
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, physicochemical characterization, toxicity and efficacy of a PEG conjugate and a hybrid PEG conjugate nanoparticle formulation of the antibiotic moxifloxacin†
Lesego L. Tshweuab,
Mohamed A. Shemis c,
Aya Abdelghanyc,
Abdullah Goudac,
Lynne A. Pilcherb,
Nicole R. S. Sibuyid,
Mervin Meyerd,
Admire Dubee and
Mohammed O. Balogun*a
c,
Aya Abdelghanyc,
Abdullah Goudac,
Lynne A. Pilcherb,
Nicole R. S. Sibuyid,
Mervin Meyerd,
Admire Dubee and
Mohammed O. Balogun*a
aBiopolymer Modification & Therapeutics Lab, Chemicals Cluster, Council for Scientific and Industrial Research, Pretoria 0001, South Africa. E-mail: mbalogun@csir.co.za; mohammedbalogun@tuks.co.za
bDepartment of Chemistry, University of Pretoria, Pretoria, South Africa
cBiochemistry & Molecular Biology Department, Theodor Bilharz Research Institute, Warak El-Hadar, Kornish El-Nile, P.O. Box 30, Imbaba, 12411-Giza, Egypt
dDST/Mintek Nanotechnology Innovation Centre, Biolabels Node, Department of Biotechnology, University of the Western Cape, Cape Town, South Africa
eInfectious Disease Nanomedicine Research Group, School of Pharmacy, University of the Western Cape, Cape Town, South Africa
First published on 26th May 2020
Abstract
Antibiotic resistance is increasing at such an alarming rate that it is now one of the greatest global health challenges. Undesirable toxic side-effects of the drugs lead to high rates of non-completion of treatment regimens which in turn leads to the development of drug resistance. We report on the development of delivery systems that enable antibiotics to be toxic against bacterial cells while sparing human cells. The broad-spectrum fluoroquinolone antibiotic moxifloxacin (Mox) was successfully conjugated to poly(ethylene glycol) (PEG) which was further encapsulated into the hydrophobic poly(ε-caprolactone) (PCL) nanoparticles (NPs) with high efficiency, average particle size of 241.8 ± 4 nm and negative zeta potential. Toxicity against erythrocytes and MDBK cell lines and drug release in human plasma were evaluated. Hemocompatibility and reduced cytotoxicity of the PEG–Mox and PCL(PEG–Mox) NPs were demonstrated in comparison to free Mox. Antimicrobial activity was assessed against drug sensitive and resistant: Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa and Klebsiella pneumoniae. The antibacterial activity of Mox was largely maintained after conjugation. Our data shows that the toxicity of Mox can be effectively attenuated while, in the case of PEG–Mox, retaining significant antibacterial activity. At the conditions employed in this study for antimicrobial activity the encapsulated conjugate (PCL(PEG–Mox) NPs) did not demonstrate, conclusively, significant antibacterial activity. These systems do, however, hold promise if further developed for improved treatment of bacterial infections.
Introduction
Since the discovery of penicillin by Sir Alexander Fleming in 1928, antibiotics have positively influenced the clinical management of bacterial infections. However, as the centenary of this first discovery approaches, major global bodies and expert groups1,2 warn that resistance to all antibiotics is occurring at a rapid and alarming rate. According to a report published by Jim O'Neill, deaths from antimicrobial resistance could rise from the current 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 10 million per annum by 2050.1 Further compounding the situation is that, for almost half a century there has been no new class of antibiotics discovered.3 Aside from over-prescription, patients often do not comply with the strict regimens for treatment due to numerous drug-related toxic side effects.4
000 to 10 million per annum by 2050.1 Further compounding the situation is that, for almost half a century there has been no new class of antibiotics discovered.3 Aside from over-prescription, patients often do not comply with the strict regimens for treatment due to numerous drug-related toxic side effects.4
To improve patient compliance to treatment and ensure drug sustainability, antibiotics must be able to discriminate efficiently between the disease-causing bacteria and the healthy human host cells. Nanoparticulate drug delivery systems have shown promise for improving antimicrobial chemotherapy through reducing systemic toxicity while retaining therapeutic effect.4 Two main options are employed for the creation of these systems – physical drug encapsulation or chemical conjugation of the drug to a polymeric carrier.5 Encapsulation involves the physical entrapment of the drug within or adsorption onto the surface of the nanoparticle (NP) which may be made up of a hydrophobic polymer.6 Hydrophobic polymers offer sustained drug release because they are slowly eroded in physiological fluids but drug leakage from the NP via diffusion or premature burst release of surface-adhered drugs during transit to the target site is often a problem.7 Conjugation addresses this issue through covalent linkage of the drug to a carrier polymer like polyethylene glycol (PEG).8,9 While conjugation offers this advantage, currently used carrier polymers are water-soluble.9,10 This could result in difficulty with transmembrane transport and a rapid renal excretion of the conjugate. Batalha et al. recently reported on an isoniazid (INH)–polymer conjugate system for the treatment of Mycobacterium tuberculosis infection.11 The delivery system involved the use of a hydrophobic polymer to which INH was linked via a pH-sensitive hydrazone bond and in which hydrophobic antibiotics could further be encapsulated. While this system worked well specifically against Mtb granulomas in zebra fish models it is not clear whether it could be as effective against a wider spectrum of bacteria. Also, it appears that the system is best suited for conjugation of water-soluble drugs while hydrophobic antibiotics will have to be encapsulated. This would imply that it is a best-fit model for combination drug delivery.
In an attempt to address the challenges of both encapsulation and chemical conjugation delivery systems we developed and investigated a polymer–drug conjugate (PEG–Moxifloxacin (Mox)) and an encapsulated hybrid of the conjugate where the PEG–Mox is further incorporated into a hydrophobic NP (poly(ε-caprolactone) (PCL)). PCL was chosen as it is relatively cheaper than other commonly used polyesters. Mox has been selected as a model antibiotic due to its broad-spectrum activity against both Gram negative and positive bacteria and toxic side effects which include ocular toxicity, cardiovascular toxicity (QT-interval prolongation that increases the risk of cardiac arrest), and tendon rupture.12 It is several orders of magnitude less soluble in water than INH and more hydrophobic than rifampicin. In this work we synthesized and extensively characterized the PEG–Mox conjugate and NPs and investigated their toxicity to cells (human erythrocytes and Madin-Darby bovine kidney (MDBK) cells) and efficacy against various Gram positive and negative bacteria.
Results and discussion
Synthesis and characterization of PEG–Mox conjugate and encapsulated NPs
Mox was reacted at 1.15 molar equivalents with PEG to give a stable conjugate with a zero-length amide linker (Fig. 1). Amide conjugates are resistant to rapid hydrolysis and biological degradation.13 In using this bond in the PEG–Mox conjugate we hoped to gain insight into the performance of the entire construct in terms of mammalian and bacterial cell toxicity.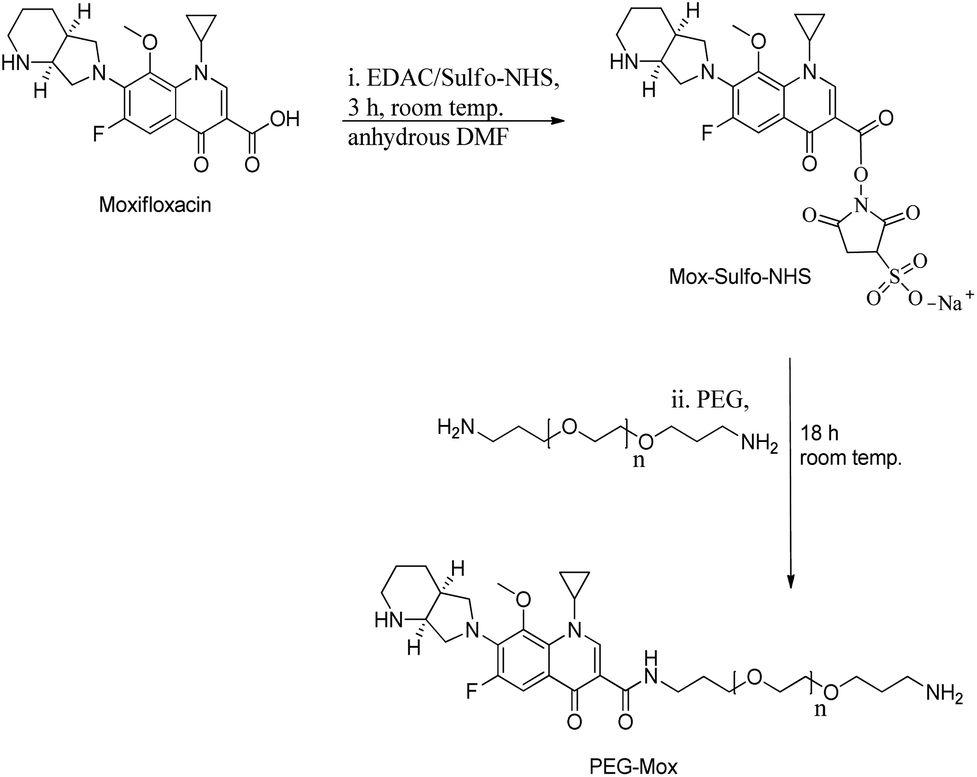 | ||
| Fig. 1 Schematic of the synthesis of the PEG–Mox conjugate using carbodiimide chemistry. | ||
The non-ionic amphiphilic PEG–Mox conjugate, i.e. a hydrophilic PEG and a hydrophobic Mox, offered a polarity that facilitated insertion into PCL NPs. The %EE of the PEG–Mox conjugate in the PCL(PEG–Mox) NPs was determined to be 60% (w/w).
NMR and FTIR analyses
Conjugation of Mox to PEG was confirmed by 1H NMR analysis. Unlike the free Mox which is insoluble in chloroform the conjugate readily dissolved in CDCl3, a property conferred by the PEG. The characteristic signals of both the Mox and PEG were observed in the spectrum of the PEG–Mox conjugate (Fig. 2). The alkenyl proton (α) was the most downfield signal appearing as a singlet at 8.63 ppm while the aromatic proton (β) appeared as a doublet at 7.61 ppm. A strong singlet attributed to the three methoxy protons (γ) was observed at 3.47 ppm. Signals of PEG include the methylene protons (ε and θ) at 3.35 ppm and 3.52 ppm, respectively. The observed broadening of the signals further confirmed the successful conjugation of Mox to PEG.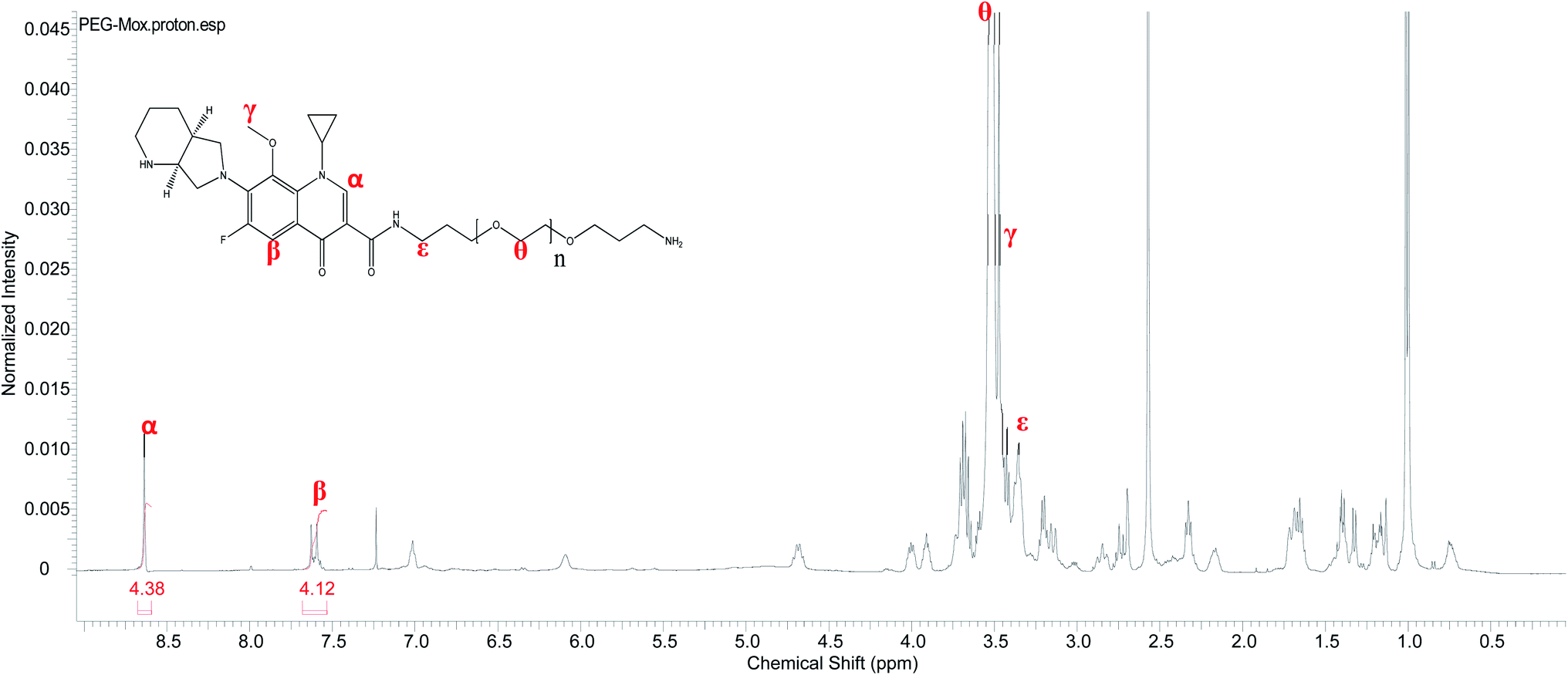 | ||
| Fig. 2 Proton NMR spectrum of PEG–Mox conjugate (CDCl3, 600 MHz). | ||
Conjugation of Mox to PEG was also confirmed by ATR-FTIR with the appearance of the amide bond signal at around 1760 cm−1 (Fig. 3). The ATR-FTIR analysis of PCL, PCL NPs and PCL(PEG–Mox) NPs showed the characteristic peak of the PCL polymer at around 1721.4 cm−1 and 1725.9 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (Fig. 3). The spectrum of pure Mox presented functional bands at 1706 cm−1 due to the carboxylic acid CO stretching vibration, C–H bonding for the substituted benzene at 1875 cm−1, stretching at 1622 cm−1, 1518 cm−1 and 1451 cm−1 due to aromatic CC, and C–N banding at 1320 cm−1.14 Weak but distinctive functional bands of the Mox moiety were also found in the spectra of PCL(PEG–Mox) NPs, suggesting that most of the Mox conjugates were encapsulated into the NPs. Slight shifts in the functional bands are ascribed to amorphous packing of the PEG–Mox during the encapsulation process.
O stretching vibration (Fig. 3). The spectrum of pure Mox presented functional bands at 1706 cm−1 due to the carboxylic acid CO stretching vibration, C–H bonding for the substituted benzene at 1875 cm−1, stretching at 1622 cm−1, 1518 cm−1 and 1451 cm−1 due to aromatic CC, and C–N banding at 1320 cm−1.14 Weak but distinctive functional bands of the Mox moiety were also found in the spectra of PCL(PEG–Mox) NPs, suggesting that most of the Mox conjugates were encapsulated into the NPs. Slight shifts in the functional bands are ascribed to amorphous packing of the PEG–Mox during the encapsulation process.
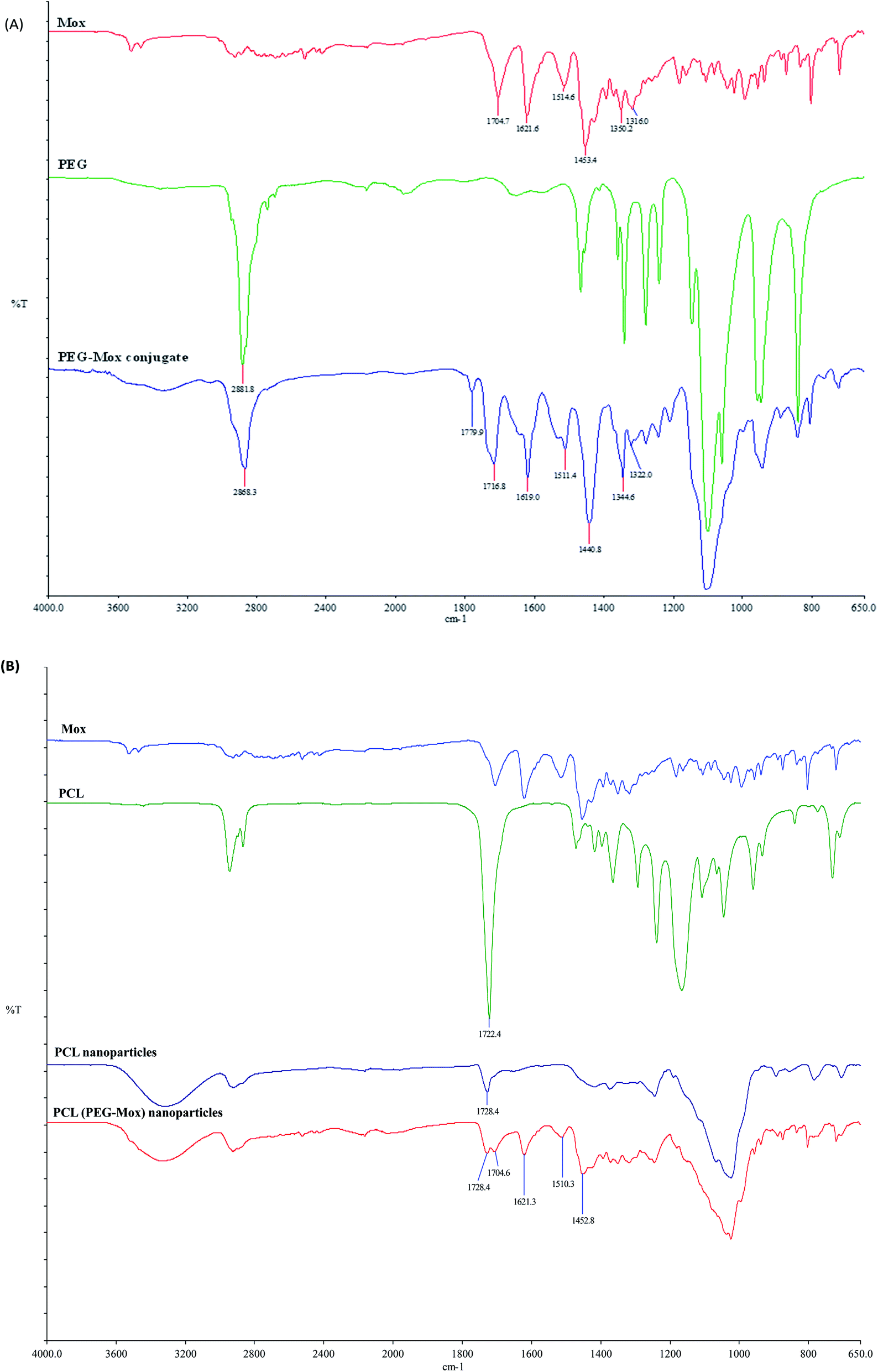 | ||
| Fig. 3 FTIR spectra of (A) Mox, PEG and PEG–Mox conjugate. (B) Mox, PCL, PCL NPs, and PCL–Mox NPs. | ||
Particle morphology, size, and zeta potential
The PEG–Mox conjugate showed two average hydrodynamic diameter distributions of 8.3 ± 4 nm and 75.7 ± 10 nm in DLS studies while the PDI was observed to be 0.191. As would be expected, due to the amphiphilicity of the construct, the PEG–Mox conjugate had a spherical morphology in TEM images (Fig. 4). All the double-emulsion-prepared NPs (PCL(PEG–Mox) and empty PCL) were spherical (Fig. 5). The PCL(PEG–Mox) NPs were 241.8 ± 4 nm in diameter, significantly larger than the 174.4 ± 10 nm of empty PCL NPs. The TEM particle sizes correlated well with the DLS hydrodynamic sizes for the conjugate and the NPs. Both PCL(PEG–Mox) and empty PCL particles were of similar zeta potentials (−23.3 ± 0.9 mV and −20.3 ± 0.4 mV) and PDIs (0.24 ± 0.01 and 0.22 ± 0.02) (Table 1). These values confirmed that stable homogeneous NPs were formed. High absolute zeta potential value is an indication of stable NPs as the electrostatic repulsion prevents particle agglomeration.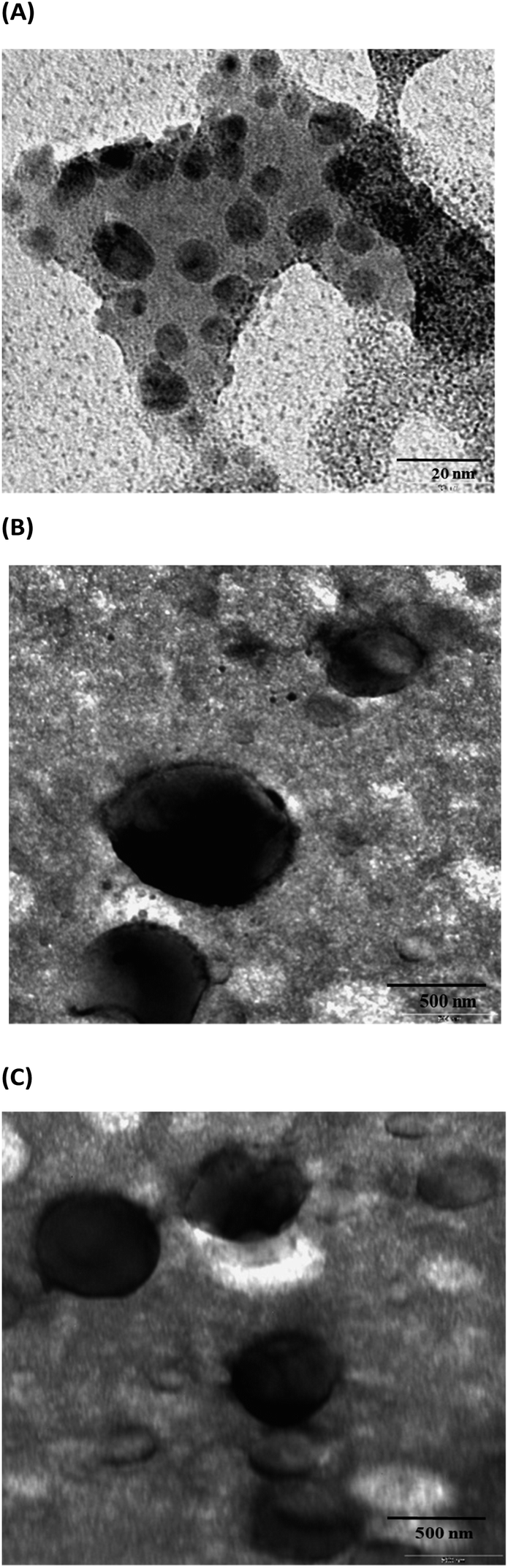 | ||
| Fig. 4 TEM images: (A) PEG–Mox conjugate, (B) PCL NPs and (C) PCL(PEG–Mox) NPs. | ||
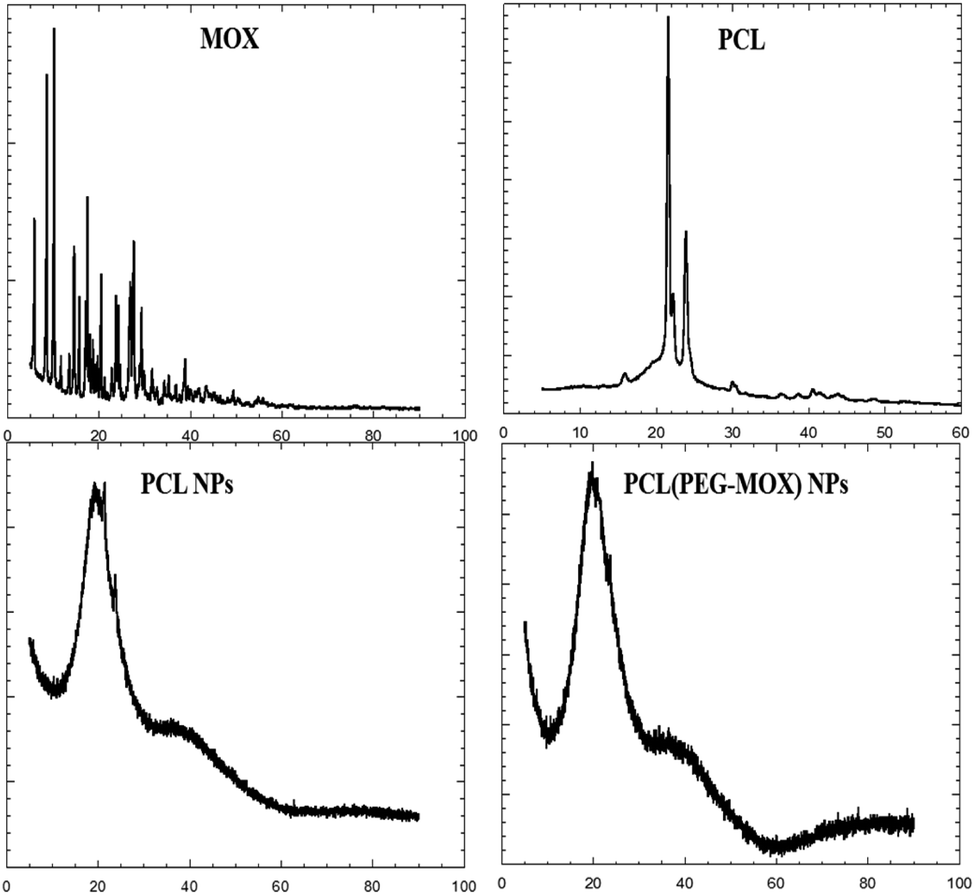 | ||
| Fig. 5 X-ray diffraction patterns of Mox, molecular PCL, PCL NPs and PCL(PEG–Mox) NPs. X-axes represent 2 theta (degree). | ||
| Formulation | Particle size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| PEG–Mox conjugate | 8.3 ± 4 & 75.7 ± 10 | 0.19 ± 0.04 | −15.2 ± 0.2 |
| PCL NPs | 174.4 ± 10 | 0.22 ± 0.02 | −20.3 ± 0.4 |
| PCL(PEG–Mox) NPs | 241.8 ± 4 | 0.24 ± 0.01 | −23.3 ± 0.9 |
XRD and TGA analyses
The physical attributes of the conjugate and NPs were also studied by XRD and TGA. The XRD patterns of the NPs (PCL NPs and PCL(PEG–Mox) NPs) presented broad signals void of any sharp peaks over the entire 2 theta (Fig. 5). This confirmed that both NPs were amorphous. On the contrary, molecular PCL showed two sharp peaks at about 21.3° and 23.9° triggered by scattering from crystalline regions. Several sharp peaks were also observed in the diffractogram of Mox. The amorphous nature of the NPs confers on them an advantage in aqueous solubility or dispersity compared to the crystalline free molecules.15The PEG–Mox conjugate and PCL(PEG–Mox) NPs possessed moisture contents similar to the PEG, Mox, PCL, and PCL NPs as indicated by an initial weight loss of between 3 to 6% in TGA experiments (Fig. 6). High moisture content provides a degradative environment that could also facilitate the outward diffusion of NP content and even microbiological contamination during storage.16 PEG rapidly decomposed between 320 to 430 °C with 89% weight loss. The PEG–Mox conjugate decomposed in a pattern more similar to the free Mox than PEG. Decomposition was initially gradual from 210 to 400 °C after the loss of water but became rapid from 400 to 435 °C. Both the empty PCL NPs and PCL(PEG–Mox) NPs showed lower thermal stability than the molecular PCL. This might be due to the NPs having a greater superficial area with thermal decomposition occurring much faster.17
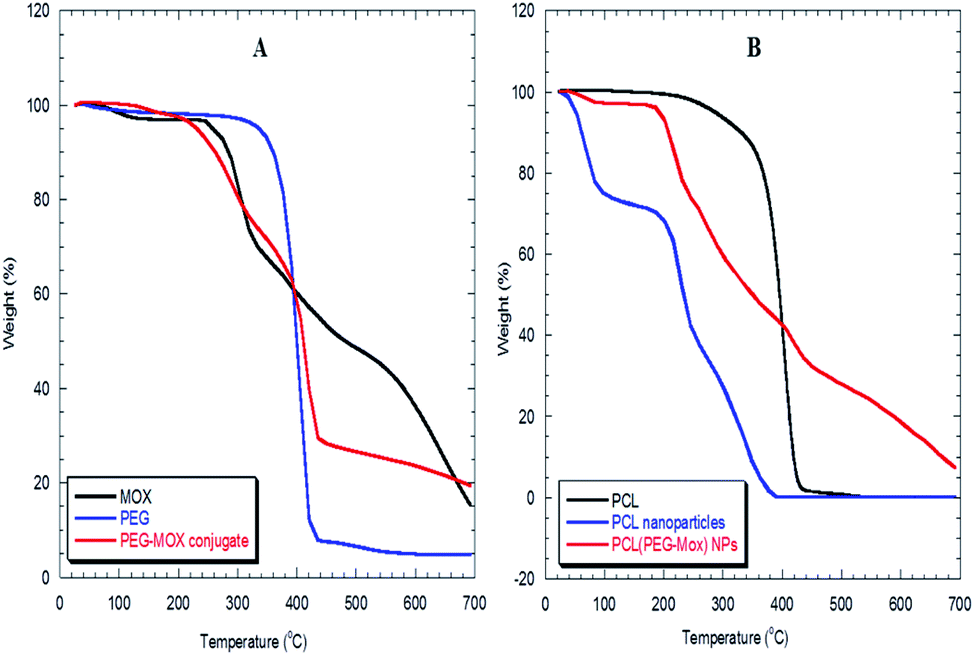 | ||
| Fig. 6 TGA thermograms of (A) Mox, PEG and PEG–Mox conjugate, (B) molecular PCL, PCL NPs and PCL(PEG–Mox) NPs. | ||
Toxicity studies
Conjugation of Mox to polysaccharides has previously been used to improve the bioavailability of the drug.18,19 In the work of Schwartz et al., Mox was conjugated to a dansylated carboxymethyl glucan which actively targeted the drug to M. tuberculosis-infected macrophages but the toxicity profile in human cells was not reported. It is therefore difficult to determine if the reported system offered an advantage over the free drug in terms of reduction in human toxicity.Hemocompatibility of the formulations
To determine the effect of incorporation into a delivery vehicle on the toxicity of Mox we evaluated the biocompatibility of the conjugate and NPs in an RBC hemolysis assay (Fig. 7). At the maximum concentration of Mox tested, 1 mg mL−1, the percentage hemolysis observed for free Mox, PEG–Mox and PCL(PEG–Mox) NPs were 89.0 ± 3.1, 16.2 ± 0.50 and 21.7 ± 1.5, respectively. It is clear that the delivery systems significantly attenuated the toxicity of Mox in the ex vivo assay and while the toxicity profile of Mox was clearly dose-dependent, that of the conjugate and the NPs were tightly restricted over the entire concentration range tested. This could translate into significant reduction in Mox systemic toxicity when used in humans.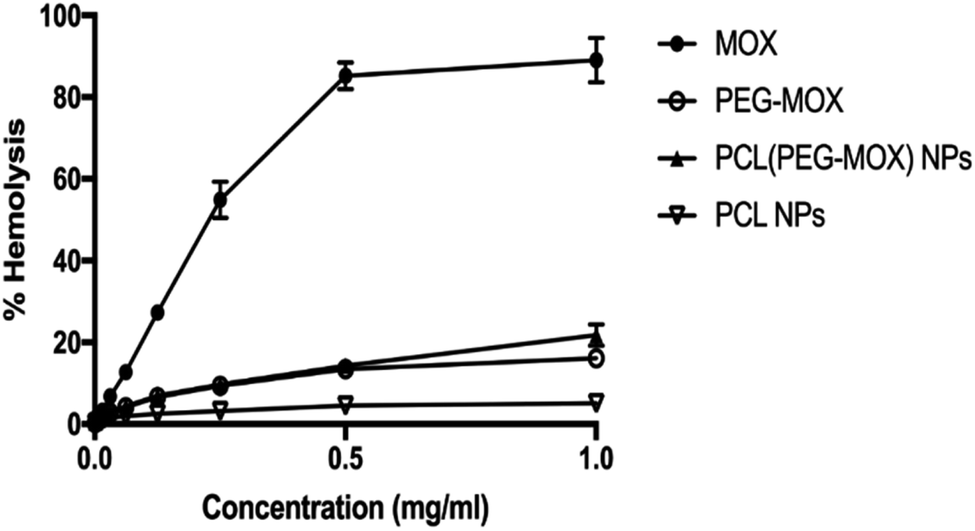 | ||
| Fig. 7 In vitro haemolysis study with Mox, PEG–Mox conjugate, PCL(PEG–Mox) NPs and empty PCL NPs incubated with human RBC suspensions for 4 h at 37 °C. Data represents the mean ± SD, n = 3. | ||
Mox release in human plasma
Mox release from PEG–Mox conjugate and the PCL(PEG–Mox) NPs was evaluated in human plasma over 96 h. There was insignificant release of Mox from the PEG–Mox conjugate and the PCL(PEG–Mox) NPs over the 96 h period for all concentrations tested, i.e. 1, 3 and 5 mg mL−1 of the PCL(PEG–Mox) NPs (Fig. 9). This was expected as the amide linkage between the PEG and Mox is not readily labile under the physiological conditions of plasma. The hydrolysis of amides is slower than that of the esters such that amides are typically excreted in the urine unchanged.20 Hydrolysis of amides would be expected within the intracellular acidic endolysosomal vesicles, which contain hydrolytic enzymes active at the low pH of about 5. Furthermore, PEG is capable of sterically preventing the approach of destabilizing plasma proteins to the PEG–Mox conjugate or PCL(PEG–Mox) NPs.21 As the analysis was for the free Mox molecule no significant release was detectable. This stability translates into improvement in the safety of Mox and further explains the low hemolysis observed for the two systems (Fig. 8). As the Mox is locked in macromolecular structures for both PEG–Mox and PCL(PEG–Mox), interactions with the cells are limited.22 These results are in close agreement with previously published data.21,23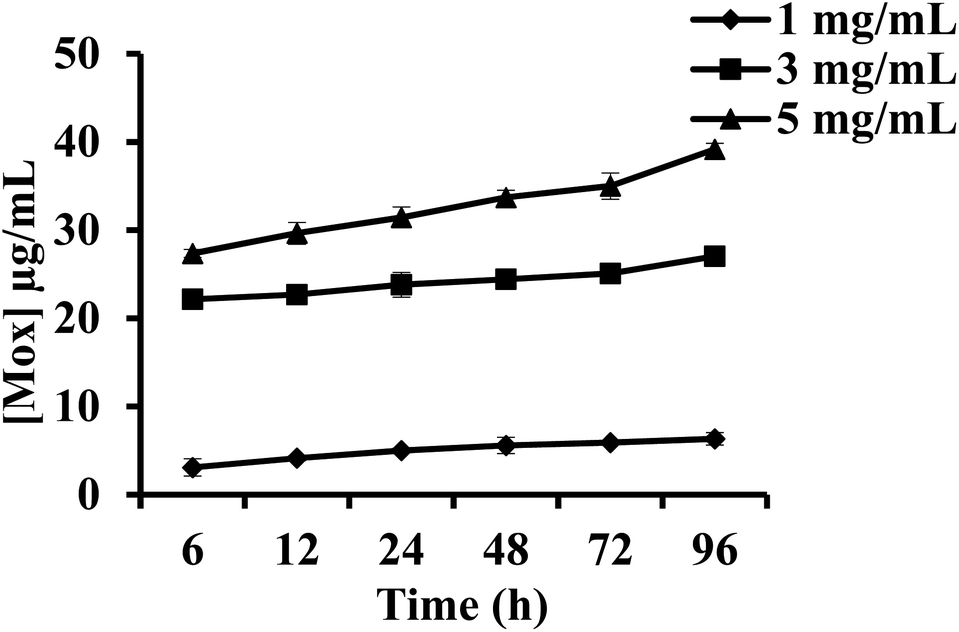 | ||
| Fig. 8 In vitro release of Mox from PCL(PEG–Mox) NPs in human plasma over 96 h at 37 °C. Data represents the mean ± SD, n = 3. | ||
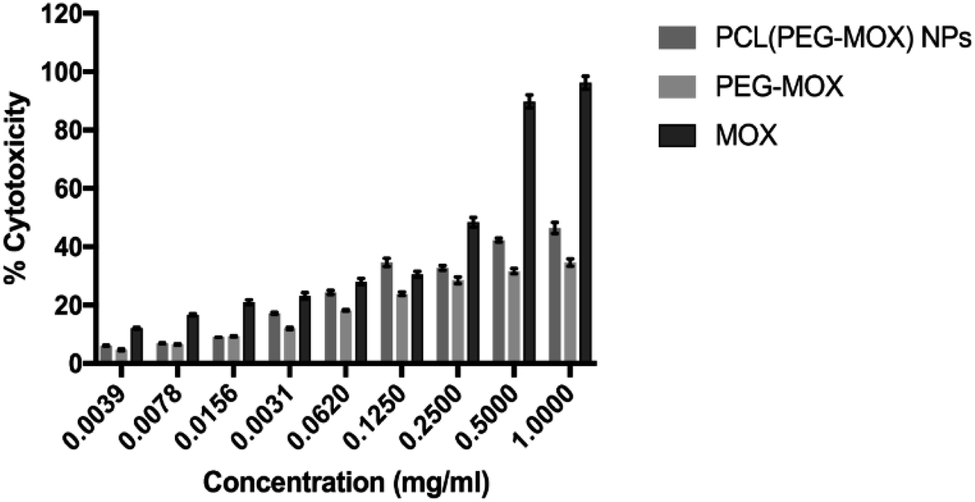 | ||
| Fig. 9 Effects of Mox, PEG–Mox conjugate and PCL(PEG–Mox) NPs on MDBK cell viability measured by MTT assay. Data represent mean ± SD, n = 3. | ||
Cytotoxicity
The cytotoxicity of the PEG–Mox conjugate and PCL(PEG–Mox) NPs in comparison to Mox was further evaluated in vitro based on cellular metabolic activity using MTT assay. MDBK cell lines were used as they have defined cell junctions, polarity and a rapid growth rate.24 In several studies MDBK cell lines have been utilized as an alternative model to Caco-2 cells.25 PCL NPs were used as negative control and there was no discernible toxicity even at 1 mg mL−1. At the highest concentration tested, i.e. 1.0 mg mL−1, PEG–Mox conjugate and PCL(PEG–Mox) NPs induced less than 50% cell mortality, while the free Mox was more than twice as toxic at ∼96.2% cell death (Fig. 9). These results correlate well with the low toxicity observed in the hemolysis assay (Fig. 7).In vitro microbiological assays
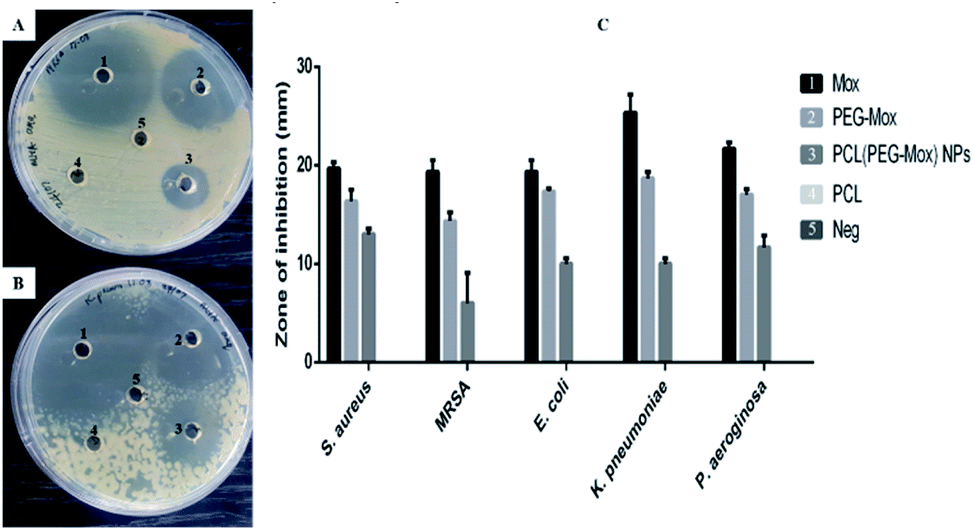 | ||
| Fig. 10 Assessing the antibacterial activity of Mox, PEG–Mox, PCL(PEG–Mox) NPs and PCL NPs on Gram negative and Gram positive clinical isolates using well diffusion method. A and B are representative agar plates showing zone of inhibition for MRSA (Gram positive) and K. pneumoniae (Gram negative), respectively. C is a bar graph combining the data (n = 3) for antibacterial testing on S. aureus, MRSA, E. coli, K. pneumoniae, and P. aeruginosa. Empty PCL NPs did not show any activity. | ||
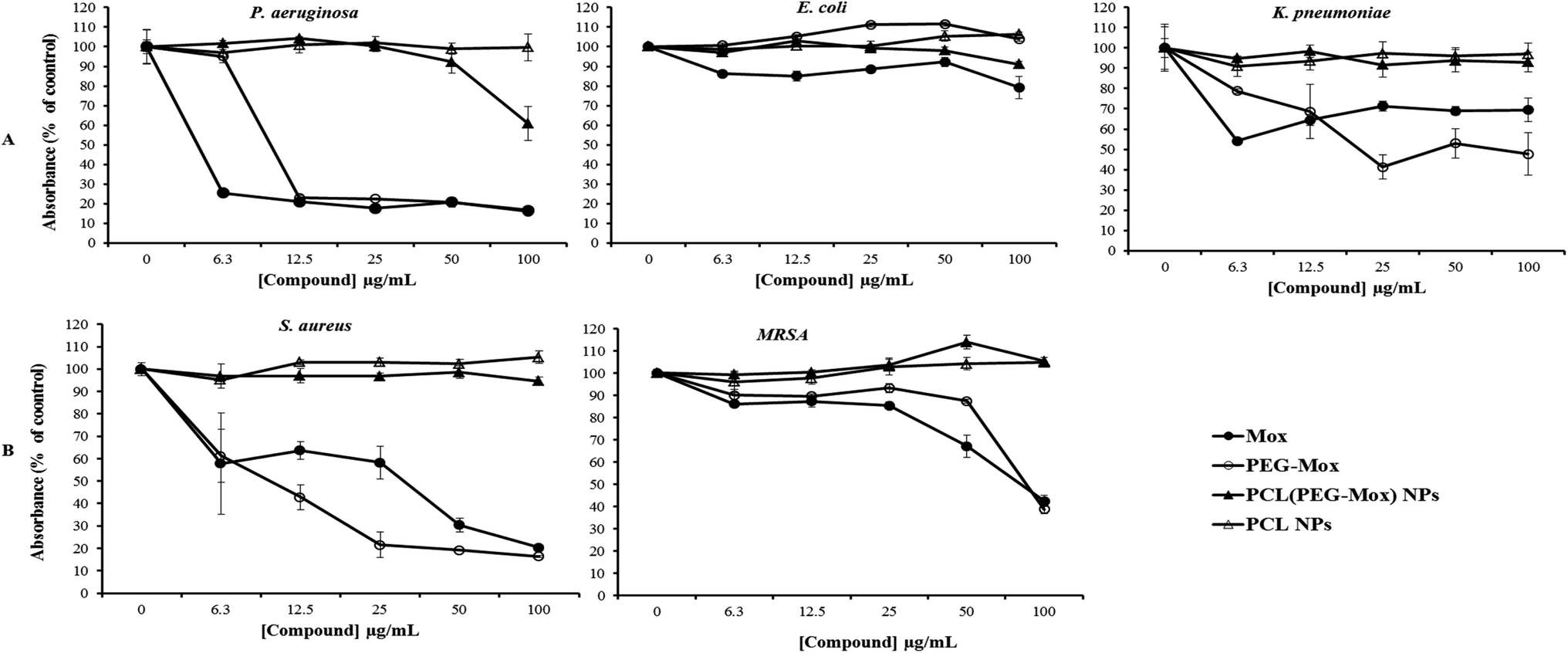 | ||
| Fig. 11 Assessing the bactericidal activity of Mox, PEG–Mox, PCL(PEG–Mox) NPs and PCL NPs using the microdilution method. Gram negative (E. coli, P. aeruginosa and K. pneumoniae) and positive (S. aureus, MRSA) clinical isolates. The bacteria were exposed to the test compounds for 24 h, after which the viability of the bacteria was assessed using the AlamarBlue® assay. (A) Gram negative (E. coli, P. aeruginosa and K. pneumoniae) and (B) Gram positive (S. aureus, MRSA) clinical isolates. * indicates significant differences at p < 0.05. | ||
The antibacterial effects of PCL(PEG–Mox) NPs were distinctly lower when compared to free Mox and PEG–Mox although a disparity was observed between the two methods used. Interestingly in the well diffusion method the PCL(PEG–Mox) NPs showed between 10 mm to 15 mm diameter rings of inhibition which is significant in comparison to inhibition zone diameters of 15 mm to 19 mm and 20 mm to 25 mm for PEG–Mox and Mox, respectively. There was no observable effect of the empty PCL carrier (Fig. 10). On the contrary, in the microdilution experiments there was no significant difference between the PCL(PEG–Mox) NPs and the empty PCL carriers, except for P. aeruginosa which showed a moderate susceptibility to the former at the highest dose (100 μg mL−1) tested (Fig. 11). All other strains were quite refractory to the PCL(PEG–Mox) NPs treatment. These findings demonstrate that formation of the polymer–drug conjugate retains the antibacterial activity of Mox, while encapsulation of the conjugate within PCL NPs reduces the effectiveness of the conjugate. This could in part be explained by the need of the PEG–Mox conjugate to first diffuse out of the NPs or, alternatively, the PCL NP should first erode, before the activity of the PEG–Mox conjugate can be observed. A better performance might be expected in vivo, due to the expected action of enzymes on breaking down the NPs (and consequently releasing the conjugate). The disparities observed between the well diffusion and microdilution data could be attributed to various factors that include, but are not limited to, diffusion efficiency of the test compound, sample polarity and concentration. Due to these limitations, microdilution assays are often considered more reliable and reproducible compared to the solid agar diffusion assays.28,29
As the threat of losing the current arsenal of antibiotics grows, research is increasingly being focused on harnessing the potentials of drug delivery systems.11,27,28 We have demonstrated here that the two main nanomedicine delivery systems technologies (polymer–drug conjugation and encapsulation) can be effectively utilized to reduce the toxicity of antibiotics, in this case Mox, to human cells. In in vitro antibacterial assays the polymer-conjugated Mox performed comparatively well with the free drug against both Gram negative and positive bacteria even while offering the possibility of an intravenous administration route due to its aqueous solubility. However, the encapsulated conjugate hybrid, PCL(PEG–Mox) NPs, did not present a definitive antibacterial performance.
Experimental
Materials
PEG (Mw = 1.5 KDa), PCL (Mw = 10 KDa), poly(vinyl alcohol) ((PVA) Mw = 13–23 KDa, 87–89% hydrolyzed), N-(ethylcarbonimidoyl)-N,N-dimethylpropane-1,3-diamine monohydrochloride (EDAC), N-hydroxysulfosuccinimide (sulfo-NHS), 2-methoxyethylmethyl (MEM), foetal bovine serum (FBS), penicillin/streptomycin, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS), N,N-dimethylformamide (DMF), ethyl acetate, and lactose monohydrate were obtained from Sigma-Aldrich (South Africa). Mox was purchased from DB Fine Chemicals (South Africa) as the chloride salt. Madin-Darby bovine kidney (MDBK) cells stock, frozen in liquid nitrogen, ATCC no. CCL-22 passage: 46 were commercially obtained from VACSERA, Giza, Egypt. Alcohol free and drug free healthy human male donors' blood was used. All aqueous experiments were carried out in freshly prepared phosphate-buffered saline ((PBS) 137 mM NaCl; 2.7 mM KCl; 4.3 mM Na2HPO4; 1.47 mM KH2PO4 adjusted to a final pH of 7.4). Ultrapure water with resistivity of 18 MΩ cm was used in all aqueous preparations. Bacterium strains were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA).Synthesis and characterization
:1 v/v) was added to the crude product and vortexed. The precipitate was collected by centrifugation (6000 rpm, 6.0 °C), washed twice again with the same solution, dissolved in PBS buffer (pH 7.4) and finally lyophilized to obtain the PEG–Mox conjugate as yellowish powder.X-ray diffraction (XRD) measurements of Mox, PCL polymer, PCL and PCL(PEG–Mox) NPs were carried out using Phillips X'Pert PRO diffractometer (PANalytical, Netherlands) under reflection–transmission mode. Samples were placed in glass sample holders and scanned from 2θ = 5° to 60°, using a beam of Cu Kα radiation of λ = 0.1542 nm, operated at 45 kV, 40 mA. The scan speed and exposure time for each sample were 0.109419° s−1 and 17 min 27 s, respectively.
Thermogravimetric (TGA) analyses of PEG and PCL polymers, Mox, PEG–Mox conjugate, PCL and PCL(PEG–Mox) NPs were carried out using a TA Instrument (TGA Q 500, USA), using balance and sample purge nitrogen gas of 30 and 60 mL min−1, respectively. Sample weights between 5.0 and 12.0 mg were used and placed into open aluminium pans. A heating rate of 10 °C min−1 was implemented, with a heating ramp from room temperature to 700 °C.
000 rpm, 10 °C) and the supernatant was analyzed at λmax = 290 nm. The %EE was calculated using the equation:where Mox0 and Moxsupernatant are the initial amount of drug in the PEG–Mox conjugate used in the production of the NPs and the amount of Mox detected in the supernatant after centrifugation of the NPs, respectively.

Toxicity assessments
where Ave.Abssample is the average absorbance of the sample, Ave.Absneg is the average absorbance of the negative control and Absmax.pos is the maximum absorbance of the positive control.
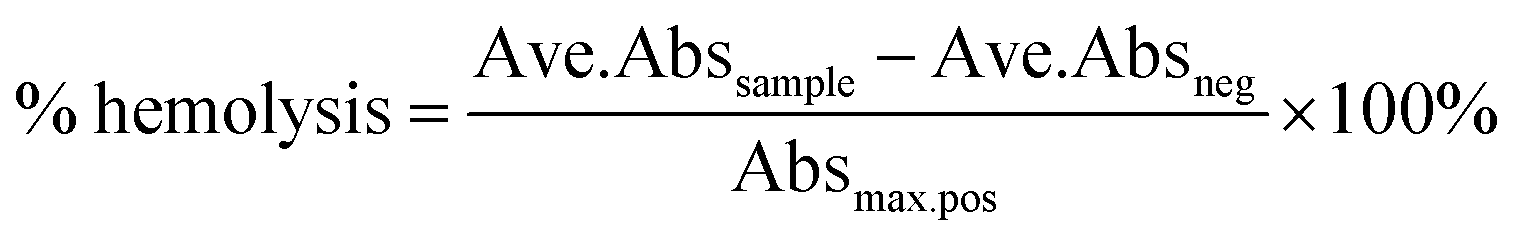
where Abssample is the absorbance of the sample, Absblank is the absorbance of the blank and Absmc is the absorbance of the control medium.
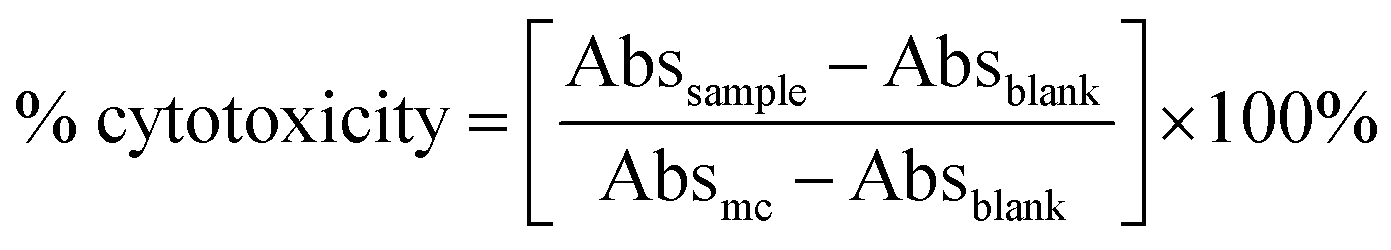 000 rpm for 5 min. The supernatant was withdrawn and analysed by HPLC (LC-10A, Shimadzu Kyoto, Japan) equipped with a SunFire column C18 (5 μm × 250 mm) and a SunFire Guard column C18 (5 μm × 20 mm) to quantify Mox. The mobile phase (acetonitrile 30%, methanol 30%, phosphate buffer 40%), was injected at a flow rate of 1 mL min−1 and elution under isocratic conditions. The λmax for detecting Mox was 290 nm. The procedure above was also applied to plasma alone to determine its corresponding chromatogram to correct for matrix interference.
000 rpm for 5 min. The supernatant was withdrawn and analysed by HPLC (LC-10A, Shimadzu Kyoto, Japan) equipped with a SunFire column C18 (5 μm × 250 mm) and a SunFire Guard column C18 (5 μm × 20 mm) to quantify Mox. The mobile phase (acetonitrile 30%, methanol 30%, phosphate buffer 40%), was injected at a flow rate of 1 mL min−1 and elution under isocratic conditions. The λmax for detecting Mox was 290 nm. The procedure above was also applied to plasma alone to determine its corresponding chromatogram to correct for matrix interference.Antimicrobial activity assessment
Two Gram-positive bacterial (Staphylococcus aureus (S. aureus, ATCC no. 25923) and methicillin-resistant S. aureus (MRSA, ATCC no. 33591) and three Gram-negative bacterial (Escherichia coli (E. coli, ATCC no. 35218), Pseudomonas aeruginosa (P. aeruginosa, ATCC no. 27853) and Klebsiella pneumoniae (K. pneumoniae, ATCC no. 13883) were studied in this work and the bacteria were handled and tested according to the guidelines set by the Clinical and Laboratory Standard Institute (CLSI, 2015). The glycerol stocks containing the selected bacteria were streaked on agar plates (31 g L−1 Nutrient agar, Merck, South Africa) and cultured for 24 h. Colonies were selected from agar plates and cultured in nutrient broth (16 g L−1 Nutrient broth, Merck, South Africa) for further tests.The antibacterial activities of the Mox, PEG–Mox conjugate, PCL(PEG–Mox) NPs and PCL NPs were evaluated against the selected bacteria using an agar well diffusion method. The organisms were sub-cultured in nutrient broth at 37 °C for 18 to 24 h. The cultures were diluted in nutrient broth to 1 × 108 CFU mL−1 (equivalent to 0.5 Mcfarland). The diluted cultures were swabbed onto nutrient agar plates. The test samples (10 μL from a 100 μg mL−1 stock solution) were added to the wells and the plates were incubated at 37 °C for 24 h. The experiments were performed in triplicates. After 24 h, the diameters of the inhibition zones around the wells were measured in millimeters.
The bactericidal effects of the test samples were also evaluated by the microdilution method using the AlamarBlue® assay.30 The active ingredient of the AlamarBlue® reagent (Invitrogen, USA), resazurin is converted to resorufin by viable cells and can be quantified by measuring the absorbance of the test sample at λmax = 570 nm. Absorbance is therefore directly proportional to the number of viable cells. Bacterial suspensions containing 1 × 108 CFU mL−1 were prepared for all the bacterial strains. These bacterial suspensions were transferred into 96-well plates, 100 μL per well. The test samples were added to the bacterial cultures and the plates were incubated at 37 °C for 24 h. The concentrations of the test samples ranged from 0 μg mL−1 to 100 μg mL−1. After 24 h treatment, the lowest concentration of the samples that inhibited visible growth of the bacteria was recorded as the MICs. Thereafter, AlamarBlue® reagent was added (10 μL per well) to each well. The plate was incubated in the dark for 3 h at 37 °C. Absorbance of each well was read at λmax = 570 and 700 nm using a POLARstar® Omega (BMG Labtech, Germany) and expressed as percentage viability (percentage of absorbance of untreated control) of the growth control.
Statistical analysis
All experiments were performed at least in triplicates and the results are expressed as mean ± standard deviation (S.D.). Data were analysed by the Student's t-test using GraphPad Prism version 8.0.0 for MacOS (GraphPad Software, La Jolla California USA). Statistical differences were considered at p < 0.05.Conclusions
This work described the synthesis of a PEG–Mox conjugate which is further encapsulated within a polymeric NP, forming a novel hybrid delivery system of the drug. These designs offer more versatility for delivery of the Mox to treat antimicrobial infections, e.g. via intravenous or inhalation routes in addition to the current oral route. Our work has shown that the toxicity of Mox to human erythrocytes and MDBK cells can be effectively attenuated while retaining significant antibacterial activity by incorporation into a polymeric delivery system. Chemical conjugation of the drug to a polymeric carrier ensured that free Mox would not be released in extracellular fluids like plasma. The system described in this work holds promise for the development of effective treatments of bacterial infections which are more tolerable by patients.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is based on a bilateral research project supported in part by the National Research Foundation of South Africa (Grant Number: 82819, 114369) and the Ministry of Higher Education and Scientific Research, Egypt (Project No. 17-2-120).References
- J. O'Neill, Tackling Drug-resistant Infections Globally: Final Report and Recommendations–The Review on Antimicrobial Resistance Chaired by Jim O'Neill, Wellcome Trust and HM Government, London, 2016 Search PubMed.
- U. Hofer, The cost of antimicrobial resistance, Nat. Rev. Microbiol., 2019, 17(1), 3 CrossRef CAS PubMed.
- E. Tacconelli, N. Magrini, G. Kahlmeter and N. Singh, Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, World Health Organization, 2017, vol. 27 Search PubMed.
- P. N. Prasad, Introduction to nanomedicine and nanobioengineering, John Wiley & Sons, 2012 Search PubMed.
- R. K. Tekade, R. Maheshwari, N. Soni, M. Tekade and M. B. Chougule, Chapter 1 - Nanotechnology for the Development of Nanomedicine, in Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes, ed. V. Mishra, P. Kesharwani, M. C. I. Mohd Amin and A. Iyer, Academic Press, 2017, pp. 3–61 Search PubMed.
- C. Wischke and S. P. Schwendeman, Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles, Int. J. Pharm., 2008, 364(2), 298–327 CrossRef CAS PubMed.
- J. M. Barichello, M. Morishita, K. Takayama and T. Nagai, Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method, Drug Dev. Ind. Pharm., 1999, 25(4), 471–476 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Polymer–drug conjugate therapeutics: advances, insights and prospects, Nat. Rev. Drug Discovery, 2019, 18(4), 273–294 CrossRef CAS PubMed.
- S. Mvango, W. M. R. Matshe, A. O. Balogun, L. A. Pilcher and M. O. Balogun, Nanomedicines for Malaria Chemotherapy: Encapsulation vs. Polymer Therapeutics, Pharm. Res., 2018, 35(12), 237 CrossRef PubMed.
- A. Duro-Castano, M. Talelli, G. Rodríguez-Escalona and M. J. Vicent, Chapter 13 - Smart Polymeric Nanocarriers for Drug Delivery, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. San Román, Woodhead Publishing, 2nd edn, 2019, pp. 439–479 Search PubMed.
- I. L. Batalha, A. Bernut, M. Schiebler, M. M. Ouberai, C. Passemar and C. Klapholz, et al., Polymeric nanobiotics as a novel treatment for mycobacterial infections, J. Controlled Release, 2019, 314, 116–124 CrossRef CAS PubMed.
- T. (Edinb), Moxifloxacin, Tuberculosis, 2008, 88(2), 127–131 CrossRef.
- R. J. Ouellette and J. D. Rawn, 12 - Amines and Amides, in Principles of Organic Chemistry, ed. R. J. Ouellette and J. D. Rawn, Elsevier, Boston, 2015, pp. 315–342 Search PubMed.
- A. Mahor, S. K. Prajapati, A. Verma, R. Gupta, A. K. Iyer and P. Kesharwani, Moxifloxacin loaded gelatin nanoparticles for ocular delivery: formulation and in vitro, in vivo evaluation, J. Colloid Interface Sci., 2016, 483, 132–138 CrossRef CAS PubMed.
- S. B. Murdande, M. J. Pikal, R. M. Shanker and R. H. Bogner, Aqueous solubility of crystalline and amorphous drugs: Challenges in measurement, Pharm. Dev. Technol., 2011, 16(3), 187–200 CrossRef CAS PubMed.
- C. Vauthier and K. Bouchemal, Methods for the Preparation and Manufacture of Polymeric Nanoparticles, Pharm. Res., 2009, 26(5), 1025–1058 CrossRef CAS PubMed.
- R. M. Mainardes, M. P. D. Gremião and R. C. Evangelista, Thermoanalytical study of praziquantel-loaded PLGA nanoparticles, Rev. Bras. Cienc. Farm., 2006, 42, 523–530 CrossRef CAS.
- Y. S. Schwartz, M. I. Dushkin, V. A. Vavilin, E. V. Melnikova, O. M. Khoschenko and V. A. Kozlov, et al., Novel conjugate of moxifloxacin and carboxymethylated glucan with enhanced activity against Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2006, 50(6), 1982–1988 CrossRef CAS PubMed.
- N. S. Abbas, M. Amin, M. A. Hussain, K. J. Edgar, M. N. Tahir and W. Tremel, Extended release and enhanced bioavailability of moxifloxacin conjugated with hydrophilic cellulose ethers, Carbohydr. Polym., 2016, 136, 1297–1306 CrossRef CAS PubMed.
- D. A. Hartman, Determination of the Stability of Drugs in Plasma, Curr. Protoc. Pharmacol., 2002, 19(1), 7.6.1–7.6.8 Search PubMed.
- H. Heiati, R. Tawashi and N. C. Phillips, Solid lipid nanoparticles as drug carriers: II. Plasma stability and biodistribution of solid lipid nanoparticles containing the lipophilic prodrug 3′-azido-3′-deoxythymidine palmitate in mice, Int. J. Pharm., 1998, 174(1), 71–80 CrossRef CAS.
- R. Duncan, P. Ferruti, D. Sgouras, A. Tuboku-Metzger, E. Ranucci and F. Bignotti, A Polymer-Triton X-100 Conjugate Capable of PH-Dependent Red Blood Cell Lysis: A Model System Illustrating the Possibility of Drug Delivery Within Acidic Intracellular Compartments, J. Drug Targeting, 1994, 2(4), 341–347 CrossRef CAS PubMed.
- K. Letchford, R. Liggins, K. M. Wasan and H. Burt, In vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticles, Eur. J. Pharm. Biopharm., 2009, 71(2), 196–206 CrossRef CAS PubMed.
- M. Trif, P. E. Florian, A. Roseanu, M. Moisei, O. Craciunescu and C. E. Astete, et al., Cytotoxicity and intracellular fate of PLGA and chitosan-coated PLGA nanoparticles in Madin–Darby bovine kidney (MDBK) and human colorectal adenocarcinoma (Colo 205) cells, J. Biomed. Mater. Res., Part A, 2015, 103(11), 3599–3611 CrossRef CAS PubMed.
- Y. Lai, P.-C. Chiang, J. D. Blom, N. Li, K. Shevlin and T. G. Brayman, et al., Comparison of In vitro Nanoparticles Uptake in Various Cell Lines and In vivo Pulmonary Cellular Transport in Intratracheally Dosed Rat Model, Nanoscale Res. Lett., 2008, 3(9), 321 CrossRef CAS.
- A. Grillon, F. Schramm, M. Kleinberg and F. Jehl, Comparative Activity of Ciprofloxacin, Levofloxacin and Moxifloxacin against Klebsiella pneumoniae, Pseudomonas aeruginosa and Stenotrophomonas maltophilia Assessed by Minimum Inhibitory Concentrations and Time-Kill Studies, PLoS One, 2016, 11(6), e0156690 CrossRef PubMed.
- S. Alven, B. A. Aderibigbe, M. O. Balogun, W. M. R. Matshe and S. S. Ray, Polymer-drug conjugates containing antimalarial drugs and antibiotics, J. Drug Delivery Sci. Technol., 2019, 53, 101171 CrossRef CAS.
- A. J. Huh and Y. J. Kwon, “Nanoantibiotics”: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era, J. Controlled Release, 2011, 156(2), 128–145 CrossRef CAS PubMed.
- L. Tshweu, L. Katata, L. Kalombo and H. Swai, Nanoencapsulation of water-soluble drug, lamivudine, using a double emulsion spray-drying technique for improving HIV treatment, J. Nanopart. Res., 2013, 15(11), 2040 CrossRef.
- G.-X. Wei, A. N. Campagna and L. A. Bobek, Effect of MUC7 peptides on the growth of bacteria and on Streptococcus mutans biofilm, J. Antimicrob. Chemother., 2006, 57(6), 1100–1109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10872f |
| This journal is © The Royal Society of Chemistry 2020 |