94 rating was V-1 and no dripping occurred. The aforementioned results showed that the flame retardancy of EP can be effectively improved by the introduction of Ni@SiO2-PA.
Table 3 Flame-retardancy parameters obtained from the LOI and UL-94 tests
| Samples |
LOI (%) |
Dripping |
UL-94 |
| EP |
25.7 |
Yes |
No rating |
| EP/Ni@SiO2-PA1.0 |
26.2 |
No |
No rating |
| EP/Ni@SiO2-PA3.0 |
26.9 |
No |
V-1 |
| EP/Ni@SiO2-PA5.0 |
28.3 |
No |
V-1 |
The influence of Ni@SiO2-PA with different contents on the flame retardancy of EP was investigated by cone calorimetry, which is an effective tool for evaluating the heat release properties of polymers, including the heat release rate (HRR), total heat release (THR), smoke production rate (SPR), and total smoke production (TSP). The heat and smoke release curves from the cone calorimetry tests are shown in Fig. 8, and the related data are summarized in Table 4. The time to ignition values of the composites were lower than those of pure EP, which may be due to the competition between the effect of thermal conductivity and the shielding performance of the external heat radiant flux.41 High peak values of HRR (1152 kW m−2, Fig. 8a) and THR (99.91 MJ m−2, Fig. 8b) were obtained for pure EP during combustion, which revealed the flammability of EP. After the incorporation of Ni@SiO2-PA, the peak value of HRR was effectively reduced, and the THR values of EP composites were significantly lower than those of pure EP. With as low as 1.0 wt% Ni@SiO2-PA, the peak values of HRR and THR that were achieved in EP/Ni@SiO2-PA here decreased by 27.2% and 24.0%, respectively. This downward trend was in agreement with the increase in additive amount. The maximum decreases in HRR and THR that were achieved with the addition of 5.0 wt% Ni@SiO2-PA accounted for 51.6% and 49.2%, respectively. This phenomenon suggested that a part of the EP matrix in the nanocomposites was shielded during combustion. The remarkable improvements in the flame retardancy of EP/Ni@SiO2-PA could be ascribed to the cooperative effect between the SiO2 and nickel phytate. Here, the supermolecular shell can catalyze the generation of P-rich char residues, and Ni2+ can catalyze the decomposition products of EP to form a stable char, thereby decreasing the combustion fuel supply and heat release.42 SiO2 can form a Si-containing polymer crosslinked network that acts as a physical barrier. The majority of deaths in fire disasters are due to the production of highly toxic smoke particles.13 The SPR and TSP curves of pure EP and its composites are shown in Fig. 8c and d, respectively. Given its specific multiaromatic structure, pure EP exhibited a high toxic smoke yield with high SPR and TSP values. The TSP value of EP/Ni@SiO2-PA5.0 was significantly decreased from 23.26 m2 for pure EP to 11.69 m2, thereby indicating that smoke production was effectively suppressed. This result exhibited the best flame retardancy of all the samples. Combined with TGA, the significant reduction in smoke release was attributed to the presence of PA, which can be thermally decomposed into pyrophosphate and polyphosphate to catalyze the formation of a cohesive and compact char layer during combustion, which delays the permeation of O and the escape of volatile degradation products. These results confirmed the suppression effect of Ni@SiO2-PA nanoparticles on the fire hazard of the EP matrix.
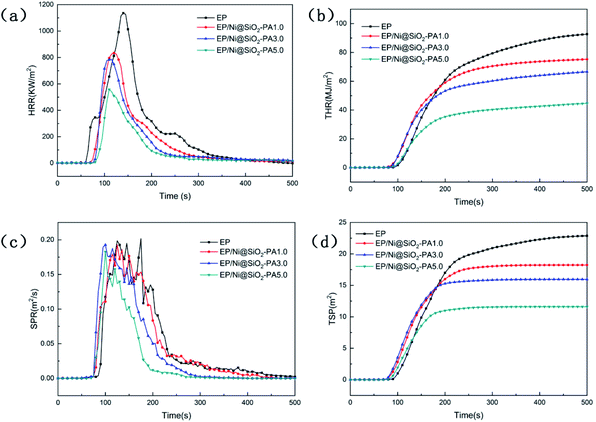 |
| | Fig. 8 HRR (a), THR (b), SPR (c), and TSP (d) vs. time curves of EP and its composites. | |
Table 4 Related results for the cone calorimeter measurements
| Samples |
TTI (s) |
TPHRR (S) |
PHRR (kW m−2) |
THR (MJ m−2) |
SP (m2) |
COP (g s−1) |
CO2P (g s−1) |
| EP |
83 |
140 |
1152 |
99.91 |
23.26 |
0.55 |
13.39 |
| EP/Ni@SiO2-PA1.0 |
71 |
117 |
839 |
75.92 |
18.23 |
0.45 |
8.10 |
| EP/Ni@SiO2-PA3.0 |
74 |
107 |
816 |
70.72 |
15.95 |
0.37 |
9.50 |
| EP/Ni@SiO2-PA5.0 |
74 |
111 |
558 |
50.76 |
11.69 |
0.34 |
9.38 |
3.4 Gas- and condensed-phase analysis
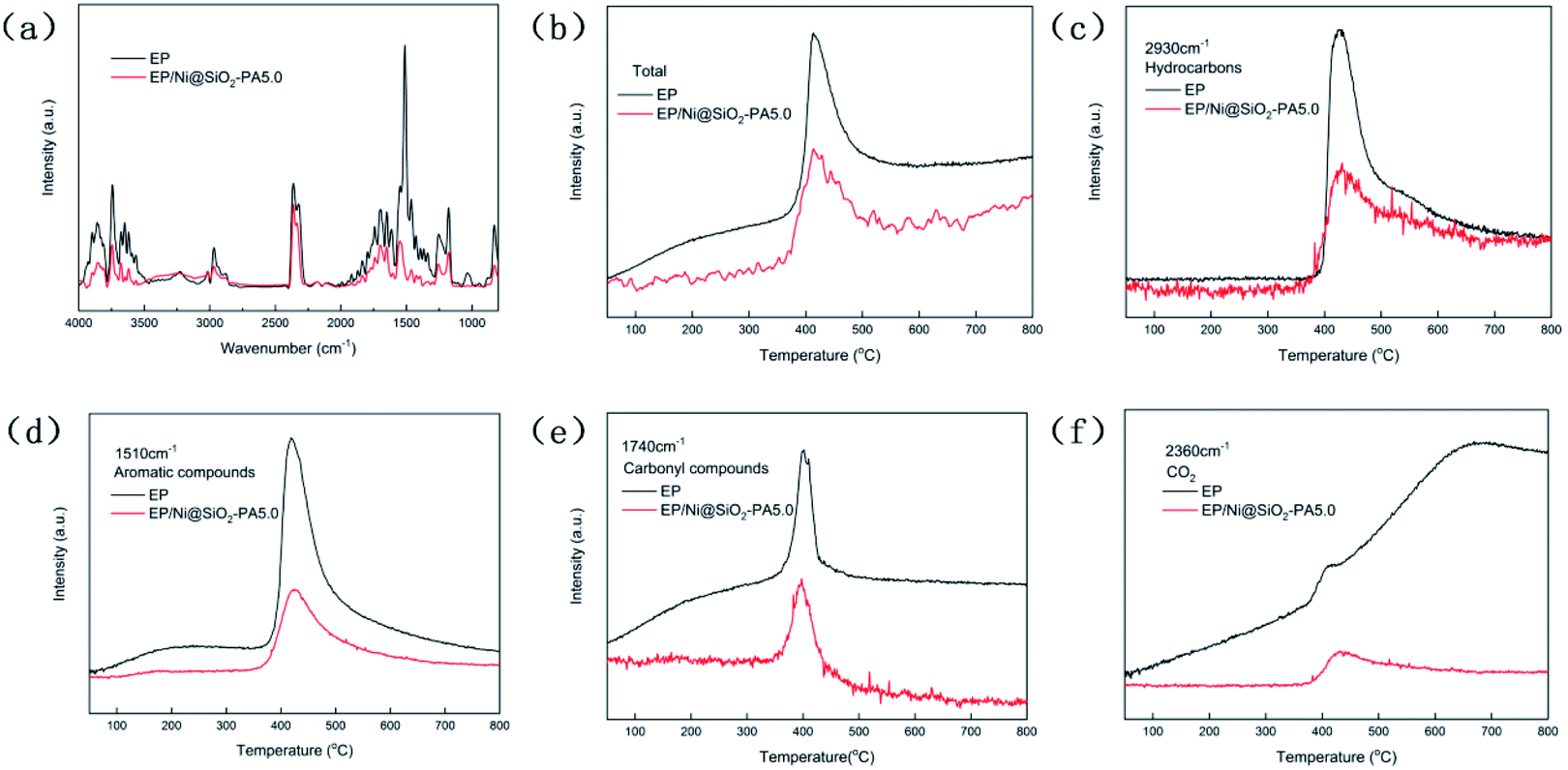
To study the decomposed volatiles of pure EP and its composites further, we performed TG-IR to track the production process of the gas volatiles at the maximum degradation, as presented in Fig. 9. The FTIR spectra of the EP composites showed similar spectra to those of pure EP, thereby indicating that the incorporation of Ni@SiO2-PA did not change the types of decomposition products (Fig. 9a). All the absorbance peaks for EP/Ni@SiO2-PA were significantly decreased compared with those of pure EP, including the total release, hydrocarbon content (2930 cm−1), aromatic compound content (1510 cm−1), carbonyl compound content (1740 cm−1), and CO2 content (2360 cm−1), as shown in Fig. 9b–f.21 This result showed that adding Ni@SiO2-PA suppressed the release of the decomposed fragments. This was attributed to the generation of a compact and cohesive char layer as a reinforcement barrier, which retarded the escape of the pyrolysis products. The reduced gases also resulted in the reduced HRR because most of the organic volatiles were flammable gases, which can act as a fuel to support the burning. This result illustrated that incorporating Ni@SiO2-PA into the EP matrix could reduce the supplementation of flammable pyrolysis products to the fire.
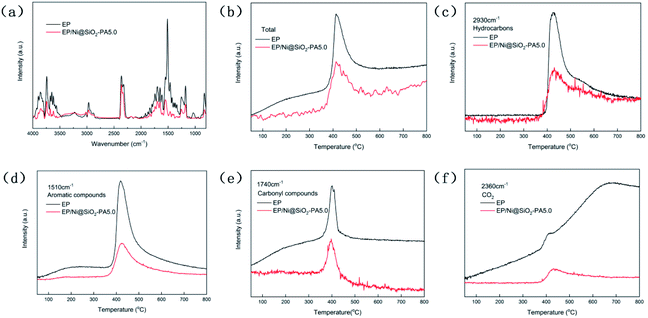 |
| | Fig. 9 (a) FTIR spectra of pyrolysis gaseous products emitted from pure EP and EP/Ni@SiO2-PA5.0 composites at maximum degradation rate; (b) total absorbance of pyrolysis products for pure EP and EP/Ni@SiO2-PA5.0 composites; (c) absorbance of pyrolysis products for pure EP and EP/Ni@SiO2-PA5.0 composites vs. temperature: hydrocarbons, (d) aromatic compounds, (e) carbonyl compounds, and (f) CO2. | |
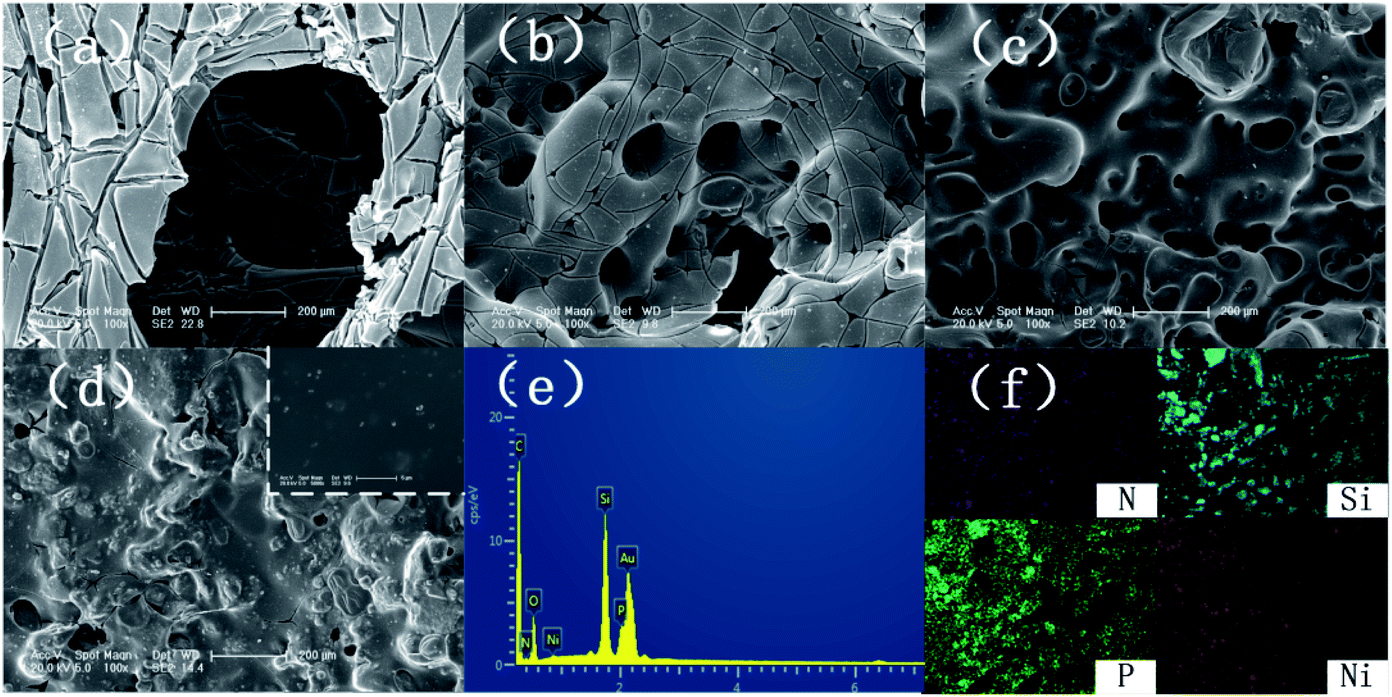
Fig. 10 presents digital photos for the residues for the pure EP and its composites after the cone calorimetry tests. Apparently, pure EP left a small amount of residue (Fig. 10a). For EP/Ni@SiO2-PA1.0, a small amount of flame retardant was insufficient to protect the matrix and did not increase the yield of the residue (Fig. 10b). With the added amount of Ni@SiO2-PA increasing, the flame-retardant EP nanocomposites produced additional char residue. For EP/Ni@SiO2-PA3.0 (Fig. 10c), an intumescence of the char layers could be observed. However, agglomerated char layers existed in the middle of the sample, which could be attributed to a relatively poor dispersion. For EP/Ni@SiO2-PA5.0 (Fig. 10d), a compact structure could be observed after the composites were burned. Then, the SEM technique coupled with EDX was used to investigate the structure and morphology of the residual char, as shown in Fig. 11. After combustion, a broken and fragile char residue was obtained for pure EP, and huge holes existed in the residual char, thereby suggesting poor thermal stability, according to its SEM image (Fig. 11a). When 1 wt% Ni@SiO2-PA was incorporated into the EP matrix, the cracks and holes significantly decreased, but they were still insufficient to protect the polymer layer inside the composites. The structural defect acted as channels for the exchange of heat, O, and gas products (Fig. 11b). For the composites incorporated with 3 wt% Ni@SiO2-PA (Fig. 11c), a bubble-like structure was observed on the resulting char. Meanwhile, the crack on the matrix disappeared. Intumescent-like bubbles can slow down the heat and mass transfer between gas and condensed phases.43 However, the presence of several ruptured bubbles revealed that the char's strength was not strong enough to withstand the expansion of the gas product.33 When the Ni@SiO2-PA content was increased to 5 wt%, a compact and rigid char residue was formed, and no evident cracks and holes were observed in its SEM image (Fig. 11d). The SiO2 nanoparticles were uniformly embedded in the matrix, as seen from high magnification. Thus, the char can act as an efficient shield for the underlying matrix. At the surface of the its residual char, N, Si, P, and Ni were detected and homogeneously distributed (Fig. 11e and f). The P-rich char residues were involved in char layer formation, which hindered the transfer of heat flow and provided good flame retardancy. Ni was deposited, thereby indicating that it participated in the charring process. Ni catalyzed the carbonization of the burning polymers and led to the formation of several types of C materials. Generally, Ni catalyzes the dehydrogenation and aromatization of the intermediate compounds, thereby forming additional protective char layers.34,36
 |
| | Fig. 10 Digital photographs of char residues after the cone calorimetry tests: (a) pure EP, (b) EP/Ni@SiO2-PA1.0, (c) EP/Ni@SiO2-PA3.0, and (d) EP/Ni@SiO2-PA5.0 composites. | |
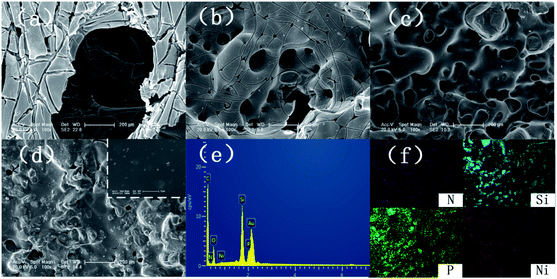 |
| | Fig. 11 SEM images of char structures obtained by cone calorimeter tests: (a) pure EP, (b) EP/Ni@SiO2-PA1.0, (c) EP/Ni@SiO2-PA3.0, and (d) EP/Ni@SiO2-PA5.0. EDX spectrum (e) and element mapping (f) of EP/Ni@SiO2-PA5.0 composites after calorimetry. | |
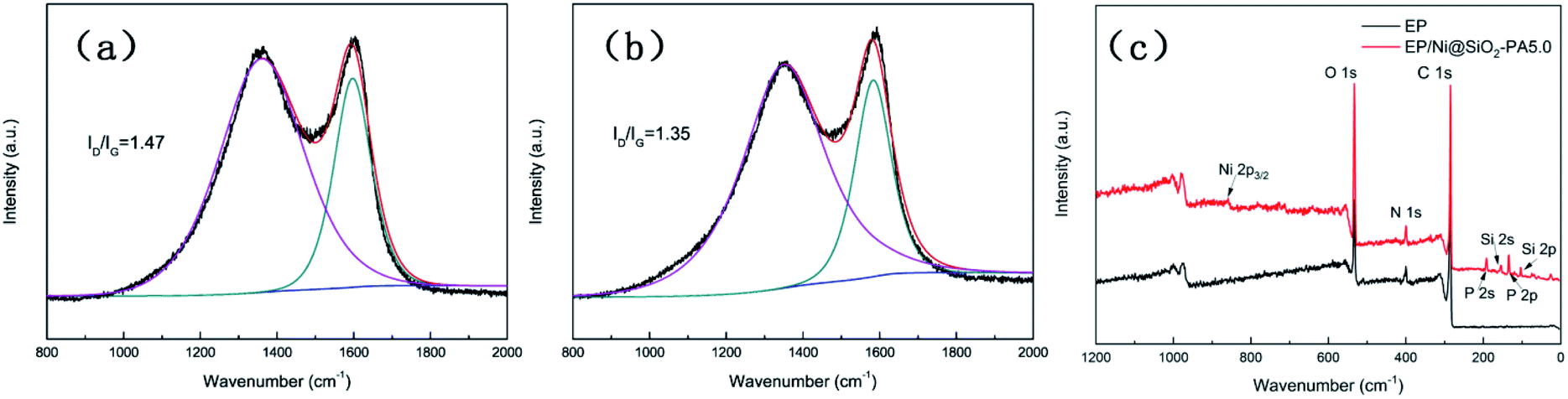
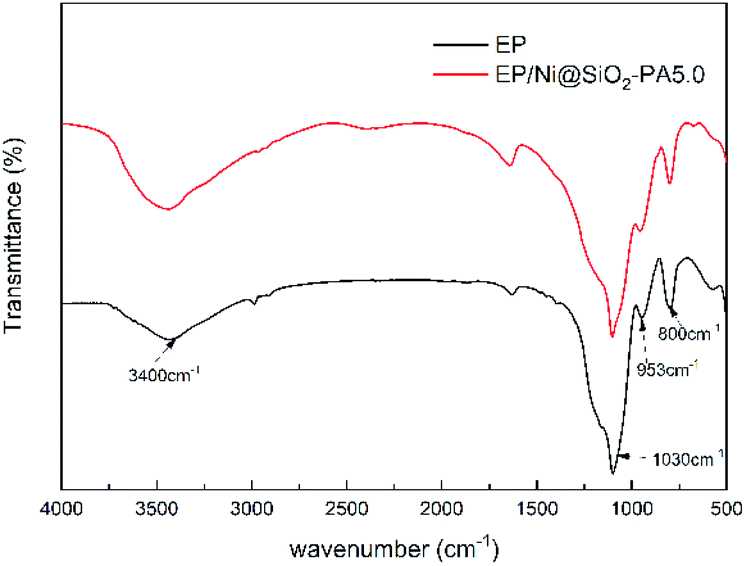
Raman spectroscopy was carried out to determine the graphitization degree of the char residues obtained from the cone calorimetry tests, as shown in Fig. 12. Raman spectra analysis revealed the presence of the D (1590 cm−1) and G bands (1361 cm−1), which were attributed to the A1g breathing vibration of the sp3-hybridized C and E2g in-plane stretching of the six-ring sp2 C, respectively.44 Here, the lower the ID/IG ratio is, the higher the graphitization degree of the residual char will be. The ID/IG ratio of pure EP was 1.47 (Fig. 12a). Nevertheless, the EP/Ni@SiO2-PA5.0 composites had a low ID/IG ratio of 1.35 (Fig. 12b), thereby indicating the formation of a char with a high degree of graphitization and a thermally stable char structure. The XPS survey scans of the residual char of pure EP and EP/Ni@SiO2-PA5.0 are shown in Fig. 12c. Si, N, P, and Ni were detected in the residual char. The intensities of the peaks responding to C 1s and O 1s in the residual char of EP/Ni@SiO2-PA5.0 were also significantly higher than those of pure EP. Fig. 13 shows the FTIR spectra of the residual char, which provide additional information on the chemical structure of the residual char. For pure EP, the peak at 3450 cm−1 was attributed to water, and the peak at approximately 1600–1500 cm−1 revealed the multiaromatic structure of the residual char. EP/Ni@SiO2-PA5.0 showed a similar char structure caused by the similar spectrum of the pure EP. However, the peaks at approximately 1102 and 1030 cm−1 were attributed to Si–O–C and Si–O–Si bonds, which were obtained from the silica crosslinked network, respectively.45 The intense peaks at approximately 1139 and 1160 cm−1 were assigned to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O–C vibrations PA groups, respectively.2,38 The phenomenon above can verify the formation of the Si crosslink network and the participation in the charing process of the composites. PA could also catalyze the generation of char and acted as a catalyst for the dehydration process, which promoted the oxidative dehydrogenation crosslinking–charring process and increased the char yield, thereby improving the flame retardancy of the EP composites.24 Meanwhile, several kinds of C materials were formed on the Ni catalyst surface. Thus, the char layer provided an effective protection, and reduced the heat and mass transfers to protect the EP matrix, which is in accordance with the TGA results.
O and P–O–C vibrations PA groups, respectively.2,38 The phenomenon above can verify the formation of the Si crosslink network and the participation in the charing process of the composites. PA could also catalyze the generation of char and acted as a catalyst for the dehydration process, which promoted the oxidative dehydrogenation crosslinking–charring process and increased the char yield, thereby improving the flame retardancy of the EP composites.24 Meanwhile, several kinds of C materials were formed on the Ni catalyst surface. Thus, the char layer provided an effective protection, and reduced the heat and mass transfers to protect the EP matrix, which is in accordance with the TGA results.
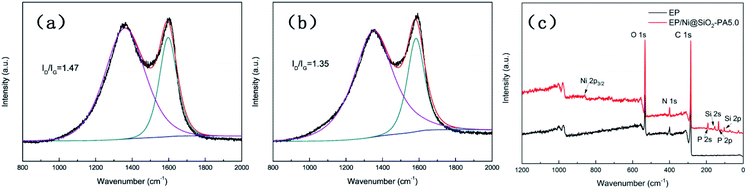 |
| | Fig. 12 Raman spectra of residual chars of (a) pure EP and (b) EP/Ni@SiO2-PA5.0 composites; (c) XPS spectra of EP and EP/Ni@SiO2-PA5.0 composite residues. | |
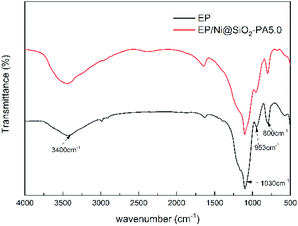 |
| | Fig. 13 FTIR spectra of the char residues of pure EP and EP/Ni@SiO2-PA5.0 composites. | |
3.5 Flame-retardancy mechanism
According to the discussion above, the added Ni@SiO2-PA could efficiently enhance the flame-retardancy properties of the EP composites. As presented in Fig. 14, when the EP/Ni@SiO2-PA composites were heated to a certain temperature, the flame retardant that was wrapped on SiO2 began to decompose. First, the N–P synergistic flame retardancy system played a major role in the gas phase. Nonflammable gases, such as N2 and NH3, formed a gas protective layer and decreased the flammable gas concentration.
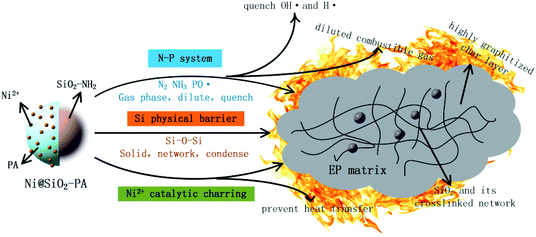 |
| | Fig. 14 Scheme of proposed flame-retardant mechanism for Ni@SiO2-PA in EP composites. | |
PA molecule also decomposed into substantial amounts of P-free radicals, such as 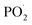 and PO˙, which react with flammable radicals (H˙ and OH˙), reduce the opportunity of combustion, and produce a quenching effect.46 Second, in the condensed phase, the crosslinking structure of silica to form a polymer network prevented further matrix decomposition. Meanwhile, the decomposition products of the supermolecular-produced phosphorous oxide, which generated P-rich intumescent char, formed stable residual chars, and restricted the formation of holes and cracks. The Ni compounds also catalyzed the carbonization of the burning polymers top and formed a compact and continuous bubble-like char layer. Thus, the physical barrier formed by Ni@SiO2-PA could protect the matrix by isolating O- and heat-transfer efficiently.
and PO˙, which react with flammable radicals (H˙ and OH˙), reduce the opportunity of combustion, and produce a quenching effect.46 Second, in the condensed phase, the crosslinking structure of silica to form a polymer network prevented further matrix decomposition. Meanwhile, the decomposition products of the supermolecular-produced phosphorous oxide, which generated P-rich intumescent char, formed stable residual chars, and restricted the formation of holes and cracks. The Ni compounds also catalyzed the carbonization of the burning polymers top and formed a compact and continuous bubble-like char layer. Thus, the physical barrier formed by Ni@SiO2-PA could protect the matrix by isolating O- and heat-transfer efficiently.
3.6 Mechanical properties
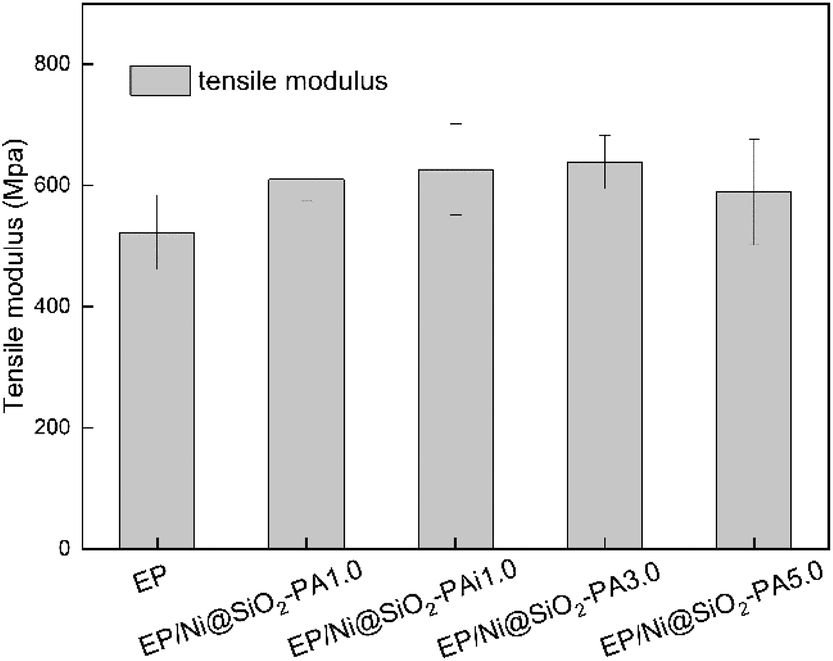
The mechanical performance of EP and its composites were evaluated with tensile testing. As shown in Fig. 15, the incorporation of Ni@SiO2-PA into the EP matrix could improve the tensile modulus at break. With the addition of 3.0 wt% Ni@SiO2-PA, the tensile modulus was increased by 22.2% compared with that of pure EP. As the incorporation further increased to 5 wt%, the tensile modulus was reduced, probably due to the increase in nanofillers, which leads to a slight agglomeration in the matrix and causes stress concentration. However, it was still higher than that of pure EP, which was sufficiently high to meet the requirements under many commercial circumstances. This result was observed because the supermolecule acted as an adhesive between the Ni@SiO2-PA and EP matrix to form strong interface interactions. The flame-retardant nanoparticles limited the movement of the matrix segment and further increased the strength of the nanocomposites.
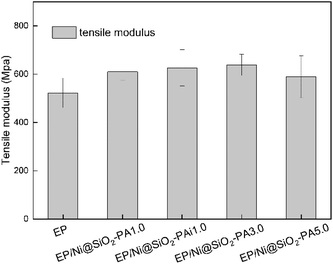 |
| | Fig. 15 Tensile modulus of pure EP and its composites. | |
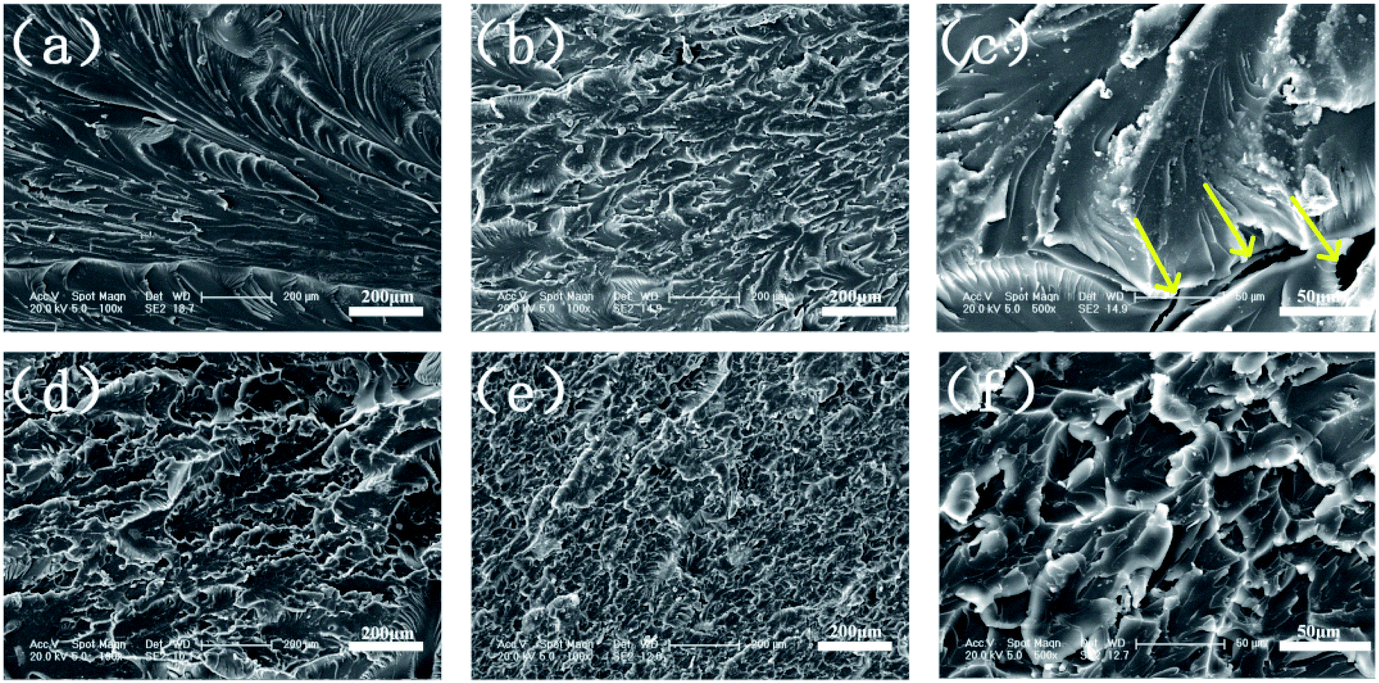
In investigating Ni@SiO2-PA dispersion in the EP matrix, the SEM images of the fracture section after tensile testing for pure EP and its composites are shown in Fig. 16. As displayed in Fig. 16a, a smooth structure and a river-like pattern with consistent directions was observed, which revealed the poor mechanical properties of the pure EP. A relatively rough fracture surface could be observed with the addition of 1 wt% SiO2 particles (Fig. 16b). However, the high-magnification SEM images (Fig. 16c) showed several holes and considerable aggregation in the EP matrix, thereby suggesting that the SiO2 nanoparticles had poor interfacial interaction. When 1 wt% Ni@SiO2-PA and 5 wt% Ni@SiO2-PA were incorporated (Fig. 16d–f), a rough and uneven fracture surface was observed due to the high compatibility between the supermolecular shell and EP matrix. These results indicated that introducing Ni@SiO2-PA could have further strong interfacial interactions with the polymer matrix.
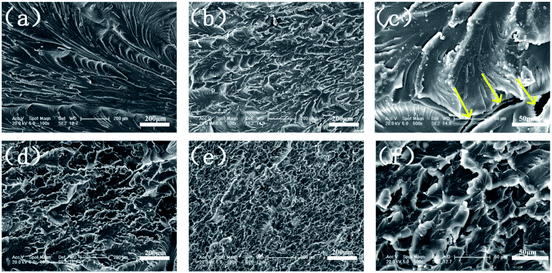 |
| | Fig. 16 SEM images of (a) pure EP, (b and c) EP/SiO21.0, (d) EP/Ni@SiO2-PA1.0, and (e and f) EP/Ni@SiO2-PA5.0 composites. | |
4. Conclusion
A facile and environmental-friendly idea was proposed to fabricate Ni@SiO2-PA by supermolecular self-assembly technology. The flame retardant with its core–shell structure was incorporated into the EP matrix to improve its thermal stability and fire resistance. The structure and morphology of Ni@SiO2-PA were characterized well by XRD, SEM, and TEM. The EP/Ni@SiO2-PA composites showed a significant improvement in thermal stability and flame retardancy. Compared with pure PE, with the addition of 5.0 wt% Ni@SiO2-PA, the PHRR, THR, and TSP decreased by 51.6%, 49.2%, and 49.7%, respectively. The TG-IR results presented that the amounts of the toxic aromatic volatiles and other volatile products from the EP decomposition were suppressed after its low addition. The TGA results of the EP composites and SEM images of the char after cone calorimetry revealed that introducing Ni@SiO2-PA into the EP matrix could efficiently increase the char residue. The significant improvements in the flame retardancy and smoke suppression were attributed to the cooperative effect between the supermolecular shell and SiO2 phase. The supermolecular shell could enhance the interaction between the nanoparticles and EP matrix to improve the flame retardancy further. This work shows the possibility of developing highly efficient and environmentally friendly flame retardants for creating high-performance EP composites.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors gratefully acknowledge the finical supports for this research from the Natural Science Foundation of Jilin Province (No. 20180101197jc) and the International Science and Technology Cooperation Programme of China (No. 20190701001GH).
Notes and references
- S. Qiu, Y. Hou, W. Xing, C. Ma, X. Zhou, L. Liu, Y. Kan, R. K. K. Yuen and Y. Hu, Chem. Eng. J., 2018, 349, 223–234 CrossRef CAS.
- J. Wang, J. Zhan, X. Mu, X. Jin, F. Chu, Y. Kan and W. Xing, J. Colloid Interface Sci., 2018, 529, 345–356 CrossRef CAS PubMed.
- S. Qiu, C. Ma, X. Wang, X. Zhou, X. Feng, R. K. K. Yuen and Y. Hu, J. Hazard. Mater., 2018, 344, 839–848 CrossRef CAS PubMed.
- B. Yu, W. Xing, W. Guo, S. Qiu, X. Wang, S. Lo and Y. Hu, J. Mater. Chem. A, 2016, 4, 7330–7340 RSC.
- S. Qiu, W. Xing, X. Feng, B. Yu, X. Mu, R. K. K. Yuen and Y. Hu, Chem. Eng. J., 2017, 309, 802–814 CrossRef CAS.
- D. Wang, K. Zhou, W. Yang, W. Xing, Y. Hu and X. Gong, Ind. Eng. Chem. Res., 2013, 52, 17882–17890 CrossRef CAS.
- T. Jiang, T. Kuila, N. H. Kim, B. C. Ku and J. H. Lee, Compos. Sci. Technol., 2013, 79, 115–125 CrossRef CAS.
- R. Jian, P. Wang, W. Duan, J. Wang, X. Zheng and J. Weng, Ind. Eng. Chem. Res., 2016, 55, 11520–11527 CrossRef CAS.
- P. Wang and Z. Cai, Polym. Degrad. Stab., 2017, 137, 138–150 CrossRef CAS.
- W. Cai, J. Wang, Y. Pan, W. Guo, X. Mu, X. Feng, B. Yuan, X. Wang and Y. Hu, J. Hazard. Mater., 2018, 352, 57–69 CrossRef CAS PubMed.
- W. Cai, X. Feng, B. Wang, W. Hu, B. Yuan, N. Hong and Y. Hu, Chem. Eng. J., 2017, 316, 514–524 CrossRef CAS.
- M. E. Shabestari, E. N. Kalali, V. J. González, D. Y. Wang, J. P. Fernández-Blázquez, J. Baselga and O. Martin, Carbon, 2017, 121, 193–200 CrossRef CAS.
- S. Wang, R. Gao and K. Zhou, J. Colloid Interface Sci., 2019, 536, 127–134 CrossRef CAS PubMed.
- H. L. Ma, H. Bin Zhang, Q. H. Hu, W. J. Li, Z. G. Jiang, Z. Z. Yu and A. Dasari, ACS Appl. Mater. Interfaces, 2012, 4, 1948–1953 CrossRef CAS PubMed.
- L. Maddalena, F. Carosio, J. Gomez, G. Saracco and A. Fina, Polym. Degrad. Stab., 2018, 152, 1–9 CrossRef CAS.
- W. Wang, H. Pan, Y. Shi, Y. Pan, W. Yang, K. M. Liew, L. Song and Y. Hu, Composites, Part A, 2016, 80, 259–269 CrossRef CAS.
- M. Hajibeygi, M. Shabanian, H. Moghanian, H. A. Khonakdar and L. Häußler, RSC Adv., 2015, 5, 53726–53735 RSC.
- A.-L. Davesne, S. Lazar, S. Bellayer, S. Qin, J. C. Grunlan, S. Bourbigot and M. Jimenez, ACS Appl. Nano Mater., 2019, 2, 5450–5459 CrossRef CAS.
- W. Cai, W. Guo, Y. Pan, J. Wang, X. Mu, X. Feng, B. Yuan, B. Wang and Y. Hu, Composites, Part A, 2018, 111, 94–105 CrossRef CAS.
- D. Wang, P. Wen, J. Wang, L. Song and Y. Hu, Chem. Eng. J., 2017, 313, 238–249 CrossRef CAS.
- S. Qiu, X. Wang, B. Yu, X. Feng, X. Mu, R. K. K. Yuen and Y. Hu, J. Hazard. Mater., 2017, 325, 327–339 CrossRef CAS PubMed.
- Z. Zhang, L. Yuan, Q. Guan, G. Liang and A. Gu, Composites, Part A, 2017, 98, 174–183 CrossRef CAS.
- K. Zhou, R. Gao and X. Qian, J. Hazard. Mater., 2017, 338, 343–355 CrossRef PubMed.
- Y. Jin, G. Huang, D. Han, P. Song, W. Tang, J. Bao, R. Li and Y. Liu, Composites, Part A, 2016, 86, 9–18 CrossRef CAS.
- Z. M. Zhu, L. X. Wang and L. P. Dong, Polym. Degrad. Stab., 2019, 162, 129–137 CrossRef CAS.
- X. Liu, S. Zou, K. Liu, C. Lv, Z. Wu, Y. Yin, T. Liang and Z. Xie, J. Power Sources, 2018, 384, 214–222 CrossRef CAS.
- H. You, J. Chen, C. Yang and L. Xu, Colloids Surf., A, 2016, 509, 91–98 CrossRef CAS.
- J. Wang, D. Zhang, Y. Zhang, W. Cai, C. Yao, Y. Hu and W. Hu, J. Hazard. Mater., 2019, 362, 482–494 CrossRef CAS PubMed.
- S. Shang, B. Yuan, Y. Sun, G. Chen, C. Huang, B. Yu, S. He, H. Dai and X. Chen, J. Colloid Interface Sci., 2019, 553, 364–371 CrossRef CAS PubMed.
- X. W. Cheng, J. P. Guan, R. C. Tang and K. Q. Liu, J. Cleaner Prod., 2016, 124, 114–119 CrossRef CAS.
- X. W. Cheng, C. X. Liang, J. P. Guan, X. H. Yang and R. C. Tang, Appl. Surf. Sci., 2018, 427, 69–80 CrossRef CAS.
- J. B. Joo, I. Lee, M. Dahl, G. D. Moon, F. Zaera and Y. Yin, Adv. Funct. Mater., 2013, 23, 4246–4254 CrossRef CAS.
- Y. Feng, C. He, Y. Wen, Y. Ye, X. Zhou, X. Xie and Y. W. Mai, J. Hazard. Mater., 2018, 346, 140–151 CrossRef CAS PubMed.
- Y. Wang, X. Yang, H. Peng, F. Wang, X. Liu, Y. Yang and J. Hao, ACS Appl. Mater. Interfaces, 2016, 8, 9925–9935 CrossRef CAS PubMed.
- B. Yuan, B. Wang, Y. Hu, X. Mu, N. Hong, K. M. Liew and Y. Hu, Composites, Part A, 2016, 84, 76–86 CrossRef CAS.
- W. C. Wei, C. Deng, S. C. Huang, Y. X. Wei and Y. Z. Wang, J. Mater. Chem. A, 2018, 6, 8643–8654 RSC.
- W. Hu, B. Yu, S. D. Jiang, L. Song, Y. Hu and B. Wang, J. Hazard. Mater., 2015, 300, 58–66 CrossRef CAS PubMed.
- B. Yu, Y. Shi, B. Yuan, S. Qiu, W. Xing, W. Hu, L. Song, S. Lo and Y. Hu, J. Mater. Chem. A, 2015, 3, 8034–8044 RSC.
- Y. Feng, J. Hu, Y. Xue, C. He, X. Zhou, X. Xie, Y. Ye and Y. W. Mai, J. Mater. Chem. A, 2017, 5, 13544–13556 RSC.
- T. Zhang, H. Yan, L. Shen, Z. Fang, X. Zhang, J. Wang and B. Zhang, RSC Adv., 2014, 4, 48285–48292 RSC.
- T. Zhang, Z. Du, W. Zou, H. Li and C. Zhang, Polym. Degrad. Stab., 2012, 97, 1716–1723 CrossRef CAS.
- B. Yuan, Y. Hu, X. Chen, Y. Shi, Y. Niu, Y. Zhang, S. He and H. Dai, Composites, Part A, 2017, 100, 106–117 CrossRef CAS.
- F. Carosio, J. Alongi and G. Malucelli, Polym. Degrad. Stab., 2013, 98, 1626–1637 CrossRef CAS.
- H. Y. Ma, L. F. Tong, Z. Bin Xu and Z. P. Fang, Adv. Funct. Mater., 2008, 18, 414–421 CrossRef CAS.
- X. Wang, W. Xing, P. Zhang, L. Song, H. Yang and Y. Hu, Compos. Sci. Technol., 2012, 72, 737–743 CrossRef CAS.
- L. Qu, C. Zhang, P. Li, X. Dai, T. Xu, Y. Sui, J. Gu and Y. Dou, RSC Adv., 2018, 8, 29816–29829 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00072h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Xueyan Dai,
Peihong Li,
Jinrui Zhang,
Shuai Luo and
Chunling Zhang
,
Xueyan Dai,
Peihong Li,
Jinrui Zhang,
Shuai Luo and
Chunling Zhang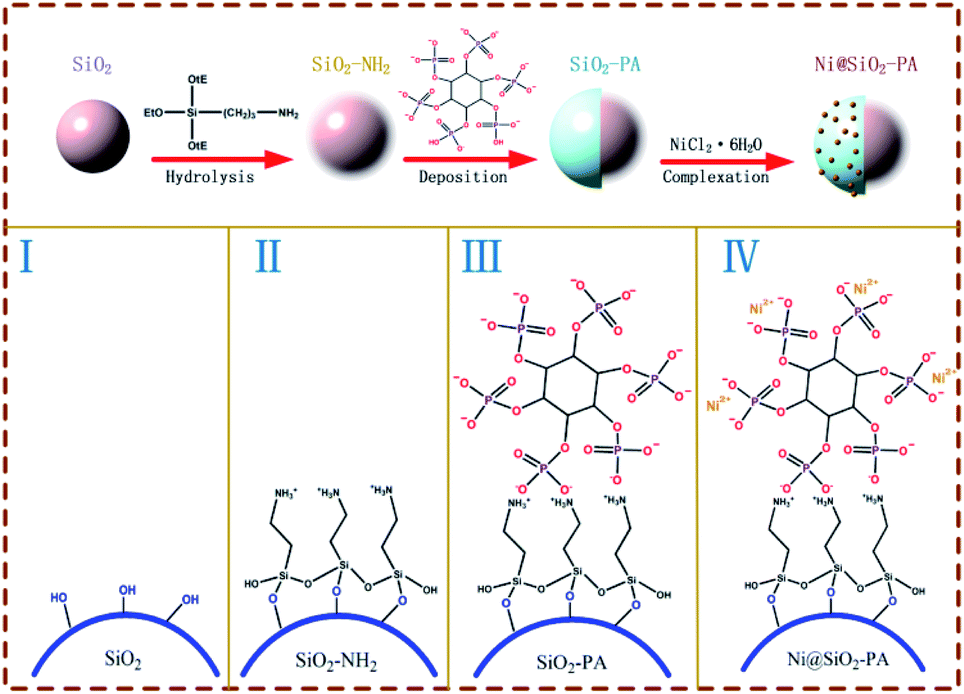





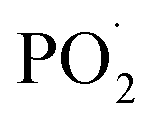 and PO˙, which react with flammable radicals (H˙ and OH˙), reduce the opportunity of combustion, and produce a quenching effect.46 Second, in the condensed phase, the crosslinking structure of silica to form a polymer network prevented further matrix decomposition. Meanwhile, the decomposition products of the supermolecular-produced phosphorous oxide, which generated P-rich intumescent char, formed stable residual chars, and restricted the formation of holes and cracks. The Ni compounds also catalyzed the carbonization of the burning polymers top and formed a compact and continuous bubble-like char layer. Thus, the physical barrier formed by Ni@SiO2-PA could protect the matrix by isolating O- and heat-transfer efficiently.
and PO˙, which react with flammable radicals (H˙ and OH˙), reduce the opportunity of combustion, and produce a quenching effect.46 Second, in the condensed phase, the crosslinking structure of silica to form a polymer network prevented further matrix decomposition. Meanwhile, the decomposition products of the supermolecular-produced phosphorous oxide, which generated P-rich intumescent char, formed stable residual chars, and restricted the formation of holes and cracks. The Ni compounds also catalyzed the carbonization of the burning polymers top and formed a compact and continuous bubble-like char layer. Thus, the physical barrier formed by Ni@SiO2-PA could protect the matrix by isolating O- and heat-transfer efficiently.