DOI:
10.1039/C9RA10556E
(Paper)
RSC Adv., 2020,
10, 12504-12510
A study on the mechanism of oxidized quinoline removal from acid solutions based on persulfate–iron systems†
Received
15th December 2019
, Accepted 11th February 2020
First published on 27th March 2020
Abstract
Quinoline (Qu) and its derivatives have been widely regarded as hazardous pollutants in the world because of their acute toxicity to humans and animals, and potential carcinogenic risks. In this study, a novel sulfate radical system co-activated by ferrous and ZVI was developed to remove Qu from acidic solutions. The optimal ratio of ferrous and ZVI in the system and the mechanism of Qu removal from acidic solutions are also explored. The ZVI can initiate activation using hydrogen ions, which are released from the reaction of Fe2+, organics and PS in acidic solutions. This may dramatically improve the overall removal efficiency of Qu. The results indicated that the initial removal rate of Qu increases from 85.8% to 92.9%. The cleavage pathway of Qu is speculated by Frontier molecular orbital (FMO) theory and verified by GC/MS analysis.
Introduction
Quinoline (Qu) and its derivatives are typical nitrogenous heterocyclic compounds. They have been widely regarded as hazardous pollutants because of their acute toxicity to humans and potential carcinogenic risks.1 It has been reported that the concentration of Qu is as high as 82 mg L−1 in the coking wastewater.2 Various methods have been developed to remove Qu and its derivatives from industrial wastewater.3,4 Although a biological treatment method is often employed, it requires much longer hydraulic retention time (HRT) due to its strong recalcitrance.4 Longer HRT not only results in larger pond size, more land area requirement, larger initial engineering project investment and higher operation cost, but also tremendously increases the concentration of Qu and its derivatives in the ambience around ponds due to their inherent high volatility.3 Compared with biological processes, advanced oxidation technology has the obvious advantages of fast reaction rate and high efficiency in removing refractory organic pollutants from complex wastewater.5,6 Recently, the rapid removal of Qu by radical oxidation of sulfate has attracted much attention in persulfate (PS)-based systems in early neutral pH wastewater due to its high redox potential and excellent selectivity.7,8 Ferrous ion is reported as one of the strongest activators in the PS-based system.4 There are mainly two main methods of using Fe to activate redox reactions in the PS-based system: directly adding in ferrous ions or zero-valent iron (ZVI) and indirectly adding in ferrous ions.9,10 In PS systems that directly add ferrous ions, the higher initial ferrous ion concentration can effectively trigger the rapid oxidation removal of Qu at the initial stage;11,12 unfortunately, ferrous ions are also strong competitors of organic pollutants at the main sites of sulfate radicals, so the Qu removal rate and efficiency are significantly reduced in the subsequent stages.13,14 In the PS-based system, with the addition of ZVI, since ZVI can continuously release the right amount of ferrous ions, the quenching effect of excess ferrous ions on persulfate radicals can be alleviated.10,15 However, the reaction rate is too slow to remove Qu from a large amount of engineering wastewater.16 The slow reaction rate can be attributed to two causes. The first factor is that the strong attraction between particles, inherent magnetic interactions and higher deposition rates cause severe aggregation of ZVI particles, and the other is that the Fe2+-releasing process from ZVI can be hindered by phase differences and particle surface oxide films.17,18
In this study, a novel PS-based system is proposed in order to remove Qu with high reaction rate and high removal rate by simultaneously adding ferrous ions and ZVI as activators of sulfate radical oxidation. To our knowledge, few information is available on the research of synergistic effects of ferrous ions and ZVI in the PS-based system. The objective of the research is to compare the Qu removal rates and the reaction rates in the three different PS-based systems activated by ferrous ions or ZVI, and co-activated by ferrous and ZVI. The ratio of ferrous ion, ZVI and PS dose in the PS-based system is also optimized according to Qu removal rate. Moreover, the mechanism of Qu removal by sulfate radical oxidation in acidic solutions in the proposed system is also explored.
Experiments
Materials
The analytical grade Qu (C9H7N), sodium persulfate (Na2S2O8), ferrous sulfate (FeSO4·7H2O), zero-valent iron (ZVI), sulfuric acid (H2SO4) and sodium hydroxide (NaOH) were purchased from Kermel Chemical Reagent Co. (Tianjin, China). Chromatographically pure methylene chloride (CH2Cl2) and methanol (CH3OH) were purchased from Shanghai Alfa Aesar (Johnson Matthey Company, USA). Water was deionized using an Aquelix 5 water system (Millipore Corp., Bedford, MA, USA). The physical and chemical characteristics and the molecular structure of Qu are shown in Table S1.†
Experimental design
A series of completely mixed reactors (CMRs) were used. Each CMR was a 250 mL amber conical flask with a screwed cap using PTFE-lined silicone as inside septa. The batch experiments were conducted at 25 °C with the initial pH of 7.0 ± 0.2. The initial Qu concentration was 0.1 mM in the 100 mL aqueous solution. The mole ratio of PS and activator was derived from eqn (1). Furthermore, 0.8 mM ZVI and 2.4 mM Fe2+ iron provide the same number of electrons to be oxidized. ZVI, Fe2+ and Fe2+/ZVI were selected as persulfate activators to compare the activation effects and oxidation rates:| | |
C9H7N + 24Fe2+ + 24S2O82− + 21H2O → 24Fe3+ + 48SO42− + NO3− + 9CO2 + 49H+
| (1) |
A 4 mL aliquot of reaction suspension was sampled from the CMR using a disposable and sterilized syringe (Shanghai kindly Co. LTD, China) and filtered through a 0.45 mm PTFE syringe filter (Thermo Scientific, Waltham, MA). The filtrate was mixed with a certain amount of quenching agent (methanol) in a 5 mL glass vial. The vial was capped and placed in a refrigerator at 4 °C until high-performance liquid chromatography (HPLC) analysis for aqueous Qu concentrations. Preliminary tests showed less than 2% loss of Qu after filtration. The control experiment showed that the average system loss of Qu was always lower than 4% of the initial concentration. For the effects of pH on the Qu oxidation, the initial pH of the Qu solution was adjusted to 3.0, 5.0, 7.0, and 9.0 by adding 0.1 M H2SO4 or NaOH; subsequently, the pH was not controlled after the start of the reaction. All experiments were conducted in triplicate.
Quantification of Qu and identification of reaction products
The aqueous Qu concentrations were quantified using a HPLC (LC-20, Shimadzu, Japan) equipped with a diode array UV detector and an XDB-C18 (250 mm × 4.6 mm, 80 Å) reverse-phase column (Agilent ZORBAX Eclipse, USA). The mobile phase was a mixture of water and methanol at a volumetric ratio of 40% to 60%. The flow rate was set at 1.0 mL min−1, and the UV detector was set at a wavelength of 313 nm. The Qu concentrations were analyzed using calibration curves of absorbance area versus concentrations. To analyze the patterns of intermediate products, a Trace 2000 gas chromatograph connected to a Thermo DSQ™ (dual stage quadrupole) mass spectrometer (DSQII, Thermo, USA) equipped with a DB-5 column (30 m × 0.25 mm × 0.25 μm) was used. Carrier gas was the ultra-pure helium at a rate of 1.0 mL min−1. The column temperature was programmed at 50 °C and held for 1 minute, increased at a rate of 10 °C min−1 until 280 °C, then increased at a rate of 5 °C min−1 until reaching 310 °C and held for 2 min. The injector and interface temperatures were set at 300 °C and 280 °C, respectively. A full-scan mode (m/z = 50–600) was used. Samples were extracted using a liquid–liquid extraction technique. A 50 mL reaction suspension was taken at 10, 20 and 30 minutes and mixed with 1 mL of 0.1 M sodium sulfide solution for quenching the free radical. Each of the samples was extracted three times with 10 mL methylene chloride. The extracts were combined and concentrated to 1 mL using a rotary evaporator followed by N2 gas purging.
Computational methods
Geometry optimization and frequency analysis were operated at the M06-2X/6-311+G (d, p) level. FMO analysis was calculated at the M06-2X/6-311+G (3df, 2p) level. Solvation Model D (SMD) was employed to mimic water solvent effects.19 The Hirshfeld charge was calculated using the Multiwfn software,20 all other calculations were performed using Gaussian 09, and the image was obtained using Gaussview 5.0. For each individual atom in a Qu molecule, the condensed dual descriptor (CDD) can be defined by eqn (2):| | |
fA(2)(r) = fA+(r) − fA−(r) = 2qAN − qAN+1 − qAN−1
| (2) |
where qA is the charge of atom A in a molecule. The positive fA(2)(r) value represents electrophilic regions, while the negative value represents nucleophilic regions. The Hirshfeld charge was exploited to evaluate the CDD values in this study.
Result and discussion
Characteristics of persulfate by ZVI and Fe2+ activation
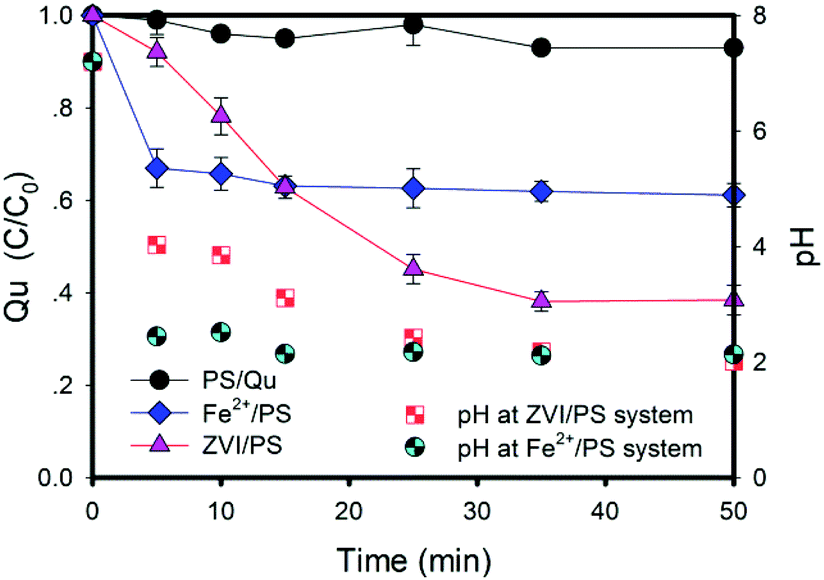
As shown in Fig. 1, sulfate radicals have good degradation effects on Qu in aqueous solutions. In a Fe2+/PS system, the degradation rate of Qu was about 33% at initial 5 min, and it increased to 38.9% at 50 min. The degradation rate of Qu was 61.6% in the ZVI/PS system at the same time. The removal rule of Qu is similar to that of similar pollutants in the Fe2+/PS system, such as carbamazepine and trichloroethylene.21,22 This process can be expressed by eqn (3)–(5):| | |
S2O82− + 2Fe2+ → 2Fe3+ + SO42− + SO4˙−, k = 2.7 × 101 m−1 s−1
| (3) |
| | |
C9H7N + SO4˙− + H2O →SO42− + NH4+ + NO3− + CmHnNOi + CO2 + H+
| (4) |
| | |
SO4˙− + 2Fe2+ → 2Fe3+ + SO42−, k = 4.6 × 109 m−1 s−1
| (5) |
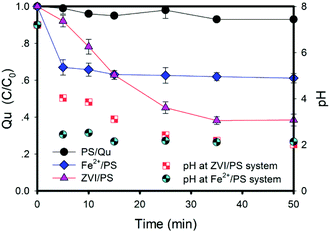 |
| | Fig. 1 Effects of different iron-based activators on Qu degradation. Conditions: [Qu]0 = 0.1 mM, [PS]0 = 2.4 mM, [ZVI]0 = 0.8 mM, [Fe2+]0 = 2.4 mM, T = 25 ± 1 °C, [pH]0 = 7.0 ± 0.2, no pH adjustment. | |
It has been reported that excessive Fe2+ ions could compete with organic pollutants for the active sites of sulfate radicals because of their reducibility.23 When highly water-soluble (26.3 g/100 g H2O at 20 °C) ferrous sulfate was added into a PS solution, PS immediately started to release sulfate radicals and the pH decreased. Due to the high oxidation potential of the sulfate radicals present in the solution (E0 = 2.6–3.1 eV), Fe2+ would preferentially react with SO4˙− rather than PS, until radicals completely disappeared. The rate constant (k) of eqn (5) was much higher than that of eqn (3). Therefore, Qu was not adequately degraded. This had been proved by the experimental results (Fig. S1a†). The oxidation reaction was considered complete when the concentration of Qu changes by less than 2%; therefore, Qu and sulfate radicals were thought to be completed in 5 min in a Fe2+/PS system.
In the ZVI/PS solution, the removal rate of Qu was up to 61.6% (Fig. 1). The decreasing mode of the Qu concentration in the ZVI/PS system was completely different from that of the Fe2+/PS system. The Qu concentration decreased significantly and stably, except for the initial reaction in the ZVI/PS system. The removal rate of Qu is shown in Fig. S1b,† the final removal percentage was higher than that in Fe2+/PS at the same electron transfer moles (such as 1.2 mM Fe2+ vs. 0.4 mM ZVI, 2.4 mM Fe2+ vs. 0.8 mM ZVI).
The dose of oxidant and activator concentrations is a critical parameter during Qu oxidation (Fig. S1a and b†). The results are summarized in Table S2.† The different molar ratios led to different Qu removal rates at the same rate of electron transfer (assuming that all iron molecules in the final state solution of the reaction exist as ferric iron). When the molar ratio of Fe2+ to PS was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the highest removal rate was achieved (44.82%). Moreover, it can be found that the excessive Fe2+ ions would quickly quench the sulfate radicals and cause the removal rate to decline, and the results can be well explained by eqn (3)–(5). In addition, the removal rate of Qu could reach 85.81% at a molar ratio of ZVI to PS of 1:1. As shown in Table S2,† the reaction rate constant (kobs) of Qu oxidation in the Fe2+/PS system was at least one order of magnitude higher than that in the ZVI/PS system. This may be ascribed to the difference between the solid and liquid phases and the passivation of oxide iron layers on the ZVI surface, which resulted in the slow Fe2+ production.24,25 Therefore, the removal rate of Qu was lower at the beginning of the reaction. However, the Fe2+ ions could be released through the slow corrosion of ZVI as the reaction proceeds according to eqn (6) and (7). ZVI could be corroded in water, and Fe2+ ions were produced in the process under both aerobic and anaerobic conditions.24 Fe2+ and PS further released sulfate radicals, oxidized Qu, and generated Fe3+ as shown in eqn (8). Fe3+ and ZVI experienced a normalization reaction to form Fe2+ as shown in eqn (9):
:1, the highest removal rate was achieved (44.82%). Moreover, it can be found that the excessive Fe2+ ions would quickly quench the sulfate radicals and cause the removal rate to decline, and the results can be well explained by eqn (3)–(5). In addition, the removal rate of Qu could reach 85.81% at a molar ratio of ZVI to PS of 1:1. As shown in Table S2,† the reaction rate constant (kobs) of Qu oxidation in the Fe2+/PS system was at least one order of magnitude higher than that in the ZVI/PS system. This may be ascribed to the difference between the solid and liquid phases and the passivation of oxide iron layers on the ZVI surface, which resulted in the slow Fe2+ production.24,25 Therefore, the removal rate of Qu was lower at the beginning of the reaction. However, the Fe2+ ions could be released through the slow corrosion of ZVI as the reaction proceeds according to eqn (6) and (7). ZVI could be corroded in water, and Fe2+ ions were produced in the process under both aerobic and anaerobic conditions.24 Fe2+ and PS further released sulfate radicals, oxidized Qu, and generated Fe3+ as shown in eqn (8). Fe3+ and ZVI experienced a normalization reaction to form Fe2+ as shown in eqn (9):
| | |
Fe0 + H2O + 0.5O2 → Fe2+ + 2OH− aerobic conditions
| (6) |
| | |
Fe0 + 2H2O → Fe2+ + 2OH− + H2 anaerobic conditions
| (7) |
| | |
C9H7N + Fe2+ + S2O82− + H2O → Fe3+ + SO42− + NH4+ + NO3− + CmHnNOi + CO2 + H+
| (8) |
It can be seen that the Fe2+ slow-release system could be formed in the solution. At the same time, more ions can accelerate the corrosion of ZVI,26,27 and the Fe2+ ions were produced continuously and incrementally. As a result, the maximum yield of sulfate radicals and the maximum degradation of Qu could be obtained. Therefore, Fe2+/PS system is the fastest yield system to produce sulfate radicals, and the ZVI/PS system is the maximum yield system in which radicals are activated by the production of Fe2+ through ZVI. How to combine their advantages will be an interesting question.
Efficiency of co-activation by ZVI and Fe2+
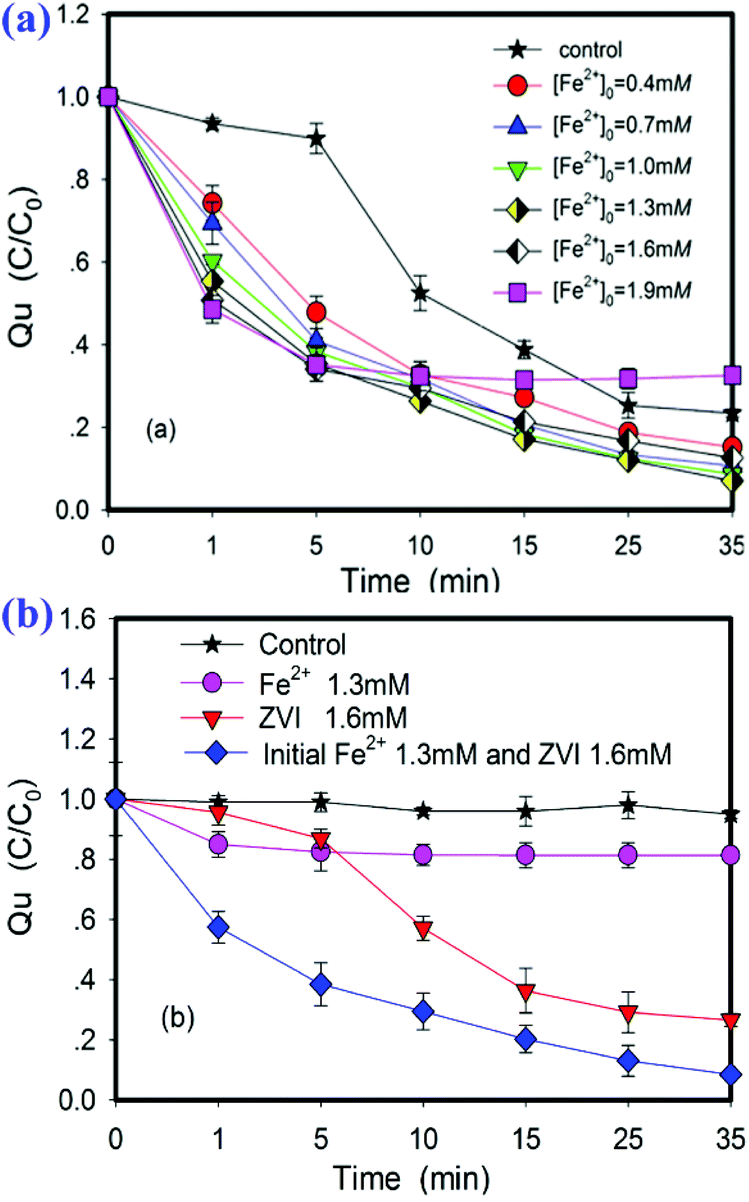
In the initial stage of the reaction, the removal rate of Qu in the ZVI/PS system is low due to the slow corrosion process of ZVI and the limited rate of Fe2+ release. In order to reduce the delay time of the reaction, a small amount of Fe2+ ions were added as an initiator at the beginning of the reaction. The experimental results are shown in Fig. 2a. When a small amount of Fe2+ was added in the initial stage of the reaction, the removal rate of Qu could be effectively improved. When the molar concentrations of adding Fe2+ were 0.4, 0.7, 1.0, 1.3, 1.6, and 1.9 mM, the removal rates were 26.38%, 30.86%, 32.65%, 37.6%, 44.87%, and 49.8% in the first 5 min, respectively. The removal rates finally were up to 85.8%, 86.4%, 90.8%, 92.9%, 85.2%, and 71.8%. The results indicated that the removal rate in the Fe2+/ZVI/PS system was significantly different from that in the Fe2+/PS and ZVI/PS systems (p < 0.05). It was clear that the combination of Fe2+ and ZVI significantly accelerated the oxidation reaction to release the sulfate radical.
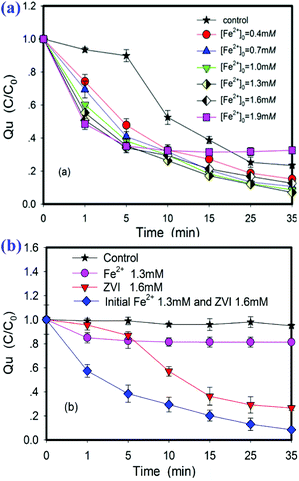 |
| | Fig. 2 (a) Effects of the initial Fe2+ concentration on the ZVI/PS system. (b) Removal rate of Qu by three different activation methods. Conditions: [Qu]0 = 0.1 mM, [PS]0 = 2.4 mM, T = 25 ± 1 °C, [pH]0 = 7.0 ± 0.2, no pH adjustment. Control: ZVI:PS:Qu = 16:24:1. | |
When the initial ferrous ion concentration was 1.3 mM, the removal rate of Qu reached highest at 35 min. Compared with the Fe2+/PS and ZVI/PS systems (Fig. 2b), the degradation rate of Qu in the Fe2+/ZVI/PS system increased by 73.5% and 7.2%, respectively. The synergistic effect of Fe2+ and ZVI was obvious within 15 min from the start of the reaction, and the Qu removal rate increased to 66% from less than 15% within 5 min. Thus, it can be seen that the value was significantly related to the initial addition of Fe2+ and the addition mode of Fe2+/ZVI combination. This is not a simple addition of the efficiency of two activators, but an increase in the removal percentage of Qu when the oxidant is unchanged and the activators are excessive. Although the co-activation of Fe2+ and ZVI can enhance the degradation, it is not enough to oxidize Qu due to its stable structure. The research shows that when ZVI particles initially entered the solution containing PS, the surface was still smooth. After the reaction was completed, the ZVI surface was obviously corroded.15,28 It has also been reported that ZVI can react with PS directly in the PS-based solution, as shown in eqn (10):
| | |
Fe0 + S2O82− → Fe2+ + 2SO42−
| (10) |
When the reaction conditions are appropriate, the reactions of eqn (6), (7) and (10) will occur simultaneously. However, the reaction rates are obviously difference. The oxidation potential of PS is much higher than that of H2O molecules and the reaction is faster. Therefore, since ZVI is a solid state, the process of S2O82− reaction with ionic state needs to break through the solid–liquid interface, and Qu removal was relatively low (less than 10%). When Fe2+ ions were added in the initial reaction, S2O82− ions in the solution will preferentially react with the ionic Fe2+ to release free radicals, Fe3+ and H+ ions (eqn (8)). The increase in H+ and Fe3+ ion concentration in the solution obviously accelerates the corrosion of ZVI, and the release of free radicals accelerates continuously; as a result, the removal rate of Qu accelerates. However, ZVI (E0 = −0.44 eV) is widely used in the removal of pollutants in water. It not only could be used as a reducing agent for high-priced toxic heavy metals,29 but also can oxidize pollutants by the ZVI/O2 system;30,31 ZVI's strong reducibility can also reduce Fe3+ ions in the solution to Fe2+ and participate in the sulfate radical generation reaction again until PS is completely consumed. Therefore, in the Fe2+/ZVI/PS system, there are at least three electron transfer processes: from Fe2+ to Fe3+, from Fe0 to Fe2+, and from Fe0 to Fe3+. The complicated electron transfer processes are considered to produce “active iron”. These electron transfer processes exhibits a positive effect on the splitting of O–O bonds in PS.
In a word, the result can be explained by four reasons. First, ZVI could be early passivated in the PS solution, and the activation is not obvious.12 The corrosion rate of ZVI can be accelerated in solutions under acidic conditions.32 Fe2+ and PS can immediately release sulfate radicals, and the pH of the solution decreased from 7.2 to 2.12 (Fig. 1). This may be due to a large of hydrogen ions released as the Qu was oxidized (eqn (7)). The accumulation of hydrogen ions and Fe3+ ions can accelerate the corrosion of ZVI, and thus Fe2+ with high activation efficiency is released quickly. Second, ZVI in an acidic solution can be oxidized by double electrons to produce H2O2. It is well known that the hydroxyl radical is a kind of radical with high redox potential (E0 = 2.7 eV). Therefore, the oxidation of pollutants is accelerated.33,34 Third, Fe2+ on the surface of ZVI and ZVI together form the binary structure of the “active iron” system, which causes a rapid electron transfer.35,36 There is the synergistic effect of Fe3+/Fe2+ and Cu2+/Cu+ cycles for peroxymonosulfate (PMS) activation.37 The rapid electron transfer on the ZVI surface exhibits ultra-high activity for PS activation and accelerates Qu oxidation. Finally, Fe2+ added at the beginning of the reaction is immediately oxidized into Fe3+, and then Fe3+ is reduced into Fe2+ by ZVI.9,10,13,14 These four factors lead to the rapid formation of Fe2+ and accelerate the start-up rate of the PS-activated reaction at the beginning reaction. In conclusion, the addition of Fe2+ is essential for triggering the ZVI/PS system in the initial state. The addition of Fe2+ in the ZVI/PS system can be used as an initiator to rapidly generate sulfate radicals at the beginning phase, and effectively accelerate the degradation of Qu. However, it should be noted that Fe2+ cannot be overdosed; otherwise, the free radical quenching will occur.
Analysis of the mechanism of sulfate radical degradation of Qu
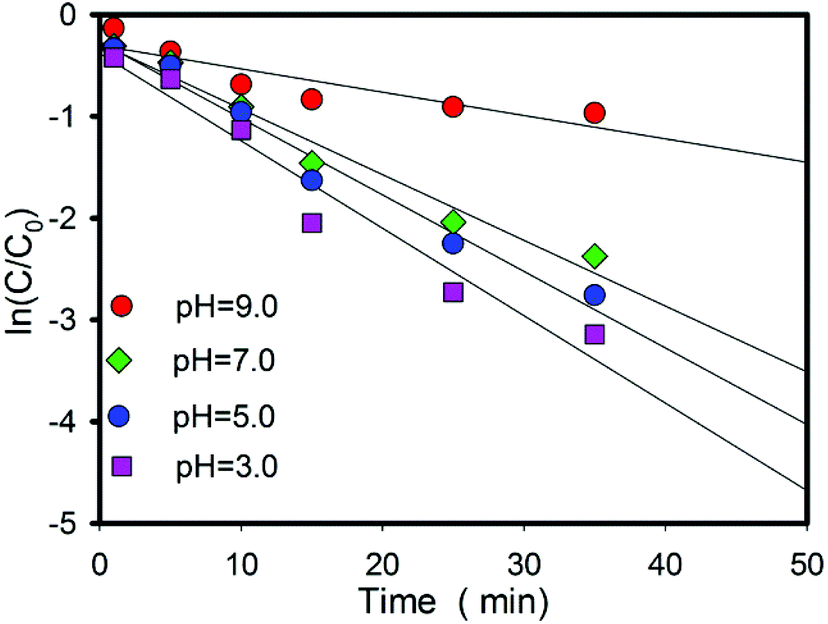
A large number of studies have found that organic pollutants are more conducive to oxidation by acid radicals in acidic solutions. However, the pH of the solution will decrease due to the continuous release of hydrogen ions of this oxidation process (eqn (4)). The influence of pH on the Fe2+/ZVI/PS system is also investigated, and the results are shown in Fig. 3. The removal efficiency of Qu increases with the decrease in pH, which is consistent with the rule of many pollutants degraded by sulfate radicals. The increase in reaction products in a solution did not inhibit the reaction but accelerated it. This result can be explained from three aspects.
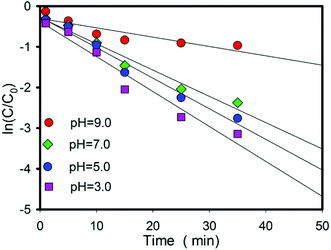 |
| | Fig. 3 Effects of the solution pH on the rates of Qu degradation. Conditions: [Qu]0 = 0.1 mM; [PS]0 = 2.4 mM; [FeSO4] = 1.3 mM; [ZVI] = 1.6 mM; T = 25 °C. | |
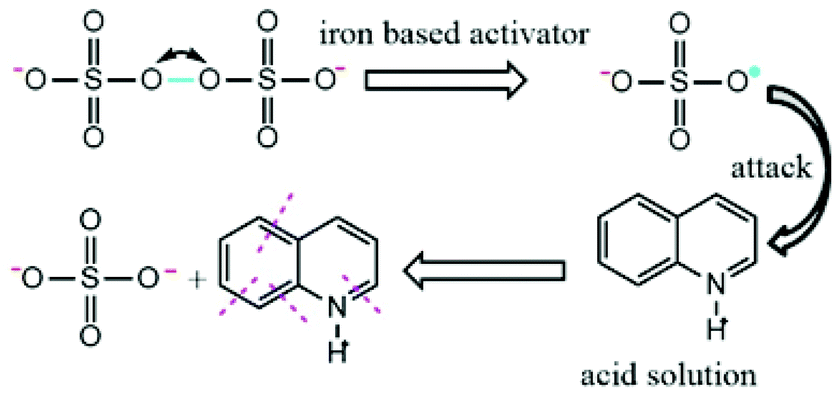
First, the advantages of sulfate radicals are high redox potential, negative charge and active single electrons (E0 = 2.65–3.1 eV).14 Luo et al. studied the kinetic of Qu oxidation by ferrate(VI), and the results indicated that Qu species was the major part at neutral and alkaline pH, and it possesses a positive charge under acidic conditions.38 Sulfate radical with negative charge is much easier to get close to Qu with a positive charge in an acidic solution. The reaction results can be represented in Fig. 4.
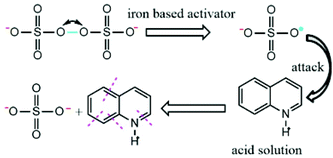 |
| | Fig. 4 Schematic of the reactions of Qu with SO4˙− in an acidic solution. | |
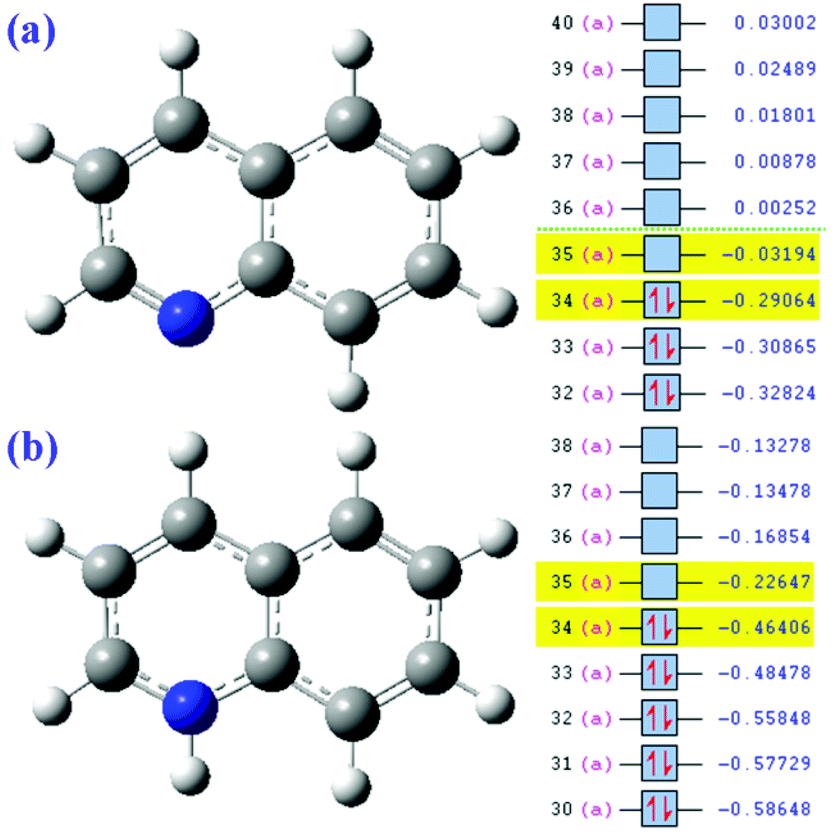
Second, the reason why Qu degrades faster under acidic conditions can also be explained by FMO theory.39 According to the FMO theory, the highest occupied molecular orbital (HOMO) is the most reactive and preferential site for electrophilic reactions. The band gaps of Qu will change after it was protonized in the acidic solution and had a positive charge (Fig. 5a and b). The relatively large band gaps and low HOMO levels imply that Qu have high stability against oxidation. By calculation, the band gaps of Qu and protonated Qu are 7.03972 eV and 6.4653 eV. Generally, the reducibility of matter is enhanced with the increase in electron cloud density and uneven distribution. It can be seen that the protonated Qu is more likely to react with sulfate radicals.
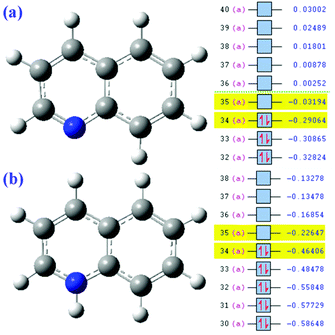 |
| | Fig. 5 Molecular orbital analysis of Qu (a) and protonated Qu (b). | |
Finally, it is reported that the orbital configuration of the free radical can be switched by the pH value in the solution, which further causes the stability to change.40 The stability of free radicals is also affected by polar effects through steric effects.41 In other words, when the pH of the solution changes, especially when the hydrogen ion increases, the protonation of the anionic or group fragment restores the regular orbital configuration; therefore, the LOMO–HOMO conversion of radicals occurs more actively in an acidic solution, and the radicals further react with protonated compounds easily. The dual descriptor (DD) method was used to describe the reactive sites of Qu. The fA(2)(r) values of each atom are listed in Table S3.† The most negative values occurred at 2C > 5C > 1C > 3C > 6C, suggesting that they are the most favorable sites for electrophilic attack by sulfate radicals. These results are highly consistent with those of the recognizable intermediate of Qu. In a word, H+ can protonate Qu and make the sulfate radical easier to react with Qu. Therefore, it is not only a reaction product, but also an important catalyst in an acidic solution.
Pathway of Qu degradation by sulfate radical
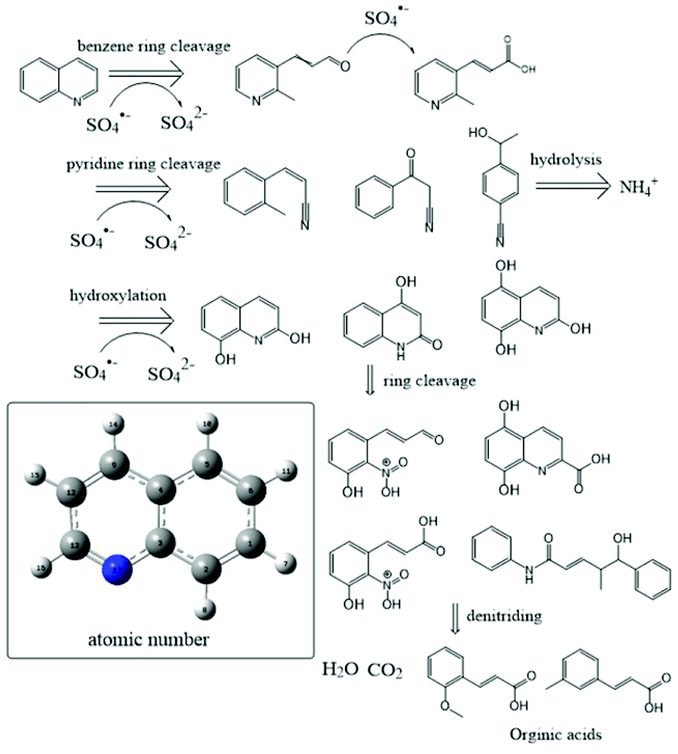
In addition to the existing Qu, ten main intermediates are identified by GC-MS, and the results are shown in Fig. 6. Total ion chromatograms from the Qu oxidation are shown in Fig. S2.† The main pathways of Qu degradation are proposed as follows: (1) a hydroxylation reaction occurred on Qu to form hydroxylated compounds such as 2,8-quinolinediol. Then, they are continuously oxidized and rings are broken, forming (or being polymerized to) new compounds containing nitro and carboxyl groups, and organic acids. (2) The cleavage of the benzene ring of Qu led to the formation of 3-pyridin-3-ylpropanal and 3-pyridine acrylic acid. (3) Some different types of nitrile compounds are produced. Then, these intermediates are further oxidized to form simple organic acids.
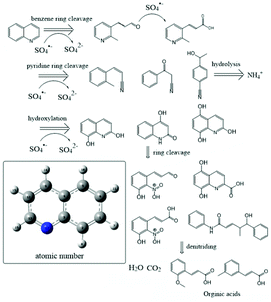 |
| | Fig. 6 Proposed degradation pathways. | |
A series of additional hydroxyl compounds are first obtained after Qu is attacked by sulphate radicals. This process is similar to Fenton and anaerobic metabolic oxidation processes (Fig. 6).42,43 Then, the pyridine ring of Qu would cleave first in most cases. Organics with benzene rings and carboxyl groups are formed after removing the nitro group. It is reported that the stable hydroxylated Qu shows lower genotoxicity and mutagenicity,44 which was consistent with the result of the biological metabolism process of wastewater containing NHCs. Furthermore, there is also a small amount of Qu, in which the benzene ring is cleaved to form an amino compound. The positions of these broken bonds are consistent with the higher negative positions of Qu given in Table S3.† The N in the Qu structure is removed as ammonia, which is beneficial for the utilization of microorganisms. The mass spectrum showed that linear alkane appeared after 30 min of oxidation (Fig. S2†). Many studies showed that the molecular structure of organics and the biodegradability have a certain relationship: chain hydrocarbons are more easily degradable than cyclic hydrocarbons; unsaturated hydrocarbons are more easily degradable than saturated hydrocarbons; and direct chain hydrocarbons are more easily degradable than branched chain hydrocarbons. After the sulfate radical oxidized Qu, the biodegradability of Qu and its intermediate is improved.
Conclusion
A novel system of PS has been developed to remove Qu from wastewater, in which Fe2+ is used as an activator. It was shown that the Fe2+/PS system had the fastest reaction rate with the optimal molar concentration ratio of 48:24:1 (Fe2+:PS:Qu). The removal rate of Qu was the most efficient by the ZVI/PS system, and the optimal molar concentration ratio of ZVI:PS:Qu was 24:24:1. In the ZVI/PS system, the removal rate of Qu could be significantly increased up to 92.9% with the initial addition of a small amount of Fe2+ as an initiator (Fe2+:ZVI:PS:Qu = 13:16:24:1). This indicated that Fe2+ could be effectively used as the initiator to accelerate the corrosion of ZVI, increase the electron frequency and dramatically enhance the overall Qu removal efficiency. The cleavage ring position of the Qu structure by sulfate radical oxidation is deduced accurately based on the FMO theory and CDD method, and the mechanism of accelerated degradation of organic pollutants under acidic conditions was explained. The results showed that co-activation by Fe0 and Fe2+ could be an effective method to improve the Qu removal by sulfate radicals, and it should be appropriate for the NHC removal.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was supported by National Natural Science Foundation of China (NSFC, No. 51608345) and the Key Research and Development (R&D) Project of Shanxi Province (No. 201603D321012).
Notes and references
- J. Chen, X. Wang, Y. Huang, S. Lv, X. Cao, J. Yun and D. Cao, Engineered Science, 2019, 5, 30–38 Search PubMed.
- X. Luo, Z. Pan, F. Pei, Z. Jin, K. Miao, P. Yang and G. Feng, J. Ind. Eng. Chem., 2018, 59, 410–415 CrossRef CAS.
- X. Shi, C. Wang, Y. Ma, H. Liu, S. Wu, Q. Shao and Z. Guo, Powder Technol., 2019, 356, 726–734 CrossRef CAS.
- D. Rameshraja, V. C. Srivastava, J. P. Kushwaha and I. D. Mall, Chem. Pap., 2018, 72, 617–628 CrossRef CAS.
- S. Guerra-Rodríguez, E. Rodríguez, D. N. Singh and J. Rodríguez-Chueca, Water, 2018, 10, 1828 CrossRef.
- M. Zhang, J. Meng, Q. Liu, S. Gu, L. Zhao, M. Dong and Z. Guo, J. Mater. Res., 2019, 34, 3050–3060 CrossRef CAS.
- D. Zhou, L. Chen, J. Li and F. Wu, Chem. Eng. J., 2018, 346, 726–738 CrossRef CAS.
- C. Huang, X. Shi, C. Wang, L. Guo, M. Dong, G. Hu and Z. Guo, Int. J. Biol. Macromol., 2019, 140, 1167–1174 CrossRef CAS PubMed.
- G. Zhen, X. Lu, L. Su, T. Kobayashi, G. Kumar, T. Zhou, K. Xu, Y. Y. Li, X. Zhu and Y. Zhao, Water Res., 2018, 134, 101 CrossRef CAS PubMed.
- L. Ran, J. P. Chen, J. Shao and M. Reinhard, Water Res., 2018, 134, 44–53 CrossRef PubMed.
- Q. Hu, N. Zhou, K. Gong, H. Liu, Q. Liu, D. Sun and Z. Guo, ACS Sustainable Chem. Eng., 2019, 7, 5912–5920 CrossRef CAS.
- L. Bu, Z. Shi and S. Zhou, Sep. Purif. Technol., 2016, 169, 59–65 CrossRef CAS.
- M. Nie, C. Yan, X. Xiong, X. Wen, X. Yang, Z. Lv and W. Dong, Chem. Eng. J., 2018, 348, 455–463 CrossRef CAS , S1385894718306971.
- Y. Y. Jin, S. P. Sun, X. Y. Yang and X. D. Chen, Chem. Eng. J., 2018, 337, 152–160 CrossRef CAS , S1385894717322167.
- J. Zeng, L. Hu, X. Tan, C. He, Z. He, W. Pan, Y. Hou and S. Dong, Catal. Today, 2917, 281, 520–526 CrossRef.
- S. Rodriguez, L. Vasquez, A. Romero and A. Santos, Ind. Eng. Chem. Res., 2014, 53, 12288–12294 CrossRef CAS.
- A. Tiraferri, K. L. Chen, R. Sethi and M. Elimelech, J. Colloid Interface Sci., 2008, 324, 71–79 CrossRef CAS PubMed.
- M. J. Duan, Z. Y. Guan, Y. W. Ma, J. Q. Wan, Y. Wang and Y. F. Qu, Chem. Pap., 2018, 72, 235–250 CrossRef CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Comput.-Aided Mol. Des., 2010, 24, 317–333 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. Rao, L. Qu, H. Yang and W. Chu, J. Hazard. Mater., 2014, 268, 23–32 CrossRef CAS PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- X. Wei, N. Gao, C. Li, D. Yang, S. Zhou and L. Lei, Chem. Eng. J., 2016, 285, 660–670 CrossRef CAS.
- S. Rodriguez, A. Santos and A. Romero, Chem. Eng. J., 2012, 213, 225–234 CrossRef CAS , S1385894716308646.
- K. M. Usher, A. H. Kaksonen and I. D. Macleod, Corros. Sci., 2014, 83, 189–197 CrossRef CAS.
- S. Rodriguez, L. Vasquez, D. Costa, A. Romero and A. Santos, Chemosphere, 2014, 101, 86–92 CrossRef CAS PubMed.
- H. Li, J. Wan, Y. Ma, M. Huang, W. Yan and Y. Chen, Chem. Eng. J., 2014, 250, 137–147 CrossRef CAS.
- C. A. Graca, L. T. Fugita, A. C. De Velosa and A. C. S. Teixeira, Environ. Sci. Pollut. Res., 2018, 25, 5474–5483 CrossRef CAS PubMed.
- X. Guo, Z. Yang, H. Dong, X. Guan, Q. Ren, X. Lv and X. Jin, Water Res., 2016, 88, 671–680 CrossRef CAS PubMed.
- S. Y. Pang, J. Jiang and J. Ma, Environ. Sci. Technol., 2011, 45, 307 CrossRef CAS PubMed.
- H. Lee, H. Lee, H. E. Kim, J. Kweon, B. D. Lee and C. Lee, J. Hazard. Mater., 2014, 265, 201–207 CrossRef CAS PubMed.
- C. Tso and Y. Shih, J. Environ. Manage., 2017, 206, 817–825 CrossRef PubMed.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 5377 CrossRef PubMed.
- S. H. Joo, A. J. Feitz, D. L. Sedlak and T. D. Waite, Environ. Sci. Technol., 2005, 39, 1263–1268 CrossRef CAS PubMed.
- J. Wan, X. Jiang, T. C. Zhang, J. Hu, D. Richter-Egger, X. Feng, A. Zhou and T. Tao, Chemosphere, 2018, 196, 153–160 CrossRef CAS PubMed.
- A. Zhihui, G. Zhiting, Z. Lizhi, H. Weiwei and Y. Jun Jie, Environ. Sci. Technol., 2013, 47, 5344–5352 CrossRef PubMed.
- W. Nie, Q. Mao, Y. Ding, Y. Hu and H. Tang, J. Hazard. Mater., 2019, 364, 59–68 CrossRef CAS PubMed.
- Z. Luo, X. Li and J. Zhai, Environ. Technol., 2015, 37, 1249–1256 CrossRef PubMed.
- K. Fukui, React. Struct. Concepts Org. Chem., 1970, 15, 1–85 Search PubMed.
- G. O. Ganna, D. L. Marshall, S. J. Blanksby and M. L. Coote, Nat. Chem., 2013, 5, 474–481 CrossRef PubMed.
- C. Anna Rita, D. Aldo and R. Fabio, J. Phys. Chem. A, 2006, 110, 10122 CrossRef PubMed.
- B. Hou, H. Han, H. Zhuang, P. Xu, S. Jia and K. Li, Bioresour. Technol., 2015, 196, 721–725 CrossRef CAS PubMed.
- N. Judith, R. Anne-Kirsten, H. Juliane and E. Adolf, Ecotoxicol. Environ. Saf., 2009, 72, 819–827 CrossRef PubMed.
- F. Pan, X. Zhong, D. Xia, X. Yin, F. Li, D. Zhao, H. Ji and W. Liu, Sci. Rep., 2017, 7, 44626 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Effects of molar ratios on the rates of Qu degradation. Total ion chromatograms form the intermediates of quinoline oxidation. Physical and chemical characteristics of Qu. See DOI: 10.1039/c9ra10556e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiuping Yue*a,
Yanqing Duana and
Zhu Raob
a,
Xiuping Yue*a,
Yanqing Duana and
Zhu Raob