
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of stapled peptides targeting phosphorylated GSK3β for regulating osteoclast differentiation†
Tairong Liu‡
ab,
Wei Cong‡c,
Lei Ye‡d,
Xike Xub,
Xiufei Liaoa,
Gang Xiec,
Zhaoxi Chengb,
Honggang Hu c,
Xiang Li*b and
Hongli Liao*a
c,
Xiang Li*b and
Hongli Liao*a
aSchool of Pharmacy, Chengdu Medical College, Chengdu 610500, P. R. China. E-mail: liaohl213@126.com
bSchoolof Pharmacy, Second Military Medical University, Shanghai 200433, P. R. China. E-mail: xiangli@smmu.edu.cn
cSchool of Translational Medicine, Shanghai University, Shanghai 200436, P. R. China
dThe First Affiliated Hospital of Shandong First Medical University, Jinan 250014, P. R. China
First published on 24th February 2020
Abstract
Glycogen synthase kinase-3β (GSK-3β), has been reported to show essential roles in osteoclast differentiation. Modeled after FRATtide, a peptide derived from a GSK-3 binding protein, here we designed and synthesized a series of stapled peptides targeting phosphorylated GSK3β, and evaluated the corresponding biological activities. The results indicated that stapled peptides with better helical contents and proteolytic stability than the linear ones showed improved biological activity in inhibiting osteoclast differentiation. Among them, FRC-2 and FRN-2 showed promising prospects for treating osteoporosis.
Introduction
Osteoporosis characterized by low bone mass and microarchitectural deterioration in bone tissue, has become an enormous and growing public health problem.1 The imbalance between resorption and formation of bone is the pathophysiological basis of osteoporosis and regulated by osteoblasts and osteoclasts, respectively.2 Bisphosphonate is currently the most widely used anti-osteoporotic drug, which inhibits the differentiation and function of osteoclasts but exhibits serious side effects.3,4 Using the exogenous molecules to regulate the specific target proteins related to osteoclast differentiation has become one of the hot topics in osteoporosis research.Previous studies have shown that the Akt/GSK3β/NFATc1 signaling pathway is a negative regulatory pathway in osteoclasts.5,6 In this pathway, a serine/threonine protein kinase, termed glycogen synthase kinase-3β (GSK-3β), can promote the transfer of NFATC1, a key osteoclast factor, from the nucleus to the cytoplasm, thus inhibiting the differentiation of osteoclasts (Fig. 1). There is a dynamic balance of mutual transformation between GSK3β and non-active GSK3β (phosphorylated GSK3β), therefore, specifically targeting and inhibiting the phosphorylation of GSK3β can promote the expression of GSK3β to a certain extent, thus achieving negative regulation of osteoclasts.
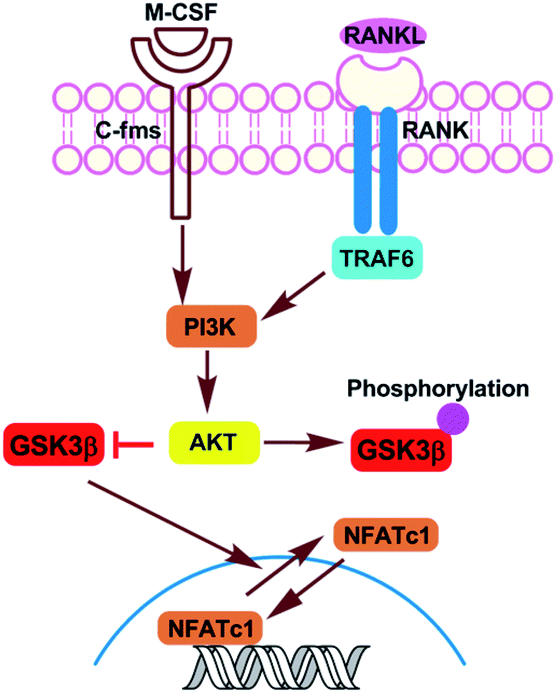 | ||
| Fig. 1 Akt-GSK3β-NFATc1 signaling pathway in osteoclasts. | ||
There exist several small-molecule GSK-3β inhibitors, such as lithium chloride and cyclic analogues of natural squamosamide.7 Of note, a series of maleimides have been discovered to show efficacy in rat osteoporosis models as GSK3β inhibitors.8 However, these small-molecule inhibitors suffer from the low selectivity and specificity. Peptide inhibitors targeting phosphorylated GSK-3β have not been reported to date, which are advantageous to small molecules in binding potency with target protein but restricted from the poor proteolytic stability and low cellular uptake.9,10 Hydrocarbon stapled peptides firstly developed by Verdine,11 with improved proteolytic stability, membrane permeability, and biological activity, have been successfully used to inhibit various protein–protein or peptide–protein interactions.9
Materials and methods
Materials
Rink Amide MBHA resin (0.3 mmol g−1 loading) was purchased from Tianjin Nankai Hecheng Science & Technology Co. Ltd (Tianjing, China). Fmoc-protected amino acids were purchased from Shanghai GL Biochem Ltd (Shanghai, China). HCTU, phenol, DIC, TIPS were obtained from adamas-beta (Shanghai, China). DMF, DCM were purchased from Wohua Chemical Co. Ltd (Shanghai, China). Ethyl ether, NMP, TFA, piperidine and other common reagent were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). The alpha modification of Eagle's medium (α-MEM), penicillin/streptomycin and fetal bovine serum (FBS) were purchased from Gibco-BRL (Gaithersburg, MD, USA). The cell counting kit-8 (CCK-8) was obtained from Dojindo Molecular Technology (Kumamoto, Kyushu, Japan). Recombinant mouse M-CSF and mouse RANKL were obtained from R&D (Minneapolis, MN, USA). The tartrate-resistant acid phosphatase (TRAP) staining kit were purchased from Joytech Bio Inc (Shaoxing, Zhejiang, China). Specific antibodies against phospho-GSK3β, GSK3β and GAPDH were obtained from Abcam (Cambridge, UK). Secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). RIPA lysis buffer, PMSF and SDS-PAGE loading buffer were purchased from Beyotime (Shanghai, China).Synthesis of the linear peptides
Rink Amide MBHA resin (500 mg, 0.3 mmol g−1 loading capacity) was swollen with DCM (5 mL) for 20 min, then treated with 20% piperidine in DMF (10 mL), followed by washing with DMF (5 times), DCM (5 times) and DMF (5 times). Fmoc-AA-OH (1 mmol), HCTU (0.9 mmol), DIEA (0.9 mmol) and DMF (6 mL) were mixed and added to the resin for 1 h to complete the coupling of the first amino acid. The deprotection, coupling and washing steps were repeated until all the amino acid residues were sequentially installed to the construct of linear peptide. The peptide-bound resin was treated with 20% piperidine in DMF (6 mL) to remove the Fmoc group from the N-terminus, followed by the addition of acetic anhydride and pyridine (6 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 20 min. Then the resin was washed with DMF (5 times), DCM (5 times), and ethyl ether (5 times). The crude peptides were cleaved from the resins by treatment with reagent B (87.5% TFA, 5% H2O, 5% phenol and 2.5% TIPS) for 4 hours at room temperature. The resultant mixture was filtered, collected the solution and TFA was evaporated by blowing with air, then precipitated the peptide with 40 mL of cold diethyl ether, and removed residual diethyl ether by air dry to obtain the crude peptide.
:1) for 20 min. Then the resin was washed with DMF (5 times), DCM (5 times), and ethyl ether (5 times). The crude peptides were cleaved from the resins by treatment with reagent B (87.5% TFA, 5% H2O, 5% phenol and 2.5% TIPS) for 4 hours at room temperature. The resultant mixture was filtered, collected the solution and TFA was evaporated by blowing with air, then precipitated the peptide with 40 mL of cold diethyl ether, and removed residual diethyl ether by air dry to obtain the crude peptide.
Synthesis of the stapled peptides
The chain elongation of the stapled peptide was carried out as described in the synthesis of the linear peptide. The couplings of N-Fmoc-α-pentene amino acid S5 were carried out over a single coupling cycle (2 h) using 2 eq. of the Fmoc protected amino acids, and the couplings of the amino acids after S5 were carried out with two cycles (1 h × 2) using the same equivalent as other amino acids. The ring-closing metathesis reaction was carried out in 1,2-dichloroethane (DCE) at room temperature using Grubbs' first-generation catalyst (10 mM). After the first round of the 2 h metathesis, we repeated the same procedure for a second round of catalyst treatment with fresh catalyst solution. Then the crude peptides were obtained as previously described.HPLC and mass spectrometry
The target compounds were purified by using Pre-HPLC SD-1 VARIAN. The purification was carried out using YMC-Pack ODS-AQ column (250 × 20 mm ID, S-5 μm, 12 nm) at a flow rate of 15 mL min−1. The target compounds were purified by gradually increasing the buffer B ratio to form the elution according to the polarities of each peptides. The purified peptides were analyzed by reverse phase HPLC (Agilent Technologies 1260 Infinity) using an analytical column (Waters “XBridge C18”, 4.6 × 150 mm, 5 μm particle size, flow rate 1.0 mL min−1, rt). Analytical injections were monitored at 214 nm. Gradient A: A linear gradient of 10% to 10% B over 5 min, then a linear gradient of 10% to 90% B over 25 min. Solution A was 0.1% TFA in water, and solution B was 0.1% TFA in MeCN. HR-Q-TOF-MS was measured on an Agilent 6538 UHD Accurate Mass Q-TOF mass spectrometer.Circular dichroism (CD) spectroscopy
Peptides were dissolved in PB (pH = 7.2) to concentrations 30 μmol L−1. The spectra were obtained on a Jasco J-715 spectropolarimeter at 20 °C. The spectra were collected using a 1 mm path-length quartz cuvette with the following measurement parameters: wavelength, 190–250 nm; step resolution, 0.1 nm; speed, 50 nm min−1; accumulations, 3; bandwidth, 1 nm. The helical content of each peptide was calculated as described in a literature based on the ellipticity of the peptide's spectrum at 245 nm and the number of amino acids in the peptide sequence.Proteolytic stability
The peptides were dissolved in PB buffer solutions (50 mM, pH = 7.4) to a final concentration of 1 mM, respectively. α-Chymotrypsin was dissolved in PBS buffer (50 mM, containing 2 mM of CaCl2, pH = 7.4) to a final concentration of 5 ng mL−1. Then the peptide solutions (1 mL) were incubated with trypsin solution (10 μL) at room temperature, which were taken at the 0 min, 10 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h and quenched with 20 μL of hydrochloric acid (1 M). The solution of the trypsin peptide fragments was monitored by HPLC at different time to determine the fraction of protease degradation.Mouse bone marrow-derived macrophages (BMMs) preparation and differentiation
All animal experiments were approved by the Institutional Animal Care and Ethics Committee of School of Life Sciences, Shanghai University (Shanghai, China) and performed in accordance with the principles and procedures of the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and the Guidelines for Animal Treatment of Shanghai University. Male C57BL/6J mice were purchased from CAVENS Laboratory (Changzhou, China). BMMs were flushed from the femur and tibia of 6 weeks-old female C57BL/6 mice and cultured in Petri dishes in α-MEM medium with 10% FBS, 1% penicillin/streptomycin, and 30 ng mL−1 of M-CSF in a 37 °C, 5% CO2 incubator until 90% confluence reached. BMMs then differentiated into osteoclasts in the presence of human RANKL and M-CSF.Cytotoxicity assay
The cytotoxic effects of peptides on BMMs were determined using CCK-8 assay. BMMs were seeded into 96-well plates at the density of 8000 cells per well in triplicate. After incubation for 24 h, fresh medium containing different dose of peptides (0, 10, 20, 40 and 80 μM) was added and incubated for another 96 h. At the indicated time, 1/10 (v/v) CCK-8 solution was added to each well and incubated for 1 h at 37 °C. The optical density (OD) was then measured at a wavelength of 450 nm (650 nm reference) on a Cell Imaging Multi-Mode Reader (BioTek, Vermont, USA).TRAP staining assay
Osteoclast formation was detected by quantifying cells positively stained by TRAP. The BMMs were seeded into a 96-well plate at a density of 8000 cells per well, in triplicate. After incubation for 24 h, BMMs were incubated in the presence of 30 ng mL−1 M-CSF, 50 ng mL−1 RANKL, and different concentrations of peptides (0, 20 and 40 μM). The fresh medium was replaced every 2 days. After osteoclastogenic inducing for 5 days, the cells were washed with PBS twice and fixed in 4% paraformaldehyde (pH 7.4) at room temperature for 10 min and then stained with commercial TRAP staining kit according to the manufacturer's instructions. TRAP-positive multinucleated (nuclei > 3) cells were counted as osteoclasts using a light microscope (Nikon, Tokyo, Japan), and the percentage of osteoclasts per well was measured using Image J software.Western blot analysis
To examine the effects of peptides on the expression of GSK3β, BMMs were seeded in six-well plates (5 × 105 cells per well). After incubation for 24 h, fresh medium containing different dose of peptides (0, 10, 20 and 40 μM) was added, and 50 ng mL−1 RANKL was added 2 h later. After 30 min, the cells were washed with PBS and lysed in RIPA buffer with added PMSF. Above samples were incubated on ice for 20 min, and centrifuged at 4 °C at 12000g, then mixed with SDS-PAGE loading buffer and heated at 100 °C for 10 min. Samples were subsequently resolved on 10% Bis–Tris acrylamide gels, and transferred to PVDF membranes. After blocked with 5% skimmed milk powder in TBS-Tween (pH 7.5, 0.15 M NaCl, 0.05 M Tris–HCl, and 0.2% Tween-20), the membranes were incubated with primary antibodies at 4 °C overnight and a corresponding secondary antibody for 90 min the next day after regular TBST rinsing. Bands were observed through the Odyssey Infrared Imaging System (LI-COR Bioscience, Lincoln, NE, USA).
Statistical analysis
The data were expressed as means SEM (standard error of the mean). The results were analyzed using Prism 7 (GraphPad Software Inc, San Diego, CA, USA) for Windows. The Student's t test was used to make comparisons between two groups. p < 0.05 indicated a significant difference between groups.Protein–protein docking
The crystal structures for FRC-0 and GSK3β (PDB ID 1GNG) were taken from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (https://www.rcsb.org). The construction of FRC-2 (stapled peptide) and pre-processing were done in the Discovery Studio 3.0 (http://www.accelrys.com) before docking and consisted of structure preparation, protonation, assignment of partial charges, and energy refinement. Protein–protein docking was performed on the ZDOCK Server 3.0.2 (http://zdock.umassmed.edu).12The following residues were pre-selected as contact points on GSK3β: L266, V267, I269, I270, F291-I296. No residues were pre-selected as contact points on the FRC-0 and FRC-2. Each docking produced 10 possible poses. For each binding region, the top-scoring pose with predicted interactions involving at least one of the selected residues was chosen for final output. Inter-residue interactions were detected and visualized in Schrödinger PyMOL (https://pymol.org).
Results and discussion
FRATtide is a peptide derived from a GSK-3 binding protein, which is discovered in Xenopus embryos.13 According to the crystal structure of phosphorylated GSK-3β bound to the FRATtide (Fig. 2A), we found that the FRATtide (FRT-0) consisted of two relatively short helical peptide sequences.14 Structural analysis showed that Pro-199, Leu-203 and Val-207 of N-terminal were the key residues interacting with GSK-3β, while the C-terminal Leu-212 and Arg-219 played the most important role. On this basis, using the N-terminal and C-terminal linear peptides as the templates, we retained the five key amino acid residues, and rationally designed a series of stapled peptides by replacing the non-interacting residues at i, i + 4 positions (N-terminal: Gln204/Leu208, His200/Gln204, Leu202/Leu206, C-terminal: Lys214/Arg218, Ala216/Leu220, Val217/His221) with (S)-N-Fmoc-2-(4-pentenyl)alanine (S5), termed as FRN-1, FRN-2, FRN-3, FRC-1, FRC-2 and FRC-3, respectively (Fig. 2B).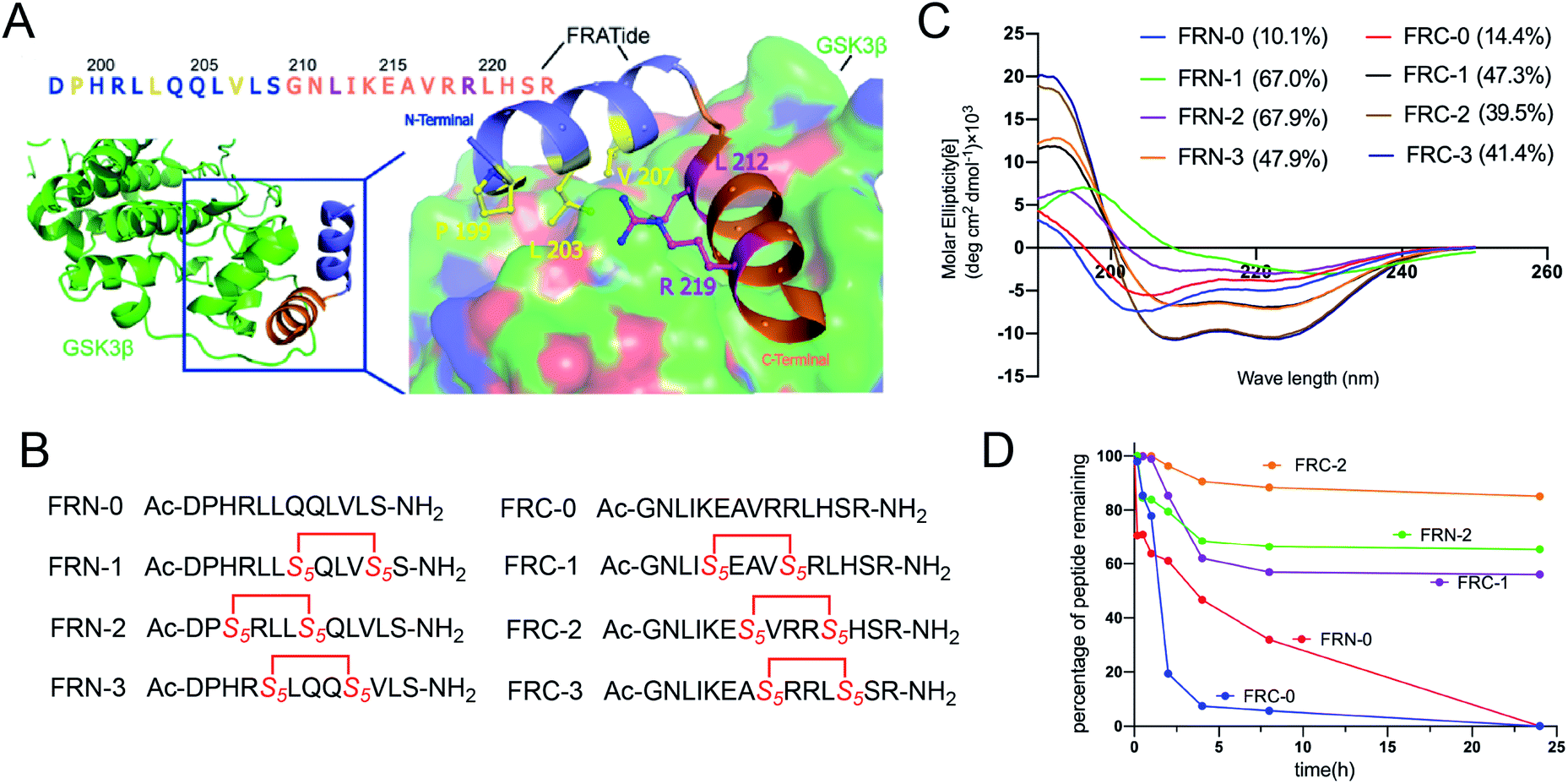 | ||
| Fig. 2 (A) Crystal structure of GSK3β bound to FRATtide (PDB ID 1GNG) with the side-chains of interacting residues shown explicitly. (B) The sequences of designed stapled peptides. (C) Circular dichroism spectra of peptides. (D) Proteolytic stability of the stapled peptides in α-chymotrypsin solution (5 ng mL−1 in 50 mM PBS buffer, pH = 7.4) at the final concentration of 0.1 mM. The percent of residual peptide was monitored by analytic HPLC. The experiments were repeated independently twice with similar results. | ||
For the synthesis of the stapled peptides, the standard Fmoc solid-phase peptide synthesis (SPPS) procedures were employed to assemble the linear sequence, followed by the cyclization of the olefin-containing peptides catalyzed by Grubbs' first-generation catalyst. The peptides were cleaved from the resin and globally deprotected with reagent B (87.5% TFA, 5% H2O, 5% phenol and 2.5% TIPS), which were purified by semi-preparative RP-HPLC and verified by HPLC and high resolution mass spectrometry (HR-MS) (Fig. S1 and S2†).
To probe structural changes induced by conformation constraint, circular dichroism (CD) spectroscopy was used to determine the helicity of stapled peptides in phosphate buffer (PB). As shown in Fig. 2C, almost all stapled peptides displayed a strong tendency to adopt a-helix on its own compared to linear peptides, of which FRN-1 and FRN-2 exhibited the highest helicity of 67% (6.7-fold of that of FRN-0). FRC-1 to FRC-3 also acquired a 3-fold enhancement compared to FRC-0. These results demonstrated that the all-hydrocarbon stapled strategy can aid the parent peptides to adopt the helical conformations.
To assess the protease stability of the stapled peptides, we measured their susceptibility towards α-chymotrypsin-mediated degradation in PBS buffer (pH 7.4) containing 2 mM of CaCl2 at room temperature, and the percentages of peptides remaining were monitored by HPLC. α-Chymotrypsin is a protease that predominantly cleaves at the carboxyl side of positively charged amino acids such as Trp and Leu. While the linear peptides were degraded to less than 30% by the enzyme within 8 h of co-incubation at room temperature, the stapled peptides were substantially more stable with a half-life of over 24 h under identical conditions (Fig. 2D). These results indicated the inherent superiority of the stapled peptides over the linear counterparts with respect to proteolytic stability.
We then examined the cytotoxicity effect of these peptides on primary bone marrow-derived macrophage (BMMs) for 96 hours. As shown in Fig. S3,† none of the peptides were cytotoxic at the dose of 40 μM. To explore whether the peptides could affect RANKL-induced osteoclastogenesis in vitro, we treated BMMs with M-CSF, RANKL, and peptides at the dose of 20 μM and 40 μM. Apparent suppression of FRC-1, FRC-2, FRC-3 and FRN-2 on the formation of mature osteoclasts could be found at the dose of 20 μM and only few mature osteoclasts could be found when treated at 40 μM (Fig. 3A and 4A). The number of mature osteoclasts and area of osteoclasts decreased in a concentration-dependent manner. The size of mature osteoclast and the number of mature osteoclast nuclei reduced dramatically in the 40 μM group compared with the control group (Fig. 3B, C, 4B and C). Among the FRC series peptides, the inhibitory effect of FRC-2 was the most significant, both in the number and area of osteoclasts. Although the inhibitory effect of FRN series peptides on osteoclasts was not as significant as that of FRC peptides in general, the higher concentration of FRN-2 showed a stronger inhibitory effect on osteoclasts. Therefore, FRN-2 and FRC-2 aroused our interest to further verify their effect on GSK3β.
 | ||
| Fig. 3 FRC stapled peptides inhibited RANKL-induced osteoclastogenesis in vitro. (A) BMMs were treated with various concentrations of various Frc peptides, M-CSF (30 ng mL−1), and RANKL (50 ng mL−1) for 5 days. Then the cells were fixed and stained for TRAP. (B and C) Osteoclast statistics of TRAP. The number and areas of TRAP multinuclear cells were determined as described in Materials and methods (*p < 0.05; **p < 0.01; ***p < 0.001). Three replicates and two independent experiments were performed. | ||
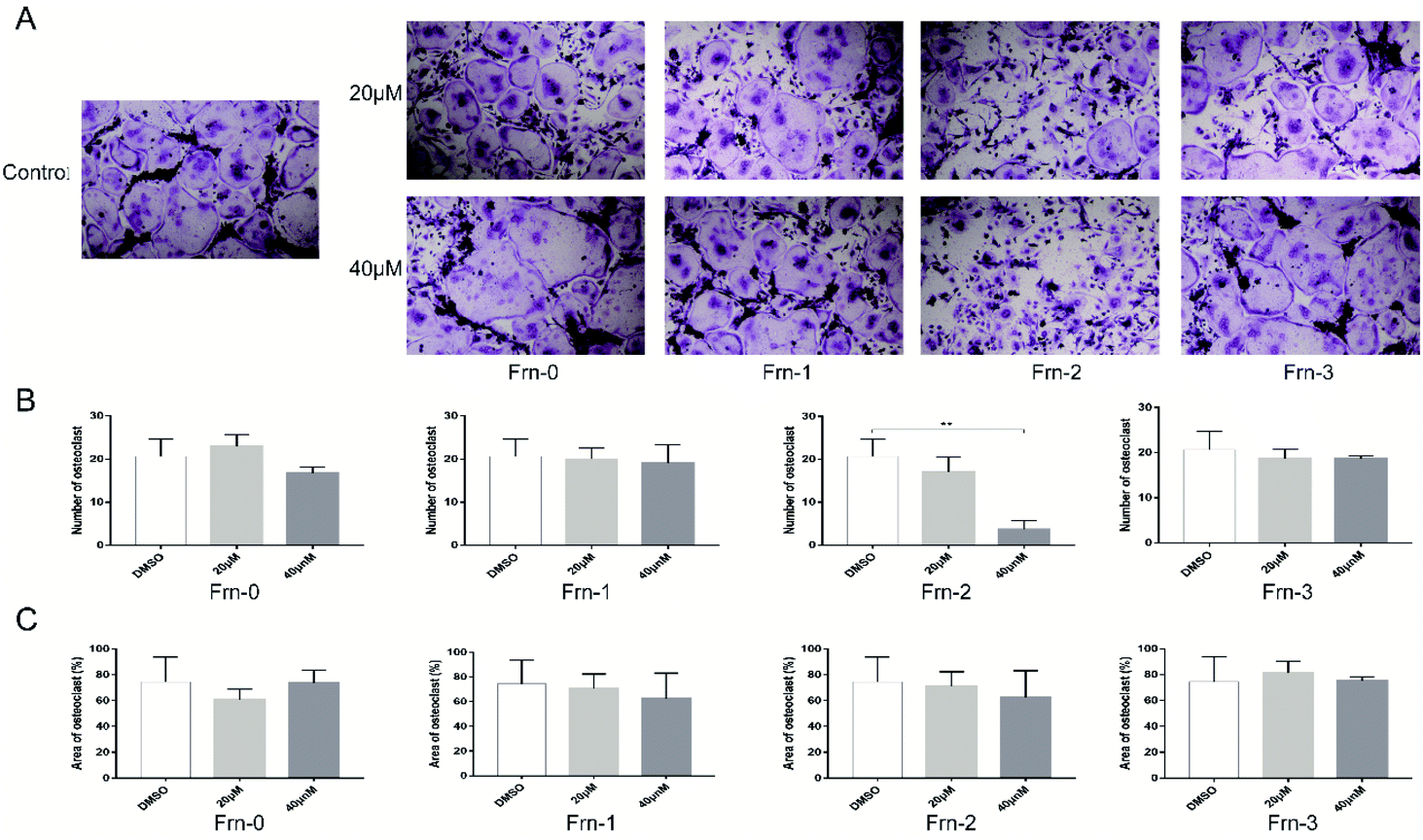 | ||
| Fig. 4 FRN peptides inhibited RANKL-induced osteoclastogenesis in vitro. (A) BMMs were treated with various concentrations of various Frn peptides, M-CSF (30 ng mL−1), and RANKL (50 ng mL−1) for 5 days. Then the cells were fixed and stained for TRAP. (B and C) Osteoclast statistics of TRAP. The number and areas of TRAP multinuclear cells were determined as described in Materials and methods (*p < 0.05; **p < 0.01; ***p < 0.001). Three replicates and two independent experiments were performed. | ||
To investigate the molecular mechanism by which FRC-2 or FRN-2 suppresses RANKL-induced osteoclastogenesis, we determined the effect of FRC-2 or FRN-2 on RANKL-induced phosphorylation of GSK3β. When RANKL stimulates the transformation of cells into osteoclasts, the expression level of phosphorylated GSK3β increases. Both FRC-2 and FRN-2 could efficiently reduce the levels of both GSK3β in dose-dependent manner (Fig. 5). Of note, the addition of FRN-2 during osteoclast differentiation reduced the phosphorylation level of GSK3β, which was more prominent at 40 μM (Fig. 5A). Western bolt analysis demonstrated that these stapled peptides regulated osteoclast differentiation by inhibiting phosphorylated GSK3β.
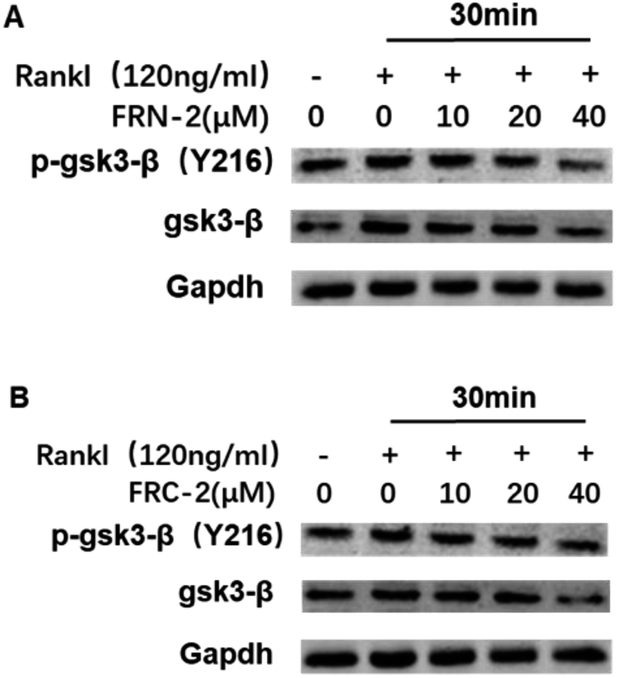 | ||
| Fig. 5 Stapled peptides inhibited RANKL-induced GSK3β phosphorylation in BMMs. BMMs were stimulated with RANKL (100 ng mL−1) for 30 min in the presence of the indicated concentrations of SPNA. Subsequently, whole cell lysates were prepared and western blot analysis performed with the indicated antibodies. The experiments were repeated independently twice with similar results. | ||
We choose FRC-2 with the best activity as the template to explore the binding mode with GSK3β. A Z-dock of the FRC-2–GSK3β complex was conducted to predict structural models of the protein–protein complexes.12,15 As shown in Fig. 6, FRC-2 could bind with GSK3β similar as FRC-0 because of the highly identification of these two sequences. Of note, the all-hydrocarbon staple of FRC-2 can confer potential enhanced affinity for the target protein through hydrophobic contacts with V262, L266, V267 and I270 of GSK3β, and this phenomenon could be observed in some cases.16 And this docking results could explain the enhanced biological activities of FRC-2 to some extent.
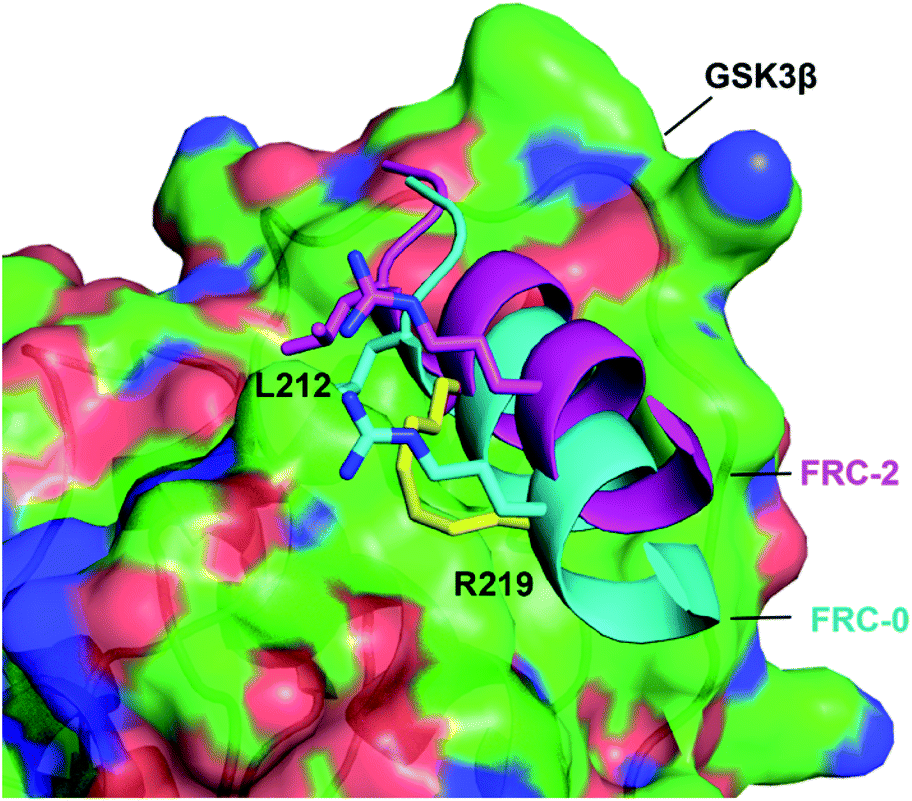 | ||
| Fig. 6 The binding mode of FRC-0 and FRC-2 with the GSK3β interface. FRC-0 was shown as cyan and FRC-2 purple. The stapling residues of FRC-2 are shown as a yellow stick. | ||
Conclusion
In summary, modeled after FRATtide which bound to phosphorylated GSK3β, six stapled peptide were rationally designed and efficiently synthesized through Fmoc-SPPS procedures and side-chain crosslink mediated by Grubbs' first-generation catalyst. Stapled peptides exhibited increased helical contents and enhanced proteolytic stability than the linear counterparts. The biological evaluation demonstrated that nearly all stapled peptides were capable of inhibiting osteoclast differentiation, among which the FRC-2 and FRN-2 had the best inhibitory effect on osteoclast both in the number and area of osteoclasts. These stapled peptides were proven to show efficiency by inhibiting phosphorylation GSK3β. Our next plan was furthermore structural optimization and biological evaluation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21807112), Youth Project in Second Military Medical University (No. 2017QN02) and Key Projects in Sichuan Province Department of Education (No. 18ZA0149).Notes and references
- S. Khosla and L. C. Hofbauer, Lancet Diabetes Endocrinol., 2017, 5, 898–907 CrossRef PubMed.
- K. E. Ensrud and C. J. Crandall, Ann. Intern. Med., 2017, 167, ITC17–ITC32 CrossRef PubMed.
- B. Z. Leder, J. N. Tsai, A. V. Uihlein, P. M. Wallace, H. Lee, R. M. Neer and S.-A. M. Burnett-Bowie, Lancet, 2015, 386, 1147–1155 CrossRef CAS.
- E. G. Lufkin, S. Sarkar, P. M. Kulkarni, A. V. Ciaccia, S. Siddhanti, J. Stock and L. Plouffe Jr, Curr. Med. Res. Opin., 2004, 20, 351–357 CrossRef CAS PubMed.
- D. G. Monroe, M. E. McGee-Lawrence, M. J. Oursler and J. J. Westendorf, Gene, 2012, 492, 1–18 CrossRef CAS PubMed.
- M. Wu, W. Chen, Y. Lu, G. Zhu, L. Hao and Y. P. Li, Nat. Commun., 2017, 8, 13700 CrossRef CAS PubMed.
- P. Duda, J. Wiśniewski, T. Wójtowicz, O. Wójcicka, M. Jaśkiewicz, D. Drulis-Fajdasz, D. Rakus, J. A. McCubrey and A. Gizak, Expert Opin. Ther. Targets, 2018, 22, 833–848 CrossRef CAS PubMed.
- L. Gong, D. Hirschfeld, Y. C. Tan, J. Heather Hogg, G. Peltz, Z. Avnur and P. Dunten, Bioorg. Med. Chem. Lett., 2010, 20, 1693–1696 CrossRef CAS PubMed.
- X. Li, Y. Zou and H.-G. Hu, Chin. Chem. Lett., 2018, 29, 1088–1092 CrossRef CAS.
- X. Li, W. D. Tolbert, H.-G. Hu, N. Gohain, Y. Zou, F. Niu, W.-X. He, W. Yuan, J.-C. Su, M. Pazgier and W. Lu, Chem. Sci., 2019, 10, 1522–1530 RSC.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- B. G. Pierce, K. Wiehe, H. Hwang, B. H. Kim, T. Vreven and Z. Weng, Bioinformatics, 2014, 30, 1771–1773 CrossRef CAS PubMed.
- C. Yost, G. H. r. Farr, S. B. Pierce, D. M. Ferkey, M. M. Chen and D. Kimelman, Cell, 1998, 93, 1031–1041 CrossRef CAS PubMed.
- B. Bax, P. S. Carter, C. Lewis, A. R. Guy, A. Bridges, R. Tanner, G. Pettman, C. Mannix, A. A. Culbert, M. J. Brown, D. G. Smith and A. D. Reith, Structure, 2001, 9, 1143–1152 CrossRef CAS PubMed.
- Y. Wu, Y. H. Li, X. Li, Y. Zou, H. L. Liao, L. Liu, Y. G. Chen, D. Bierer and H. G. Hu, Chem. Sci., 2017, 8, 7368–7373 RSC.
- S. Baek, P. S. Kutchukian, G. L. Verdine, R. Huber, T. A. Holak, K. W. Lee and G. M. Popowicz, J. Am. Chem. Soc., 2011, 134, 103–106 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00008f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |