
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTherapeutic lipid-coated hybrid nanoparticles against bacterial infections
Lai Jiang†
a,
Hiang Wee Lee† a and
Say Chye Joachim Loo*abc
a and
Say Chye Joachim Loo*abc
aSchool of Materials Science & Engineering, Nanyang Technological University, 50 Nanyang Ave, Singapore 639798. E-mail: joachimloo@ntu.edu.sg; scloo@hsph.harvard.edu
bSingapore Centre for Environmental Life Sciences Engineering, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551
cHarvard T.H. Chan School of Public Health, 677 Huntington Ave, Boston, MA 02115, USA
First published on 27th February 2020
Abstract
One of the most important health concerns in society is the development of pathogen-causing nosocomial infections. Since the first discovery of antibiotics, bacterial infections have been highly treatable. However, with evolution and the nondiscretionary usage of antibiotics, pathogens have also found new ways to survive the onslaught of antibiotics by surviving intracellularly or through the formation of obstinate biofilms, and through these, the outcomes of regular antibiotic treatments may now be unsatisfactory. Lipid-coated hybrid nanoparticles (LCHNPs) are the next-generation core–shell structured nanodelivery system, where an inorganic or organic core, loaded with antimicrobials, is enveloped by lipid layers. This core–shell structure, with multifarious decorations, not only improves the loading capabilities of therapeutics but also has the potential to improve therapeutic delivery, especially for targeting biofilm-based and intracellular bacterial infections. Although there has been significant interest in the development of LCHNPs, they have yet to be widely exploited for bacterial infections. In this review, we will provide an overview on the latest development of LCHNPs and the various approaches in synthesizing this nano-delivery system. In addition, a discussion on future perspectives of LCHNPs, in combination with other novel anti-bacterial technologies, will be provided towards the end of this review.
1. Introduction
Microbial infection is one of the biggest threats to global public health today due to the rapid evolution of antibiotic resistant strains.1–3 Increasingly, multidrug resistant pathogenic strains of bacteria are now routinely isolated from all parts of the world.4–7 Pathogenic microorganisms, such as Enterococcus, Staphylococcus and Pseudomonas, are closely related species that are responsible for a wide variety of common bacterial infections. However, the widespread non-discretionary usage of antibiotics over the last 70 years has driven the emergence and prevalence of bacteria that exhibit drug resistant characteristics. This makes infectious diseases, which were once easily treatable, fatal again. In the United States alone, antibiotic resistance has led to more than two million infections and about 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year, resulting in $20 million of excess medical spending and $35 billion in lost productivity annually.8–10 In fact, it was recently projected that antibiotic resistant pathogens would cause more than 10 million deaths worldwide per year by 2050.11 For instance, the eradication of certain bacterial infections, such as methicillin-resistant Staphylococcus aureus (MRSA), still remains a challenge. Often, this is attributed not only to the complex pathogenic mechanisms in subverting the host's immune system, but also the cellular barriers and the formation of a biofilm that prevents antibiotics from reaching the foci of infection.12,13 Without doubt, addressing drug resistant and tolerant microbes is now a major global health challenge, and this calls for an urgent need to revisit approaches that can effectively control bacterial infections, either through novel formulations, delivery systems or through the discovery of more effective next generation antibiotics.
000 deaths each year, resulting in $20 million of excess medical spending and $35 billion in lost productivity annually.8–10 In fact, it was recently projected that antibiotic resistant pathogens would cause more than 10 million deaths worldwide per year by 2050.11 For instance, the eradication of certain bacterial infections, such as methicillin-resistant Staphylococcus aureus (MRSA), still remains a challenge. Often, this is attributed not only to the complex pathogenic mechanisms in subverting the host's immune system, but also the cellular barriers and the formation of a biofilm that prevents antibiotics from reaching the foci of infection.12,13 Without doubt, addressing drug resistant and tolerant microbes is now a major global health challenge, and this calls for an urgent need to revisit approaches that can effectively control bacterial infections, either through novel formulations, delivery systems or through the discovery of more effective next generation antibiotics.
Over the last few decades, drug-loaded nanoparticles, as therapeutic delivery platforms, have been extensively developed and evaluated to overcome the inherent shortcomings of current antibiotic treatments.14–19 By protecting and delivering the antibiotic to the intended site of action, such delivery platforms are hypothesized to enhance effectiveness of the antibiotics, while potentially mitigating the emergence of drug-tolerant microbes. Of the myriad nano-delivery systems that have been reported for this purpose, most efforts have revolved around the use of liposomes,20–22 polymeric nanoparticles,23–25 dendrimers,26–28 and inorganic nanoparticles (Fig. 1).29–31 These studies have demonstrated that antimicrobial molecules loaded into such delivery nanoparticles, either through physical encapsulation or chemical conjugation, exhibited an improved pharmacokinetic profile and therapeutic index, as compared to their free drug counterparts. Nevertheless, there are also drawbacks with delivery systems, especially those that are composed of a single material (non-hybrid), such as low drug loading efficiency, premature drug release, and rapid clearance of these nanoparticles mediated by the mononuclear phagocyte system (MPS).32,33 To overcome these challenges, hybrid nanoparticles that tap on the advantages of two or more material systems are therefore preferred. For instance, a hybrid nanoparticle may consist of a core polymer that encapsulates the drug, while its surface may be coated or functionalized with another material to avoid the MPS. Such hybrid delivery system, through a coating layer, could also suppress any initial drug release, while allowing for targeting of the infection site.
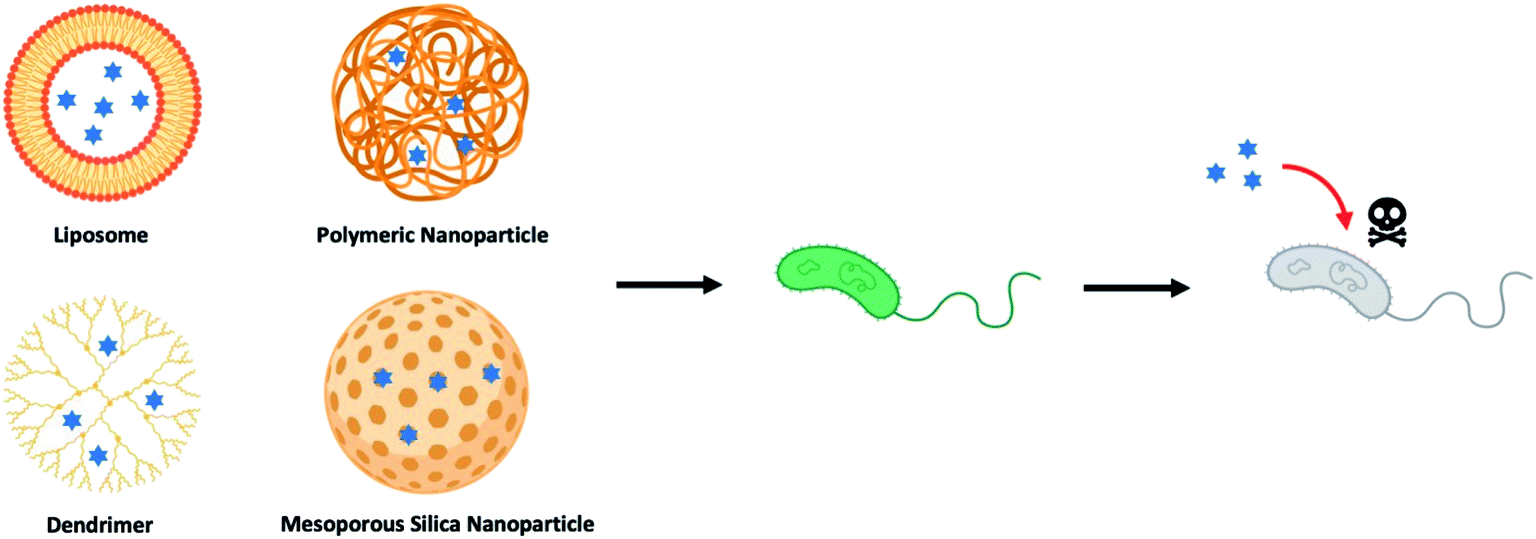 | ||
| Fig. 1 Illustration of different types of nanoparticle-based drug delivery system to against bacterial strains (blue hexagrams represent antibiotics and other therapeutic antimicrobials). | ||
One novel progeny of a hybrid delivery system, that has been gaining much attention within the scientific community in the recent years,34–36 is the lipid-coated hybrid nanoparticle (LCHNP). LCHNPs can be described as composing of an organic or inorganic nanoparticulate core that is coated with single or multiple layers of lipids – constituting of simple lipids (i.e. DOTAP), compound lipids (i.e. phospholipids), or derived lipids (i.e. cholesterol). Possessing a core–shell architecture, the LCHNP exhibit promising attributes as a versatile and robust therapeutic, such as a stable structural integrity with good biocompatibility, holding the ability to encapsulate lipophilic and hydrophilic therapeutic agents, can be designed for controlled release, and flexibility for functionalization to suit a specific application.
Earlier scientific reviews on lipid-polymer hybrid nanoparticles chiefly focus on the preparation of LCHNPs, either organic or inorganic cores, and their applications in drug and gene delivery specifically for cancer therapy.37–41 As a contrast, this review aims to provide an up-to-date overview on the fabrication approaches of LCHNPs, before embarking on exploring how LCHNPs potentially can be exploited, as a therapeutic platform, for both intracellular and biofilm-mediated microbial infections. As a conclusion, this review will highlight the challenges and future perspectives of LCHNPs against infectious diseases.
2. Strategies to synthesize LCHNPs
As inferred from its name, LCHNPs consist of two or more material systems as building blocks (Fig. 2), whereby the core is an organic or inorganic nanoparticle that is enveloped by a lipid layer. In some hybrid nanoparticulate systems, a polymeric/inorganic mesoporous core is chosen because of their excellent capabilities in encapsulating antibiotics. In other hybrid systems, a metallic core is preferred because of its other antimicrobial capabilities that are non-drug-related, i.e. ROS-generation, hyperthermia, etc. Payloads (i.e. antibiotics, antimicrobial peptides, RNA, etc.) can be encapsulated or associated in either the core or lipid layer, or, in some cases, both, depending on the construct of the hybrid nanoparticle. The lipid layer behaves as a molecular barrier that mitigates the loss of the entrapped agents (i.e. drugs) during fabrication while protecting the core from degradation in any physiological environment.42 Given the unique structure of this hybrid system, this review shall commence by highlighting some key strategies in the preparation of LCHNPs. In general, fabrication of LCHNPs can be achieved through two distinctive approaches: (1) a multi-step process whereby the nanoparticle core and lipid shell are prepared separately;43–46 or (2) a single-step process.47–50 The latter is often obtained through a one-pot emulsion–solvent–evaporation (ESE) or through a nanoprecipitation method. In this review, only the most widely employed approaches will be discussed, along with several recent representative examples of each fabrication method.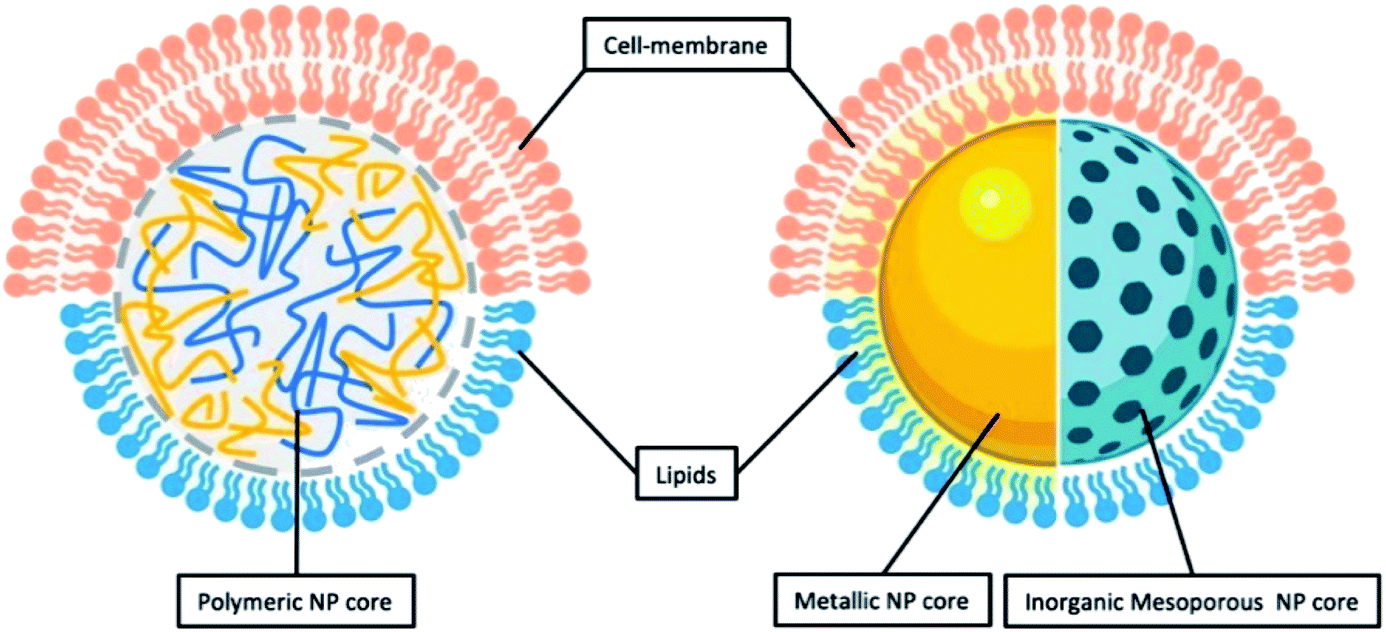 | ||
| Fig. 2 General illustration of core–shell structure of LCHNP demonstrating either polymeric NP or inorganic NP could be coated with either lipids or cell-membrane. | ||
2.1. Multi-step fabrication approaches
| Core | Shell | Loaded agents | Remarks |
|---|---|---|---|
| Fusion of NPs and liposomal vesicles | |||
| PLGA NP | DOTAP | pDNA | The delivery system increased transfection efficiencies compared to free DNA. It is the first time to present NP carrying IL-12 to be efficient in gene delivery to liver cancer cells in the presence of a very high concentration of serum51 |
| CHOL-grafted PAMAM NP | DOTAP, DOPE, CHOL | siRNA | The core–shell nanoparticle carrying siRNA exhibited stronger downregulation of EGFR protein expression level in MCF-7 cells in vitro and the greatest inhibition on tumour growth in vivo44 |
| PLGA NP | DOTAP, CHOL | BSA | The lipid shell not only promoted in vitro cellular uptake of hybrid NPs, improved the stability and protected the integrity of the hybrid structure during long- term storage52 |
| PLGA NP | DOTAP, CHOL | HspX/EsxS protein | This delivery system has a good potential to enhance immune responses against HspX EsxS antigen after subcutaneous immunization of BALB/c mice53 |
| Silver NP | POPS | — | The coating presented excellent inhibition of Gram-positive and Gram-negative bacteria growth and reduced inflammatory response from bone marrow derived macrophages54 |
| Gold NPs | PC, PS | — | LCHNP-based methods allow for highly sensitive detection of protein-induced membrane apposition under conditions that minimize large-scale aggregation55 |
| MgP NP | DOTAP, CHOL | CAT | This LCHNP carrying CAT protected MCF-7 cells from lethal level of exogenous H2O2 and lowered the ROS levels in EA.hy 926 cells56 |
| PS NP | DOTAP, CHOL, DOPC, and DSPE-PEG-2000 | — | The lipid envelopes enable the diffusion of LCHNPs into the mucus layer and promote the interaction of LCHNPs with a bacterial biofilm57 |
|
|||
| Re-hydration from lipid thin-film | |||
| CS-TPP NP | Egg PL, CHOL and DPPE | Moxifloxacin | The mean residence time, area under the curve, apparent permeability coefficient of LCHNPs were up to 6.74-fold, 4.29-fold, and 3.29-fold higher than commercials45 |
| CS NP | Egg PL, RHL, CHOL and DSPE-PEG-2000 | CLR | The CLR-loaded LCHNP exhibited excellent eradicating ability to H. pylori biofilm58 |
| PLGA NP | DSPC, DSPE-PEG-Mal and CHOL | ATRA | It firstly reported that LCHNP conjugated with CD133 aptamers showed a sustained release of ATRA, presented efficiently and specifically ATRA delivery to osteosarcoma initiating cells, and achieved superior therapeutic efficacy59 |
| MSN | PL and CHOL | HCPT | This HCPT-LCHNP had stronger inhibitory effects on hepatocellular carcinoma cells compared with free HCPT, providing an effective strategy for delivering poorly-soluble anticancer drugs60 |
| MSN | Soybean PL, CHOL and PEG-2000 | PTX, CUR | The co-delivery LCHNP system enabled the intravenous administration of hydrophobic drugs, and sustained released trend showed strong cytotoxic effect against breast cancer cells61 |
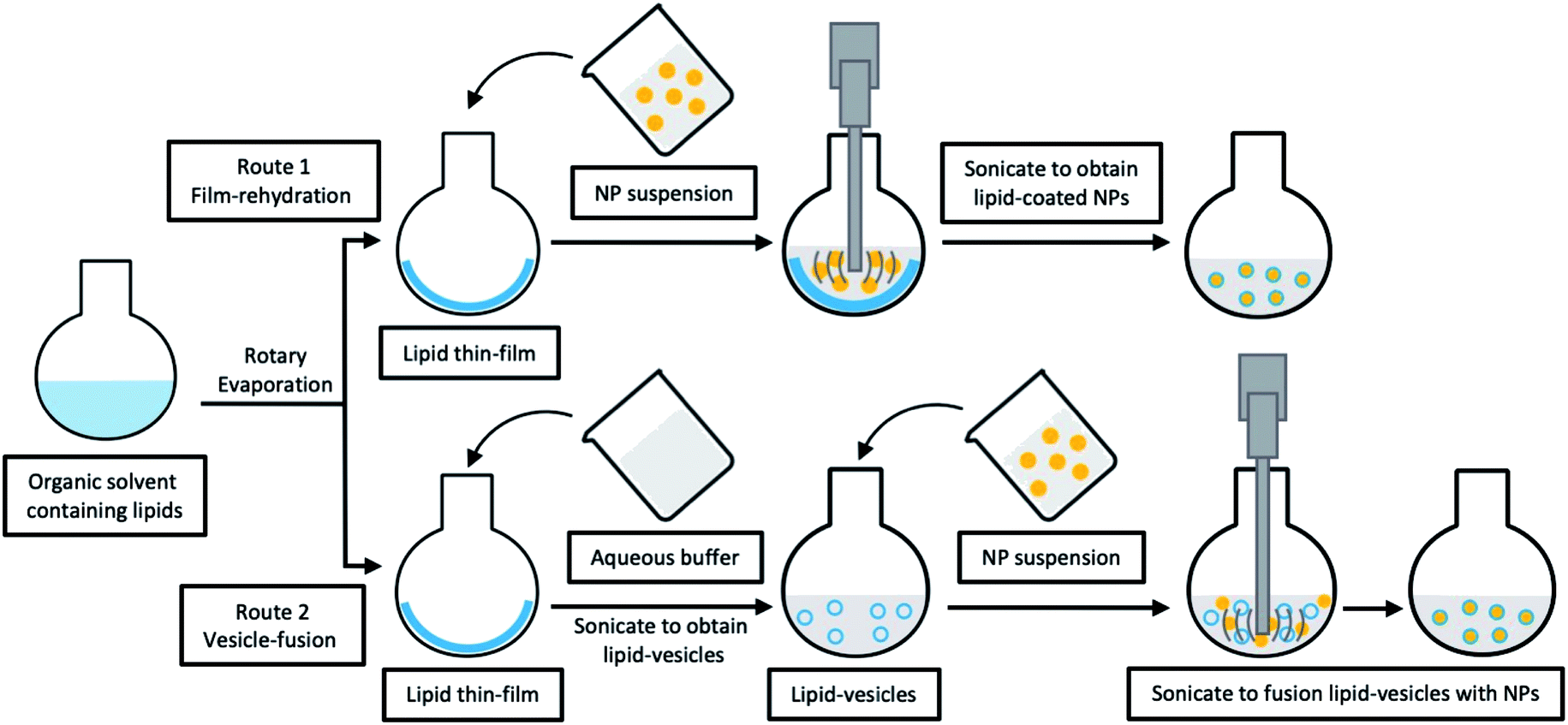 | ||
| Fig. 3 Schematic illustration of typical multi-step fabrications for LCHNPs. Route 1, film-rehydration method, where the core–shell structure is obtained through direct hydration of the lipid thin film within the NP dispersion; route 2, vesicle-fusion method, where the core–shell structure is obtained by fusing the preformed NP into lipid-vesicles. | ||
Unlike vesicle-fusion and film-rehydration methods, the preparation of cell-membrane coated nanoparticles involves an additional step of extracting membrane vesicles from cells (Fig. 4).71 Another point for consideration is that cell membrane extraction procedures are highly dependent on the cell source.72 For instance, to obtain bioactive cell membrane from nucleus-free cells (e.g. Red Blood Cells), the cells have to be first isolated from whole blood, which are then lysed with either a hypotonic treatment or a repeated freeze-thaw process, before extruding through polycarbonate membranes using nanosized pores to obtain purified vesicles. It is noted that the extraction and purification of membrane vesicles from eukaryotes (e.g. leukocytes), compared with nucleus-free cells, are more complicated. The harvested cells are then lysed through hypotonic treatment combined with mechanical membrane destruction, followed by the removal of the intracellular components (e.g. nucleus) by discontinuous sucrose gradient centrifugations. The final ideal vesicles are finally obtained by washing and extrusion. A list of different nanoparticulate cores coated with cell-membrane from various source cells is shown in Table 2. From this list, this recently developed strategy has demonstrated to be effective for surface functionalization of nanoparticles.
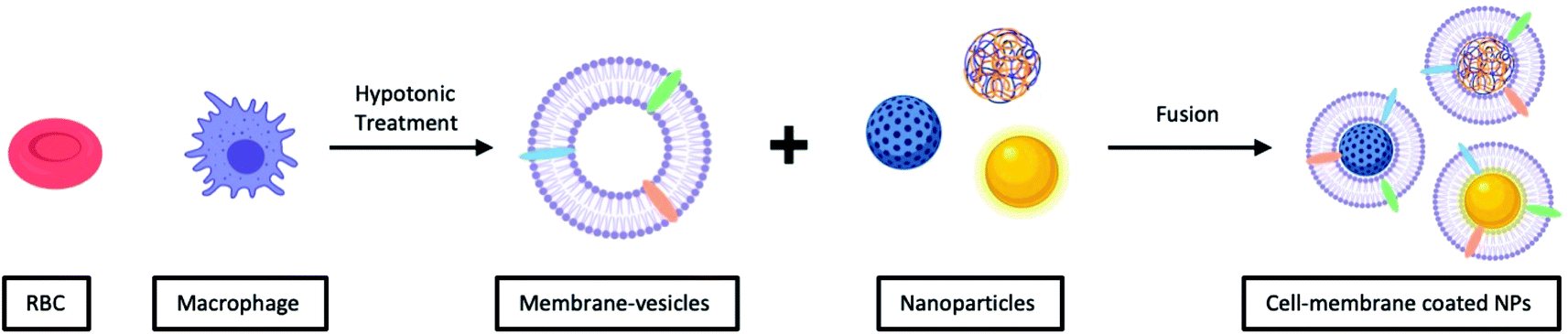 | ||
| Fig. 4 Typical preparation of cell-membrane coated NPs. Cell-membrane-derived vesicles are collected from either nucleus-free cells or eukaryotes and then coated onto various types of NPs. | ||
| Core | Source cell | Loaded agents | Remarks |
|---|---|---|---|
| PLGA NP | Red blood cell | DOX | The LCHNP efficiently deliver DOX to solid tumour sites for significantly increased tumour growth inhibition compared with conventional free drug treatment73 |
| PLGA NP | Platelets | RAP | The plates-coated NP displayed 4.98-fold greater radiant efficiency than control in atherosclerotic arterial trees, indicating its effective homing to atherosclerotic plaques in vivo. NP loading RAP significantly attenuated the progression of atherosclerosis and stabilized atherosclerotic plaques74 |
| PLGA NP | Gastric epithelial cell (AGS cell) | CLR | The AGS-NPs preferentially accumulate on the H. pylori surfaces in vitro. The CLR-loaded AGS-NPs demonstrate superior therapeutic efficacy in a mouse model of H. pylori infection than free CLR75 |
| PCL and Pluronic F68 NP | 4T1 Breast cancer cell | PTX | The LCHNP achieved specific targeting of homotypic tumours or metastatic nodules in the lung. It further demonstrated a remarkable anti-tumour and metastasis efficacy, not only in the orthotopic transplantation tumour models but also in the advanced metastasis mice models76 |
| PPC8, PPiP NP | Mouse peritoneal macrophage | PTX | The designed macrophage-membrane-coated nanoparticle showed enhanced penetration efficiency on tumour cells, the loaded PTX was quickly released from the nanoparticles in response to the endosome pH. It exhibited an enhanced therapeutic effect inherited from both membrane-derived tumour homing and step-by-step controlled drug release77 |
| PCPDTBT and PEG-b-PPG-b-PEG NP | Activated fibroblast cell | — | The cell-membrane coated NP specifically target cancer-associated fibroblasts, leading to enhanced tumour accumulation. It generates enhanced cytotoxic heat and single oxygen to exert combinational photo-thermal and photo-dynamic therapy78 |
| Gold NP | Escherichia coli | — | The bacterial membrane-coated NPs induced rapid activation and maturation of dendritic cells in the lymph nodes of vaccinated mice. It generated strong Th1 and Th17 biased cell responses against the source bacteria79 |
| Gold nanocage | 4T1 Breast cancer cell | DOX | The LCHNP exhibit a stimuli-release of DOX under the hyperthermia and a high cell-specific targeting of the 4T1 cells in vitro. The therapy with about 98.9% and 98.5% inhibiting rates of the tumour volume and metastatic nodules is observed in the 4T1 orthotopic mammary tumour models80 |
| Gold–silver nanocage | Macrophage | — | The membrane-coated nano-system targets bacteria more efficiently. It retained at the infection site and showed greatly prolonged circulation time and excellent biocompatibility81 |
| MSNC | Macrophage | DOX | The MSNC greatly increase the loading capacity for DOX. The macrophage-coated MSNC not only offer camouflage function, but also provide active targeting ability due to surface proteins82 |
2.2. Singe-step fabrication approaches
Multi-step approaches are limited by the separate preparation of the core nanoparticles and lipid vesicles/cell membrane vesicles, and are likely to be less cost effective. In addition, there is always the possibility of drug leakages, especially for hydrophilic agents, during the assembly process. To improve on these drawbacks, single-step approaches have been improvised to synthesize LCHNPs, of which several of such approaches will be highlighted here.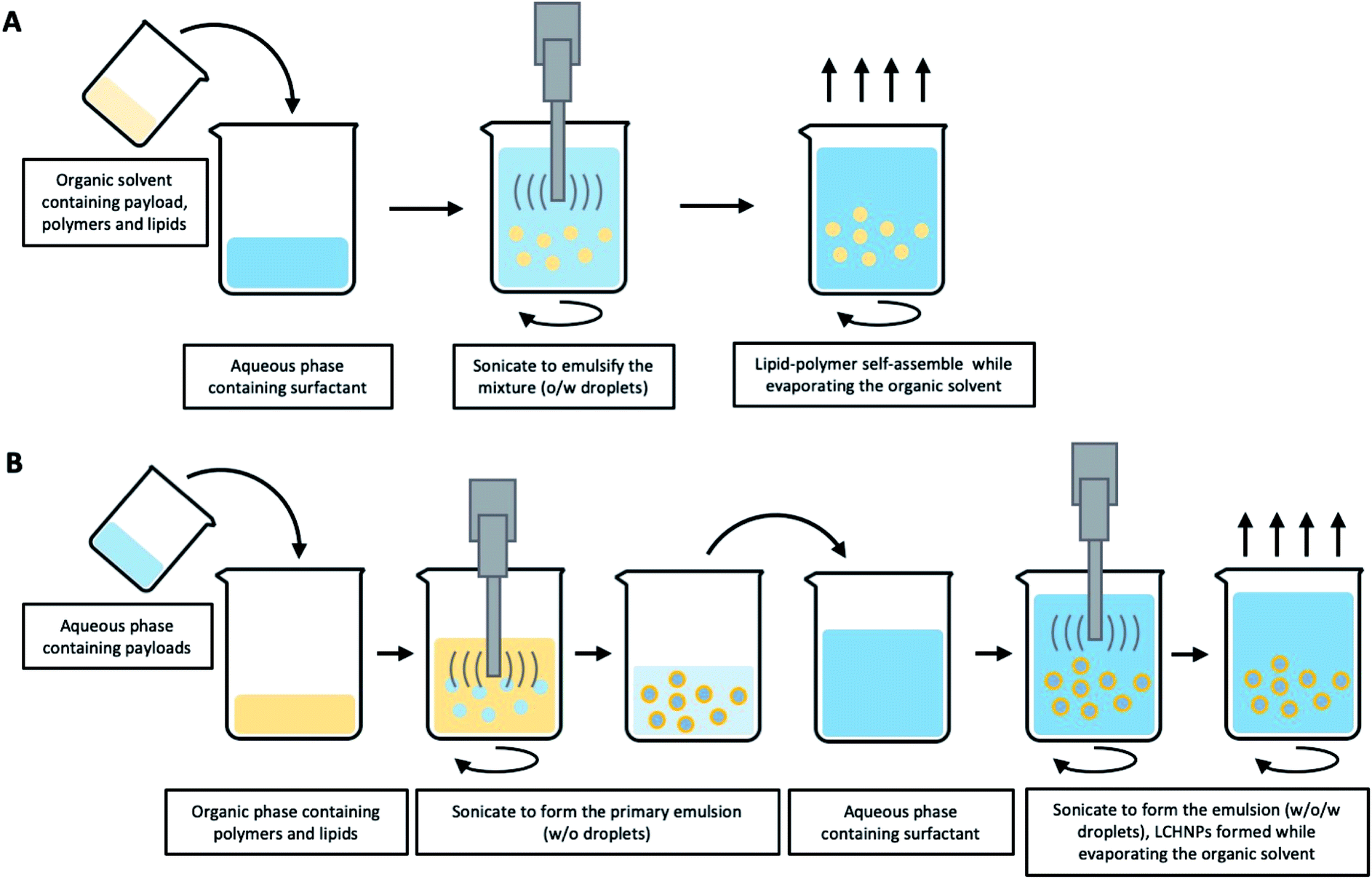 | ||
| Fig. 5 Schematic illustration of typical emulsification-solvent-evaporation method for LCHNPs. (A) An o/w single emulsion formulation is suitable for loading hydrophobic agents, where the emulsion is formed when the organic phase (polymer, lipid and payload) is emulsified with an aqueous phase (surfactant). (B) A w/o/w double emulsion is used for the entrapment of the hydrophilic payloads, where the aqueous solution (hydrophilic agents) is firstly emulsified with an organic solvent (polymer and lipid) to form a primary w/o emulsion, then a w/o/w double emulsion is generated when the primary emulsion is emulsified again with an aqueous phase (surfactant). | ||
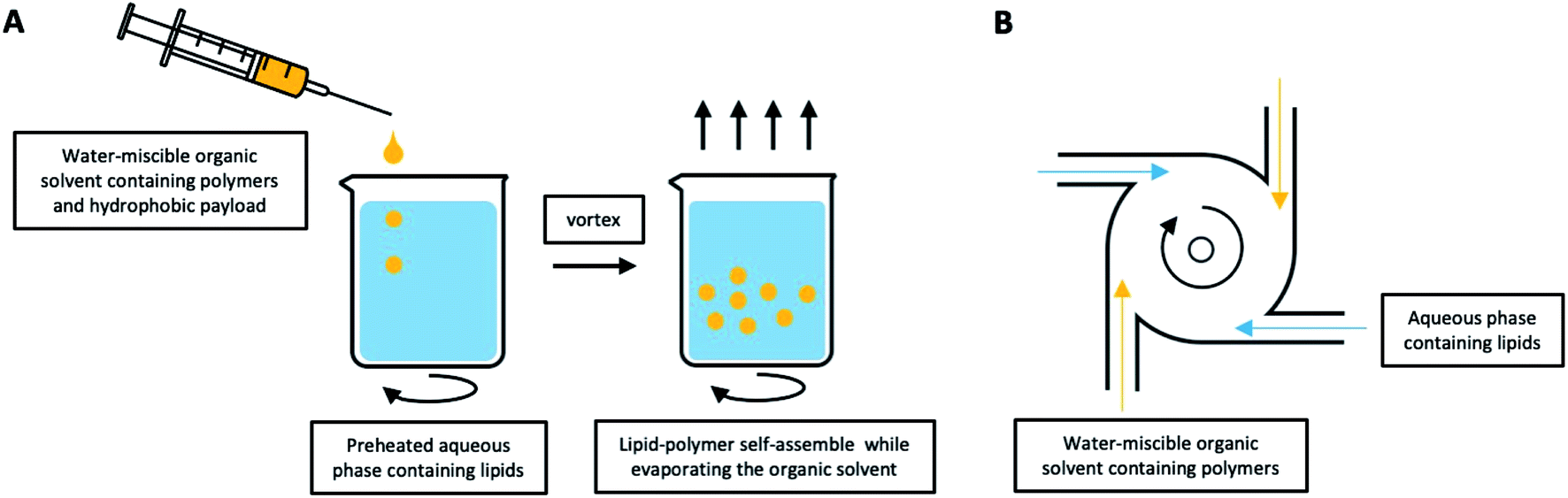 | ||
| Fig. 6 LCHNPs preparation using nanoprecipitation. (A) Schematic illustration of typical nanoprecipitation. The polymer-contained organic solution is dropwise added into the preheated lipid-contained aqueous phase under continuous stirring, the lipids will self-assemble onto the NPs. (B) An improved nanoprecipitation method using MIVRs for large-scale production of LCHNPs. | ||
:POPS (70:30) liposomes and propanethiol to freshly synthesize AuNP, to produce a hybrid lipid bilayer in a single step. The thiol groups of the propanethiol molecules form a first coating layer on the AuNP surface, followed by the self-assembling of POPC and POPS phospholipids to form a second amphiphilic layer.55In summary, the multi-step methods in preparing LCHNPs are more widely reported and are generally employed in lab-scale research. However, the drawbacks in preparing lipids/cell-membrane vesicles and NPs separately, renders this method far from efficient in terms of time and resources and this could possibly limit the clinical translation of these promising LCHNPs for treating infectious diseases. Although the one-step method for preparing LCHNPs overcomes the shortcomings of the multi-step processes, it is still unable to meet the large-scale production requirements for clinical usage.
3. LCHNPs as therapeutic delivery system against bacteria-derived diseases
Bacteria can become resistant or highly tolerant to antibiotics through different mechanisms.100 While antibiotic resistance is typically caused by inherited mutations stemming from diverse molecular mechanisms, non-inherited antibiotic tolerance is induced by phenotypic switching (e.g. reduced growth rate/metabolism) of a subpopulation of bacteria surviving in biofilms and/or within the host cells.101,102 Exploiting host cells as a shield against antibiotic appears to be one of the most effective evasion strategy. In fact, many severe, chronic and recurrent infections are mediated by biofilm bacteria or pathogens that persist intracellularly.103,104 The persistence of intracellular pathogens is not solely due to metabolic dormancy, but that intracellular lifestyle shield the pathogens from antibiotics owing to poor drug penetration and retention, as well as decreased intracellular drug activity.105 Such a tactic is commonly employed by many pathogens responsible for healthcare-associated and community-acquired infections, such as Mycobacterium tuberculosis, Escherichia coli, Listeria monocytogenes, Salmonella enterica, Enterococcus faecalis, Staphylococcus aureus, etc., in both wound and surgical-related infections.103,106,107 This mechanism is particularly relevant since most antibiotics are precluded from entering or are poorly retained by the host cells. Even if compounds do penetrate the host cells, they will either be rapidly degraded by the lysosomal acids or enzymes within the endolysosomes, or their concentration does not reach therapeutic levels for sufficient duration.108,109 When residing in the host cells, intracellular bacteria may also reduce their growth rate to lower their susceptibility to antibiotics.103,110 All these make eradication of intracellular pathogens extremely challenging. Of greater clinically significance is that intracellular pathogens can propagate inside phagocytic (i.e. macrophages, neutrophils, etc.) and non-phagocytic cells (i.e. fibroblasts, enterocytes, keratinocytes, hepatocytes, etc.),108 leading to the spread of infection even during antibiotic therapy.103,111 As such, the shielding effect conferred by the host cells has not only led to numerous chronic and recurrent infections, such as tuberculosis, urinary tract infections, pulmonary infections, endocarditis, atopic dermatitis, etc., but also contributed to the enrichment of antibiotic resistant strains during prolonged antibiotic treatment.1043.1. Intracellular bacterial infections
Regular pathogenic bacteria usually would be recognized and engulfed by professional phagocytes (i.e. macrophages), kept in a bubble-like structure – phagosome within the cytoplasm. This bacteria-harbouring phagosome then fuses with a lysosome, becoming a “phagolysosome”, where the lysosome provides enzymes and drastically lowers the internal pH to break down the contents (Fig. 7A).112 However, certain intracellular bacterial pathogens, such as Mycobacterium tuberculosis,113,114 Salmonella enterica,115 and Klebsiella pneumoniae,116 develop various defence mechanisms to escape the phagocytosis pathways or remain viable in the phagolysosomes, which is the case for infectious diseases, such as tuberculosis,117 listeriosis,118 and salmonellosis.119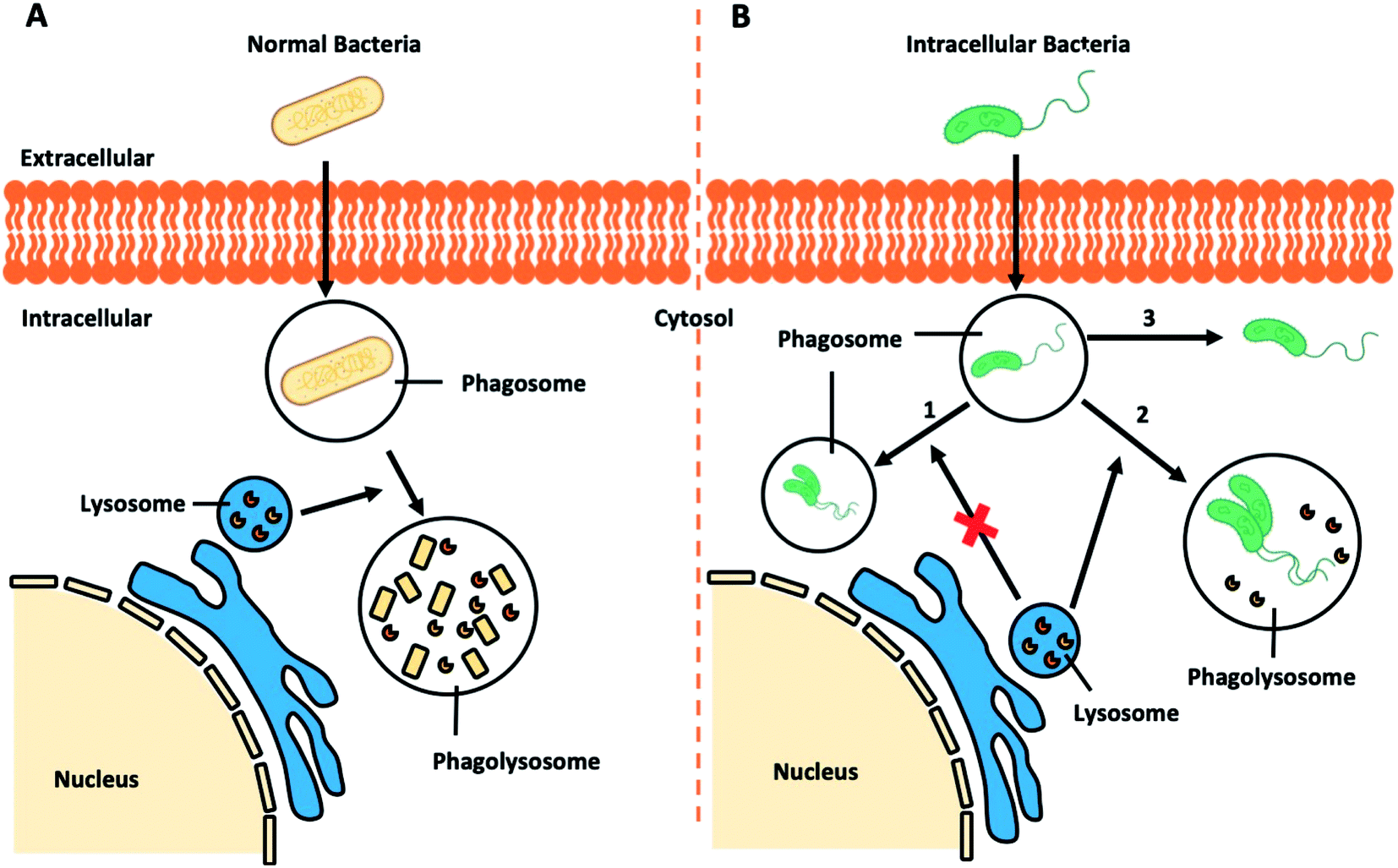 | ||
| Fig. 7 Schematic illustration of (A) bacteria-involved phagocytosis and (B) resistance mechanisms developed by certain intracellular pathogens. | ||
Recent research demonstrated that Klebsiella pneumoniae can promote the activation of Akt to arrest phagosome maturation, avoiding fusion into lysosomes, thus creating a Klebsiella-containing vacuole to survival intracellularly (Fig. 7B, route 1).120 Similar mechanisms are also observed in the phagocytosis pathways of Salmonella typhimurium and Mycobacterium tuberculosis.121 Previously considered as an extracellular bacterium, Staphylococcus aureus has also been shown to invade and survive in either professional or non-professional phagocytes, including keratinocytes, endothelial cells, epithelial cells, fibroblast and osteoblasts.122–125 The adhesion of Staphylococcus aureus to the host cell surface could lead to cytoskeletal rearrangement, allowing Staphylococcus aureus to move into cells and even replicate within the acidic phagolysosome (Fig. 7B, route 2).126 Other studies report that other bacteria could escape from the phagocytic vacuole into the cytosol.127 In the case of intracellular Listeria monocytogenes infection, pores can be formed in the vacuole membrane to release phospholipases to disrupt the vacuole membrane (Fig. 7B route 3). Once in the cytosol, Listeria monocytogenes will induce the polymerization of host actin filaments and the force generated by actin polymerization allow them to move into neighbouring cells resulting in a productive infection.128 Similar routes to infection is adopted by Shigella flexneri,129,130 Salmonella enterica,131 and Mycobacterium tuberculosis.132
Thus, while professional phagocytes not only serve to eradicate intracellular pathogens, ironically it also provides a reservoir of latent infection representing a significant barrier to antibiotic treatment. Phagocytes harbouring such pathogens, acting as “Trojan Horses”, allow those intracellular pathogens to establish secondary infection foci, thus causing recurrent systemic infections.133,134 In such a situation, a delivery system, e.g. LCHNPs, that allows antibiotics to target intracellular infections would serve to be extremely beneficial. Theoretically, LCHNPs can either passively accumulate at the infection foci, or actively target the pathogen and macrophages. More importantly, upon macrophage phagocytosis, the nanoparticle shares the same pathway as pathogens, thus allowing the payloads to be delivered into the infected cells, thereby enhancing the penetration and permitting the release of therapeutics within these cells. To achieve this, the lipid coating on drug-loaded nanoparticles has emerged as an important step to not only promote colloidal stability, and to also provide a means for intracellular delivery of the payload. Diverse LCHNPs have been developed and employed recently for the delivery of therapeutics to intracellular pathogens.
:0 PEG-2000 PE, using a lipid film-rehydration method and further modified with UBI29–41 (LU). The Gen@MSN-LU was assembled by vesicle-fusion method of these two components (Fig. 8A). The core–shell structure would prevent premature release of gentamicin and its shell, once at the infection foci, will be degraded by bacterial toxins to achieve rapid release of gentamicin (Fig. 8B). In their subsequent antimicrobial studies, the Gen@MSN-LU efficiently targeted Staphylococcus aureus from in vitro studies, and effectively inhibited bacteria growth in an intracellular-infected in vivo mouse model.
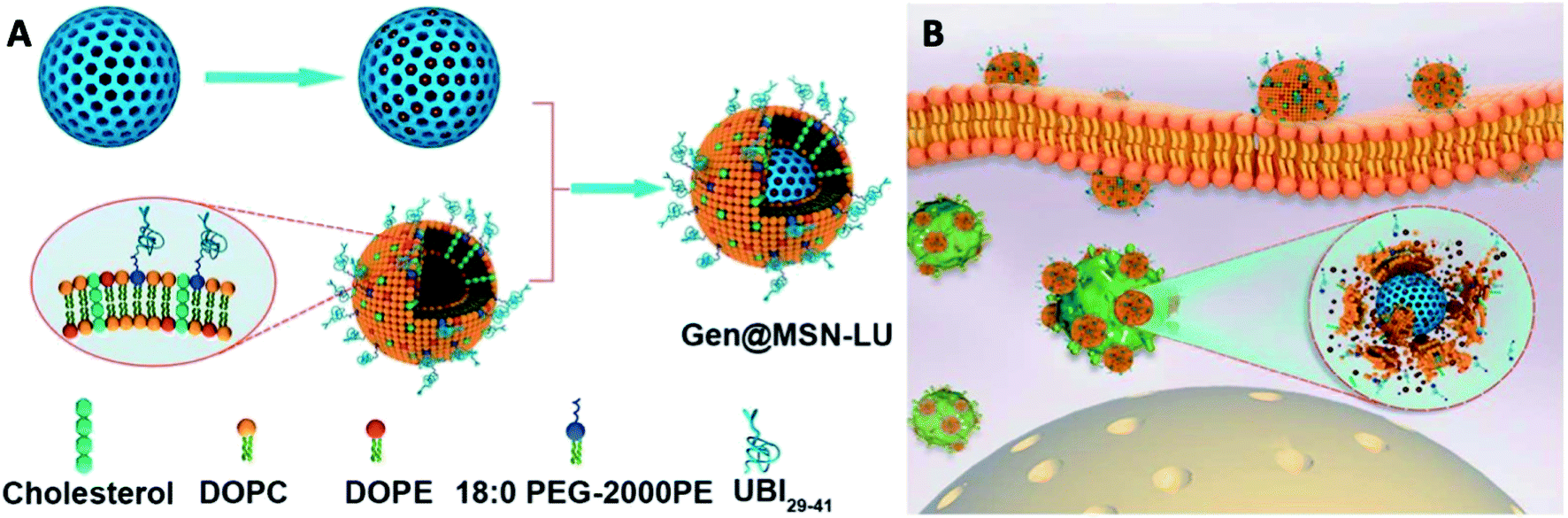 | ||
| Fig. 8 (A) Fabrication route of Gen@MSN-LU. (B) Schematics of possible antimicrobial mechanism in phagocytic cells, where the liposomes outer layer can be degraded by the bacterium-secreted toxins, leading to the gentamicin release (adapted with permission from Yang et al., 2018.135 Copyright © 2018, American Chemical Society). | ||
Naked gene therapeutics, such as small interfering RNA (siRNA), are proving to be highly promising but are limited by their short half-lives, and they need to be localized sub-cellularly to be functional. In view of this, a variety of nanoparticle delivery vehicles are explored to protect these oligonucleotides for intracellular delivery. Despite the use of delivery systems, it was reported more than 70% of siRNA payload would still be excreted extracellularly by cells, with most degraded in lysosomes; and typically, only less than 2% of the administered siRNA could escape the early endosomal pathway to potentially undergo RNA interference. In order to increase the quantity of RNA delivered, Kim et al. presented fusogenic liposomal-coated porous silicon nanoparticles, functionalized with a targeting peptide, for siRNA delivery (Fig. 9).136 Prepared with DMPC, DSPE-PEG, and DOTAP, the fusogenic thin-film was rehydrated within an aqueous calcium chloride solution containing pre-synthesized siRNA-loaded porous silicon nanoparticles. The final fusogenic liposomal siRNA-pSiNPs were obtained by subsequent co-extrusion through polycarbonate membrane. The fusogenic liposome, through membrane fusion mechanism, made it possible for hydrophilic payloads siRNA to be released directly from the nanoparticulate core into the cell cytoplasm. This dramatically increased the probability that siRNAs could reach the perinuclear region to undergo RNA interference, by entirely avoiding endocytosis. Their study demonstrated in vivo gene silencing immunotherapy for intracellular infection using a LCHNP delivery system. This macrophage-targeting fusogenic liposomal siRNA-pSiNPs provided a significantly high siRNA knockdown efficiency in vitro, which subsequently showed a strong therapeutic efficacy in terms of full recovery from a lethal dosage against intracellular Staphylococcus aureus infection in mice.
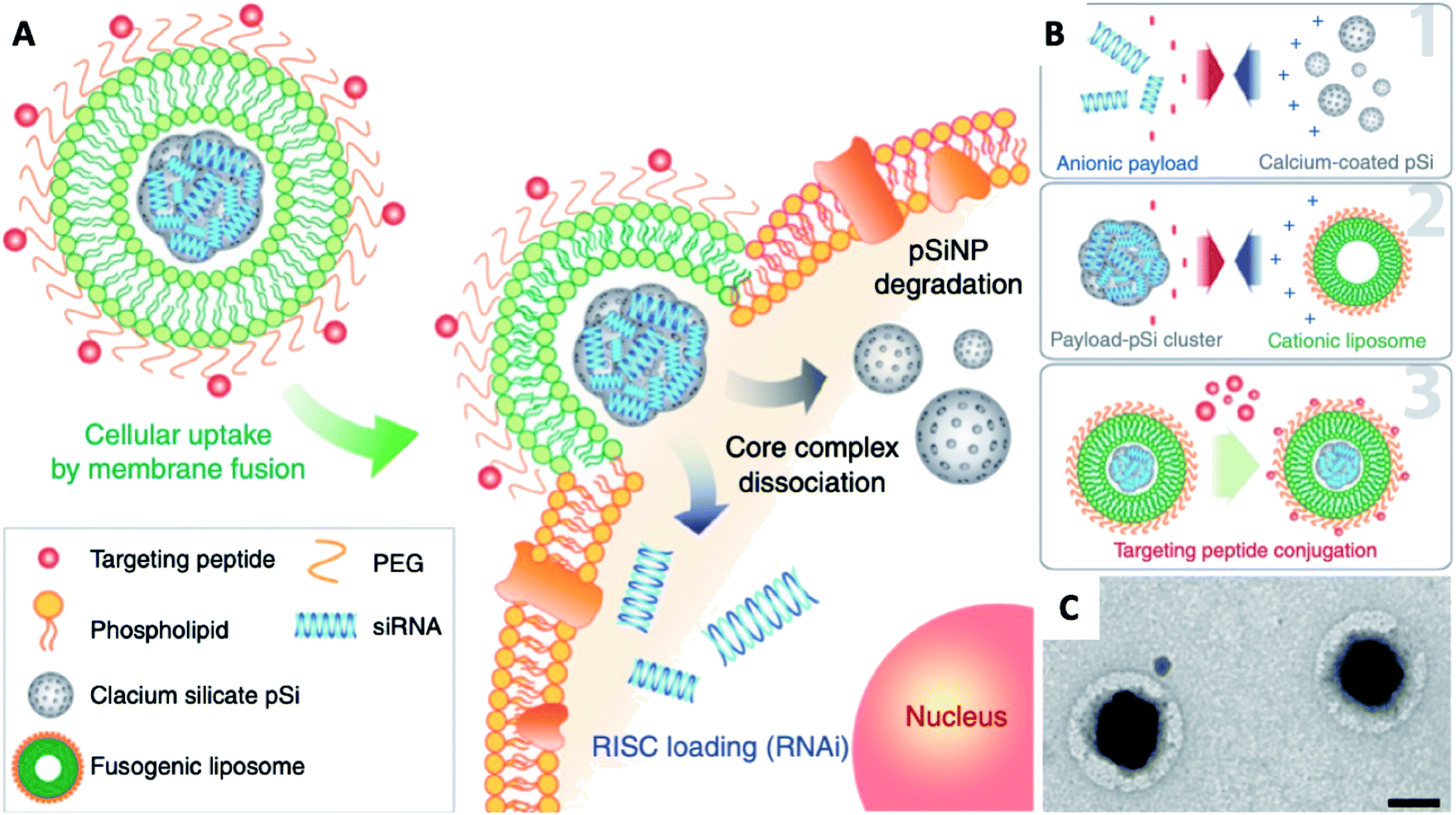 | ||
| Fig. 9 Fusogenic pSi NP system. (A) Schematic showing mode of action of the fusogenic pSiNP. (B) Schematic showing nanoparticle synthesis, including (1) siRNA encapsulating into MSNs; (2) coating with cationic liposome; and (3) conjugation of targeting peptides. (C) TEM image of final LCHNP constructs (reprinted with permission from Kim et al., 2018.136 Copyright © 2018, Springer Nature). | ||
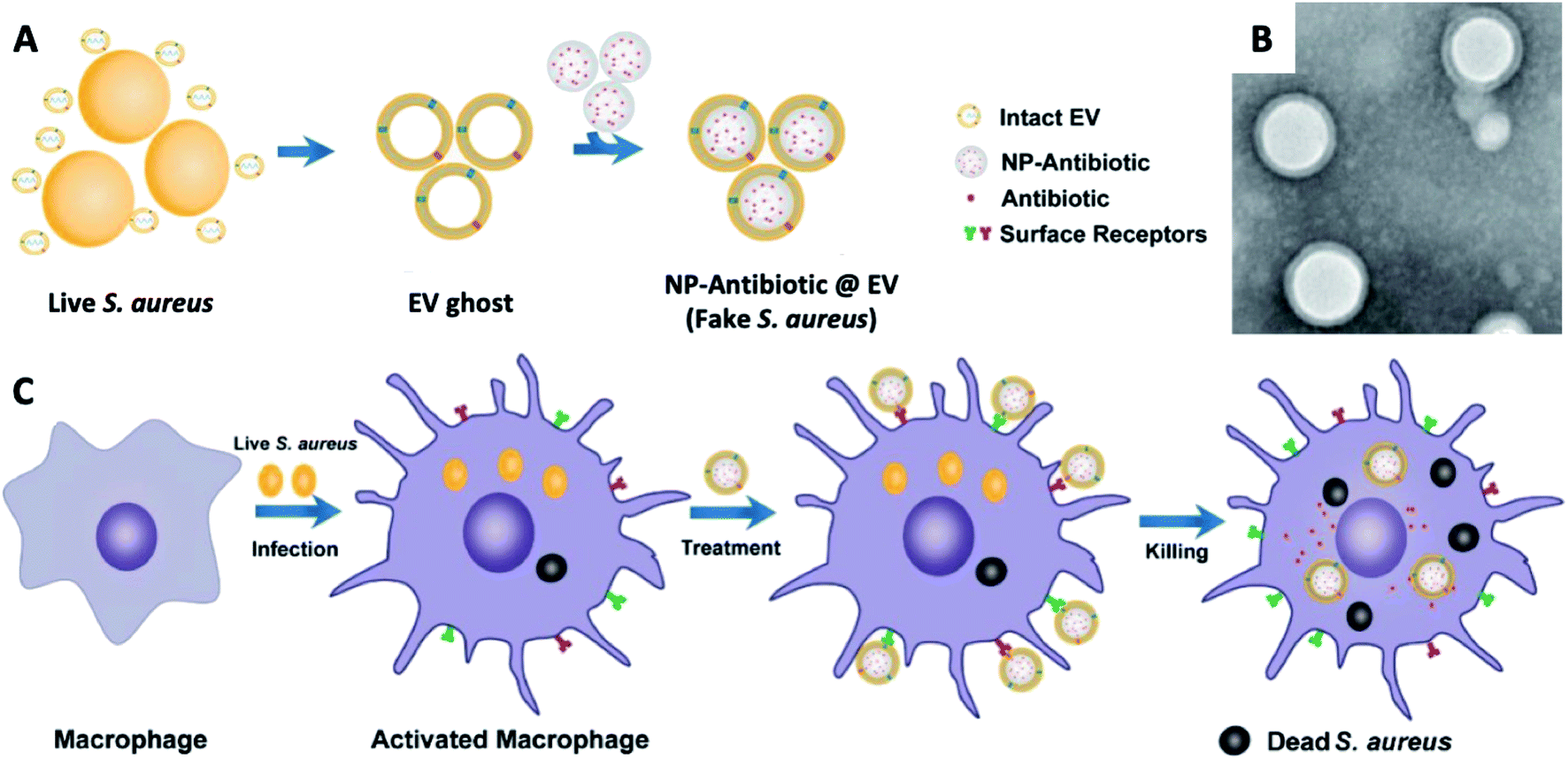 | ||
| Fig. 10 (A) Schematic illustration on the preparation of NP@EV, by fusing the derived membrane vesicle of the extracellular vesicles secreted by Staphylococcus aureus over the surface of NPs. (B) TEM of Staphylococcus aureus – EV coated NPs. (C) Exposure of a macrophage to a Staphylococcus aureus leads to phagocytosis and consequent antigen presentation, and re-exposure of the phagocyte to the same pathogen type (Staphylococcus aureus – EV coated NPs) leads to rapid pathogen detection and clearance (adapted with permission from Gao et al., 2019.137 Copyright © 2019, American Chemical Society). | ||
3.2. Bacterial biofilms
Biofilms refer to a sessile community of bacteria embedded within a matrix of secreted extracellular polymeric substances (EPS), such as polysaccharides, proteins, enzymes and DNA, which are attached on surfaces and the compact EPS layer can protect the bacteria residing within it.138 Furthermore, it not only acts as a barrier to retard the penetration of antibiotics, but also allow pathogens to resist the actions of the host immune system. The failure of existing strategies makes it essential to develop novel treatments to treat biofilm infections. Thus, improving antibiotic delivery is a cornerstone for preventing biofilm-mediated infections. Among the various types of nano-delivery systems, LCHNPs possess huge advantages as the lipid layer presents a charged surface, and protects the payload from either being inactivated by the acidic microenvironment or degraded by extracellular enzymes.More recently, Li et al. developed a novel LCHNP to overcome both biofilm and mucus layer obstruction (Fig. 11).58 To be more specific, in their work, chitosan nanoparticle, employed as the core, was coated with a mixed lipid layer containing rhamnolipids and clarithromycin as the shell. The core–shell structure was obtained by using a film-rehydration method, followed by further modification with DSPE-PEG2000 to improve hydrophilicity. Combined with rhamnolipids and clarithromycin chitosan, these LCHNPs could disrupt biofilm architecture, remove EPS, prevent reformation of biofilm by effectively inhibiting bacterial adhesion and eliminate pathogens, thus exhibiting an excellent eradicating ability to Helicobacter pylori biofilm.
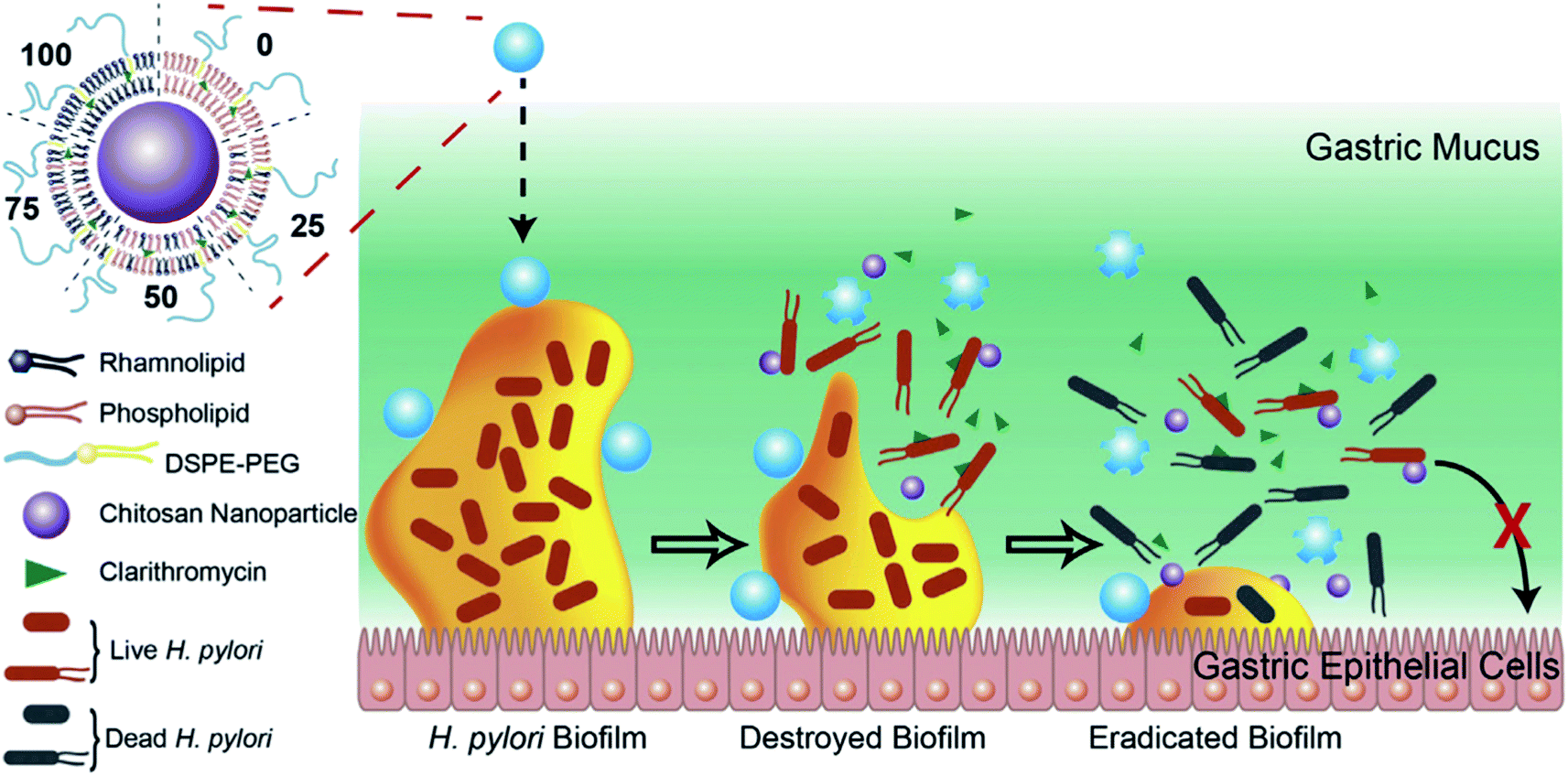 | ||
| Fig. 11 Schematic illustration of the structure of LCHNPs and the process of eradicating bacterial Helicobacter pylori biofilm by LCHNPs (reprinted from Li et al., 2019,58 Copyright (2019), with permission from Elsevier). | ||
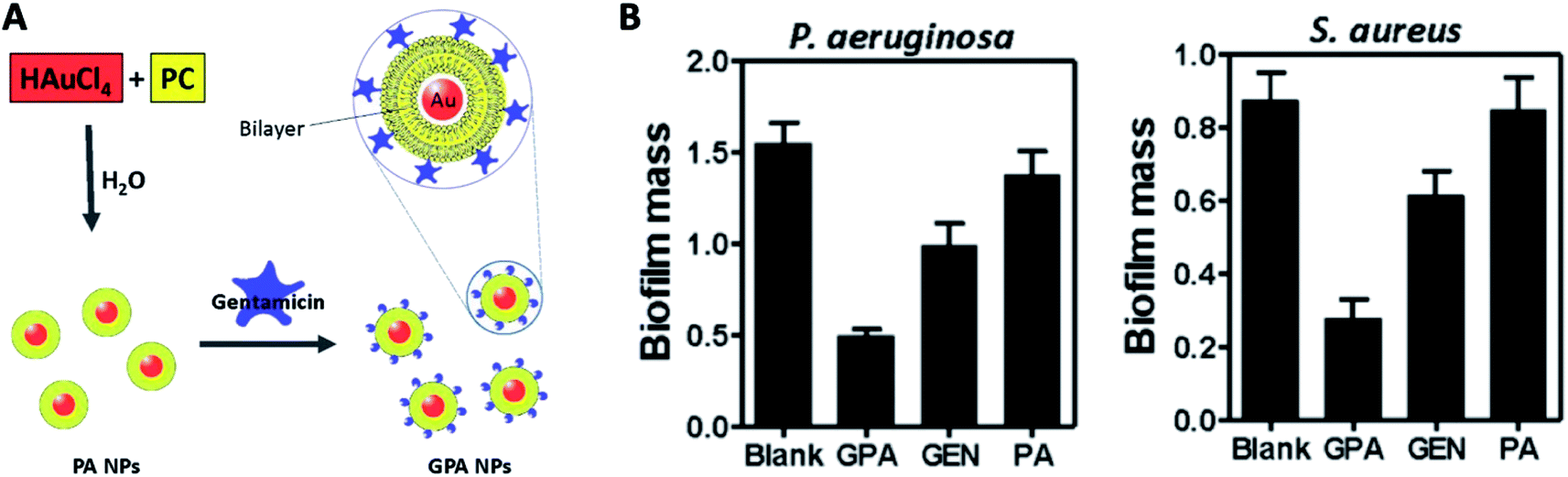 | ||
| Fig. 12 (A) Schematic of the procedure for preparing GPA NPs. (B) Crystal violet assay for biofilm biomass to assess the anti-biofilm activity of GPA against Pseudomonas aeruginosa and Staphylococcus aureus (adapted with permission from Mu et al., 2016.139 Copyright © 2016, Springer Nature). | ||
4. Challenges and perspectives
4.1. Challenges for current nanoparticle formulations
Many of these nano-delivery systems, notably LCHNPs, have demonstrated significantly enhanced bactericidal activity compared to free drugs, particularly against intracellular bacterial strains and biofilms. In addition to delivering existing antibiotics, LCHNPs with inorganic and metallic materials also possess potent antimicrobial properties. However, despite of their outstanding potential, there has not been any clinical translations arising from this technology, not only because of their complex constituents but also the many barriers faced by this nano-delivery system. Some issues that need to be addressed include understanding the interactions of nanoparticles with biological systems, solving the issues of nanoparticle aggregation in physiological fluids, and characterizing these nanoparticles under physiological conditions.Scalable manufacturing remains as one of the biggest challenges faced by nanoparticle therapies. The scale-up production of single-material systems or formulations, such as liposomes and polymeric nanoparticles loaded with an active pharmaceutical ingredient, have already been successfully achieved and are widely utilized by the pharmaceutical industry. The rapid development of new biomaterials, complex nanostructures and novel synthesis approaches will continue to pose new challenges to the scalable production of these technologies. In recent years, Particle Replication In Non-wetting Templates (PRINT), an advanced unique soft lithography particle moulding process, has been gaining interest.140–143 The PRINT process enables the specific design and large-scale synthesis of well-defined micro- and nanoparticles with uniformed size and shape. In addition, a double usage of spray-drying technique serves as a promising large-scale method to assemble lipids onto nanoparticle surfaces. Accompanied with these advanced techniques, the large-scale production of LCHNPs with acceptable batch-to-batch variation that is free of contamination could be achieved.
Given the usage of LCHNPs in the biological environment, it is therefore of great importance to evaluate its performance under relevant biological conditions. Usually, given limited research resources, in vitro evaluation of delivery systems on cells/bacterial cells is reported. However, in vitro studies by introducing nanoparticles onto planktonic bacteria often lack the complexities of a true biological environment.144 For clinical translation, the use of appropriate animal models to assess in vivo performances, such as tolerability, biodistribution, toxicity, and potential side effects, are often mandatory.145 In studies on bacterial infection, such models are highly diverse, usually employing either patient- or laboratory-derived bacteria strains infected zebrafish, Galleria mellonella larvae and mouse models. However, even these in vivo models do not fully replicate the complexity of a human infection, whereby the reticuloendothelial system (RES) would play a prominent role in the biodistribution of nanoparticles.69,146 For instance, to prolong blood circulation half-lives of nanoparticles to target extracellular pathogens, or to target intracellular bacteria, proper strategies must be adopted in designing ‘stealth’ nanoparticles to be less susceptible to the efficient RES and thus improving targeting capabilities.
4.2. Perspectives of LCHNPs
Surface functionalisation of LCHNPs with targeting ligands (i.e. antibodies) has the potential to significantly enhance delivery efficiency.152–154 A key concept in this active targeting strategy for infections is that the pathogens and the LCHNPs tend to accumulate in the same phagocytes, which could be exemplified by the current interest in antibody nanoparticle conjugates (ANCs).155,156 ANCs represent a relatively new approach that builds on the success and potential of both antibody antibiotic conjugates (AACs) and nano-technological delivery systems. AACs, consisting of monoclonal antibodies specifically targeted to pathogens with potent antibiotics, have been successfully developed for the treatment of Staphylococcus aureus infection.111 Conceptually ANCs are similar to AACs in that the antibodies can be used to specifically target pathogens or cells, which could significantly improve the possibilities to deliver encapsulated therapeutic cargoes to the right places.
The diagnostic application of magnetic nanoparticles (MNPs) has now been examined in a range of inflammatory pathologies. For example, Hoerr et al. labelled Staphylococcus aureus with iron oxide nanoparticles before in vivo inoculation, allowing real-time visualisation of the morphology of the infected organs and the resultant host inflammatory response.157 Interestingly, such MNPs with their superior superparamagnetic qualities hold the potential to be manipulated by an external magnetic field and be developed into active delivery systems.158,159 However, their application is strongly limited by the high cytotoxicity which is often associated with MNPs. Lipids are biocompatible molecules with an amphiphilic structure, emerging as perfect candidates to produce nano-biointerfaces by coating the MNPs surface, which improves the MNPs stability toward aggregation and reduces the related cytotoxic effects. More importantly, the lipid outer layers are the best candidates to encapsulate or load amphiphilic antibiotics as a protection to avoid undesired release. Thus, there is strong potential in combining diagnostic function, active targeting, and therapeutic delivery ability together in an antibiotic-loaded lipid-coated MNP formulation, which could be further exploited for pathogen imaging and antimicrobial therapy, known as antimicrobial nano-theranostics. Such proposed nano-theranostic anti-infection strategy is an interesting direction holding the potential to be personalized nanomedicines for pathogenic infections in the future.
To successfully develop LCHNPs as anti-biofilm therapeutics, it is essential to understand the roles that each signalling pathway plays in the biofilm environment. The c-Di-GMP and (p)ppGPP signalling pathways are hypothesized to play an important role in promoting biofilm formation, virulence, antibiotic and stress resistances.170–173 However, there remain more to be uncovered on the exact mechanisms of these signalling pathways and how to disrupt these pathways. Moreover, different pathogenic strains may differ in signalling pathways that are dominant in regulating biofilm formation and antibiotic resistance. In this aspect, LCHNPs have the potential to act as carriers to deliver anti-biofilm compounds, such as the QS inhibitors mentioned earlier, to the biofilm infection site.
From these perspectives, we can definitely predict numerous opportunities for LCHNPs in antimicrobial therapies, and hopefully these new approaches will bring the LCHNPs closer to the clinic. In conclusion, challenges faced by this novel delivery system remain; but advances in manufacturing and a more detailed understanding of their antimicrobial properties will promote LCHNPs to be a feasible proposition for the treatment of seemingly untreatable pathogen-causing infections in the near future.
List of abbreviations
| Akt | Protein kinase B |
| ATRA | All-trans retinoic acid |
| BSA | Bovine serum albumin |
| CAT | Catalase |
| CFU | Colony formed units |
| CHOL | Cholesterol |
| CLR | Clarithromycin |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CS | Chitosan |
| CUR | Curcumin |
| DMPC | 14:0 PC; 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DOPC | 18:1 PC; 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPE | 18:1 PE; 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| DOTAP | 18:1 TAP; 1,2-dioleoyl-3-trimethylammonium-propane |
| DOX | Doxorubicin |
| DSPC | 18:0 PC; 1,2-distearoyl-sn-glycero-3-phosphocholine |
| DSPE | 18:0 PE; 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine |
| EPR | Enhanced permeation and retention |
| ESE | Emulsion-solvent-evaporation |
| EVs | Extracellular vesicles |
| HCPT | Hydroxycamptothecin |
| LCHNPs | Lipid-coated hybrid nanoparticles |
| LPC | Lysophosphocholine |
| MgP | Magnesium phosphate |
| MIVRs | Multi-inlet vortex reactors |
| MNPs | Magnetic nanoparticles |
| MPS | Mononuclear phagocyte system |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| MSN | Mesoporous silica nanoparticle |
| MSNC | Mesoporous silica nanocapsules |
| NAC | N-Acetyl cysteine |
| NP | Nanoparticle |
| PA | Phosphatidylcholine |
| PAMAM | Poly(amidoamine) |
| PCL | Poly(caprolactone) |
| PCPDTBT | Poly(cyclopentadithiophenealt-benzothiadiazole) |
| PE | Phosphatidylethanolamine |
| PEG | Polyethylene glycol |
| PPG | Poly(propylene glycol) |
| PEI | Polyethylenimine |
| PL | Phospholipid |
| PLGA | Poly(D,L-lactide-co-glycolide) |
| POPC | 16:0-18:1 PC; 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPS | 16:0-18:1 PS; 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine |
| Particle replication in non-wetting templates | |
| PS | Polystyrene |
| PTX | Paclitaxel |
| RAP | Rapamycin |
| RES | Reticuloendothelial system |
| RHL | Rhamnolipid |
| TPP | Tripolyphosphate sodium |
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to acknowledge the financial support from the Singapore Centre for Environmental Life Sciences Engineering (SCELSE) (MOE/RCE: M4330019.C70), Ministry of Education AcRF-Tier 1 grant (RG19/18), Agri-Food & Veterinary Authority of Singapore (APF LCK102), Biomedical Research Council (BMRC) – Therapeutics Development Review (TDR-G-004-001), NTU-HSPH grant (NTU-HSPH 17002), and the Bill and Melinda Gates Foundation (OPP1199116).References
- F. Prestinaci, P. Pezzotti and A. Pantosti, Antimicrobial resistance: a global multifaceted phenomenon, Pathog. Glob Health, 2015, 109(7), 309–318 CrossRef PubMed.
- C. L. Ventola, The antibiotic resistance crisis: part 1: causes and threats, P & T, 2015, 40(4), 277–283 Search PubMed.
- S. H. Podolsky, The evolving response to antibiotic resistance (1945–2018), Palgrave Commun., 2018, 4(1), 124 CrossRef.
- S. Baker, N. Thomson, F. X. Weill and K. E. Holt, Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens, Science, 2018, 360(6390), 733–738 CrossRef CAS PubMed.
- D. Brown, Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void?, Nat. Rev. Drug Discovery, 2015, 14(12), 821–832 CrossRef CAS PubMed.
- M. Exner, S. Bhattacharya, B. Christiansen, J. Gebel, P. Goroncy-Bermes, P. Hartemann, P. Heeg, C. Ilschner, A. Kramer, E. Larson, W. Merkens, M. Mielke, P. Oltmanns, B. Ross, M. Rotter, R. M. Schmithausen, H.-G. Sonntag and M. Trautmann, Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria?, GMS Hyg. Infect. Control., 2017, 12, Doc05 Search PubMed.
- M. Mhondoro, N. Ndlovu, D. Bangure, T. Juru, N. T. Gombe, G. Shambira, P. Nsubuga and M. Tshimanga, Trends in antimicrobial resistance of bacterial pathogens in Harare, Zimbabwe, 2012–2017: a secondary dataset analysis, BMC Infect. Dis., 2019, 19(1), 746 CrossRef PubMed.
- B. Li and T. J. Webster, Bacteria antibiotic resistance: New challenges and opportunities for implant-associated orthopedic infections, J. Orthop. Res., 2018, 36(1), 22–32 Search PubMed.
- C. f. D. C. a. Prevention, Antibiotic Resistance Threats in the United States, 2019. Services, U. S. D. o. H. a. H., Atlanta, 2019 Search PubMed.
- D. W. Nelson, J. E. Moore and J. R. Rao, Antimicrobial resistance (AMR): significance to food quality and safety, Food Qual. Saf., 2019, 3(1), 15–22 CrossRef CAS.
- J. O'Neill, Review on Antimicrobial Resistance Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations, https://amr-review.org/sites/default/files/AMRReviewPaper-Tacklingacrisisforthehealthandwealthofnations_1.pdf Search PubMed.
- G. Gebreyohannes, A. Nyerere, C. Bii and D. B. Sbhatu, Challenges of intervention, treatment, and antibiotic resistance of biofilm-forming microorganisms, Heliyon, 2019, 5(8), e02192 CrossRef PubMed.
- S. Singh, S. K. Singh, I. Chowdhury and R. Singh, Understanding the Mechanism of Bacterial Biofilms Resistance to Antimicrobial Agents, Open Microbiol. J., 2017, 11, 53–62 CrossRef CAS PubMed.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z. G. She, D. Kim, A. P. Mann, T. Molder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy, Nat. Biomed. Eng., 2018, 2(2), 95–103 CrossRef CAS.
- M. Kumar, A. Curtis and C. Hoskins, Application of Nanoparticle Technologies in the Combat against Anti-Microbial Resistance, Pharmaceutics, 2018, 10(1), 11 CrossRef PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, Nano based drug delivery systems: recent developments and future prospects, J. Nanobiotechnol., 2018, 16(1), 71 CrossRef PubMed.
- P. V. Baptista, M. P. McCusker, A. Carvalho, D. A. Ferreira, N. M. Mohan, M. Martins and A. R. Fernandes, Nano-Strategies to Fight Multidrug Resistant Bacteria-“A Battle of the Titans”, Front. Microbiol., 2018, 9, 1441 CrossRef PubMed.
- H. A. Hemeg, Nanomaterials for alternative antibacterial therapy, Int. J. Nanomed., 2017, 12, 8211–8225 CrossRef CAS PubMed.
- L. Wang, C. Hu and L. Shao, The antimicrobial activity of nanoparticles: present situation and prospects for the future, Int. J. Nanomed., 2017, 12, 1227–1249 CrossRef CAS PubMed.
- R. Catania, O. Maguire, C. Moore, F. H. Falcone, W. Chan, G. Mantovani, S. Stolnik and A. Huett, Functionalised liposomal formulations for delivery of antibiotic agents, Access Microbiology, 2019, 1(1A) DOI:10.1099/acmi.ac2019.po0507.
- M. Alhariri, A. Azghani and A. Omri, Liposomal antibiotics for the treatment of infectious diseases, Expert Opin. Drug Delivery, 2013, 10(11), 1515–1532 CrossRef CAS PubMed.
- Z. Drulis-Kawa and A. Dorotkiewicz-Jach, Liposomes as delivery systems for antibiotics, Int. J. Pharm., 2010, 387(1–2), 187–198 CrossRef CAS PubMed.
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Nanoparticle-based local antimicrobial drug delivery, Adv. Drug Delivery Rev., 2018, 127, 46–57 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics, ACS Nano, 2012, 6(5), 4279–4287 CrossRef CAS PubMed.
- W. S. Cheow and K. Hadinoto, Antibiotic Polymeric Nanoparticles for Biofilm-Associated Infection Therapy, in Microbial Biofilms: Methods and Protocols, ed. G. Donelli, Springer New York, New York, NY, 2014, pp. 227–238 Search PubMed.
- M. Kalomiraki, K. Thermos and N. A. Chaniotakis, Dendrimers as tunable vectors of drug delivery systems and biomedical and ocular applications, Int. J. Nanomed., 2016, 11, 1–12 CrossRef CAS PubMed.
- M. A. Mintzer, E. L. Dane, G. A. O'Toole and M. W. Grinstaff, Exploiting dendrimer multivalency to combat emerging and re-emerging infectious diseases, Mol. Pharmaceutics, 2012, 9(3), 342–354 CrossRef CAS PubMed.
- C. A. Omolo, R. S. Kalhapure, N. Agrawal, M. Jadhav, S. Rambharose, C. Mocktar and T. Govender, A hybrid of mPEG-b-PCL and G1-PEA dendrimer for enhancing delivery of antibiotics, J. Controlled Release, 2018, 290, 112–128 CrossRef CAS PubMed.
- C. A. Cheng, T. Deng, F. C. Lin, Y. Cai and J. I. Zink, Supramolecular Nanomachines as Stimuli-Responsive Gatekeepers on Mesoporous Silica Nanoparticles for Antibiotic and Cancer Drug Delivery, Theranostics, 2019, 9(11), 3341–3364 CrossRef CAS PubMed.
- Z. Li, D. L. Clemens, B. Y. Lee, B. J. Dillon, M. A. Horwitz and J. I. Zink, Mesoporous Silica Nanoparticles with pH-Sensitive Nanovalves for Delivery of Moxifloxacin Provide Improved Treatment of Lethal Pneumonic Tularemia, ACS Nano, 2015, 9(11), 10778–10789 CrossRef CAS PubMed.
- A. M. Mebert, C. Aimé, G. S. Alvarez, Y. Shi, S. A. Flor, S. E. Lucangioli, M. F. Desimone and T. Coradin, Silica core–shell particles for the dual delivery of gentamicin and rifamycin antibiotics, J. Mater. Chem. B, 2016, 4(18), 3135–3144 RSC.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nanoparticle Uptake: The Phagocyte Problem, Nano Today, 2015, 10(4), 487–510 CrossRef CAS PubMed.
- C. Li, J. Wang, Y. Wang, H. Gao, G. Wei, Y. Huang, H. Yu, Y. Gan, Y. Wang, L. Mei, H. Chen, H. Hu, Z. Zhang and Y. Jin, Recent progress in drug delivery, Acta Pharm. Sin. B, 2019, 9(6), 1145–1162 CrossRef PubMed.
- K. M. Vargas and Y. S. Shon, Hybrid lipid-nanoparticle complexes for biomedical applications, J. Mater. Chem. B, 2019, 7(5), 695–708 RSC.
- S. Krishnamurthy, R. Vaiyapuri, L. Zhang and J. M. Chan, Lipid-coated polymeric nanoparticles for cancer drug delivery, Biomater. Sci., 2015, 3(7), 923–936 RSC.
- N. Tahir, M. T. Haseeb, M. A. Madni, F. Parveen, M. Khan, S. Khan, N. Jan and A. Khan, Lipid Polymer Hybrid Nanoparticles: A Novel Approach for Drug Delivery, Role of Novel Drug Delivery Vehicles in Nanobiomedicine, 2019 Search PubMed.
- R. J. C. Bose, R. Ravikumar, V. Karuppagounder, D. Bennet, S. Rangasamy and R. A. Thandavarayan, Lipid–polymer hybrid nanoparticle-mediated therapeutics delivery: advances and challenges, Drug Discovery Today, 2017, 22(8), 1258–1265 CrossRef CAS PubMed.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, Lipid and polymer nanoparticles for drug delivery to bacterial biofilms, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS PubMed.
- K. Hadinoto, A. Sundaresan and W. S. Cheow, Lipid–polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review, Eur. J. Pharm. Biopharm., 2013, 85(3), 427–443 CrossRef CAS PubMed.
- B. Mandal, H. Bhattacharjee, N. Mittal, H. Sah, P. Balabathula, L. A. Thoma and G. C. Wood, Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform, Nanomedicine, 2013, 9(4), 474–491 CrossRef CAS PubMed.
- A. Mukherjee, A. K. Waters, P. Kalyan, A. S. Achrol, S. Kesari and V. M. Yenugonda, Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: state of the art, emerging technologies, and perspectives, Int. J. Nanomed., 2019, 14, 1937–1952 CrossRef CAS PubMed.
- A. M. El-Toni, M. A. Habila, J. P. Labis, Z. A. Alothman, M. Alhoshan, A. A. Elzatahry and F. Zhang, Design, synthesis and applications of core–shell, hollow core, and nanorattle multifunctional nanostructures, Nanoscale, 2016, 8(5), 2510–2531 RSC.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery, Biomaterials, 2009, 30(8), 1627–1634 CrossRef CAS PubMed.
- L.-Y. Gao, X.-Y. Liu, C.-J. Chen, J.-C. Wang, Q. Feng, M.-Z. Yu, X.-F. Ma, X.-W. Pei, Y.-J. Niu, C. Qiu, W.-H. Pang and Q. Zhang, Core-Shell type lipid/rPAA-Chol polymer hybrid nanoparticles for in vivo siRNA delivery, Biomaterials, 2014, 35(6), 2066–2078 CrossRef CAS PubMed.
- D. Liu, Y. Lian, Q. Fang, L. Liu, J. Zhang and J. Li, Hyaluronic-acid-modified lipid-polymer hybrid nanoparticles as an efficient ocular delivery platform for moxifloxacin hydrochloride, Int. J. Biol. Macromol., 2018, 116, 1026–1036 CrossRef CAS PubMed.
- B. Mandal, H. Bhattacharjee, N. Mittal, H. Sah, P. Balabathula, L. A. Thoma and G. C. Wood, Core–shell-type lipid–polymer hybrid nanoparticles as a drug delivery platform, Nanomedicine, 2013, 9(4), 474–491 CrossRef CAS PubMed.
- V. Dave, R. B. Yadav, K. Kushwaha, S. Yadav, S. Sharma and U. Agrawal, Lipid-polymer hybrid nanoparticles: Development & statistical optimization of norfloxacin for topical drug delivery system, Bioact. Mater., 2017, 2(4), 269–280 CrossRef PubMed.
- C. Wang, L. Su, C. Wu, J. Wu, C. Zhu and G. Yuan, RGD peptide targeted lipid-coated nanoparticles for combinatorial delivery of sorafenib and quercetin against hepatocellular carcinoma, Drug Dev. Ind. Pharm., 2016, 42(12), 1938–1944 CrossRef CAS PubMed.
- X.-Z. Yang, S. Dou, Y.-C. Wang, H.-Y. Long, M.-H. Xiong, C.-Q. Mao, Y.-D. Yao and J. Wang, Single-Step Assembly of Cationic Lipid–Polymer Hybrid Nanoparticles for Systemic Delivery of siRNA, ACS Nano, 2012, 6(6), 4955–4965 CrossRef CAS PubMed.
- M. Zhang, J. He, W. Zhang and J. Liu, Fabrication of TPGS-Stabilized Liposome-PLGA Hybrid Nanoparticle Via a New Modified Nanoprecipitation Approach: In Vitro and In Vivo Evaluation, Pharm. Res., 2018, 35(11), 199 CrossRef PubMed.
- S. Díez, I. Miguéliz and C. Tros de Ilarduya, Targeted cationic poly(D,L-lactic-co-glycolic acid) nanoparticles for gene delivery to cultured cells, Cell. Mol. Biol. Lett., 2009, 14(2), 347 Search PubMed.
- Y. Hu, R. Hoerle, M. Ehrich and C. Zhang, Engineering the lipid layer of lipid-PLGA hybrid nanoparticles for enhanced in vitro cellular uptake and improved stability, Acta Biomater., 2015, 28, 149–159 CrossRef CAS PubMed.
- F. Khademi, A. Sahebkar, M. Fasihi-Ramandi and R. A. Taheri, Induction of strong immune response against a multicomponent antigen of Mycobacterium tuberculosis in BALB/c mice using PLGA and DOTAP adjuvant, APMIS, 2018, 126(6), 509–514 CrossRef CAS PubMed.
- S. Taheri, A. Cavallaro, S. N. Christo, P. Majewski, M. Barton, J. D. Hayball and K. Vasilev, Antibacterial Plasma Polymer Films Conjugated with Phospholipid Encapsulated Silver Nanoparticles, ACS Biomater. Sci. Eng., 2015, 1(12), 1278–1286 CrossRef CAS.
- D. J. Hamilton, M. D. Coffman, J. D. Knight and S. M. Reed, Lipid-Coated Gold Nanoparticles and FRET Allow Sensitive Monitoring of Liposome Clustering Mediated by the Synaptotagmin-7 C2A Domain, Langmuir, 2017, 33(36), 9222–9230 CrossRef CAS PubMed.
- Y. Fang, M. Vadlamudi, Y. Huang and X. Guo, Lipid-Coated, pH-Sensitive Magnesium Phosphate Particles for Intracellular Protein Delivery, Pharm. Res., 2019, 36(6), 81 CrossRef PubMed.
- F. Wan, T. Nylander, S. N. Klodzinska, C. Foged, M. Yang, S. G. Baldursdottir and H. M. Nielsen, Lipid Shell-Enveloped Polymeric Nanoparticles with High Integrity of Lipid Shells Improve Mucus Penetration and Interaction with Cystic Fibrosis-Related Bacterial Biofilms, ACS Appl. Mater. Interfaces, 2018, 10(13), 10678–10687 CrossRef CAS PubMed.
- P. Li, X. Chen, Y. Shen, H. Li, Y. Zou, G. Yuan, P. Hu and H. Hu, Mucus penetration enhanced lipid polymer nanoparticles improve the eradication rate of Helicobacter pylori biofilm, J. Controlled Release, 2019, 300, 52–63 CrossRef CAS PubMed.
- K. Gui, X. Zhang, F. Chen, Z. Ge, S. Zhang, X. Qi, J. Sun and Z. Yu, Lipid-polymer nanoparticles with CD133 aptamers for targeted delivery of all-trans retinoic acid to osteosarcoma initiating cells, Biomed. Pharmacother., 2019, 111, 751–764 CrossRef CAS PubMed.
- X. Liu, M. Li, W. Yuan, Y. Liu, Y. Wang and Y. Wang, Lipid-coated mesoporous silica nanoparticles of hydroxycamptothecin for sustained release and cancer therapy, Pharmazie, 2018, 73(8), 447–453 CAS.
- J. Lin, Q. Cai, Y. Tang, Y. Xu, Q. Wang, T. Li, H. Xu, S. Wang, K. Fan, Z. Liu, Y. Jin and D. Lin, PEGylated Lipid bilayer coated mesoporous silica nanoparticles for co-delivery of paclitaxel and curcumin: Design, characterization and its cytotoxic effect, Int. J. Pharm., 2018, 536(1), 272–282 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Cell Membrane Coating Nanotechnology, Adv. Mater., 2018, 30(23), e1706759 CrossRef PubMed.
- Z. Chai, X. Hu and W. Lu, Cell membrane-coated nanoparticles for tumor-targeted drug delivery, Sci. China Mater., 2017, 60(6), 504–510 CrossRef CAS.
- J. Sherwood, J. Sowell, N. Beyer, J. Irvin, C. Stephen, A. J. Antone, Y. Bao and L. M. Ciesla, Cell-membrane coated iron oxide nanoparticles for isolation and specific identification of drug leads from complex matrices, Nanoscale, 2019, 11(13), 6352–6359 RSC.
- W. Gao and L. Zhang, Coating nanoparticles with cell membranes for targeted drug delivery, J. Drug Targeting, 2015, 23(7–8), 619–626 CrossRef CAS PubMed.
- J. Jin, B. Krishnamachary, J. D. Barnett, S. Chatterjee, D. Chang, Y. Mironchik, F. Wildes, E. M. Jaffee, S. Nimmagadda and Z. M. Bhujwalla, Human Cancer Cell Membrane-Coated Biomimetic Nanoparticles Reduce Fibroblast-Mediated Invasion and Metastasis and Induce T-Cells, ACS Appl. Mater. Interfaces, 2019, 11(8), 7850–7861 CrossRef CAS PubMed.
- G. M. Cooper, The cell: a molecular approach, ASM Press, Washington, D.C, 2nd edn, 2000 Search PubMed.
- A. Cox, P. Andreozzi, R. Dal Magro, F. Fiordaliso, A. Corbelli, L. Talamini, C. Chinello, F. Raimondo, F. Magni, M. Tringali, S. Krol, P. Jacob Silva, F. Stellacci, M. Masserini and F. Re, Evolution of Nanoparticle Protein Corona across the Blood–Brain Barrier, ACS Nano, 2018, 12(7), 7292–7300 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- B. T. Luk and L. Zhang, Cell membrane-camouflaged nanoparticles for drug delivery, J. Controlled Release, 2015, 220(Pt B), 600–607 CrossRef CAS PubMed.
- M. Wu, W. Le, T. Mei, Y. Wang, B. Chen, Z. Liu and C. Xue, Cell membrane camouflaged nanoparticles: a new biomimetic platform for cancer photothermal therapy, Int. J. Nanomed., 2019, 14, 4431–4448 CrossRef CAS PubMed.
- Y. Zhai, J. Su, W. Ran, P. Zhang, Q. Yin, Z. Zhang, H. Yu and Y. Li, Preparation and Application of Cell Membrane-Camouflaged Nanoparticles for Cancer Therapy, Theranostics, 2017, 7(10), 2575–2592 CrossRef CAS PubMed.
- B. T. Luk, R. H. Fang, C.-M. J. Hu, J. A. Copp, S. Thamphiwatana, D. Dehaini, W. Gao, K. Zhang, S. Li and L. Zhang, Safe and Immunocompatible Nanocarriers Cloaked in RBC Membranes for Drug Delivery to Treat Solid Tumors, Theranostics, 2016, 6(7), 1004–1011 CrossRef CAS PubMed.
- Y. Song, Z. Huang, X. Liu, Z. Pang, J. Chen, H. Yang, N. Zhang, Z. Cao, M. Liu, J. Cao, C. Li, X. Yang, H. Gong, J. Qian and J. Ge, Platelet membrane-coated nanoparticle-mediated targeting delivery of Rapamycin blocks atherosclerotic plaque development and stabilizes plaque in apolipoprotein E-deficient (ApoE(-/-)) mice, Nanomedicine, 2019, 15(1), 13–24 CrossRef CAS PubMed.
- P. Angsantikul, S. Thamphiwatana, Q. Zhang, K. Spiekermann, J. Zhuang, R. H. Fang, W. Gao, M. Obonyo and L. Zhang, Coating Nanoparticles with Gastric Epithelial Cell Membrane for Targeted Antibiotic Delivery against Helicobacter pylori Infection, Adv. Ther., 2018, 1(2), 1800016 CrossRef PubMed.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen, W. Gu, P. Zhang, Z. Zhang, H. Yu, S. Wang and Y. Li, Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Homotypic Tumors, Adv. Mater., 2016, 28(43), 9581–9588 CrossRef CAS.
- Y. Zhang, K. Cai, C. Li, Q. Guo, Q. Chen, X. He, L. Liu, Y. Zhang, Y. Lu, X. Chen, T. Sun, Y. Huang, J. Cheng and C. Jiang, Macrophage-Membrane-Coated Nanoparticles for Tumor-Targeted Chemotherapy, Nano Lett., 2018, 18(3), 1908–1915 CrossRef CAS.
- J. Li, X. Zhen, Y. Lyu, Y. Jiang, J. Huang and K. Pu, Cell Membrane Coated Semiconducting Polymer Nanoparticles for Enhanced Multimodal Cancer Phototheranostics, ACS Nano, 2018, 12(8), 8520–8530 CrossRef CAS PubMed.
- W. Gao, R. H. Fang, S. Thamphiwatana, B. T. Luk, J. Li, P. Angsantikul, Q. Zhang, C.-M. J. Hu and L. Zhang, Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles, Nano Lett., 2015, 15(2), 1403–1409 CrossRef CAS PubMed.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen, W. Gu, Z. Zhang, H. Yu, P. Zhang, S. Wang and Y. Li, Cancer Cell Membrane-Coated Gold Nanocages with Hyperthermia-Triggered Drug Release and Homotypic Target Inhibit Growth and Metastasis of Breast Cancer, Adv. Funct. Mater., 2017, 27(3), 1604300 CrossRef.
- C. Wang, Y. Wang, L. Zhang, R. J. Miron, J. Liang, M. Shi, W. Mo, S. Zheng, Y. Zhao and Y. Zhang, Pretreated Macrophage-Membrane-Coated Gold Nanocages for Precise Drug Delivery for Treatment of Bacterial Infections, Adv. Mater., 2018, 30(46), 1804023 CrossRef PubMed.
- M. Xuan, J. Shao, L. Dai, Q. He and J. Li, Macrophage Cell Membrane Camouflaged Mesoporous Silica Nanocapsules for In Vivo Cancer Therapy, Adv. Healthcare Mater., 2015, 4(11), 1645–1652 CrossRef CAS PubMed.
- Y. Wang, P. Li, T. Truong-Dinh Tran, J. Zhang and L. Kong, Manufacturing Techniques and Surface Engineering of Polymer Based Nanoparticles for Targeted Drug Delivery to Cancer, Nanomaterials, 2016, 6(2), 26 CrossRef.
- C. Dhand, N. Dwivedi, X. J. Loh, A. N. Jie Ying, N. K. Verma, R. W. Beuerman, R. Lakshminarayanan and S. Ramakrishna, Methods and strategies for the synthesis of diverse nanoparticles and their applications: a comprehensive overview, RSC Adv., 2015, 5(127), 105003–105037 RSC.
- R. L. McCall and R. W. Sirianni, PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS, J. Visualized Exp., 2013,(82), 51015 Search PubMed.
- E. Sah and H. Sah, Recent Trends in Preparation of Poly(lactide-co-glycolide) Nanoparticles by Mixing Polymeric Organic Solution with Antisolvent, J. Nanomater., 2015, 2015, 22 CrossRef.
- M. Kumar, R. S. Bishnoi, A. K. Shukla and C. P. Jain, Techniques for Formulation of Nanoemulsion Drug Delivery System: A Review, Prev. Nutr. Food Sci., 2019, 24(3), 225–234 CrossRef PubMed.
- R. Jc Bose, S. Lee and H. Park, Lipid polymer hybrid nanospheres encapsulating antiproliferative agents for stent applications, J. Ind. Eng. Chem., 2016 Search PubMed.
- J.-S. Baek, C. H. Tan, N. K. J. Ng, Y. P. Yeo, S. A. Rice and S. C. J. Loo, A programmable lipid-polymer hybrid nanoparticle system for localized, sustained antibiotic delivery to Gram-positive and Gram-negative bacterial biofilms, Nanoscale Horiz., 2018, 3(3), 305–311 RSC.
- C. Dima and S. Dima, Water-in-oil-in-water double emulsions loaded with chlorogenic acid: release mechanisms and oxidative stability, J. Microencapsulation, 2018, 35(6), 584–599 CrossRef CAS.
- U. Bilati, E. Allémann and E. Doelker, Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles, Eur. J. Pharm. Sci., 2005, 24(1), 67–75 CrossRef CAS.
- J. Zhuang, R. H. Fang and L. Zhang, Preparation of particulate polymeric therapeutics for medical applications, Small Methods, 2017, 1(9) CrossRef CAS PubMed.
- P. Ahmaditabar, A. A. Momtazi-Borojeni, A. H. Rezayan, M. Mahmoodi, A. Sahebkar and M. Mellat, Enhanced Entrapment and Improved in Vitro Controlled Release of N-Acetyl Cysteine in Hybrid PLGA/Lecithin Nanoparticles Prepared Using a Nanoprecipitation/Self-Assembly Method, J. Cell. Biochem., 2017, 118(12), 4203–4209 CrossRef CAS PubMed.
- Z. Liu, M. Ramezani, R. O. Fox, J. C. Hill and M. G. Olsen, Flow Characteristics in a Scaled-up Multi-inlet Vortex Nanoprecipitation Reactor, Ind. Eng. Chem. Res., 2015, 54(16), 4512–4525 CrossRef CAS.
- A. Bokare, A. Takami, J. H. Kim, A. Dong, A. Chen, R. Valerio, S. Gunn and F. Erogbogbo, Herringbone-Patterned 3D-Printed Devices as Alternatives to Microfluidics for Reproducible Production of Lipid Polymer Hybrid Nanoparticles, ACS Omega, 2019, 4(3), 4650–4657 CrossRef CAS PubMed.
- Y. Shi, J. C. Cheng, R. O. Fox and M. G. Olsen, Measurements of turbulence in a microscale multi-inlet vortex nanoprecipitation reactor, J. Micromech. Microeng., 2013, 23(7), 075005 CrossRef.
- R. H. Fang, K. N. H. Chen, S. Aryal, C.-M. J. Hu, K. Zhang and L. Zhang, Large-Scale Synthesis of Lipid–Polymer Hybrid Nanoparticles Using a Multi-Inlet Vortex Reactor, Langmuir, 2012, 28(39), 13824–13829 CrossRef CAS PubMed.
- A. Luchini, C. Irace, R. Santamaria, D. Montesarchio, R. K. Heenan, N. Szekely, A. Flori, L. Menichetti and L. Paduano, Phosphocholine-decorated superparamagnetic iron oxide nanoparticles: defining the structure and probing in vivo applications, Nanoscale, 2016, 8(19), 10078–10086 RSC.
- A. Luchini, G. D'Errico, S. Leone, Z. Vaezi, A. Bortolotti, L. Stella, G. Vitiello and L. Paduano, Structural organization of lipid-functionalized-Au nanoparticles, Colloids Surf., B, 2018, 168, 2–9 CrossRef CAS PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Molecular mechanisms of antibiotic resistance, Nat. Rev. Microbiol., 2015, 13(1), 42–51 CrossRef CAS PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Bacterial persistence as a phenotypic switch, Science, 2004, 305(5690), 1622–1625 CrossRef CAS PubMed.
- A. Brauner, O. Fridman, O. Gefen and N. Q. Balaban, Distinguishing between resistance, tolerance and persistence to antibiotic treatment, Nat. Rev. Microbiol., 2016, 14(5), 320–330 CrossRef CAS PubMed.
- G. Rollin, X. Tan, F. Tros, M. Dupuis, X. Nassif, A. Charbit and M. Coureuil, Intracellular Survival of Staphylococcus aureus in Endothelial Cells: A Matter of Growth or Persistence, Front. Microbiol., 2017, 8, 1354 CrossRef PubMed.
- B. P. Conlon, Staphylococcus aureus chronic and relapsing infections: Evidence of a role for persister cells: An investigation of persister cells, their formation and their role in S. aureus disease, BioEssays, 2014, 36(10), 991–996 CrossRef PubMed.
- S. Xie, Y. Tao, Y. Pan, W. Qu, G. Cheng, L. Huang, D. Chen, X. Wang, Z. Liu and Z. Yuan, Biodegradable nanoparticles for intracellular delivery of antimicrobial agents, J. Controlled Release, 2014, 187, 101–117 CrossRef CAS PubMed.
- Y. Pei and Y. Yeo, Drug delivery to macrophages: Challenges and opportunities, J. Controlled Release, 2016, 240, 202–211 CrossRef CAS PubMed.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Urinary tract infections: epidemiology, mechanisms of infection and treatment options, Nat. Rev. Microbiol., 2015, 13(5), 269–284 CrossRef CAS PubMed.
- H. Zazo, C. I. Colino and J. M. Lanao, Current applications of nanoparticles in infectious diseases, J. Controlled Release, 2016, 224, 86–102 CrossRef CAS PubMed.
- D. Lebeaux, A. Chauhan, S. Letoffe, F. Fischer, H. de Reuse, C. Beloin and J. M. Ghigo, pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms, J. Infect. Dis., 2014, 210(9), 1357–1366 CrossRef PubMed.
- S. Helaine, J. A. Thompson, K. G. Watson, M. Liu, C. Boyle and D. W. Holden, Dynamics of intracellular bacterial replication at the single cell level, Proc. Natl. Acad. Sci., 2010, 107(8), 3746 CrossRef CAS PubMed.
- S. M. Lehar, T. Pillow, M. Xu, L. Staben, K. K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. L. Hazenbos, J. H. Morisaki, J. Kim, S. Park, M. Darwish, B. C. Lee, H. Hernandez, K. M. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. P. Lyssikatos, M. Strandh, K. Koefoed, P. S. Andersen, J. A. Flygare, M. Wah Tan, E. J. Brown and S. Mariathasan, Novel antibody-antibiotic conjugate eliminates intracellular S. aureus, Nature, 2015, 527(7578), 323–328 CrossRef CAS PubMed.
- R. S. Flannagan, G. Cosio and S. Grinstein, Antimicrobial mechanisms of phagocytes and bacterial evasion strategies, Nat. Rev. Microbiol., 2009, 7(5), 355–366 CrossRef CAS PubMed.
- W. Zhai, F. Wu, Y. Zhang, Y. Fu and Z. Liu, The Immune Escape Mechanisms of Mycobacterium Tuberculosis, Int. J. Mol. Sci., 2019, 20(2), 340 CrossRef PubMed.
- D. Mahamed, M. Boulle, Y. Ganga, C. Mc Arthur, S. Skroch, L. Oom, O. Catinas, K. Pillay, M. Naicker, S. Rampersad, C. Mathonsi, J. Hunter, E. B. Wong, M. Suleman, G. Sreejit, A. S. Pym, G. Lustig and A. Sigal, Intracellular growth of Mycobacterium tuberculosis after macrophage cell death leads to serial killing of host cells, eLife, 2017, 6 CAS.
- L. Diacovich, L. Lorenzi, M. Tomassetti, S. Meresse and H. Gramajo, The infectious intracellular lifestyle of Salmonella enterica relies on the adaptation to nutritional conditions within the Salmonella-containing vacuole, Virulence, 2017, 8(6), 975–992 CrossRef CAS PubMed.
- J. A. Bengoechea and J. Sa Pessoa, Klebsiella pneumoniae infection biology: living to counteract host defences, FEMS Microbiol. Rev., 2018, 43(2), 123–144 CrossRef PubMed.
- C. Bussi and M. G. Gutierrez, Mycobacterium tuberculosis infection of host cells in space and time, FEMS Microbiol. Rev., 2019, 43(4), 341–361 CrossRef PubMed.
- D. A. Portnoy, V. Auerbuch and I. J. Glomski, The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity, J. Cell Biol., 2002, 158(3), 409–414 CrossRef CAS PubMed.
- M. G. Pucciarelli and F. García-Del Portillo, Salmonella Intracellular Lifestyles and Their Impact on Host-to-Host Transmission, Microbiol. Spectrum, 2017, 5(4) DOI:10.1128/microbiolspec.mtbp-0009-2016.
- V. Cano, C. March, J. L. Insua, N. Aguiló, E. Llobet, D. Moranta, V. Regueiro, G. P. Brennan, M. I. Millán-Lou, C. Martín, J. Garmendia and J. A. Bengoechea, Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes, Cell. Microbiol., 2015, 17(11), 1537–1560 CrossRef CAS PubMed.
- G. Weiss and U. E. Schaible, Macrophage defense mechanisms against intracellular bacteria, Immunol. Rev., 2015, 264(1), 182–203 CrossRef CAS PubMed.
- F. Hanses, A. Kopp, M. Bala, C. Buechler, W. Falk, B. Salzberger and A. Schäffler, Intracellular Survival of Staphylococcus aureus in Adipocyte-Like Differentiated 3T3-L1 Cells Is Glucose Dependent and Alters Cytokine, Chemokine, and Adipokine Secretion, Endocrinology, 2011, 152, 4148–4157 CrossRef CAS PubMed.
- A. Lacoma, V. Cano, D. Moranta, V. Regueiro, D. Dominguez-Villanueva, M. Laabei, M. Gonzalez-Nicolau, V. Ausina, C. Prat and J. A. Bengoechea, Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line, Virulence, 2017, 8(8), 1761–1775 CrossRef CAS PubMed.
- C. Garzoni and W. L. Kelley, Return of the Trojan horse: intracellular phenotype switching and immune evasion by Staphylococcus aureus, EMBO Mol. Med., 2011, 3(3), 115–117 CrossRef CAS PubMed.
- M. A. Reott Jr, S. L. Ritchie-Miller, J. Anguita and M. C. Hudson, TRAIL expression is induced in both osteoblasts containing intracellular Staphylococcus aureus and uninfected osteoblasts in infected cultures, FEMS Microbiol. Lett., 2008, 278(2), 185–192 CrossRef.
- M. Fraunholz and B. Sinha, Intracellular Staphylococcus aureus: live-in and let die, Front. Cell. Infect. Microbiol., 2012, 2, 43 Search PubMed.
- R. L. Lamason and M. D. Welch, Actin-based motility and cell-to-cell spread of bacterial pathogens, Curr. Opin. Microbiol., 2017, 35, 48–57 CrossRef CAS PubMed.
- G. A. Dabiri, J. M. Sanger, D. A. Portnoy and F. S. Southwick, Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly, Proc. Natl. Acad. Sci. U. S. A., 1990, 87(16), 6068–6072 CrossRef CAS PubMed.
- N. Mellouk and J. Enninga, Cytosolic Access of Intracellular Bacterial Pathogens: The Shigella Paradigm, Front. Cell. Infect. Microbiol., 2016, 6, 35 Search PubMed.
- H. Agaisse, Molecular and Cellular Mechanisms of Shigella flexneri Dissemination, Front. Cell. Infect. Microbiol., 2016, 6(29) DOI:10.3389/fcimb.2016.00029.
- J. H. Brumell, P. Tang, M. L. Zaharik and B. B. Finlay, Disruption of the Salmonella-containing vacuole leads to increased replication of Salmonella enterica serovar typhimurium in the cytosol of epithelial cells, Infect. Immun., 2002, 70(6), 3264–3270 CrossRef CAS PubMed.
- S. V. Jamwal, P. Mehrotra, A. Singh, Z. Siddiqui, A. Basu and K. V. Rao, Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism, Sci. Rep., 2016, 6, 23089 CrossRef CAS PubMed.
- F. H. Santiago-Tirado and T. L. Doering, False friends: Phagocytes as Trojan horses in microbial brain infections, PLoS Pathog., 2017, 13(12), e1006680 CrossRef PubMed.
- A. Zlotkin, S. Chilmonczyk, M. Eyngor, A. Hurvitz, C. Ghittino and A. Eldar, Trojan Horse Effect: Phagocyte-Mediated Streptococcus iniae Infection of Fish, Infect. Immun., 2003, 71(5), 2318 CrossRef CAS PubMed.
- S. Yang, X. Han, Y. Yang, H. Qiao, Z. Yu, Y. Liu, J. Wang and T. Tang, Bacteria-Targeting Nanoparticles with Microenvironment-Responsive Antibiotic Release To Eliminate Intracellular Staphylococcus aureus and Associated Infection, ACS Appl. Mater. Interfaces, 2018, 10(17), 14299–14311 CrossRef CAS PubMed.
- B. Kim, H.-B. Pang, J. Kang, J.-H. Park, E. Ruoslahti and M. J. Sailor, Immunogene therapy with fusogenic nanoparticles modulates macrophage response to Staphylococcus aureus, Nat. Commun., 2018, 9(1), 1969 CrossRef.
- F. Gao, L. Xu, B. Yang, F. Fan and L. Yang, Kill the Real with the Fake: Eliminate Intracellular Staphylococcus aureus Using Nanoparticle Coated with Its Extracellular Vesicle Membrane as Active-Targeting Drug Carrier, ACS Infect. Dis., 2019, 5(2), 218–227 CrossRef CAS PubMed.
- R. Nazir, M. R. Zaffar and I. Amin, Bacterial biofilms: the remarkable heterogeneous biological communities and nitrogen fixing microorganisms in lakes, in Freshwater Microbiology, ed. S. A. Bandh, S. Shafi and N. Shameem, Academic Press, 2019, ch. 8, pp. 307–340 Search PubMed.
- H. Mu, J. Tang, Q. Liu, C. Sun, T. Wang and J. Duan, Potent Antibacterial Nanoparticles against Biofilm and Intracellular Bacteria, Sci. Rep., 2016, 6, 18877 CrossRef CAS PubMed.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. Tian, M. E. Napier, P. D. Pohlhaus, J. P. Rolland and J. M. DeSimone, Delivery of Multiple siRNAs Using Lipid-Coated PLGA Nanoparticles for Treatment of Prostate Cancer, Nano Lett., 2012, 12(1), 287–292 CrossRef CAS PubMed.
- A. Beletskii, A. Galloway, S. Rele, M. Stone and F. Malinoski, Engineered PRINT® nanoparticles for controlled delivery of antigens and immunostimulants, Hum. Vaccines Immunother., 2014, 10(7), 1908–1913 CrossRef CAS.
- J. Xu, D. H. C. Wong, J. D. Byrne, K. Chen, C. Bowerman and J. M. DeSimone, Future of the Particle Replication in Nonwetting Templates (PRINT) Technology, Angew. Chem., Int. Ed., 2013, 52(26), 6580–6589 CrossRef CAS PubMed.
- X. Fu, J. Cai, X. Zhang, W.-D. Li, H. Ge and Y. Hu, Top-down fabrication of shape-controlled, monodisperse nanoparticles for biomedical applications, Adv. Drug Delivery Rev., 2018, 132, 169–187 CrossRef CAS PubMed.
- K. Hegde, S. K. Brar, M. Verma and R. Y. Surampalli, Current understandings of toxicity, risks and regulations of engineered nanoparticles with respect to environmental microorganisms, Nanotechnol. Environ. Eng., 2016, 1(1), 5 CrossRef.
- S. Hua, M. B. C. de Matos, J. M. Metselaar and G. Storm, Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization, Front. Pharmacol., 2018, 9, 790 CrossRef PubMed.
- S.-D. Li and L. Huang, Nanoparticles evading the reticuloendothelial system: role of the supported bilayer, Biochim. Biophys. Acta, 2009, 1788(10), 2259–2266 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard Jr, Nanoparticle-based targeted drug delivery, Exp. Mol. Pathol., 2009, 86(3), 215–223 CrossRef CAS PubMed.
- J. Y. Oh, H. S. Kim, L. Palanikumar, E. M. Go, B. Jana, S. A. Park, H. Y. Kim, K. Kim, J. K. Seo, S. K. Kwak, C. Kim, S. Kang and J.-H. Ryu, Cloaking nanoparticles with protein corona shield for targeted drug delivery, Nat. Commun., 2018, 9(1), 4548 CrossRef PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanoparticle PEGylation for imaging and therapy, Nanomedicine, 2011, 6(4), 715–728 CrossRef CAS PubMed.
- S. R. Bandara, T. G. Molley, H. Kim, P. A. Bharath, K. A. Kilian and C. Leal, The structural fate of lipid nanoparticles in the extracellular matrix, Mater. Horiz., 2020 Search PubMed.
- S. W. Choi, W. S. Kim and J. H. Kim, Surface Modification of Functional Nanoparticles for Controlled Drug Delivery, J. Dispersion Sci. Technol., 2003, 24(3–4), 475–487 CrossRef CAS.
- D. Sun, J. Chen, Y. Wang, H. Ji, R. Peng, L. Jin and W. Wu, Advances in refunctionalization of erythrocyte-based nanomedicine for enhancing cancer-targeted drug delivery, Theranostics, 2019, 9(23), 6885–6900 CrossRef PubMed.
- F. U. Din, W. Aman, I. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Forming next-generation antibody–nanoparticle conjugates through the oriented installation of non-engineered antibody fragments, Chem. Sci., 2018, 9(1), 79–87 RSC.
- M. C. Johnston and C. J. Scott, Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy, Drug Discovery Today: Technol., 2018, 30, 63–69 CrossRef PubMed.
- M. M. Billingsley, R. S. Riley and E. S. Day, Antibody-nanoparticle conjugates to enhance the sensitivity of ELISA-based detection methods, PLoS One, 2017, 12(5), e0177592 CrossRef PubMed.
- V. Hoerr, L. Tuchscherr, J. Hüve, N. Nippe, K. Loser, N. Glyvuk, Y. Tsytsyura, M. Holtkamp, C. Sunderkötter, U. Karst, J. Klingauf, G. Peters, B. Löffler and C. Faber, Bacteria tracking by in vivo magnetic resonance imaging, BMC Biol., 2013, 11(1), 63 CrossRef PubMed.
- D. Singh, J. M. McMillan, A. V. Kabanov, M. Sokolsky-Papkov and H. E. Gendelman, Bench-to-bedside translation of magnetic nanoparticles, Nanomedicine, 2014, 9(4), 501–516 CrossRef CAS PubMed.
- K. Wu, D. Su, J. Liu, R. Saha and J. P. Wang, Magnetic nanoparticles in nanomedicine: a review of recent advances, Nanotechnology, 2019, 30(50), 502003 CrossRef PubMed.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems, Chem. Soc. Rev., 2016, 45(5), 1457–1501 RSC.
- S. F. Medeiros, A. M. Santos, H. Fessi and A. Elaissari, Stimuli-responsive magnetic particles for biomedical applications, Int. J. Pharm., 2011, 403(1), 139–161 CrossRef CAS.
- P. Meers, M. Neville, V. Malinin, A. W. Scotto, G. Sardaryan, R. Kurumunda, C. Mackinson, G. James, S. Fisher and W. R. Perkins, Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections, J. Antimicrob. Chemother., 2008, 61(4), 859–868 CrossRef CAS PubMed.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-C. Wang, B. Sun and J. Wang, Lipase-Sensitive Polymeric Triple-Layered Nanogel for “On-Demand” Drug Delivery, J. Am. Chem. Soc., 2012, 134(9), 4355–4362 CrossRef CAS PubMed.
- M. A. B. Shabbir, M. Z. Shabbir, Q. Wu, S. Mahmood, A. Sajid, M. K. Maan, S. Ahmed, U. Naveed, H. Hao and Z. Yuan, CRISPR-cas system: biological function in microbes and its use to treat antimicrobial resistant pathogens, Ann. Clin. Microbiol. Antimicrob., 2019, 18(1), 21 CrossRef PubMed.
- J. R. Strich and D. S. Chertow, CRISPR-Cas Biology and Its Application to Infectious Diseases, J. Clin. Microbiol., 2019, 57(4), e01307 CAS.
- D. Rath, L. Amlinger, A. Rath and M. Lundgren, The CRISPR-Cas immune system: biology, mechanisms and applications, Biochimie, 2015, 117, 119–128 CrossRef CAS PubMed.
- F. Hille and E. Charpentier, CRISPR-Cas: biology, mechanisms and relevance, Philos. Trans. R. Soc., B, 2016, 371(1707), 20150496 CrossRef PubMed.
- C. A. Lino, J. C. Harper, J. P. Carney and J. A. Timlin, Delivering CRISPR: a review of the challenges and approaches, Drug Delivery, 2018, 25(1), 1234–1257 CrossRef CAS PubMed.
- Y. K. Kang, K. Kwon, J. S. Ryu, H. N. Lee, C. Park and H. J. Chung, Nonviral Genome Editing Based on a Polymer-Derivatized CRISPR Nanocomplex for Targeting Bacterial Pathogens and Antibiotic Resistance, Bioconjugate Chem., 2017, 28(4), 957–967 CrossRef CAS PubMed.
- U. Romling and C. Balsalobre, Biofilm infections, their resilience to therapy and innovative treatment strategies, J. Intern. Med., 2012, 272(6), 541–561 CrossRef CAS PubMed.
- Y. Itoh, X. Wang, B. J. Hinnebusch, J. F. Preston 3rd and T. Romeo, Depolymerization of beta-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms, J. Bacteriol., 2005, 187(1), 382–387 CrossRef CAS PubMed.
- R. M. Corrigan, J. C. Abbott, H. Burhenne, V. Kaever and A. Gründling, c-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress, PLoS Pathog., 2011, 7(9), e1002217 CrossRef CAS PubMed.
- L. D. Christensen, M. van Gennip, T. H. Jakobsen, M. Alhede, H. P. Hougen, N. Hoiby, T. Bjarnsholt and M. Givskov, Synergistic antibacterial efficacy of early combination treatment with tobramycin and quorum-sensing inhibitors against Pseudomonas aeruginosa in an intraperitoneal foreign-body infection mouse model, J. Antimicrob. Chemother., 2012, 67(5), 1198–1206 CrossRef CAS PubMed.
Footnote |
| † L. J. and H. W. L. contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |