
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on strategies for the fabrication of graphene fibres with graphene oxide
Fei Yin
a,
Jianchen Hu
 *a,
Zhenglin Honga,
Hui Wanga,
Gang Liub,
Jun Shenc,
Hsing-Lin Wangd and
Ke-Qin Zhang
*a
*a,
Zhenglin Honga,
Hui Wanga,
Gang Liub,
Jun Shenc,
Hsing-Lin Wangd and
Ke-Qin Zhang
*a
aNational Engineering Laboratory for Modern Silk, College of Textile and Clothing Engineering, Soochow University, Suzhou 215123, China. E-mail: hujianchen@suda.edu.cn; kqzhang@suda.edu.cn
bShanghai Institute of Spacecraft Equipment, Shanghai 200240, China
cShanghai Key Laboratory of Special Artificial Microstructure Materials and Technology, School of Physics Science and Engineering, Tongji University, Shanghai 200092, China
dDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China
First published on 5th February 2020
Abstract
Graphene fibres have been recognized as ideal building blocks to make advanced, macroscopic, and functional materials for a variety of applications. Direct fabrication of graphene fibres with ideal graphene sheets is still far from reality due to the weak intermolecular bonding between graphene sheets. In contrast, the construction of graphene oxide fibres by following a reduction process is a common compromise. The self-assembly of graphene oxide is an effective strategy for the continuous fabrication of graphene fibre. Different fabrication strategies endow graphene fibres with different performances. Over the past decade, various studies have been carried out into integrating graphene oxide nanosheets into graphene fibres. In this review, we summarize the assembly methods of graphene fibres and compare the mechanical and electrical performances of the graphene fibres fabricated by different strategies. Also the influence of the fabrication strategy on mechanical performance is discussed. Finally, the expectation of macroscopic graphene fibres in the future is further presented.
 Fei Yin | Fei Yin is a PhD candidate of the College of Textile and Clothing Engineering in Soochow University. She received her Master's degree in materials engineering from University of Jinan. Her current research focuses on functionalization of fibre materials and exploring their applications based on their thermal, electrical and mechanical properties. |
 Jianchen Hu | Jianchen Hu is an associated professor in National Engineering Laboratory for Modern Silk and College of Textile and Clothing Engineering of Soochow University. His current research is focused on electrical application of fibre materials in fabric based wearable devices, especially the synergistic effect of carbon based fibres and metals. |
 Ke-Qin Zhang | Ke-Qin Zhang has been a professor in National Engineering Laboratory for Modern Silk and College of Textile and Clothing Engineering of Soochow University since 2009. He received his PhD degree from Nanjing University in 2000. His research interests include fabrication, modification and functionalization of fibres in different scales, as well as the applications of fibre materials in wearable devices. |
Introduction
Graphene, a two-dimensional (2D) hexagonal monolayer of sp2-hybridized carbon atoms, is attracting great interest owing to its excellent electrical and thermal conductivity, and mechanical properties.1–4 These novel properties mean that graphene has potential applications in aerospace, aviation, and the electronic, mechanical, and chemical industries. Single-layer graphene was first obtained by the mechanical exfoliation (“Scotch-tape” method) of bulk graphite1 and by epitaxial chemical vapor deposition.5 High-quality graphene could be obtained by these methods, but they were not effective for large-scale manufacturing in which the main problem was to overcome the interlayer van der Waals force. Chemical methods, in which intercalation, chemical derivatization, thermal expansion, chemical reduction, the use of surfactants, or some combination were usually used, were effective approaches for exfoliating graphite to obtain graphene sheets in higher production.6–9 But in this process, strong oxidizing agents were firstly used to yield graphene oxide (GO) and subsequently the as-fabricated GO was reduced and named reduced graphene oxide (RGO).10–12 On the one hand, the potential applications of graphene raise respective requirements about the material's features, including but not limited to the shape, size or dimensions. On the other hand, the limitation of producing ideal graphene restricts the fabrication of graphene to arbitrary shapes. These challenges have motivated a lot of research into integrating the significant properties of graphene into graphene-based macroscopic structures. As a representative example, many researchers focused on graphene fibres (GFs) since the common characteristics of fibres, such as mechanical flexibility, as well as high tensile strength (σ) and Young's modulus (E), enable graphene to be used in micro-actuators, supercapacitors and other flexible devices. However, the design and fabrication of one-dimensional (1D) macroscopic GFs still pose challenges. Ideally, GFs should be made by crimping perfect graphene films, but this is ruled out by the lack of graphene films of suitable size. In this situation, the arrangement of smaller graphene sheets in the 1D direction to fabricate GFs has emerged as a more reasonable strategy. In addition to the large-scale preparation of graphene, other problems need to be overcome to make graphene into GFs. Firstly, graphene is a material with a typical 2D nanostructure which has weak intermolecular bonding, so it is hard to obtain a stable attachment by direct layer stacking. Moreover, it is hard to form a uniform dispersion or assembly of graphene flakes in uniform solution, and it is also impossible for graphene sheets to be uniformly and orderly arranged along the fibre axis.12 These properties make the subsequent processing of graphene solution difficult. To fabricate GFs in easy ways, other strategies were proposed, for which GO was a good candidate in this context. GO, usually used as an intermediate for synthesizing graphene, unlike other 2D materials, has abundant oxygen functional groups on its basal planes so that a good dispersion in water and other polar organic solvents can be achieved.13–16 Brodie firstly fabricated GO in 1859 by adding a portion of potassium chlorate to a slurry of graphite in fuming nitric acid (HNO3).17 In 1898, Staudenmaier improved this method and realized the production of highly oxidized GO in a single reaction vessel.18 In 1958, Hummers et al. reported a method, in which the graphite was oxidized by potassium permanganate (KMnO4) and sodium nitrate (NaNO3) in concentrated sulfuric acid (H2SO4), which is called the Hummers' method and is the mostly commonly used today.19 Marcano et al. presented an improved Hummers' method to prepare GO sheets of large size.20 Jia et al. thermally expanded graphite at 1050 °C for 15 s as a pretreatment and then used HNO3 and concentrated H2SO4 to intercalate graphite at room temperature for 24 h.21 Liu et al. added ammonium persulfate ((NH4)2S2O8) to concentrated H2SO4 as a pretreatment for graphite to form a porous structure and easily obtained large-sized GO sheets.22 These methods can be classified as being of the same type as that based on tearing bulk graphite into flakes: that is, a “top-down” process. In contrast, “bottom-up” methods also exist, by manipulating chemical groups. For the application of graphene in ionic sieves of high selectivity and transparency and in unconventional electronic devices, researchers have designed functionalized nanopores in graphene.23,24 These nanopores were formed in the graphene monolayers by ion etching or local oxidation. The chemical components of the graphene on the functionalized nanopore edges were similar to GO. The advantages of GO, such as easy processing and scalable production at low cost, enable it to be a precursor for assembly into macroscopic graphene-based functional materials.25,26 In addition, GO can be easily reduced to graphene using several methods, which provides the possibility of solution processing of GFs.27 Among the methods for obtaining graphene from GO, the chemical reduction method is favorable, because it can be scalable in production. Fabrication of graphene oxide fibres (GOFs) first, followed by reduction into the graphene state is the most commonly used method in forming GFs.Inspired by the liquid crystalline behaviours of carbon nanotubes in the formation of 1D macroscopic materials, Xu et al. demonstrated the liquid crystalline behaviours of GO in water and found that the nematic phase of GO sheets could evolve into a lamellar phase when the GO concentration is increased to 30 mg mL−1.28 This work opened up the field of fabrication of 1D macroscopic GFs with GO. On the basis of finding GO liquid crystals (LCs), they first utilized the traditional wet-spinning technique to produce macroscopic fibres by injecting GO solution into a coagulation bath, and used a chemical reduction method to obtain GFs with σ of ∼140 MPa and electrical conductivity of ∼250 S cm−1.29 Recently, various methods and techniques have been explored for the assembly of macroscopic fibres by GO with excellent electrical conductivity and strength, such as microfluidics, electrophoretic method, twisting 2D GO films, and other novel methods. Xin et al. adopted the microfluidic technique to fabricate GOFs with neatly stacked GO sheets and subsequent thermal reduction. The formed GFs showed superior electrical and thermal conductivities and mechanical strength. The σ of the macroscopic graphene belt they obtained reached 1.9 (±0.1) GPa, and the E was 309 ± 16 GPa.30 With the increasing research interest and industrial need for macroscopic GFs, it is important to figure out the relationship between the fabrication process and the properties achieved. In this review, we introduce the assembly strategies for GOFs and compare the performances of the fibres after reduction. Finally, we present the expectations for 1D macroscopic graphene materials in the future.
Assembly strategies for 1D macroscopic GFs
Wet-spinning with GO in LC phase
The formation of lyotropic LCs is one of the most important approaches to producing macroscopic functional materials such as fibres. The aspect ratio (or width/thickness ratio) and sufficient dispersibility/solubility are the two major factors determining the formation of GO LCs. Xu et al. prepared a highly soluble single-layered GO with a high aspect ratio to study the possible liquid crystalline behaviour of GO.28 Fig. 1 shows the polarized-light optical microscopy (POM) images of GO aqueous dispersions between crossed polarizers. With an increase in the maximum mass fraction (fm), the stable birefringence spread whole dispersions and displays vivid Schlieren texture, also the typical texture of nematic phases have changed to parallel-banded structures. This is direct evidence for the liquid crystalline behaviour of GO.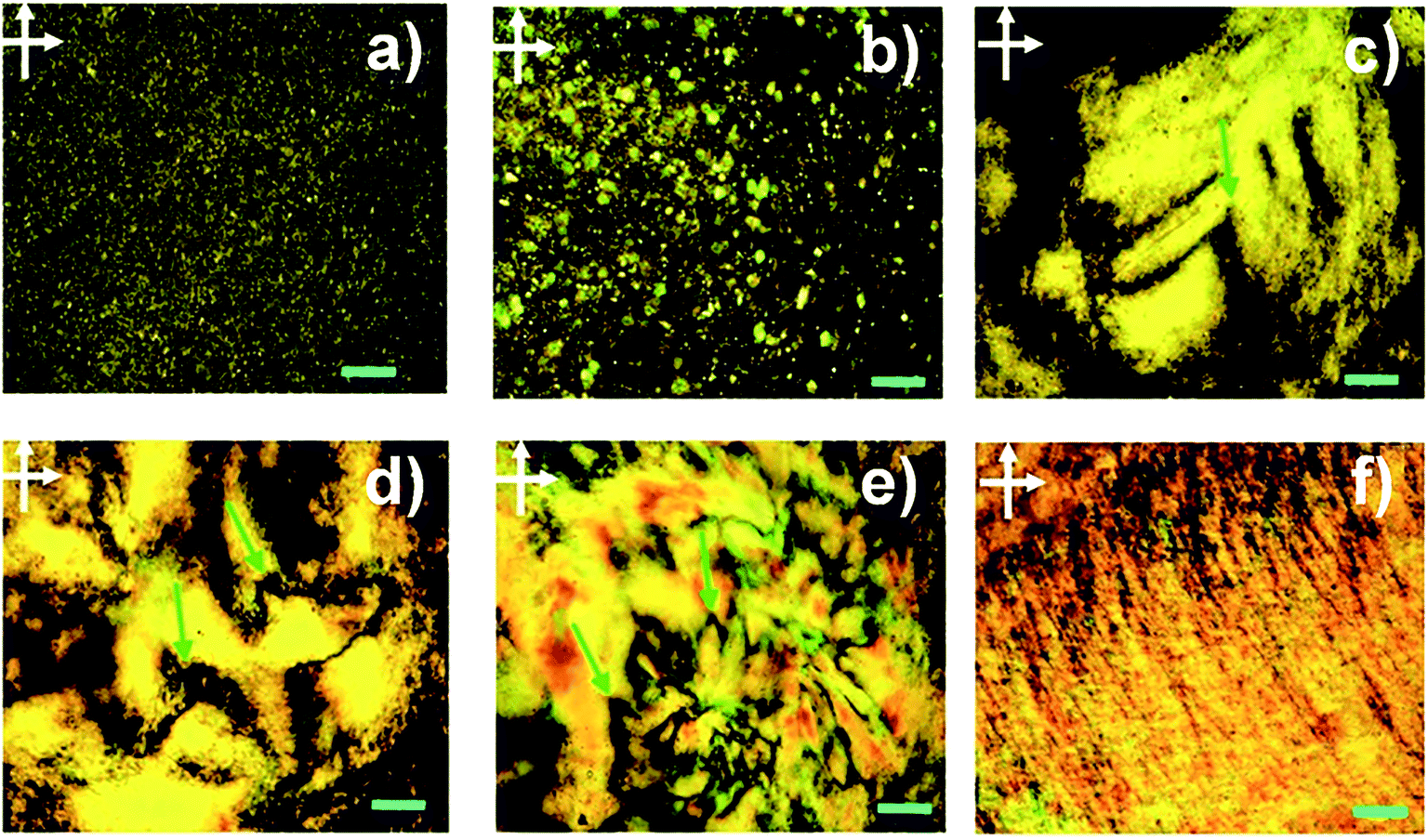 | ||
| Fig. 1 POM images between crossed polarizers of GO aqueous dispersions in planar cells with fm of 5 × 10−3, 1 × 10−3, 3 × 10−3, 5 × 10−3, 8 × 10−3, and 1.0 × 10−2 (from (a) to (f)). The scale bar represents 200 μm.28 Reproduced with permission from ref. 28. Copyright 2011, American Chemical Society. | ||
In 2011, Xu et al. spun continuous GFs from GO LCs via traditional wet-spinning technology for the first time.29 As shown in Fig. 2a, the concentrated GO LC dispersions were injected into NaOH/methanol solution. The alignment of assembled GO sheets (Fig. 2b and c) inherited from the intrinsic lamellar order of the LCs provided strong interactions between contacted sheets that were responsible for the strong mechanical strength and flexibility of the fibres. GFs were obtained by chemical reduction of GOFs in hydroiodic acid. The reduced graphene oxide fibres (RGOFs) have similar flexibility to GOFs, and can be woven into a tight knot (Fig. 2d) and can be woven together with cotton threads (Fig. 2e). Cong et al. used cetyltrimethyl ammonium bromide (CTAB) solution as a coagulation bath instead of NaOH/methanol solution to trigger the GO sheets to assemble in a highly ordered manner to improve the performance of the final GFs.31
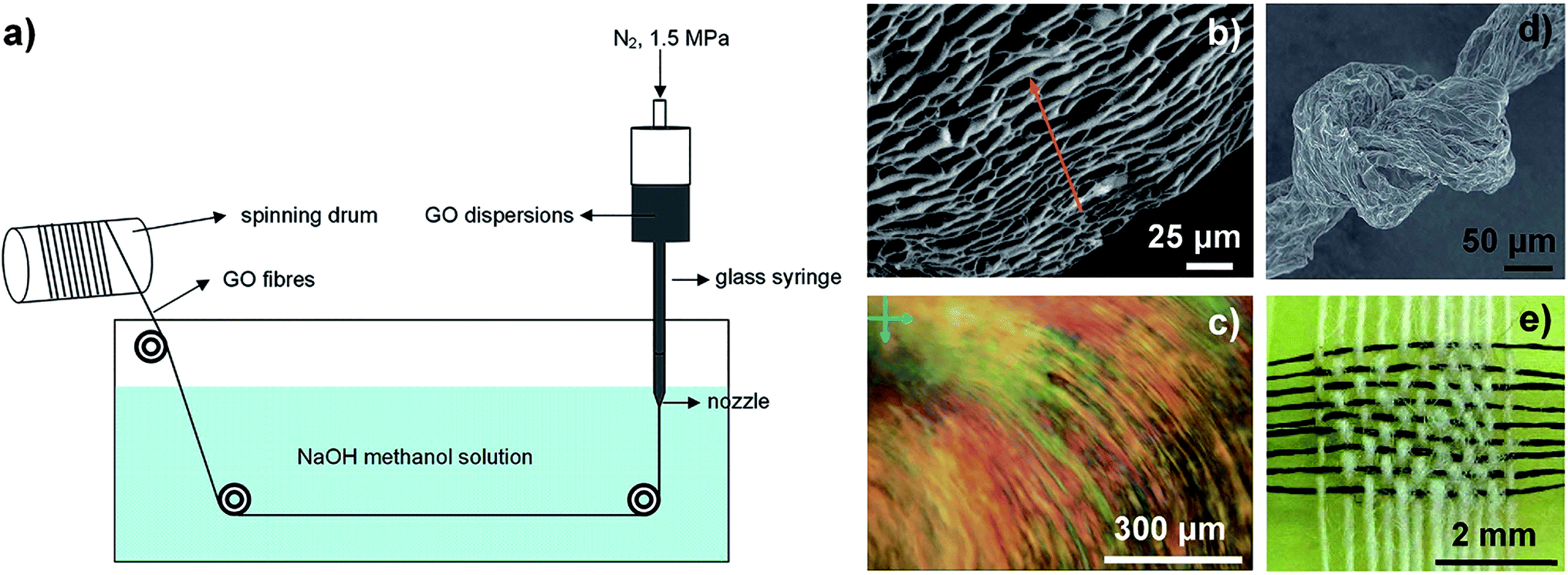 | ||
| Fig. 2 (a) Schematic illustration of wet-spinning technology. (b and c) Side-view scanning electron microscopy (SEM) image and POM image for GO chiral LCs. (d) A tight knot of GFs. (e) A mat of GFs (horizontal) woven together with cotton threads (vertical).29 Reproduced with permission from ref. 29. Copyright 2011, Springer Nature. | ||
The large-scale availability of GO with its liquid crystalline behaviour has promoted efforts to create graphene-based fibres.28,29,32–36 Composite fibres consisting of graphene, carbon nanotubes (CNTs), and poly(ethylene alcohol) (PVA) presented superior behaviour because of the synergistic effect between CNTs and graphene, but limited electrical conductivity due to the presence of polymer.37 Compared to graphene-based composite fibres, GFs exhibited higher electrical conductivity but lower strength. In order to improve the strength while maintaining good conductivity, giant graphene oxide (GGO) sheets with extremely high aspect ratios (Fig. 3a–c) were used to form LCs.38 The SEM image (Fig. 3d and e) reveals that GGO fibres had fewer defective edges, and a highly ordered arrangement of GGO sheets was realized to make high strength performance fibres. The GGO sheets significantly improved the σ of the GFs (up to 501.5 MPa). This means that the GFs reached a mechanical performance which was an order of magnitude better. For a better understanding of the processing–structure–property relationship and further improvement in the properties of GFs, Chen et al. fabricated a series of GFs by changing the size of the GO sheets, spinning conditions, fibre density and orientation, respectively.39 Their results revealed that the mechanical and electrical properties of GFs can be significantly influenced by the alignment of the graphene sheets, inter-sheet interactions and defects or packing density in the fibres. This work showed that high-performance GFs can be achieved by using large-sized graphene sheets, making highly aligned structures and eliminating as many defects as possible. According to this research, Cao et al. chose two sizes of GO sheets (30 and 17 μm) as spinning dopes with the same viscosity and injected them into the CTAB coagulation bath.40 The results showed that the σ of the GOFs assembled from sheets with a lateral size of 30 μm was 267 MPa. This was higher than those of fibres assembled from sheets with a lateral size of 17 μm (192 MPa). After chemical reduction, the σ of GFs assembled from graphene sheets with a lateral size of 30 μm had been enhanced to 365 MPa. This result also further proved the above conclusion that the size of the graphene sheets induced an enhancement in the mechanical property of GFs.
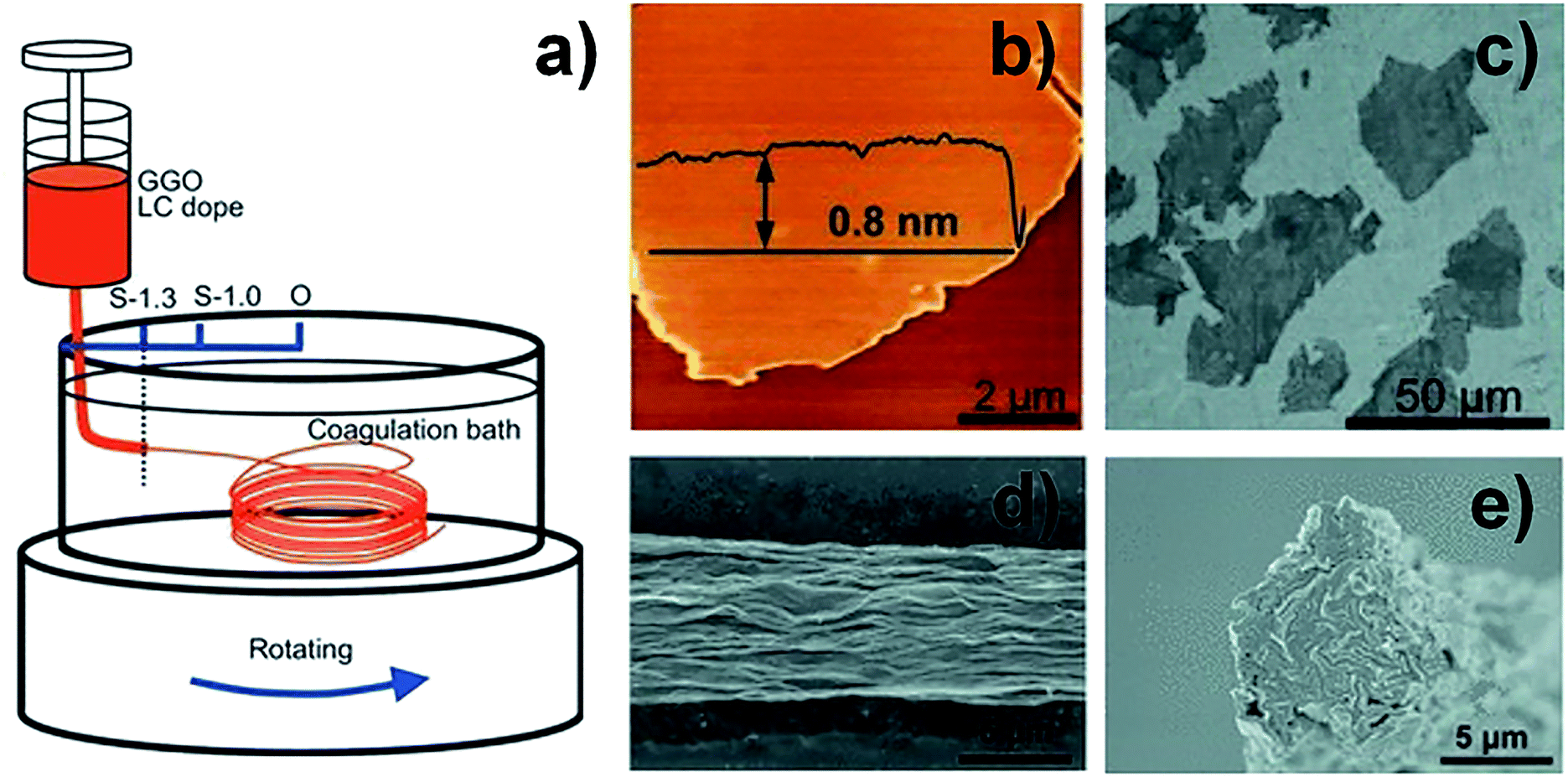 | ||
| Fig. 3 (a) Schematic illustration of wet-spinning for GOFs with GGO sheets. (b) Atomic force microscope (AFM) image of a GGO sheet. (c) SEM image of a GGO sheet. (d) Surface SEM image of a GGO fibre. (e) Cross-section SEM image of a GGO fibre.39 Reproduced with permission from ref. 38. Copyright 2013, Wiley-VCH. | ||
Coaxial two-capillary spinning technology is a development of traditional wet-spinning technology. A coaxial two-capillary spinneret was fabricated by inserting a stainless steel needle into a branched glass tube with a capillary tip. Fig. 4a is a schematic illustration of the coaxial two-capillary spinning process. The GO hollow fibres (GO-HFs), as shown in Fig. 4b, were continuously fabricated while the stainless steel needle was connected to the coagulation bath and the glass tube was filled with GO suspension through the branch. By replacing the fluid in the stainless steel needle with compressed air, a necklace-like structure of GO-HFs (Fig. 4c) can be obtained.41 This coaxial two-capillary spinning technology is also suitable for the fabrication of polymer-wrapped GFs.42 Fig. 5a shows the spinning process of polymer-wrapped GFs. A GO suspension and sodium carboxymethyl cellulose (CMC) aqueous solutions were chosen as the inner and outer fluid, respectively. The fabricated fibres featured GO sheets ordered along the fibre axis as core with CMC well retained as the shell (Fig. 5b–d). After treatment by chemical reduction, the fibres were used to fabricate a yarn supercapacitor (YSC). The resultant polymer-wrapped graphene yarn supercapacitor (Fig. 5e and f) was flexible enough to make a knot (Fig. 5g). Graphene macroscopic fibres fabricated from GO LCs via wet-spinning technology have certain defects at different scales, so that the properties of the fibres are limited.
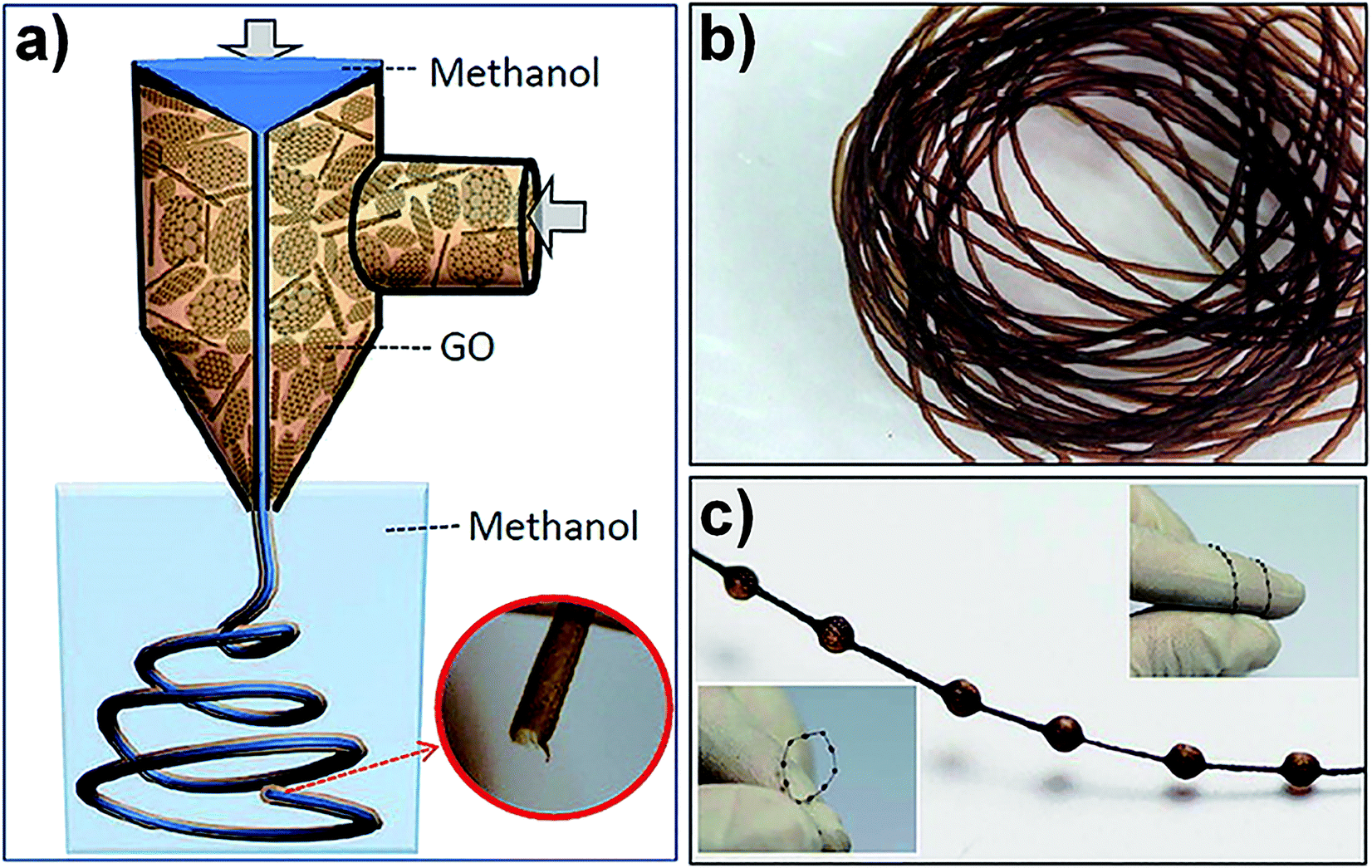 | ||
| Fig. 4 (a) Schematic illustration of coaxial two-capillary spinning technology. Photograph of GO hollow fibre (b) and GO necklace-like hollow fibre (c).41 Reproduced with permission from ref. 41. Copyright 2013, American Chemical Society. | ||
 | ||
| Fig. 5 (a) Schematic illustration of coaxial two-capillary spinning technology. (b–d) SEM images of CMC@GO core–shell fibres. SEM images of the cross-section (e) and side-view (f) of a polymer-wrapped graphene YSC. Inset of (f) is the schematic illustration of YSC. (g) SEM image of a polymer-wrapped graphene YSC knot.42 Reproduced with permission from ref. 42. Copyright 2014, Springer Nature. | ||
In order to reduce the defects generated in the production process, Xin et al. designed an “intercalated” structure in which GO sheets of large size formed a highly aligned backbone and GO sheets of small size filled the spaces and voids between GO sheets of large size without altering the high degree of orientation and alignment of the sheets (Fig. 6a). After thermal annealing at 1800 °C, the σ of intercalated GF (Fig. 6b–d) was improved to 1080 ± 61 MPa, but the GF of pure large-sized sheets was in the range of 616 to 823 MPa.43 Xu et al. presented full-scale synergetic defect engineering technology of GFs. A schematic illustration of the production process, which consists of two main stages, is shown in Fig. 6e. SEM images of the obtained GF (Fig. 6f–h) reveal the high degree of orientation of the graphene sheets. After graphitization at 3000 °C, the GO fibres were transformed into GFs and an average σ of 1.78 ± 0.15 GPa was achieved.44
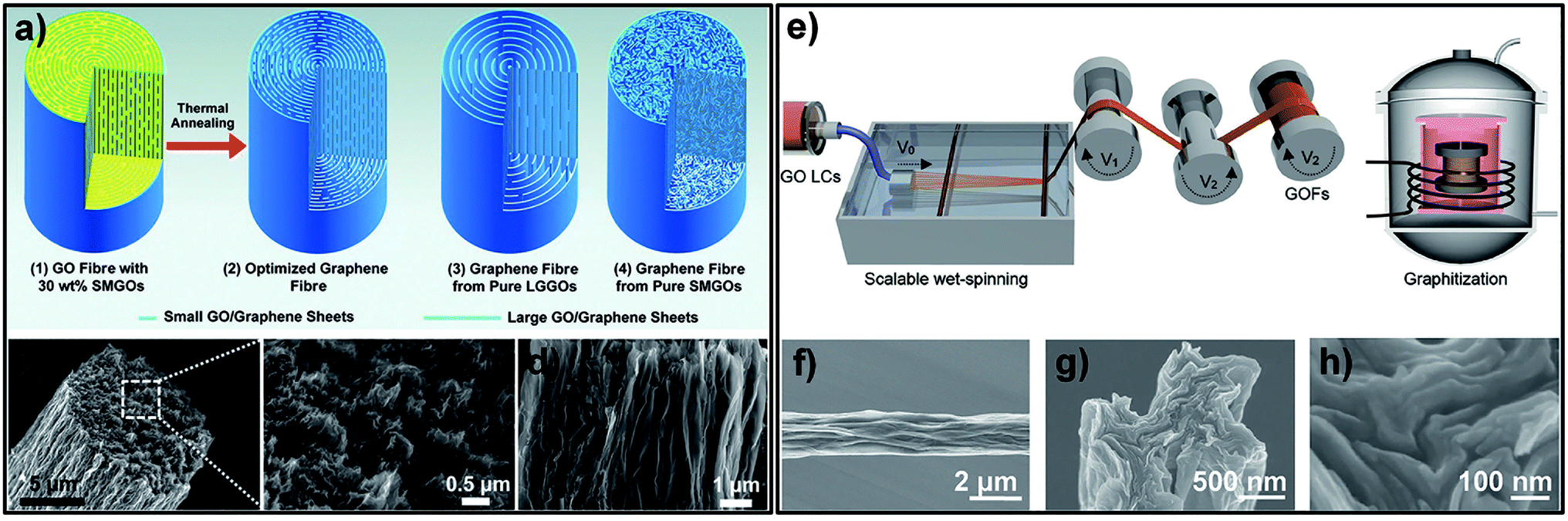 | ||
| Fig. 6 (a) Schematic of the “intercalated” structure of GFs. SEM images of tilted (b), transverse (c) and longitudinal (d) section views of the obtained GF.43 Reproduced with permission from ref. 43. Copyright 2015, American Association for the Advancement of Science. (e) Schematic illustration of wet-spinning of GOFs with continuous stretching in two stages, followed by graphitization to transform the GOFs into GFs. surface (f) and cross-section (g) and (h) SEM images of the obtained GF.44 Reproduced with permission from ref. 44. Copyright 2016, Wiley-VCH. | ||
Recently, a novel technology, microfluidics, which has been employed to controllably fabricate GOFs and graphene-based composite fibres, was developed.30,45 A microfluidic chip was designed to simulate a spider's liquid spinning process. The prototype, a Nephila clavipes gland (Fig. 7a), consisted of a tail and an ampulla for the secretion of spidroin into liquid crystal. The ampulla transported the liquid crystal into a conical funnel and finally into a narrowing S-shaped duct. Generally, spiders use internal shearing of a liquid crystal dope to produce silk fibres. Inspired by this liquid spinning process, Pan et al. have designed a microfluidic chip, including an injection pipe, a cushion chamber, a hyperbolic-type funnel, and a linear-shearing channel to simulate the construction of the spinning process (Fig. 7b). After colloidal liquid crystals were pumped into the microchannel, microfibres could be obtained (Fig. 7c). A microsupercapacitor was assembled from the obtained microfibres and showed great flexibility under bending (Fig. 7d and e).45 For a better understanding of the process of GO flow through the microchannel, Xin et al. fabricated a series of microchannels with different geometries.30 When GO sheets are flowing in channels with different cross-sectional geometries, such as tubular, anisotropic or thin and flat, the orientations of the sheets are different. The 2D GO sheets became curved and distorted when they were flowing in a tubular channel. The obtained GOFs show a random orientation of sheets with heavy wrinkles and defective voids, and the order parameter measured by wide-angle X-ray scattering (WAXS) was 0.75. In contrast, GO sheets flowing in an anisotropic flat channel showed good arrangement. The flattened sheets were assembled into a layer structure of a GO belt and the order parameter was increased to 0.86. To further improve the orientation of the GO sheets, a thin flat channel was used to produce the GO belt. A highly ordered and compact structure of the GO belt was obtained and the order parameter increased to 0.94. Due to the significantly improved sheet alignment and orientation order, the graphene belts obtained from a thin flat channel followed by thermal annealing at 1800 °C achieved high σ (1.9 ± 0.1 GPa) and E (309 ± 16 GPa). These values were much higher than those of GFs obtained from a tubular channel or anisotropic flat channel. This result shows the potential application of microfluidic design in the fabrication of GFs with high mechanical and electrical properties.
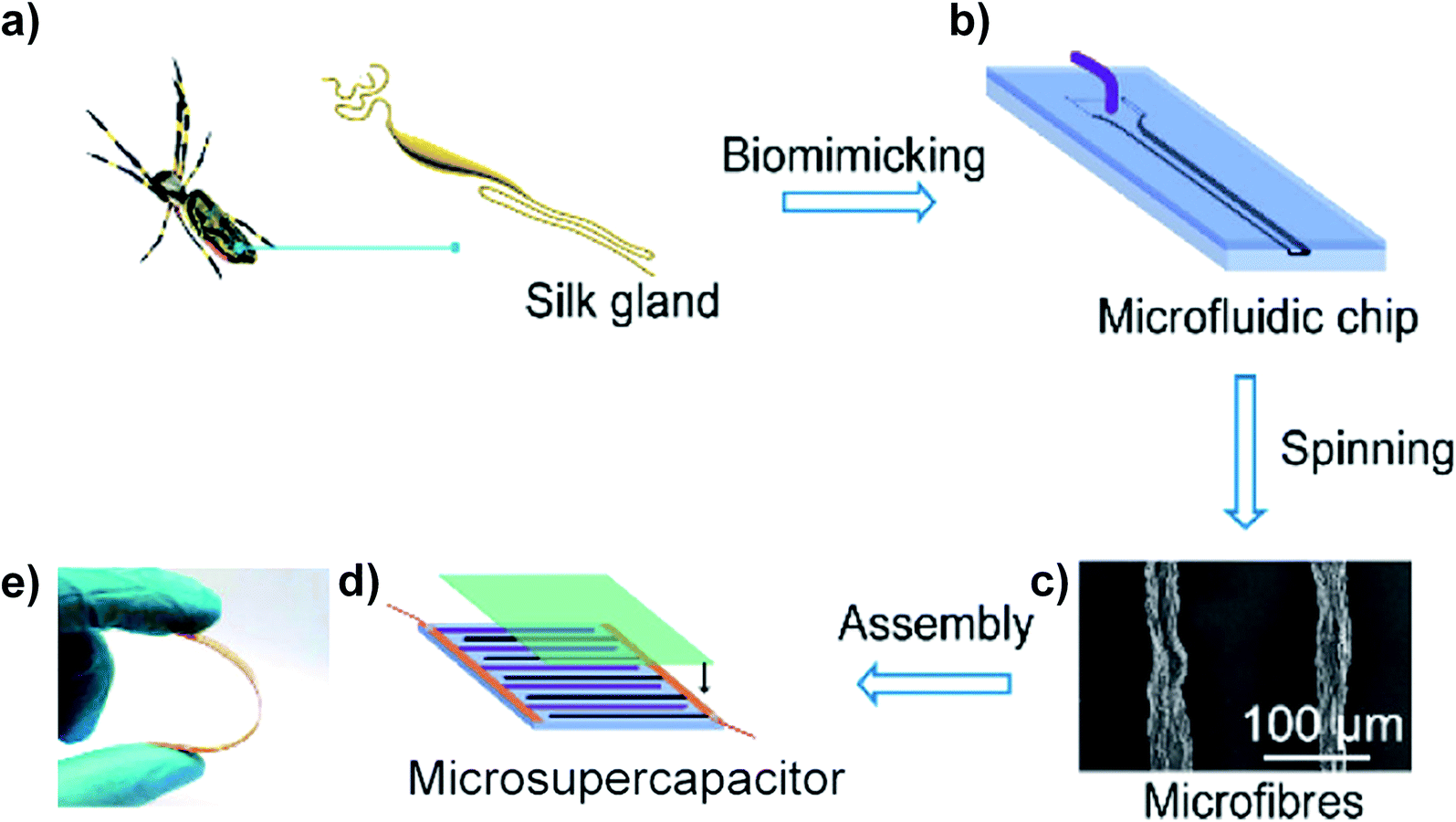 | ||
| Fig. 7 (a) Major silk gland of a Nephila clavipes. (b) The microfluidic chip designed to simulate the liquid spinning process of spiders. (c) SEM image of the obtained microfibres. (d) Schematic assembly of a microsupercapacitor. (e) Photographs of the microsupercapacitor under bending.45 Reproduced with permission from ref. 45. Copyright 2018, American Chemical Society. | ||
Dimensionally confined hydrothermal method
The hydrothermal method is effective for assembling dispersed GO nanosheets randomly into 3D cross-linked porous network structures. Dong et al. proposed a facile one-step dimensionally confined hydrothermal strategy to produce GFs. The aqueous GO suspension was injected into a glass pipeline with 0.4 mm inner diameter. After sealing up the two ends of the glass pipeline, it was baked at 230 °C for 2 h. In this way, a GF matching the pipe geometry was produced (Fig. 8a and b). The fabricated GFs exhibited high flexibility which can be designed into different shapes by bending, such as simple round, triangular, quadrilateral, tetrahedral graphene springs with strong elasticity (Fig. 8c–g).46 | ||
| Fig. 8 Surface (a) and cross-section (b) SEM images of GF which were fabricated by a one-step dimensionally confined hydrothermal strategy. (c and d) The handmade planar and 3d geometric structures of GFs, respectively. (e–g) The spring of GF at free, stretched, and compressed status respectively.46 Reproduced with permission from ref. 46. Copyright 2012, Wiley-VCH. | ||
In order to further improve the mechanical properties of GFs, Sheng et al. used highly interconnected graphene ribbons (IGRs) and graphene nanosheets (GNs) to produce cross-linking and synergistic effects in the graphene hybrid fibres.47 The graphene sheet/ribbon hybrid fibres were prepared by a “spraying–rapid freezing” approach, which is shown in Fig. 9a. The interconnected graphene ribbons were embedded in the skeleton of graphene sheets and formed a twisted, folded surface structure (Fig. 9b–d). Compared to pure GFs, the σ of graphene hybrid fibres was increased to 223 MPa. Ma et al. fabricated carbon-capped reduced graphene oxide (RGO@C) fibres via a dimensionally confined hydrothermal method (Fig. 10a). The RGO@C fibres were prepared by assembling polydopamine (PDA) capped GO (GO@PDA) into fibres and further pyrolyzing the interfacing PDA into graphite-like carbon. Fig. 10b–d suggest that GO and PDA were assembled with a uniform linear morphology and highly aligned, close-packed lamellar microstructures. As a result, the σ of RGO@C fibres was improved to 724 MPa.48
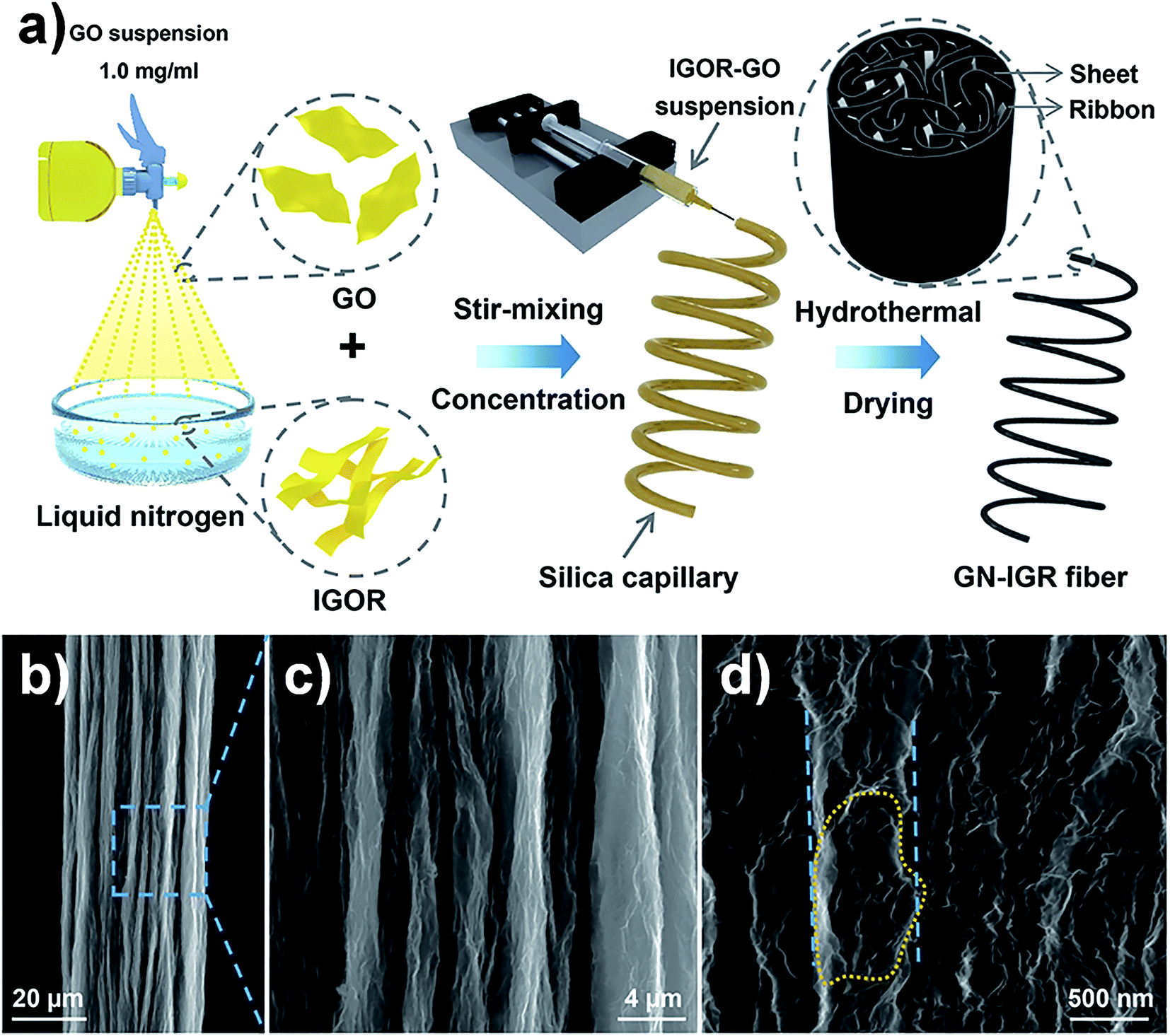 | ||
| Fig. 9 (a) Graphical illustration for the formation process of graphene sheet/ribbon hybrid fibres. (b–d) SEM images of the GN-IGR fibre. The dotted circle shows that the graphene ribbons were bound by the sheets, and the parallel dotted lines show that the width of the bundled ribbons was about 600 nm.47 Reproduced with permission from ref. 47. Copyright 2017, Elsevier Ltd. | ||
 | ||
| Fig. 10 (a) Schematic illustration of the preparation process of the RGO@C fibres via dimensionally confined hydrothermal method. (b and c) Surface SEM images of the RGO@C fibres (inset in (b) is the corresponding photograph of RGO@C fibre). (d) SEM image of the cross-section of RGO@C fibre.48 Reproduced with permission from ref. 48. Copyright 2018, Wiley-VCH. | ||
Electrophoretic self-assembly method
Reduced graphene oxide nanoribbon (GONR) fibres were fabricated by using an electrophoretic self-assembly method without any polymer or surfactant. Jang et al. used a graphitic tip as a positive electrode immersed in a chemically reduced GONR colloidal solution in a Teflon vessel coated with the counter electrode. The distance between the immersed tip and the counter electrode was 5 mm, and the constant voltage ranged from 1 to 2 V. The obtained reduced GONR fibres were made of numerous nanoribbons which were densely packed with each other. After chemical reduction and annealing treatment at 200 °C, the fibres showed giant current density and a low threshold electric field, even though they were robust against a harsh environment with a high current level for a long time.49Assisted assembly from 2D graphene/GO films method
Zhu et al. utilized 2D graphene film to fabricate GF directly.50 Firstly they obtained the 2D graphene films via a vapour deposition (CVD) method. After substrate etching, the free-standing graphene film floated on a water surface but scrolled up on an organic surface and tended to sink into the organic solvent (e.g. ethanol or acetone). Drawn out from the organic solvent by tweezers, the huddled graphene shrank into a fibre-like structure and dried to give a porous and crumpled structure. GFs self-assembled from 2D graphene films, with different morphologies and pore structures, can be obtained by controlling the surface tension at different evaporation rates of different organic solvents.51–53 Further research realized a graphene-based woven fabric assembled from intersecting graphene micro-ribbons grown on woven copper mesh via a CVD method.54 This graphene-based woven fabric can be transferred to a polydimethylsiloxane substrate to form a composite film.Inspired by the method of assisted assembly from 2D graphene films, Cruz-Silva et al. presented a novel method to assemble GFs from 2D GO films. Firstly, 2D large-area GO film was prepared by bar coating water-based dispersions of GO followed by drying under environmental conditions. The obtained GO film was well-ordered and a slightly wrinkled structure was developed that conferred additional ductility, where the wrinkles probably acted as “springs” (Fig. 11a). Then the film was converted into 1D GO macroscopic fibre by film scrolling (Fig. 11b). The typical dry-scrolled fibre showed well-ordered areas of alternate stacks of GO sheets and voids (Fig. 11c and d) and displayed great knotability (Fig. 11e–i). After thermal annealing at 2000 °C, the GFs showed high electrical conductivity up to 416 S cm−1.55
 | ||
| Fig. 11 (a) SEM image of 2D GO film showing the corrugated structure which provides ductility as springs. (b) SEM image of a thin GO film being scrolled into a GOF. (c and d) Cross-section and higher magnification SEM image of the obtained fibre. surface SEM images of (e) typical dry-scrolled GOF, and GOF with (f) quadruple overhand knot, (g) single knot, (h) self-coiled homochiral yarn, (i) neat two-ply yarn.55 Reproduced with permission from ref. 55. Copyright 2014, American Chemical Society. | ||
Performances of 1D macroscopic GFs
As mentioned above, the first continuous GFs spun by traditional wet-spinning technology showed a σ of 140 MPa with a tensile strain of 5.8%.29 Compared to the superior properties of ideal graphene (σ, 130 GPa, E, 1.1 TPa),1–4 lots of work is still needed to improve the performances of GFs. Based on reported research into GFs, the performance of GFs was significantly determined by the microstructure which was affected by the preparation method (as shown in Table 1).| Fibre type | Year | Preparation method | Strength (MPa) | Modulus (GPa) | Conductivity (S cm−1) | References |
|---|---|---|---|---|---|---|
| Pure graphene fibre | 2011 | Wet-spinning | 140 | 7.7 | 250 | 29 |
| 2012 | Wet-spinning | 182 | 8.7 | 35 | 31 | |
| 2012 | Dimensionally confined hydrothermal | 180 | N/A | N/A | 46 | |
| 2012 | Dimensionally confined hydrothermal, treated at 800 °C | 420 | N/A | 10 | 46 | |
| 2012 | Electrophoretic self-assembly | N/A | N/A | 0.37 | 49 | |
| 2012 | Electrophoretic self-assembly, treated at 800 °C | N/A | N/A | 179 | 49 | |
| 2013 | Wet-spinning | 208.7 ± 11.4 | 7.9 ± 0.3 | 210 | 39 | |
| 2013 | Wet-spinning, large size of GO sheets | 360.1 ± 12.7 | 12.8 ± 0.8 | 320 | 39 | |
| 2013 | Wet-spinning | 183 ± 25 | 11 ± 2.4 | 2.21 | 56 | |
| 2013 | Wet-spinning, large size of GO sheets, treated at 800 °C | 214 ± 38 | 47 ± 8.1 | 294 | 57 | |
| 2013 | Wet-spinning, large size of GO sheets | 501.5 | 11.2 | 410 | 38 | |
| 2014 | Wet-spinning, large size of GO sheets | 365 | 21 | 270 | 40 | |
| 2015 | Wet-spinning, 70% large size and 30% small size of GO sheets, treated at 1800 °C | 1080 ± 61 | 135 ± 8 | 2210 ± 60 | 43 | |
| 2016 | Wet-spinning, large size of GO sheets, graphitization at 3000 °C | 1450 | 282 | 8000 | 44 | |
| 2017 | Dimensionally confined hydrothermal, GN-IGR fibre | 223 | N/A | N/A | 47 | |
| 2019 | Microfluidic, large size of GO sheets, treated at 1800 °C | 1900 ± 100 | 309 ± 16 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 ± 1700 400 ± 1700 |
30 | |
| Graphene-based composite fibre | 2012 | Wet-spinning, hyperbranched polyglycerol (HPG) | 125 ± 10 | 8.2 ± 2.2 | 2.4 × 10−3 | 58 |
| 2013 | Wet-spinning, PVA | 199 | 17.1 | 3.5 | 59 | |
| 2013 | Wet-spinning, PAN | 452 | 8.31 | N/A | 60 | |
| 2013 | Wet-spinning, poly glycidyl methacrylate (PGMA) | 440 ± 60 | N/A | 1.86 | 61 | |
| 2013 | Wet-spinning, large size of GO sheets, Ag | N/A | N/A | 930 | 62 | |
| 2014 | Coaxial two-capillary spinning, CMC | 73–116 | N/A | 70 | 42 | |
| 2016 | Wet-spinning, large size of GO sheets, SA | 784.9 | 58 | 35.76 | 63 | |
| 2016 | Wet-spinning, large size of GO sheets, graphitization at 3000 °C, FeCl3 | N/A | N/A | 7.7 × 104 | 64 | |
| 2016 | Wet-spinning, large size of GO sheets, graphitization at 3000 °C, Br2 | N/A | N/A | 1.5 × 105 | 64 | |
| 2016 | Wet-spinning, large size of GO sheets, graphitization at 3000 °C, K | N/A | N/A | 2.24 × 105 | 64 | |
| 2018 | Dimensionally confined hydrothermal, PDA | 724 | 35 | 660 | 48 | |
| 2018 | Microfluidic, MoS2 | N/A | N/A | 327 | 45 |
To further improve the properties of GFs, three aspects should be considered: (1) increasing the orientation of graphene sheets along the fibre axis; (2) adjusting the defects within the GFs; and (3) enhancing the interlayer interaction between the graphene sheets.36,39 Stretching and fluidic-assisted assembly are effective in increasing the orientation and alignment of graphene sheets to form fibres with compact macrostructures.30,45
Defect control is an important part of the fabrication of fibres. Large-sized GO sheets were used to decrease the defects, including sheet boundaries, voids and impurities, within the GFs and to enhance the σ.38,40,56,57 Alternatively, doping MoS2, K, FeCl3, Br2, Ni, or Ag into the interlayers of GO sheets plays a role in increasing defects but increasing the carrier density to enhance the electrical conductivity of GFs.45,62,64–66 Graphene-based composite fibres were designed to enhance the interlayer interaction between graphene sheets. Hydrogen bonding (HB), covalent bonding (CB) and ionic bonding (IB) have been demonstrated to be effective ways to develop the interlayer interaction. Functional groups containing oxygen on the surface of GO enabled interaction with hyperbranched molecules (HPG), PVA, polyacrylonitrile (PAN), poly(glycidyl methacrylate) (PGMA), sodium alginate (SA), or polydopamine (PDA) by means of HB, and also introduce an IB interaction with Ca2+ and CTAB.31,39,40,48,54,59–61,63 As a direct result, the toughness of the fibres increased. Synergistic interactions between IB and CB, IB and π–π stacking were effective for the enhancement of mechanical and electrical performances of graphene-based composite fibres.67–69 Thermal annealing treatment at high temperature was a more effective strategy for enhancing the performances of GFs due to the decrease in defects.30,43,45
Conclusions and prospects
Over the past decade, researchers have made many breakthroughs in the synthesis and application of GFs. GFs possess the common characteristics of fibres, such as the flexibility needed for textiles, while maintaining their uniqueness, such as low cost, light weight, and ease of functionalization. These remarkable features of GFs endow them with prominent advantages over common carbon fibres and metal wires for the development of unconventional, lightweight, flexible devices, especially in fibre shape for wearable electronics. With functionalized GFs, graphene-based materials have exhibited fast, reversible, controllable shape changes in response to environmental stimuli, such as pH or moisture. These excellent performances mean that graphene-based materials have potential applications in robots, sensors and memory chips. This has promoted the development of smart systems and devices. With the increasing demand for 1D foundation materials in industry and life, many methods are being developed to fabricate 1D macroscopic GFs with high σ and electrical conductivity. According to reported research, GFs show good mechanical and electrical properties, but there are still some problems, such as the interconnected junctions among the graphene fibres being extremely weak mechanically, and low conductivity in comparison with pristine graphene, because the weaving or knitting process cannot provide the most intimate connection, which might cause serious problems when these graphene-based materials are embedded in insulating fabrics, which would lead to the performance of the resulting fabric not meeting practical requirements. Moreover, large-scale production of GFs is still limited. In order to improve the properties, the processing–structure–property relationship of GFs has to be considered. Furthermore, it also provides guidance for the design and improvement of GFs from the processing techniques. Although great progress has been made to improve the performances of GFs, there is still a huge gap compared with the intrinsic properties of ideal graphene. More effective assembly technology and improvements to the existing technology to obtain high-performance GFs and functionalization of GFs are and will continue to be the focus of pioneering research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0204600), the Natural Science Foundation of China (51873134, 51603135), the Key Field Research and Development Program of Guangdong Province (2019B010941001), the Natural Science Foundation of Jiangsu Province of China (BK20161253) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_1912).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 669–671 CrossRef PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. c. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. d. Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- N. Behabtu, J. R. Lomeda, M. J. Green, A. L. Higginbotham, A. Sinitskii, D. V. Kosynkin, D. Tsentalovich, A. N. Parra-Vasquez, J. Schmidt, E. Kesselman, Y. Cohen, Y. Talmon, J. M. Tour and M. Pasquali, Nat. Nanotechnol., 2010, 5, 406–411 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolos, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. L. Higginbotham, J. R. Lomeda, A. B. Morgan and J. M. Tour, ACS Appl. Mater. Interfaces, 2009, 1, 2256–2261 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. Tao, N. Kong, X. Ji, Y. Zhang, A. Sharma, J. Ouyang, B. Qi, J. Wang, N. Xie, C. Kang, H. Zhang, O. C. Farokhzad and J. S. Kim, Chem. Soc. Rev., 2019, 48, 2891–2912 RSC.
- S. C. Dhanabalan, B. Dhanabalan, X. Chen, J. S. Ponraj and H. Zhang, Nanoscale, 2019, 11, 3046–3101 RSC.
- J. He, L. Tao, H. Zhang, B. Zhou and J. Li, Nanoscale, 2019, 11, 2577–2593 RSC.
- S. Meng, T. Kong, W. Ma, H. Wang and H. Zhang, Small, 2019, 15, e1902691 CrossRef PubMed.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 14, 249–259 Search PubMed.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- W. Hummers Jr and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitsk, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Jia, C.-M. Kan, X. Lin, X. Shen and J.-K. Kim, Carbon, 2014, 77, 244–254 CrossRef CAS.
- Y. Liu, X. Wu, Y. Tian, X. Zhou, B. Yu, Q. Zhang, R. Du, Q. Fu and F. Chen, Carbon, 2019, 146, 618–626 CrossRef CAS.
- D. Gunlycke and C. T. White, Phys. Rev. Lett., 2011, 106, 136806 CrossRef CAS PubMed.
- K. Sint, B. Wang and P. Král, J. Am. Chem. Soc., 2008, 130, 16448–16449 CrossRef CAS PubMed.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef PubMed.
- D. Kim, S. J. Yang, Y. S. Kim, H. Jung and C. R. Park, Carbon, 2012, 50, 3229–3232 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- X. Zhen and G. Chao, ACS Nano, 2011, 5, 2908–2915 CrossRef.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef.
- G. Xin, W. Zhu, Y. Deng, J. Cheng, L. T. Zhang, A. J. Chung, S. De and J. Lian, Nat. Nanotechnol., 2019, 14, 168–175 CrossRef CAS.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Sci. Rep., 2012, 2, 613 CrossRef.
- S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J.-K. Kim, Adv. Funct. Mater., 2011, 21, 2978–2988 CrossRef CAS.
- B. Dan, N. Behabtu, A. Martinez, J. S. Evans, D. V. Kosynkin, J. M. Tour, M. Pasquali and I. I. Smalyukh, Soft Matter, 2011, 7, 11154 RSC.
- F. Guo, F. Kim, T. H. Han, V. B. Shenoy, J. Huang and R. H. Hurt, ACS Nano, 2011, 5, 8019–8025 CrossRef CAS.
- J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., Int. Ed., 2011, 50, 3043–3047 CrossRef CAS.
- Y. Liu, Z. Xu, W. Gao, Z. Cheng and C. Gao, Adv. Mater., 2017, 29, 1606794 CrossRef PubMed.
- M. K. Shin, B. Lee, S. H. Kim, J. A. Lee, G. M. Spinks, S. Gambhir, G. G. Wallace, M. E. Kozlov, R. H. Baughman and S. J. Kim, Nat. Commun., 2012, 3, 650 CrossRef.
- Z. Xu, H. Sun, X. Zhao and C. Gao, Adv. Mater., 2013, 25, 188–193 CrossRef CAS.
- L. Chen, Y. He, S. Chai, H. Qiang, F. Chen and Q. Fu, Nanoscale, 2013, 5, 5809–5815 RSC.
- J. Cao, Y. Zhang, C. Men, Y. Sun, Z. Wang, X. Zhang and Q. Li, ACS Nano, 2014, 8, 4325–4333 CrossRef CAS PubMed.
- Y. Zhao, C. Jiang, C. Hu, Z. Dong, J. Xue, Y. Meng, N. Zheng, P. Chen and L. Qu, ACS Nano, 2013, 7, 2406–2412 CrossRef CAS PubMed.
- L. Kou, T. Huang, B. Zheng, Y. Han, X. Zhao, K. Gopalsamy, H. Sun and C. Gao, Nat. Commun., 2014, 5, 3754 CrossRef CAS PubMed.
- G. Xin, T. Yao, H. Sun, S. M. Scott, D. Shao, G. Wang and J. Lian, Science, 2015, 349, 1083–1087 CrossRef CAS PubMed.
- Y. Liu, Z. Xu, J. Zhan, P. Li and C. Gao, Adv. Mater., 2016, 28, 7941–7947 CrossRef CAS PubMed.
- H. Pan, D. Wang, Q. Peng, J. Ma, X. Meng, Y. Zhang, Y. Ma, S. Zhu and D. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 10157–10164 CrossRef CAS PubMed.
- Z. Dong, C. Jiang, H. Cheng, Y. Zhao, G. Shi, L. Jiang and L. Qu, Adv. Mater., 2012, 24, 1856–1861 CrossRef CAS PubMed.
- L. Sheng, T. Wei, Y. Liang, L. Jiang, L. Qu and Z. Fan, Carbon, 2017, 120, 17–22 CrossRef CAS.
- T. Ma, H. L. Gao, H. P. Cong, H. B. Yao, L. Wu, Z. Y. Yu, S. M. Chen and S. H. Yu, Adv. Mater., 2018, 30, e1706435 CrossRef PubMed.
- E. Y. Jang, J. Carretero-Gonzalez, A. Choi, W. J. Kim, M. E. Kozlov, T. Kim, T. J. Kang, S. J. Baek, D. W. Kim, Y. W. Park, R. H. Baughman and Y. H. Kim, Nanotechnology, 2012, 23, 235601 CrossRef PubMed.
- X. Li, T. Zhao, K. Wang, Y. Yang, J. Wei, F. Kang, D. Wu and H. Zhu, Langmuir, 2011, 27, 12164–12171 CrossRef CAS PubMed.
- D. D. Nguyen, S. Suzuki, S. Kato, B. D. To, C. C. Hsu, H. Murata, E. Rokuta, N.-H. Tai and M. Yoshimura, ACS Nano, 2015, 9, 3206–3214 CrossRef CAS.
- M. Xiao, T. Kong, W. Wang, Q. Song, D. Zhang, Q. Ma and G. Cheng, Adv. Funct. Mater., 2015, 25, 6165–6172 CrossRef CAS.
- X. Shao, A. Srinivasan, Y. Zhao and A. Khursheed, Carbon, 2016, 110, 378–383 CrossRef CAS.
- X. Li, P. Sun, L. Fan, M. Zhu, K. Wang, M. Zhong, J. Wei, D. Wu, Y. Cheng and H. Zhu, Sci. Rep., 2012, 2, 395 CrossRef.
- R. Cruz-Silva, A. Morelos-Gomez, H.-i. Kim, H.-k. Jang, F. Tristan, S. Vega-Diaz, L. P. Rajukumar, A. L. Elias, N. Perea-Lopez, J. Suhr, M. Endo and M. Terrones, ACS Nano, 2014, 8, 5959–5967 CrossRef CAS PubMed.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, R. L. Shepherd, J. Chen, S. Aminorroaya-Yamini, K. Konstantinov, A. I. Minett, J. M. Razal and G. G. Wallace, Adv. Funct. Mater., 2013, 23, 5345–5354 CrossRef CAS.
- C. Xiang, C. C. Young, X. Wang, Z. Yan, C. C. Hwang, G. Cerioti, J. Lin, J. Kono, M. Pasquali and J. M. Tour, Adv. Mater., 2013, 25, 4592–4597 CrossRef CAS PubMed.
- X. Hu, Z. Xu and C. Gao, Sci. Rep., 2012, 2, 767 CrossRef.
- L. Kou and C. Gao, Nanoscale, 2013, 5, 4370–4378 RSC.
- Z. Liu, Z. Xu, X. Hu and C. Gao, Macromolecules, 2013, 46, 6931–6941 CrossRef CAS.
- X. Zhao, Z. Xu, B. Zheng and C. Gao, Sci. Rep., 2013, 3, 3164 CrossRef.
- Z. Xu, Z. Liu, H. Sun and C. Gao, Adv. Mater., 2013, 25, 3249–3253 CrossRef CAS.
- X. Hu, S. Rajendran, Y. Yao, Z. Liu, K. Gopalsamy, L. Peng and C. Gao, Nano Res., 2016, 9, 735–744 CrossRef CAS.
- Z. Xu, Y. Liu, X. Zhao, L. Peng, H. Sun, Y. Xu, X. Ren, C. Jin, P. Xu, M. Wang and C. Gao, Adv. Mater., 2016, 28, 6449–6456 CrossRef CAS PubMed.
- G. Wu, P. Tan, X. Wu, L. Peng, H. Cheng, C.-F. Wang, W. Chen, Z. Yu and S. Chen, Adv. Funct. Mater., 2017, 27, 1702493 CrossRef.
- W. Ma, Y. Liu, S. Yan, T. Miao, S. Shi, Z. Xu, X. Zhang and C. Gao, Nano Res., 2018, 11, 741–750 CrossRef CAS.
- Y. Cheng, J. Peng, H. Xu and Q. Cheng, Adv. Funct. Mater., 2018, 28, 1800924 CrossRef.
- Y. Zhang, J. Peng, M. Li, E. Saiz, S. E. Wolf and Q. Cheng, ACS Nano, 2018, 12, 8901–8908 CrossRef CAS PubMed.
- S. Wan, Y. Chen, Y. Wang, G. Li, G. Wang, L. Liu, J. Zhang, Y. Liu, Z. Xu, A. P. Tomsia, L. Jiang and Q. Cheng, Matter, 2019, 1, 389–401 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |