
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme encapsulation by protein cages
Soumyananda Chakraborti†
 a,
Ting-Yu Lin
b,
Sebastian Glatt
b and
Jonathan G. Heddle
*a
a,
Ting-Yu Lin
b,
Sebastian Glatt
b and
Jonathan G. Heddle
*a
aBionanoscience and Biochemistry Laboratory, Malopolska Centre of Biotechnology, Jagiellonian University, Krakow 30-387, Poland. E-mail: jonathan.heddle@uj.edu.pl
bMax Planck Research Group, Malopolska Centre of Biotechnology, Jagiellonian University, Krakow 30-387, Poland
First published on 1st April 2020
Abstract
Protein cages are hollow protein shells with a nanometric cavity that can be filled with useful materials. The encapsulating nature of the cages means that they are particularly attractive for loading with biological macromolecules, affording the guests protection in conditions where they may be degraded. Given the importance of proteins in both industrial and all cellular processes, encapsulation of functional protein cargoes, particularly enzymes, are of high interest both for in vivo diagnostic and therapeutic use as well as for ex vivo applications. Increasing knowledge of protein cage structures at high resolution along with recent advances in producing artificial protein cages means that they can now be designed with various attachment chemistries on their internal surfaces – a useful tool for cargo capture. Here we review the different available attachment strategies that have recently been successfully demonstrated for enzyme encapsulation in protein cages and consider their future potential.
 Soumyananda Chakraborti | Soumyananda Chakraborti (Soumya) is currently an assistant professor at Amrita Vishwa Vidyapeetham Centre for Nanoscience and Molecular Medicine. Before joining his present position he was a member of Bionanoscience and Biochemistry Laboratory headed by Professor Jonathan Heddle. His present research objective is to develop DNA/protein based functional nano-materials. Soumya obtained his PhD from the Bose Institute, India where he studied protein nanoparticle interactions. He has authored more than 30 publications. His major research areas are protein engineering, protein nanoparticle interaction, chemical biology, DNA-origami, synthetic biology and nano-biotechnology. |
 Ting-Yu Lin | Ting-Yu Lin obtained her PhD from the department of Clinical Laboratory Sciences and Medical Biotechnology, National Taiwan University, Taipei, Taiwan. Her interests are in nucleic acid and nucleic acid-regulatory proteins. She was recently awarded the Taiwanese Ministry of Science Technology postdoctoral fellowship, which supports her working at the Max Planck Laboratory led by Dr Sebastian Glatt. |
 Sebastian Glatt | Sebastian Glatt, born in Austria, studied genetics and microbiology at the University of Vienna. After his PhD thesis in molecular oncology at the pharmaceutical industry, he moved to EMBL Heidelberg and transformed from a cell biologist into a protein/RNA biochemist and structural biologist. Since September 2015 he leads an independent Max Planck Research Group at the Malopolska Centre of Biotechnology (MCB), the postgraduate research centre for life science of the Jagiellonian University in Krakow, Poland. He is also deputy director for scientific affairs at the MCB and head of the “National Cryo-EM facility” at the neighbouring Solaris synchrotron. |
 Jonathan G. Heddle | Jonathan Heddle completed his PhD in biochemistry at the University of Leicester. After a postdoctoral fellowship at Yokohama City University and two PI positions at Tokyo Institute of Technology and RIKEN he moved to his current position at the Malopolska Centre of Biotechnology where he leads the Bionanoscience and Biochemistry Laboratory. He aims to understand and build artificial and natural bionano machines with a focus on protein and nucleic acid structures. This includes designed DNA origamis and novel protein cages. He hopes to continue to advance both basic developments and applications of these structures in the future. |
1 Introduction
Nanoscale structures consisting of a protein shell surrounding a central cavity are known as protein cages. Such structures can be used for protective encapsulation of cargoes which may otherwise be degraded. Nature has exploited this admirably, most obviously in the case of viruses. These typically have an approximately spherical protein shell, called a capsid, that can protect a nucleic acid genome from external degradation machineries. The virus capsid does an excellent job of shielding its genome and specifically and efficiently delivering it to target cells where it can be released and copied. Viruses then, act as the gold standard exemplar of the potential of protein cages. Indeed, viruses have been modified to take advantage of these capabilities, most notably where the viral genome has been replaced with other nucleic acids of interest for gene therapy. Currently a number of virus vectors are being tested for potential use in this way1 and adeno-associated virus mediated transgene expression of human coagulation factor FVIII, as a treatment of hemophilia B has met with some success.2However, for therapeutic3,4 or industrial5 use, a more radical redesign is required to expand the library of possible cargoes beyond nucleic acids, to include proteins. This is because, in simple terms, for any biological process it is usually proteins (i.e. enzymes, hormones etc.) that carry out the bulk of the important functionality. Thus, for example, it is desirable to be able to deliver proteins to cells for therapy. Delivering a protein rather than its respective coding information may also have the added benefit of avoiding unwanted side effects associated with the latter including deregulation and horizontal gene transfer. Proteins, in the form of enzymes, are also used in many industrial processes.6 Examples include manganese peroxidase (MnP) for biodegradation of phenolic compounds and amidase for degradation of nitrile-containing waste.7
Typically, protein cages are highly stable and retain their structure at, for proteins, extremely high temperatures. In addition, by nature of their constrained volume, cargoes are maintained physically close to one another and cannot diffuse away. These are favourable characteristics in several scenarios. For example, in in vivo therapeutics, “naked” proteins may not be used in many cases as they would not survive in the body long enough to reach the target site, being for example degraded by proteases, denatured or cleared. Under these circumstances, a thermostable protein shell can act as a protective “armour”. This stabilisation effect may also be beneficial for enzymes used as industrial catalysts: if a higher temperature favours non-enzymatic parts of the process, the ability of an enzyme to also function at such higher temperatures may lead to an increased overall efficiency. In addition, having multiple enzymes that are involved in a reaction physically close to each other could conceivably increase local concentrations and reaction rates. Apart from these potential applied uses, putting proteins into cages in a tuneable manner which controls their density may allow the investigation of more basic research questions such as the effects of protein crowding.8
These perceived benefits have led to the development of protein cages able to house other proteins in their cavity. One way this can be achieved is by modification of existing natural cage proteins. In this review, we focus on recent studies that have successfully demonstrated enzyme encapsulation using protein cages. Given that there are many different kinds of protein cages available, three main categories of encapsulation methods are summarized, namely (i) electrostatic interactions, (ii) affinity and (iii) genetic fusion (Table 1).
| Cage | Cargo | Cage size (nm) | Encapsulation method | Reference |
|---|---|---|---|---|
| a CA: human carbonic anhydrase II.b RA, KE: evolved artificial enzymes ((retro-) aldolase RA95.5-8F and Kemp eliminase HG3.17).c RuBisCO: ribulose-1,5-bisphosphate carboxylase/oxygenase.d GOx: glucose oxidase; GCK: gluconokinase. | ||||
| AfFtn | SOD | 12 | Maleimide-mediated conjugation | 63 |
| AfFtn | CAa, RAb, KEb | 12 | Electrostatic/supercharging | 27 |
| AfFtn | Luciferase | 12 | Fusion to AfFtn monomer | 81 |
| TmFtn | Lysozyme | 12 | Electrostatic/diffusion | 8 |
| Horse spleen ferritin | ATHase | 12 | Electrostatic/diffusion | 29 |
| AaLS-13 | TEVp, RuBisCOc, CAa, APEX2 | 40 | Electrostatic/supercharging | 18–20 |
| CCMV | GOxd, GCKd | 28 | Electrostatic/SS-nucleotide | 13 |
| ELP-CCMV | Lipase CalB | 28 | SrtA-mediated coupling | 42 |
| ELP-CCMV | T4 lysozyme | 28 | SrtA-mediated coupling | 43 |
| MS2 phage | TnaA & FMO | 28 | SpyTag/SpyCatcher | 49 |
| MS2 phage | GFP-neg, PhoA-neg | 28 | Electrostatic/negative peptide | 11 |
| HBV (Cp149) | β-Glucosidase | 32, 36 | Ca2+-mediated binding | 52 |
| Bacterial encapsulin | DyP | 23 | Unique anchoring sequence | 61 and 62 |
| P22 | CelB, Hyd-1 | ∼60 | Fusion/scaffold protein tagging | 74 and 75 |
| P22 | CYP | ∼60 | Fusion/scaffold protein tagging | 76 |
| Vault | MnP | ∼34 | Fusion to INT domain | 82 |
2 Electrostatic interactions
Electrostatic interactions rely on the interior of the protein cage and the protein cargo carrying opposing charges and is one of the most common methods for protein cargo encapsulation. Natural packaging of genetic material inside viruses for example utilizes this type of interaction. This can be exploited and viruses devoid of their genetic cargo (called virus like particles, VLP) constitute the bulk of protein cages used for protein encapsulation to date. Early demonstrations of protein encapsulation employing proven or probable electrostatic interactions were shown for a number of cages including cowpea chlorotic mottle virus (CCMV),9 lumazine synthase (LS)10 and MS2 bacteriophage.11 In recent years, the number of enzyme cargoes encapsulated via electrostatic effects has significantly expanded (Fig. 1).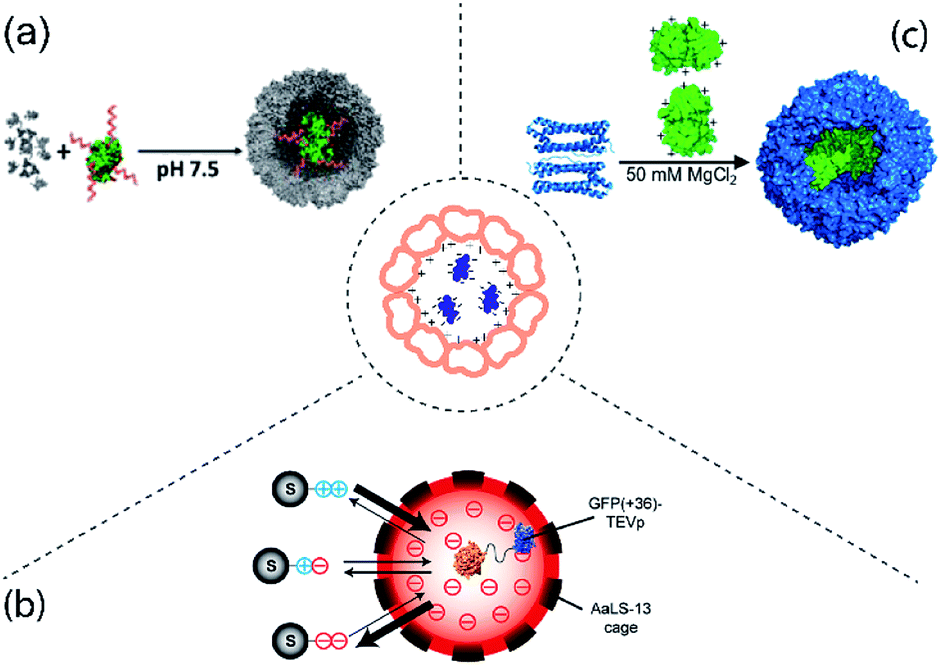 | ||
| Fig. 1 Examples of protein cargo encapsulation inside protein cages mediated by electrostatic interactions. Centre: Concept of electrostatically-mediated cargo encapsulation. (a) Attachment of DNA (red) to gluconokinase (green) provides sufficient negative charge to facilitate encapsulation in CCMV (grey). Figure reproduced with permission from ref. 13 (https://pubs.acs.org/doi/10.1021/jacs.6b10948). (b) AaLS-13 has an internal negative charge allowing capture of TEV protease (TEVp) tagged with positively supercharged GFP. By altering the charge of the tag on the substrates (S) their entry into the cage and therefore cleavage by the protease is controlled. Figure reproduced with permission from ref. 19 (https://pubs.acs.org/doi/full/10.1021/jacs.7b11210). (c) Mixing of individual TmFtn subunits (left, blue cartoon representation) and supercharged GFP (green, surface representation) results in encapsulation of GFP in the assembled ferritin cage (right, blue surface representation) under favourable salt conditions. The image was adapted and modified with permission from ref. 8 (https://pubs.acs.org/doi/full/10.1021/acsnano.7b07669). Further permissions related to the material excerpted should be directed to the ACS. | ||
2.1 CCMV
Work on CCMV was an early example showing that a large protein could be encapsulated inside a protein cage.12 Later it was shown that cargoes can be efficiently loaded inside CCMV using electrostatic interactions between the highly positively charged N-terminus of coat protein (CP) and the overall negative surface charged cargo.9,13 However, one of the major drawbacks of this method is the overall cargo loading capacity. Using diffusion-controlled electrostatics, a rather weak interaction force, usually results in packing density of cargo proteins per cage well below the theoretically achievable maximum based on volume calculations. To increase the strength of the electrostatic interaction between the partners, the cargo encapsulation method was further improved.13 In this new approach, cargo protein was coated with highly negatively charged polyanionic templates such as single stranded DNA, and then assembled with positively charged CCMV coat protein (Fig. 1a). This not only led to an improvement in loading capacity but also allowed capturing of two sequentially active enzymes inside a single CCMV. This permitted a complete enzymatic reaction cascade to take place inside the cage.132.2 Lumazine synthase
LS is an enzyme involved in riboflavin (vitamin B2) biosynthesis.14 LS from Aquifex aeolicus (AaLS-wt) possess an unique cage-like quaternary architecture, and has been developed as a biotechnology tool including extensive protein cargo encapsulations based on electrostatic interactions.15 To facilitate electrostatic interactions for cargo loading, the lumen of AaLS-wt was engineered to generate AaLS-neg, a variant with high internal negative charge. AaLS-neg was shown capable of efficiently capturing a deca-arginine tagged GFP.10 This variant was used to encapsulate a toxic protease from human immunodeficiency virus (HIV) tagged with a deca-arginine, protecting the E. coli host cells from its toxic action.16 Interestingly, the fact that encapsulation of the cargo was required for host cell survival in this case meant that the system could be used to evolve cages with greater cargo capturing capability, yielding the new variant termed AaLS-13. The concept of using a positively charged tag to direct cargo into the LS cavity was developed further with the use of a so-called supercharged GFP carrying a high positive charge (+36GFP). This could be loaded into assembled AaLS-13 cages with high efficiency. Additionally, the interaction with +36GFP accelerates cage assembly of the pentameric capsomer units.17 +36GFP could also be used as a fusion tag, which when appended to a number of different enzymes was able to direct their encapsulation.18–22 Amongst these, an engineered ascorbate peroxidase (APEX2) was proven to perform constrained polymerization of compound 3,3-diaminobenzidine (DAB) inside the LS cage20 while encapsulation of Tobacco Etch Virus (TEV) protease allowed construction of a substrate-sorting nanoreactor (Fig. 1b).2.3 Ferritin
Ferritin is an iron storage protein found in almost all organisms except yeast.23 Like many cage proteins, ferritins spontaneously assemble and are stable, requiring relatively harsh conditions for disassembly.24,25 For capturing or releasing active proteins which may often be quite fragile, this represents a challenge. Some archaeal ferritins offer a potential solution as they show reversible assembly in response to relatively mild changes in salt concentration (salt-dependency). This is seen for example in ferritin from Archaeoglobus fulgidus (AfFtn) which also shows an unusual symmetry (tetrahedral, rather than octahedral as seen in most other ferritins).23,26 This advantage along with the fact that AfFtn has a negatively charged interior was exploited to encapsulate enzymes tagged with +36GFP.27One possible disadvantage of AfFtn is that it has 4 large holes in its surface23,26 which are potentially large enough for proteases to enter and degrade guests or for smaller protein cargoes to be released. This problem was partially solved by using ferritin from Thermotoga maritima (TmFtn), which was also found to be salt-dependent but had octahedral symmetry and lacked the large holes. TmFtn was shown able to encapsulate +36GFP (Fig. 1c) and protein cargoes were protected from proteases. In fact, encapsulated lysozyme showed increased activity, most likely due to protein crowding effects.8 The fact that TmFtn containing active enzyme can be assembled into a superlattice in the presence of gold nanoparticles was exploited to generate an enzymically active superlattice.28
Another early demonstration of protein encapsulation in ferritin was performed using horse spleen ferritin. It was shown that this ferritin can incorporate an artificial transfer hydrogenase (ATHase) during reassembly of the cage triggered by pH changes.29 Given no covalent or engineered affinity interactions between cargo and cage, electrostatic effects likely play a role.29
3 Encapsulation via affinity methods
The affinity strategy (Fig. 2) is mainly achieved by genetic engineering of cage proteins to create specific interactions with selected cargoes. An example is complementary coiled–coiled sequences on cage and guest proteins.30,31 There are also naturally occurring examples of affinity interactions between cage and cargoes which can be mimicked for protein encapsulation.32,33 Some recent examples are explained in detail in this section.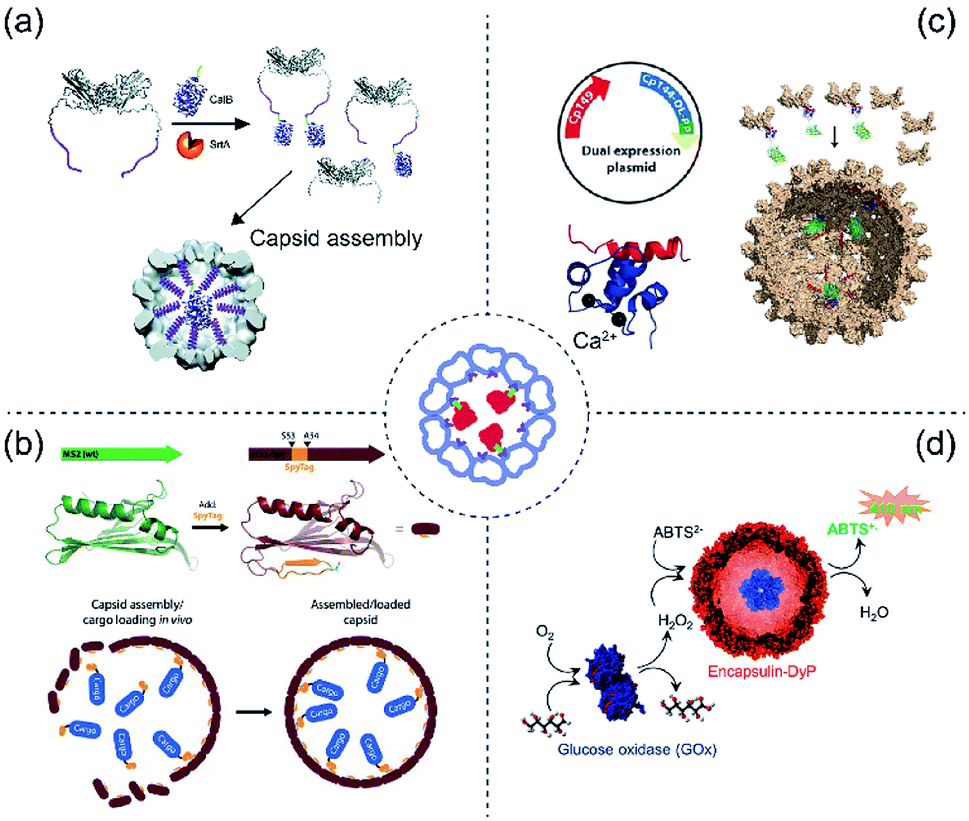 | ||
| Fig. 2 Scheme of protein cargo encapsulation inside protein cages by affinity methods. Centre: Concept of affinity-based cargo encapsulation. (a) SrtA-conjugation-mediated mechanism in CCMV. Glycine-ELP (purple) was fused to the N-terminus of CCMV whereas the CalB cargo protein was fused to the sorting peptide (LPETG, green). The image was reproduced from ref. 42 with permission from The Royal Society of Chemistry. (b) SpyTag and SpyCatcher-mediated mechanism in MS2. The wild type MS2 (green) was genetically engineered to produce an version (brick red) having the SpyTag (orange) inserted between S53 and A54. The cargo protein (blue) was fused with SpyCatcher. The image was reproduced with permission from ref. 49 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) A dual expression system produces a truncated capsid protein from HBV either not fused (Cp149, red) or fused (Cp144-DL-PP, blue) to C20W for construction of mosaic capsids (top left). In the presence of Ca2+ ions (black) the α-helical C20W binds TR2C (bottom left). GFP fused to TR2C can be encapsulated into the capsid via the C20W–TR2C interaction (right). The image was used with permission from ref. 52 Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) DyP-E (blue) can be encapsulated in encapsulin. In tandem with GOx this forms a catalytic cascade producing green-coloured ABTS radicals in the presence of glucose. The image was used with permission from ref. 61 (https://doi.org/10.1021/acsnano.7b07669). Further permissions related to the figure should be directed to the ACS. | ||
3.1 SrtA-mediated coupling
Sortase A (SrtA) is a 206 amino acid long transpeptidase found in Gram-positive bacteria which is involved in catalysing the attachment of proteins to the cell wall.34 The enzyme is able to specifically recognize a LPXTG cell wall sorting signal (where X can be any amino acid).35,36 It cleaves the bond between tyrosine and glycine residues to form the LPXT–SrtA intermediate. Subsequently this intermediate joins to the amino group of a glycine residue on host pentaglycine crossbridges via an amide bond, resulting in anchoring of the surface protein to the host cell wall. The sortase A and LPXTG peptide system was firstly identified in Staphylococcus aureus as part of the pathogen infection mechanism.37,38The SrtA system has clear biotechnology potential in attaching together two discrete proteins including protein cage/cargo combinations.39 This was demonstrated using elastin-like-polypeptide (ELP)40 modified CCMV (ELP-CCMV)41 where ELP facilitates capsid assembly at pH 7.5 triggered by high salt concentration42 (Fig. 2a). The resulting salt-induced icosahedral ELP-CCMV capsid has a diameter of 20–30 nm (triangular number (T) = 1). To exploit the sortase reaction, an additional glycine was introduced at the N-termini of ELP-CCMV capsid proteins (G-ELP-CCMV).42 Meanwhile, a SrtA recognition site peptide was genetically appended to the C-terminus of cargo proteins. The G-ELP-CCMV was then covalently linked to the cargo proteins equipped with the peptide in the presence of SrtA to form the covalently coupled product (cargo-LPETG-ELP-CCMV). The co-assembly of Candida Antarctica lipase B-ELP-CCMV (CalB-ELP-CCMV) and G-ELP-CCMV in capsids was demonstrated and showed that a maximum of two copies of the CalB enzyme can be encapsulated in the ELP-CCMV capsid cavity. This low loading efficiency is probably caused by steric constraints.42
A second generation of ELP-CCMV was further developed using the sortase-based encapsulation method to load cargo proteins without the need for high salt conditions (∼2 M NaCl).43 This strategy used metal ions to stabilize the assembly process and bypass the high salt concentration effect. Here, a hexahistidine tag was added to ELP-CCMV at the N-terminus and the presence of divalent nickel ions resulted in local clustering44 of the ELP-CCMV capsid proteins to subsequently trigger the assembly at pH 7.5 without the necessity for high salt concentrations. Using this approach, it is possible to encapsulate four copies of T4-lysozyme despite the fact that it has a cationic surface charge.
One disadvantage noted in this study of the second generation of ELP-CCMV is that they cannot be disassembled upon demand as would be expected by the addition of EDTA which presumably would chelate the nickel ions to disrupt the interaction. A possible explanation is that the action of EDTA could be hampered by the crowding effect inside the cavity.43
3.2 SpyTag/SpyCatcher targeting
SpyTag/SpyCatcher is one of the most robust protein biotechnology tools for protein ligation45 which has been further developed for affinity purification.46 Extracellular proteins of Gram-positive bacteria such as pilins and adhesin, contain spontaneously formed isopeptide bonds that help stabilize protein structures.47 Such a strategy is found, for example, within immunoglobulin-like domains CnaB1 or CnaB2. SpyTag (13 aa) and SpyCatcher (116 aa) were generated by splitting the CnaB2 domain of the fibronectin-binding protein FbaB from Streptococcus pyogenes. This peptide-protein pair can spontaneously form a covalently-linked complex. The system has been widely used for bioconjugation.48The system was introduced to bacteriophage MS2 capsid to encapsulate multiple enzymes for cascade reactions with possible applications for use as a nanoreactor49 (Fig. 2b). The SpyTag sequence (GSGAHIVMVDAYKPTKGSG) was inserted into the MS2 capsid protein (insertion site: S53A54) to produce MS2-Spy. This insertion does not affect the capsid assembly (outer diameter = 27 nm, T = 3). Meanwhile, cargo proteins were genetically fused to the SpyCatcher tag peptide which allowed the cargo to be covalently linked to the interior of MS2 capsid during the co-expression and co-purification process. The resulting modified MS2-Spy forms into two products; elongated nanotubes and spheres, however, only the sphere capsid contains the SpyCatcher-tagged cargo. Encapsulation of two-enzymes was demonstrated: pyridoxal phosphate (PLP)-dependent tryptophanase (TnaA) and nicotinamide adenine dinucleotide phosphate (NADPH)-dependent monooxygenase (FMO), responsible for biosynthesis of the commercially relevant dye Indigo.
As an additional test, the two enzymes were further constructed in a polycistronic operon and their order (TnaA then FMO or FMO then TnaA) was tested to obtain optimal expression and encapsulation. Having the TnaA ORF prior to the FMO ORF was found to result in better production of the dye. Enzyme encapsulation not only helped with increased indigo production but also stabilized the enzymes to retain activity for up to seven days comparing to free enzymes.
3.3 Calcium-mediated cargo-loading and release
Hepatitis B virus (HBV)-VLP primarily forms icosahedral capsids with 120 core protein homodimers (T = 4)50 along with a small population of T = 3 icosahedral symmetry (90 homodimers).51 The assembly domain (Cp149) can be expressed in E. coli and also self-assembles into capsids morphologically equivalent to wild type HBV-VLP.50 Protein cargoes can be packaged into HBV-VLP via fusion to its C-terminus or via non-specific loading during self-assembly. This Cp149 capsid forming property was used to achieve efficient and specific cargo encapsulation via a Ca2+-dependent method52 (Fig. 2c). The loading mechanism is based on the Ca2+-mediated binding53 (nanomolar affinity) between the C-terminal domain of calmodulin (TR2C) and an α-helical sequence derived from the membrane plasma Ca2+ pump (C20W).Introducing C20W to the C-terminus of Cp149 while fusing the cargo proteins to TR2C facilitates encapsulation during self-assembly in vitro. The model cargo protein was a β-glucosidase from Clostridium cellulovorans (CcBglA) which still retains its activity while being encapsulated inside the capsid. Two advantages of Ca2+-mediated guest loading are (i) calcium ions are biocompatible, with lower toxicity compared to many other metal ions. (ii) The interaction is reversible, meaning that the cargo can be unloaded from the interior surface. This detachment of cargo can be achieved either by the addition of EDTA or dialysis against decalcified buffer.
3.4 Spontaneous cargo loading
Spontaneous guest cargo loading of protein cages via natural targeting peptides has been demonstrated in LS54,55 and some bacterial microcompartments (BMCs), such as Salmonella enterica ethanolamine utilization bacterial microcompartment32,33 or encapsulins.56Encapsulins are protein-based nanocompartment organelles found in prokaryotes.57 They share a similar protein fold to the Hong Kong 97-like virion, which is one of the most well-characterized protein fold architectures in virus capsids.58 Encapsulins form icosahedral particles (T = 1) 23 nm in diameter and are known to be used in nature as containers for protein cargoes.32,56 There are many other encapsulins found in different bacteria and archaea.57,59,60 Here we specifically discuss the encapsulin from Brevibacterium linens which has recently been demonstrated to encapsulate enzymes61,62 with the natural cargo being a dye-decolorizing peroxidase (DyP) (Fig. 2d). The mechanism of cargo capture is via an interaction between the unique anchoring sequence at the C-terminus of DyP and a defined region of the encapsulin interior surface.
The natural DyP cargo was replaced with a foreign cargo which was fused to the DyP C-terminal anchoring sequence.61,62 Teal fluorescence protein (TFP) was used and cargo loading efficiency was 10–12 TFP per capsid. In the case of the original cargo, DyP, only one copy locates in each encapsulin even though the encapsulation capacity (2.6 × 106 Å3) can accommodate considerably more though shape and oligomerization state of the cargo may limit loading.
DyP-loaded encapsulin can be fixed on a glass surface without affecting enzyme activity which constitutes an advantage for biosensor applications.61 For in vivo applications, B. linens encapsulin has other advantages, such as lysine residues on its outer surface that are available for chemical modification.
3.5 Maleimide-mediated cargo conjugation
The classical way of achieving encapsulation in ferritin is by electrostatic interactions (see Section 2.3). However, an alternative approach has been demonstrated employing conjugation using AfFtn.63 Cargo loading is specifically controlled by maleimide-mediated conjugation between AfFtn and target protein. The assembly of AfFtn is dependent on the ionic strength of the buffer. This allows the cargo protein to be conjugated onto AfFtn first under phosphate-buffered saline conditions (PBS with 0.15 M NaCl) followed by assembly of AfFtn nanocages in high salt buffer (1 M NaCl). The formed nanocages then undergo a crosslinking procedure to stabilise the cage and are conjugated with antibodies against plasmalemmal vesicle-associated protein (Plvap) for endothelial drug targeting. This research aimed to deliver superoxidase dismutase (SOD) to caveolae, the flask-shaped invaginations of plasma membranes abundant in the pulmonary endothelium as an anti-inflammatory agent. Because the internal diameter of caveolae stomata apertures is considered to be less than 30 nm, AfFtn cages (outer diameter = 10–12 nm, with an 8 nm diameter cavity) are sufficiently small for targeted delivery. SOD-carrying-antibody-modified-AfFtn cages were confirmed to be delivered into cells and mostly located in endosomes and lysosomes. Subsequent animal studies have shown that the enzyme-containing antibody-modified-AfFtn provides protection against lipopolysaccharide-induced inflammation.4 Gene fusion
This section deals with protein cargo encapsulation utilizing direct genetic fusion either to protein cage monomers or some auxillary subunits necessary for assembly. Genetic fusion is normally carried out in such a way that the cargo should face the internal lumen of the cage (Fig. 3). This method is advantageous compared to other methods in terms of achieving an efficient encapsulation ratio and retention of the cargo.64 One of the major challenges is to control the extent of cargo loading inside protein cage, as potentially every monomer will carry a cargo, possibly leading to steric clashes and inhibition of cage formation. A patchwork assembly method addresses this problem by mixing cargo-fused monomer with unmodified monomer in different ratios to generate cages with different numbers of cargo in different protein cages, such as CCMV and AaLS.65,66 There are several instances of protein cages known where cargo encapsulation is carried out using genetic fusion.67 Among these perhaps the most notable is the P22 bacteriophage cage VLP where protein cargo encapsulation has almost exclusively been achieved using genetic fusion.68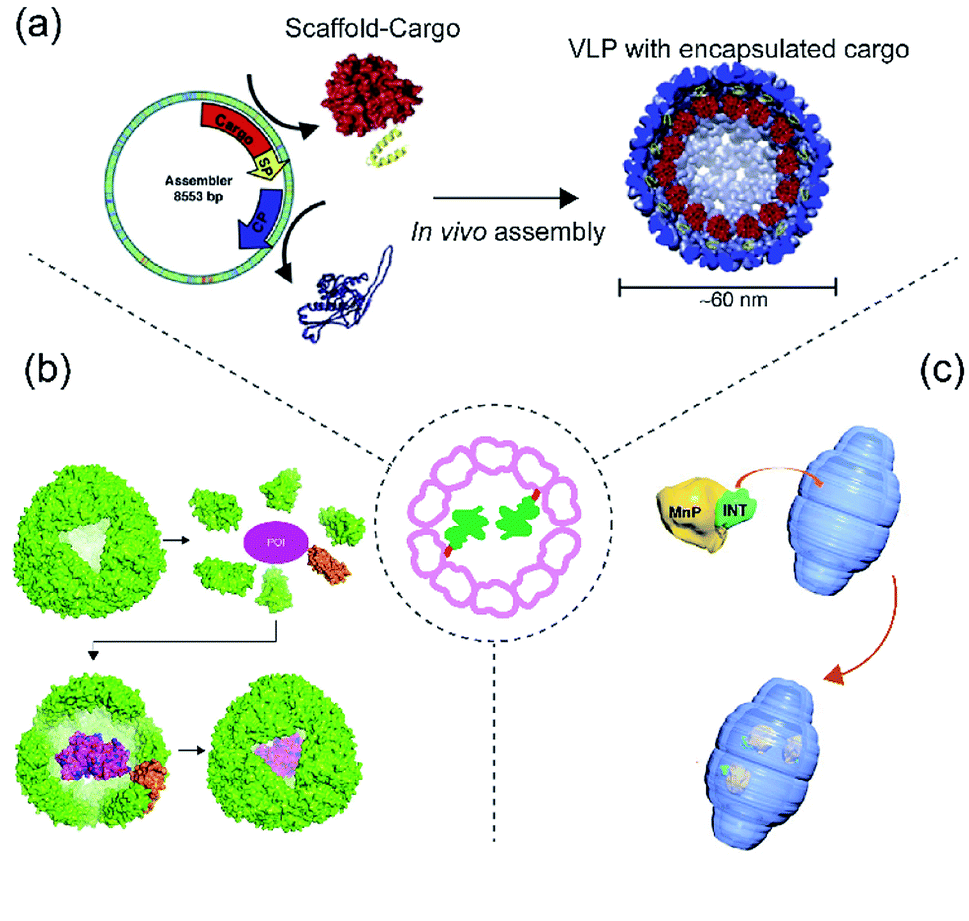 | ||
| Fig. 3 Scheme of protein cargo encapsulation inside cage by fusion methods. Centre: Concept of fusion-based cargo encapsulation. (a) Schematic showing P22 VLP assembly from individual subunits (coat and scaffolding protein), scaffold protein is first genetically modified (fused) with cargo protein and then co-expressed and assembled in presence of coat protein to generate the cage inside E. coli. The image was reproduced from ref. 80 with permission from Elsevier. (b) Schematic showing ferritin-fused with cargo and encapsulation of the cargo during assembly.81 (c) Schematic showing encapsulation of MnP in vault via its fusion with the INT domain. Reprinted (adapted) with permission from ref. 82 (https://doi.org/10.1021/acsnano.5b04073). Copyright (2015) American Chemical Society. | ||
4.1 Bacteriophage P22
Bacteriophage P22 is a double stranded DNA virus belonging to the Podoviridae family and infects the bacterium Salmonella typhimurium.69 Phage P22 assembles into a T = 7 icosahedral capsid (known as a “procapsid”) with a 50 nm central cavity into which DNA is subsequently packaged.69,70 Assembly of the CP requires the presence of an additional assembly chaperone “scaffold protein” (SP). P22 VLP is structurally highly similar to the procapsid (PC) form of the bacteriophage and is normally formed by 420 CP and 100–300 SPs. SPs attach to the capsid inner wall mainly by electrostatic interaction. Protein cargo encapsulation inside P22 is then achieved via fusion of cargoes to SP wherein modified cargo-SP, unmodified-SP and coat protein are co-expressed and assembled inside E. coli (Fig. 3a). Using this approach, several studies have demonstrated P22 as the carrier for direct cargo encapsulation.71 Examples of encapsulated proteins include fluorescence protein (GFP and mCherry),72 alcohol dehydrogenase D (AdhD),68 β-glycosidase (CelB) (a truncated P22 was employed to form the protein cages),73,74 NiFe hydrogenase (Hyd-1)75 and cytochrome P450 (CYP).76 It is also possible to encapsulate several different enzymes inside a single P22 capsid and to use them to perform a cascade reaction with a good efficiency.77 Such a reaction has also been demonstrated in P22 which is itself arranged into a higher order, superlattice structure.64,78–804.2 Ferritin
Although ferritin is known for encapsulation via electrostatic interactions, AfFtn can also be genetically modified in order to accommodate several protein cargoes via fusion. GFP, Renilla luciferase, and truncated versions of horseradish peroxidase have all been fused to the ferritin C-terminus leading to their presence in the cavity after ferritin assembly81 (Fig. 3b). Unexpectedly, it was observed that the protein cargo can be easily released from the cage by mild alteration of pH followed by a cleavage reaction. In addition, the encapsulated cargo appears resistant to thermal, chaotropic, and proteolytic stress and in some cases appeared to result in several-fold increase in protein folding efficiency.814.3 Vault
Vault is another example of protein cage where cargo loading was achieved using direct genetic fusion82 (Fig. 3c). Vault is a large (roughly 13 MDa) protein assembly with dimensions of approximately 67 × 40 × 40 nm. It is found in almost all eukaryotes and possesses a unique architecture. Its main physiological function in eukaryotes is still unknown. Almost all known vaults consist of one major protein component known as the major vault protein (MVP), with minor components including poly(ADP-ribose) polymerase (VPARP), telomerase and small non-translated RNA.83 Structural and biophysical studies have shown that MVP and VPARP interact through involvement of a particular domain known as interaction domain (INT) present on VPARP. The domain consists of 161 amino acids and resides on the C-terminus of VPARP. As VPARP binding to MVP is facilitated by the INT domain, it was initially predicted and later demonstrated that it can be used as a tag for direct protein capture inside of the vault particle.84 Because of its unique architecture and its large cavity size, the vault cage has been widely used for cargo encapsulation of numerous molecules including therapeutics, small molecules and polymers.83,85 Recently it was used to load MnP for bioremediation. The enzyme was packaged inside vault by genetically fusing it with INT-domain, and then encapsulation was achieved via high affinity interaction between the INT domain and vault interior.825 Conclusions and perspectives
The growing library of molecular tools allowing capture of cargoes within protein cages will widen the possible combinations of cages and contents that can be paired together. In turn, this will lead to an increased breadth of potential applications for enzyme-filled cages. Combined with modifications to the exterior of protein cages this may allow the development of cages able to deliver therapeutic enzymes to particular tissues and cells. It is notable that all of the above-described examples are guest loading into (modified) naturally occurring protein cages. The structure of natural protein cages may have intrinsic limits to the extent or type of modification that can be carried out. In recent years, artificial protein cages that do not exist in nature have been designed and realised in a range of sizes86–88 with unusual protein–protein interfaces and triggerable disassembly properties.89–91 Little work has yet been carried out in adding protein cargoes to these artificial cages, but their programmability should allow bespoke functionalities. In addition, artificial cages will allow the use of all the capture strategies outlined above as well as further capture strategies which may not be compatible with a given natural cage. As natural and artificial protein cages carrying useful enzyme cargoes are developed further we may expect to see them integrated into more sophisticated ensembles with a suite of capabilities ensuring desired stability and positioning of reacting components for industrial catalysis. Alternatively they may be used for therapeutic applications targeting and release of the cargo with correct localisation and timing.Conflicts of interest
JGH is named as an inventor on a patent application related to protein-cage assembly construction. He is also the founder of and holds equity in nCage Therapeutics LLC, which aims to commercialise protein cages for therapeutic applications.Acknowledgements
We thank Dr Yusuke Azuma for his constructive comments and discussions. This work was supported by a National Science Centre (NCN, Poland) grant no. 2016/20/W/NZ1/00095 (Symfonia-4) to J. G. H., the Homing program (Homing/2016-2/14; T.-Y. L.), the Homing program (Homing/2017-3/22; S. C) from the Foundation for Polish Science and the Ministry of Science and Technology (MOST, Taiwan) fellowship funding (108-2917-I-564-043-; T.-Y. L.).References
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef PubMed.
- H. T. Spencer, B. E. Riley and C. B. Doering, Haemophilia, 2016, 22, 66–71 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 Search PubMed.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS PubMed.
- M. V. Arbige, J. K. Shetty and G. K. Chotani, Trends Biotechnol., 2019, 37, 1355–1366 CrossRef CAS PubMed.
- R. Singh, M. Kumar, A. Mittal and P. K. Mehta, 3 Biotech, 2016, 6, 174 CrossRef PubMed.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stepien, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef CAS PubMed.
- L. Sanchez-Sanchez, R. D. Cadena-Nava, L. A. Palomares, J. Ruiz-Garcia, M. S. Koay, J. J. Cornelissen and R. Vazquez-Duhalt, Enzyme Microb. Technol., 2014, 60, 24–31 CrossRef CAS PubMed.
- F. P. Seebeck, K. J. Woycechowsky, W. Zhuang, J. P. Rabe and D. Hilvert, J. Am. Chem. Soc., 2006, 128, 4516–4517 CrossRef CAS PubMed.
- J. E. Glasgow, M. A. Asensio, C. M. Jakobson, M. B. Francis and D. Tullman-Ercek, ACS Synth. Biol., 2015, 4, 1011–1019 CrossRef CAS PubMed.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. Sommerdijk, A. E. Rowan, P. C. Christianen, J. C. Maan, B. J. Verduin, J. J. Cornelissen and R. J. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef CAS PubMed.
- M. Brasch, R. M. Putri, M. V. de Ruiter, D. Luque, M. S. Koay, J. R. Caston and J. J. Cornelissen, J. Am. Chem. Soc., 2017, 139, 1512–1519 CrossRef CAS PubMed.
- A. Bacher, S. Eberhardt, M. Fischer, K. Kis and G. Richter, Annu. Rev. Nutr., 2000, 20, 153–167 CrossRef CAS PubMed.
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC.
- B. Worsdorfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef PubMed.
- B. Worsdorfer, Z. Pianowski and D. Hilvert, J. Am. Chem. Soc., 2012, 134, 909–911 CrossRef CAS PubMed.
- R. Frey, S. Mantri, M. Rocca and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 10072–10075 CrossRef CAS PubMed.
- Y. Azuma, D. L. V. Bader and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 860–863 CrossRef CAS PubMed.
- R. Frey, T. Hayashi and D. Hilvert, Chem. Commun., 2016, 52, 10423–10426 RSC.
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., Int. Ed., 2016, 55, 1531–1534 CrossRef CAS PubMed.
- Y. Azuma and D. Hilvert, in Protein Scaffolds, ed. A. K. Udit, Humana Press, New York, NY, 2018, pp. 39–55 Search PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Boker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- K. Yoshizawa, Y. Mishima, S. Y. Park, J. G. Heddle, J. R. Tame, K. Iwahori, M. Kobayashi and I. Yamashita, J. Biochem., 2007, 142, 707–713 CrossRef CAS PubMed.
- K. W. Pulsipher and I. J. Dmochowski, Isr. J. Chem., 2016, 56, 660–670 CrossRef CAS.
- S. Tetter and D. Hilvert, Angew. Chem., Int. Ed. Engl., 2017, 56, 14933–14936 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2012, 8, 52–56 CrossRef PubMed.
- M. Hestericová, T. Heinisch, M. Lenz and T. R. Ward, Dalton Trans., 2018, 47, 10837–10841 RSC.
- I. J. Minten, L. J. A. Hendriks, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771–17773 CrossRef CAS PubMed.
- I. J. Minten, V. I. Claessen, K. Blank, A. E. Rowan, R. J. M. Nolte and J. J. L. M. Cornelissen, Chem. Sci., 2011, 2, 358–362 RSC.
- M. B. Quin, S. A. Perdue, S.-Y. Hsu and C. Schmidt-Dannert, Appl. Microbiol. Biotechnol., 2016, 100, 9187–9200 CrossRef CAS PubMed.
- S. Choudhary, M. B. Quin, M. A. Sanders, E. T. Johnson and C. Schmidt-Dannert, PLoS One, 2012, 7, e33342 CrossRef CAS PubMed.
- H. Ton-That, S. K. Mazmanian, K. F. Faull and O. Schneewind, J. Biol. Chem., 2000, 275, 9876–9881 CrossRef CAS PubMed.
- O. Schneewind, P. Model and V. A. Fischetti, Cell, 1992, 70, 267–281 CrossRef CAS PubMed.
- A. P. A. Hendrickx, W. J. B. van Wamel, G. Posthuma, M. J. M. Bonten and R. J. L. Willems, J. Bacteriol., 2007, 189, 8321–8332 CrossRef CAS PubMed.
- S. K. Mazmanian, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- O. Schneewind, A. Fowler and K. Faull, Science, 1995, 268, 103–106 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- M. B. van Eldijk, J. C. Wang, I. J. Minten, C. Li, A. Zlotnick, R. J. Nolte, J. J. Cornelissen and J. C. van Hest, J. Am. Chem. Soc., 2012, 134, 18506–18509 CrossRef CAS PubMed.
- L. Schoonen, R. J. M. Maas, R. J. M. Nolte and J. C. M. van Hest, Tetrahedron, 2017, 73, 4968–4971 CrossRef CAS.
- L. Schoonen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2016, 8, 14467–14472 RSC.
- L. Schoonen, S. Maassen, R. J. M. Nolte and J. C. M. van Hest, Biomacromolecules, 2017, 18, 3492–3497 CrossRef CAS PubMed.
- M. B. van Eldijk, L. Schoonen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Small, 2016, 12, 2476–2483 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- I. N. A. Khairil Anuar, A. Banerjee, A. H. Keeble, A. Carella, G. I. Nikov and M. Howarth, Nat. Commun., 2019, 10, 1734 CrossRef PubMed.
- H. J. Kang, F. Coulibaly, F. Clow, T. Proft and E. N. Baker, Science, 2007, 318, 1625–1628 CrossRef CAS PubMed.
- S. C. Reddington and M. Howarth, Curr. Opin. Chem. Biol., 2015, 29, 94–99 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, Chembiochem, 2016, 17, 1931–1935 CrossRef CAS PubMed.
- S. Singh and A. Zlotnick, J. Biol. Chem., 2003, 278, 18249–18255 CrossRef CAS PubMed.
- A. Zlotnick, N. Cheng, J. F. Conway, F. P. Booy, A. C. Steven, S. J. Stahl and P. T. Wingfield, Biochemistry, 1996, 35, 7412–7421 CrossRef CAS PubMed.
- R. Lizatovic, M. Assent, A. Barendregt, J. Dahlin, A. Bille, K. Satzinger, D. Tupina, A. J. R. Heck, S. Wennmalm and I. Andre, Angew. Chem., Int. Ed. Engl., 2018, 57, 11334–11338 CrossRef CAS PubMed.
- T. Vorherr, P. James, J. Krebs, A. Enyedi, D. J. McCormick, J. T. Penniston and E. Carafoli, Biochemistry, 2002, 29, 355–365 CrossRef PubMed.
- Y. Azuma, R. Zschoche and D. Hilvert, J. Biol. Chem., 2017, 292, 10321–10327 CrossRef CAS PubMed.
- X. Han and K. J. Woycechowsky, Biochemistry, 2017, 56, 6211–6220 CrossRef CAS PubMed.
- C. Cassidy-Amstutz, L. Oltrogge, C. C. Going, A. Lee, P. Teng, D. Quintanilla, A. East-Seletsky, E. R. Williams and D. F. Savage, Biochemistry, 2016, 55, 3461–3468 CrossRef CAS PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Günther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CrossRef CAS PubMed.
- N. G. Abrescia, D. H. Bamford, J. M. Grimes and D. I. Stuart, Annu. Rev. Biochem., 2012, 81, 795–822 CrossRef CAS PubMed.
- H. Moon, J. Lee, J. Min and S. Kang, Biomacromolecules, 2014, 15, 3794–3801 CrossRef CAS PubMed.
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed.
- R. M. Putri, C. Allende-Ballestero, D. Luque, R. Klem, K. A. Rousou, A. Liu, C. H. Traulsen, W. F. Rurup, M. S. T. Koay, J. R. Caston and J. Cornelissen, ACS Nano, 2017, 11, 12796–12804 CrossRef CAS PubMed.
- J. Snijder, O. Kononova, I. M. Barbu, C. Uetrecht, W. F. Rurup, R. J. Burnley, M. S. Koay, J. J. Cornelissen, W. H. Roos, V. Barsegov, G. J. Wuite and A. J. Heck, Biomacromolecules, 2016, 17, 2522–2529 CrossRef CAS PubMed.
- V. V. Shuvaev, M. Khoshnejad, K. W. Pulsipher, R. Y. Kiseleva, E. Arguiri, J. C. Cheung-Lau, K. M. LeFort, M. Christofidou-Solomidou, R. V. Stan, I. J. Dmochowski and V. R. Muzykantov, Biomaterials, 2018, 185, 348–359 CrossRef CAS PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- W. F. Rurup, F. Verbij, M. S. T. Koay, C. Blum, V. Subramaniam and J. J. L. M. Cornelissen, Biomacromolecules, 2014, 15, 558–563 CrossRef CAS PubMed.
- Y. Azuma, M. Herger and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 558–561 CrossRef CAS PubMed.
- D. Diaz, A. Care and A. Sunna, Genes, 2018, 9, 370 CrossRef PubMed.
- D. P. Patterson, P. E. Prevelige and T. Douglas, ACS Nano, 2012, 6, 5000–5009 CrossRef CAS PubMed.
- M. M. Suhanovsky and C. M. Teschke, Virology, 2015, 479–480, 487–497 CrossRef CAS PubMed.
- G. C. Lander, L. Tang, S. R. Casjens, E. B. Gilcrease, P. Prevelige, A. Poliakov, C. S. Potter, B. Carragher and J. E. Johnson, Science, 2006, 312, 1791–1795 CrossRef CAS PubMed.
- D. P. Patterson, in Protein Scaffolds, ed. A. K. Udit, Humana Press, New York, NY, 2018, pp. 11–24 Search PubMed.
- A. O'Neil, P. E. Prevelige, G. Basu and T. Douglas, Biomacromolecules, 2012, 13, 3902–3907 CrossRef PubMed.
- D. P. Patterson, B. Schwarz, K. El-Boubbou, J. van der Oost, P. E. Prevelige and T. Douglas, Soft Matter, 2012, 8, 5000–5009 Search PubMed.
- K. McCoy, E. Selivanovitch, D. Luque, B. Lee, E. Edwards, J. R. Castón and T. Douglas, Biomacromolecules, 2018, 19, 3738–3746 CrossRef CAS PubMed.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2015, 8, 179–185 CrossRef PubMed.
- L. Sánchez-Sánchez, A. Tapia-Moreno, K. Juarez-Moreno, D. P. Patterson, R. D. Cadena-Nava, T. Douglas and R. Vazquez-Duhalt, J. Nanobiotechnol., 2015, 13, 66 CrossRef PubMed.
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2013, 9, 359–365 CrossRef PubMed.
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige, B. Lee and T. Douglas, ACS Nano, 2017, 12, 942–953 CrossRef PubMed.
- K. McCoy, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2018, 12, 3541–3550 CrossRef CAS PubMed.
- E. Selivanovitch and T. Douglas, Curr. Opin. Virol., 2019, 36, 38–46 CrossRef CAS PubMed.
- S. Deshpande, N. D. Masurkar, V. M. Girish, M. Desai, G. Chakraborty, J. M. Chan and C. L. Drum, Nat. Commun., 2017, 8, 1442 CrossRef PubMed.
- M. Wang, D. Abad, V. A. Kickhoefer, L. H. Rome and S. Mahendra, ACS Nano, 2015, 9, 10931–10940 CrossRef CAS PubMed.
- A. Muñoz-Juan, A. Carreño, R. Mendoza and J. L. Corchero, Pharmaceutics, 2019, 11 CrossRef PubMed.
- V. A. Kickhoefer, Y. Garcia, Y. Mikyas, E. Johansson, J. C. Zhou, S. Raval-Fernandes, P. Minoofar, J. I. Zink, B. Dunn, P. L. Stewart and L. H. Rome, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4348–4352 CrossRef CAS PubMed.
- B. C. Ng, M. Yu, A. Gopal, L. H. Rome, H. G. Monbouquette and S. H. Tolbert, Nano Lett., 2008, 8, 3503–3509 CrossRef CAS PubMed.
- E. Golub, R. H. Subramanian, J. Esselborn, R. G. Alberstein, J. B. Bailey, J. A. Chiong, X. Yan, T. Booth, T. S. Baker and F. A. Tezcan, Nature, 2020, 578, 172–176 CrossRef CAS PubMed; N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. Andre, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- A. D. Malay, N. Miyazaki, A. Biela, S. Chakraborti, K. Majsterkiewicz, I. Stupka, C. S. Kaplan, A. Kowalczyk, B. Piette, G. K. A. Hochberg, D. Wu, T. P. Wrobel, A. Fineberg, M. S. Kushwah, M. Kelemen, P. Vavpetic, P. Pelicon, P. Kukura, J. L. P. Benesch, K. Iwasaki and J. G. Heddle, Nature, 2019, 569, 438–442 CrossRef CAS PubMed.
- M. Imamura, T. Uchihashi, T. Ando, A. Leifert, U. Simon, A. D. Malay and J. G. Heddle, Nano Lett., 2015, 15, 1331–1335 CrossRef CAS PubMed.
- A. D. Malay, J. G. Heddle, S. Tomita, K. Iwasaki, N. Miyazaki, K. Sumitomo, H. Yanagi, I. Yamashita and Y. Uraoka, Nano Lett., 2012, 12, 2056–2059 CrossRef CAS PubMed.
Footnote |
| † Current address: Centre for Nanosciences Amrita Institute of Medical Sciences, Amrita Vishwa Vidyapeetham AIMS Ponekkara P. O., Kochi, Kerala – 682041, India. |
| This journal is © The Royal Society of Chemistry 2020 |