
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors governing the competition between group IA and IB cations for monensin A: a DFT/PCM study
Todor Dudev *a,
Diana Cheshmedzhievaa,
Radoslava Dimitrovab,
Peter Dorkovc and
Ivayla Pantcheva*b
*a,
Diana Cheshmedzhievaa,
Radoslava Dimitrovab,
Peter Dorkovc and
Ivayla Pantcheva*b
aLaboratory of Computational Chemistry and Spectroscopy, Faculty of Chemistry and Pharmacy, “St. Kl. Ohridski” University of Sofia, 1, J. Bourchier Blvd., 1164 Sofia, Bulgaria. E-mail: ohtttd@chem.uni-sofia.bg; Tel: +359-2-8161323
bLaboratory of Biocoordination and Bioanalytical Chemistry, Faculty of Chemistry and Pharmacy, “St. Kl. Ohridski” University of Sofia, 1, J. Bourchier Blvd., 1164 Sofia, Bulgaria. E-mail: ipancheva@chem.uni-sofia.bg; Tel: +359 2 8161446
cBiovet Ltd., Research & Development Department, Peshtera, Bulgaria
First published on 4th February 2020
Abstract
The affinity of monensin A to bind monovalent metal cations was evaluated by means of density functional theory (DFT) combined with polarizable continuum model (PCM) computations. The effect of various factors on complex formation between the monensinate A anion and group IA and IB metal ions was assessed. Competition between Na+ taken as a reference and monovalent metal cations was estimated using the Gibbs free energy for substituting the ligand-bound Na+ with its rival ions in the process [M+-solution] + [Mon−Na+] → [Mon−M+] + [Na+-solution] (M+ = Li+, K+, Rb+, Cs+, Cu+, Ag+ and Au+). The calculations revealed that the decrease in size of the cations accompanied by an increase of their accepting ability enhances the metal selectivity towards ligand donor atoms. In the gas-phase the affinity of monensinate A decreases in the order Cu+ > Li+ > Na+ > Au+ > Ag+ > K+ > Rb+ > Cs+. The complex formation can be manipulated by changing the solvent used. The polyether ionophore selectively binds Na+ ions in polar solvents but could become Li+ or Cu+-selective in low-polarity solvents.
Introduction
Monensic acid A (monensin A, MonH) belongs to the group of natural polyether ionophores (PI) – a class of highly active molecules with pronounced biological efficacy. They are polyether polyalcohol monocarboxylic acids varying in molecular weight from 700 to 1000 g mol−1.1–3 The carboxylic moiety is placed at the “head” of the polyether chain while at least one hydroxyl group is located at the “tail” of the molecule. Alkyl substituents bound to the polyether chain are oriented externally ensuring the overall lipophilic character of the ligands. Polyether oxygens are placed inward forming a hydrophilic cavity able to host water molecules or monovalent metal ions.The structure of monensic acid monohydrate (MonH·H2O) and its complexes with monovalent metal cations [Mon−M+] (M+ = Li+, Na+, K+, Rb+, Ag+) has been evaluated by X-ray crystallography on single crystals.4–11 Data revealed that monensin reacts as a carboxylate monoanion (Mon−) and the metal cations are coordinated by six oxygens of various origin. No participation of carboxylate group in the internal sphere of the metal ions was observed. Complexation of M+ ions leads to the formation of a pseudochelate structure secured by a number of “head-to-tail” hydrogen bonds.
By wrapping monovalent metal ions as overall neutral compounds, monensin is able to transfer them through the lipid membranes into the intracellular space where dissociation of cations occurs thus disturbing metal homeostasis. Changes in the gradients of physiologically essential metal ions such as K+ and Ca2+ induce a cascade of energy-consuming processes which lead to ultimate cell death. Monensin A is used as coccidiostat against Eimeria spp. but also possesses antibacterial, antiviral and promising antitumor properties.12–17 It was observed that inclusion of monovalent metal ions into the structure of the ligand influences it's in vitro and in vivo activity.18–25
The striking feature of PI is their selectivity towards monovalent metal cations despite similarities between all of them from structural point of view and biological mode of action. An extensive study on affinity of monensin A to bind monovalent metal cations was performed in protic and dipolar aprotic solvents, in heterogeneous systems (water-organic phase) as well as in model biomembranes.26–40 Experimental data (cyclovoltammetry, microcalorimetry, potentiometry)26–40 revealed that monensinate complexes of IA cations are very stable with sharp stability maximum for sodium complex [Mon−Na+]. The observed selectivity was explained in terms of cavity – cation-size dependence and negative charge of ligand anion, introducing direct electrostatic stabilization. Generally, affinity of monensinate A anion toward alkali metal ions in polar solvents decreases in the order of Na+ > K+ > Rb+ > Cs+ although variation was observed in some cases depending on the reaction phase/medium and metal cations studied.
Thus, Henderson et al.26 reported affinity order of Na+ > K+ ≈ Li+ > Rb+ > Cs+ in membranes and liquid crystals, confirmed by studies of Ashton and Stenrauf27 in model transport systems (Na+ > K+ > Li+ > Rb+). A slightly different sequence was evaluated by Cornelius et al.29 using biomembranes: Na+ > K+ > Tl+ > Rb+ > Cs+. Hoogerheide and Popov31 studied affinity of monensinate anion to bind monovalent metal ions in anhydrous methanol, and detected decreased selectivity of the ligand in the order Ag+ > Na+ > Tl+ > K+ > Rb+ > Cs+ > Li+, whereas later Mimouni et al.36 observed some inversion between particular pairs of ions in methanol and biphasic systems. Furthermore, Forbes et al.41 studied the gas-phase behaviour of monensin A towards IA metal ions to assess the maximum stability of [Mon−K+] as compared to other monovalent metal cations.
Although a substantial body of information has been accumulated on the monensin chemistry/biochemistry, the key factors governing its metal affinity are still not well defined. Here, we endeavor to shed light on this issue by employing a combination of density functional theory (DFT) calculations and polarizable continuum model (PCM) computations. The free energies of complex formation between monensinate anion and group IA and IB metal cations are evaluated and the role of various factors on the process (metal ion radius, its charge accepting power, and dielectric properties of the medium) is assessed. Note that our aim is to evaluate reliable trends of changes in the thermodynamic quantities rather than reproducing their absolute values. The approach adopted has proven quite successful in deciphering the mechanism of metal affinity/selectivity in a number of chemical/biological systems such as cyclodextrins,42 enzymes,43 signaling proteins,44 and ion channels.45
Methods
Reaction modeled
The competition between the group IA and IB cations and Na+ (taken as a reference) in monensinate A can be expressed in terms of the Gibbs free energy for substituting the ligand-bound Na+ with its rival cations:| [M+-solution] + [Mon−Na+] → [Mon−M+] + [Na+-solution] | (1) |
DFT/PCM calculations
All calculations were performed using Gaussian 09 package of programs.46 All the structures were fully optimized in the gas phase at the B3LYP/6-31+G(d,p) level of theory yielding the respective electronic energies, Eelect of the studied species. SDD basis set and effective core potential were used for heavier metal ions in the series such as Rb+, Cs+, Ag+ and Au+. This combination of method/basis set was chosen for the present calculations as it proved reliable in reproducing the experimental structural parameters of [Mon−Na+] complex used as reference in this work. The optimized geometry of [Mon−Na+] complex (Fig. 1) is compared with the one determined from X-ray diffraction.9 The interatomic distances between the oxygen atoms in monensinate ion and the sodium cation as well as selected bond angles are given in Table 1. As can be seen from the data in Table 1, the structure obtained from DFT computations is very close to that determined by X-ray diffraction. The comparison between calculated structural parameters and available experimental data shows quantitative agreement between theory and experiment for the geometry of [Mon−Na+] complex (Table 1).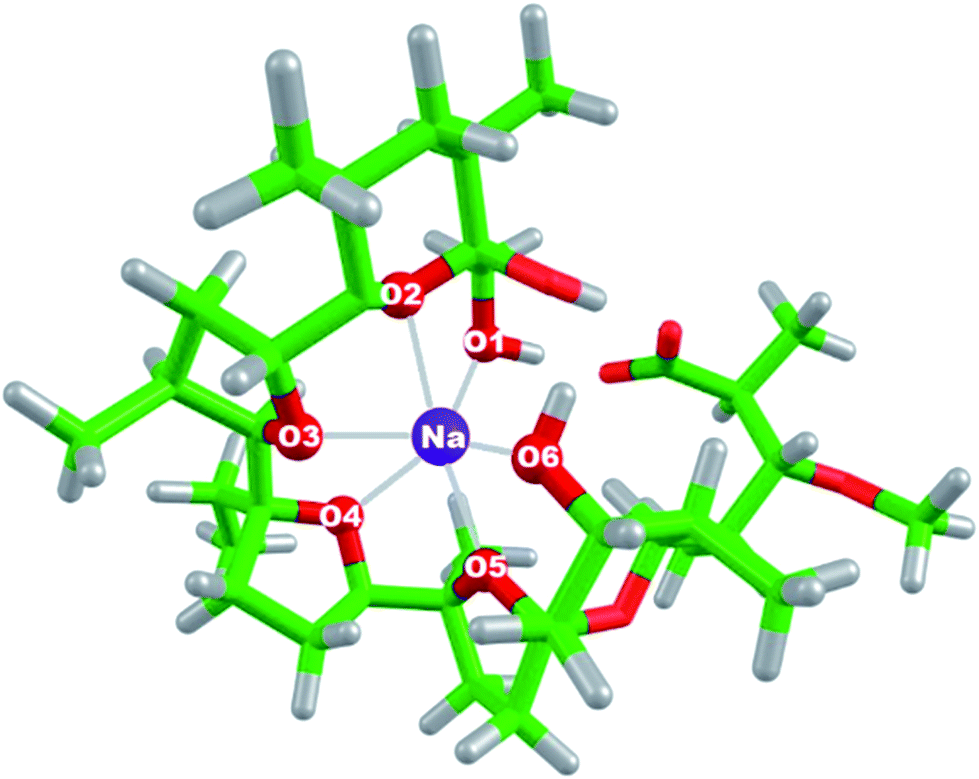 | ||
| Fig. 1 Optimized geometry of [Mon−Na+] complex at the B3LYP/6-31+G(d,p) level of theory. Color scheme: C – green, O – red, H – light grey, Na – purple. | ||
| Bond length/bond angle | [Mon−Na+] | |
|---|---|---|
| Exp9 | Calcda | |
| a Optimization at the B3LYP/6-31+G(d,p) level of theory. | ||
| Na–O1 | 2.34 | 2.34 |
| Na–O2 | 2.33 | 2.37 |
| Na–O3 | 2.60 | 2.60 |
| Na–O4 | 2.45 | 2.45 |
| Na–O5 | 2.41 | 2.46 |
| Na–O6 | 2.38 | 2.34 |
| Na–O average | 2.42 | 2.42 |
| O1–Na–O2 | 68.2 | 68.6 |
| O2–Na–O5 | 173.3 | 163.5 |
| O1–Na–O5 | 117.3 | 122.8 |
| O1–Na–O6 | 113.5 | 111.4 |
| O5–Na–O6 | 76.2 | 75.5 |
Vibrational analysis was performed at the same level of theory. No imaginary frequency was found for any of the optimized structures indicating a local minimum of the potential energy surface. The vibrational frequencies were used to obtain the thermal energies, including the zero-point energy, ET, and entropy, S. These values were employed in evaluating the gas-phase Gibbs free energies for reaction (2), ΔG1, at room temperature, T = 298.15 K, according to (3):
| [M+] + [Mon−Na+] → [Mon−M+] + [Na+] | (2) |
| ΔG1 = ΔEelect + ΔET − TΔS, | (3) |
| ΔGε = ΔG1 + ΔGsolvε([Mon−M+]) + ΔGsolvε([Na+-solution]) − ΔGsolvε([Mon−Na+]) − ΔGsolvε([M+-solution]) | (4) |
Notably, the experimentally determined solvation free energies for the metal cations in aqueous solution (where available) were used in the calculations. For other cations and solvents estimated values were employed where the dependencies in the theoretically evaluated quantities in combination with the experimental values were used to determine the respective ΔGsolvε([M+/Na+-solution]) (Table 2). Such a methodology (employing thermodynamic cycle with experimental solvation free energies of some species) has proven quite accurate in reproducing the experimental thermodynamic data.48
| Metal ion | ΔG78 (exp)50 | ΔG32/ΔG78 (calc) | ΔG32 (est) | ΔG4/ΔG78 (calc) | ΔG4 (est) | ΔG2/ΔG78 (calc) | ΔG2 (est) |
|---|---|---|---|---|---|---|---|
| a Estimated from the experimental solvation energy of Ag+ and the respective ratio between the calculated ΔGs of Cu+ and Ag+.b Estimated from the experimental solvation energy of Ag+ and the respective ratio between the calculated ΔGs of Au+ and Ag+. | |||||||
| Li+ | −123.5 | 0.974 | −120.3 | 0.768 | −94.8 | 0.560 | −69.2 |
| Na+ | −98.3 | 0.968 | −95.2 | 0.763 | −75.0 | 0.554 | −54.5 |
| K+ | −80.8 | 0.960 | −77.5 | 0.757 | −61.1 | 0.545 | −44.0 |
| Rb+ | −76.6 | 0.971 | −74.4 | 0.766 | −58.7 | 0.557 | −42.7 |
| Cs+ | −71.0 | 0.967 | −68.7 | 0.764 | 54.2 | 0.554 | −39.3 |
| Cu+ | −148.7a | 0.978 | −145.4 | 0.770 | −114.5 | 0.562 | −83.5 |
| Ag+ | −114.5 | 0.975 | −111.6 | 0.768 | −88.0 | 0.560 | −64.1 |
| Au+ | −124.8b | 0.977 | −121.9 | 0.768 | −95.9 | 0.561 | −70.0 |
The basis set superposition error for the type of exchange reactions modeled by reaction (1) was found to be insignificant49 and was thus not considered in the present calculations.
Results and discussion
Group IA
Alkali metals form spherical monovalent cations whose ionic radius gradually increases with increasing the atomic number (Table 3). The ion charge density decreases in the same direction thus weakening the ligand affinity of the metal and rendering the bulkiest Cs+ the weakest Lewis' acid (with lowest ligand affinity) in the group.| Metal ion | Ionic radius51 (Å) | Average M–O distance in [Mon−M+] (Å) | Charge transfer to the metal (e) in M+–(CH3OCH3)a,b | Charge transfer to the metal (e) in M+–(CH3OH)a,c |
|---|---|---|---|---|
| a This work. Calculated from the Hirshfeld population analysis at the B3LYP/6-31+G(d,p) level.b CH3OCH3 is used as a model for the ether group in monensin A.c CH3OH is used as a model for the hydroxyl moiety in monensin A. | ||||
| Li+ | 0.76 (VI) | 2.26 | 0.286 | 0.260 |
| Na+ | 1.02 (VI) | 2.42 | 0.215 | 0.198 |
| K+ | 1.38 (VI) | 2.81 | 0.166 | 0.152 |
| Rb+ | 1.52 (VI) | 2.89 | 0.142 | 0.132 |
| Cs+ | 1.67 (VI) | 3.08 | 0.129 | 0.119 |
| Cu+ | 0.77 (VI) | 2.44 | 0.360 | 0.338 |
| Ag+ | 1.15 (VI) | 2.56 | 0.300 | 0.281 |
| Au+ | 1.37 (VI) | 2.71 | 0.412 | 0.381 |
Structures of the fully optimized metal complexes with monensinate A anion are given in Fig. 2. The metal cation binds to ether and hydroxyl oxygens. The metal coordination numbers are 6 in agreement with the experimental observations.4–6,8–12 The average M+–O bond length increases in the same direction (spanning the range of 2.28–3.08 Å) which is compatible with the increased ionic radius of the metal species (Table 3). This result implies that the monensinate A ligand is relatively flexible and its internal cavity, unlike that of other “rigid” macrocycles (e.g. crown ethers), can fluctuate in size adapting to the spatial requirements of the guest cation.
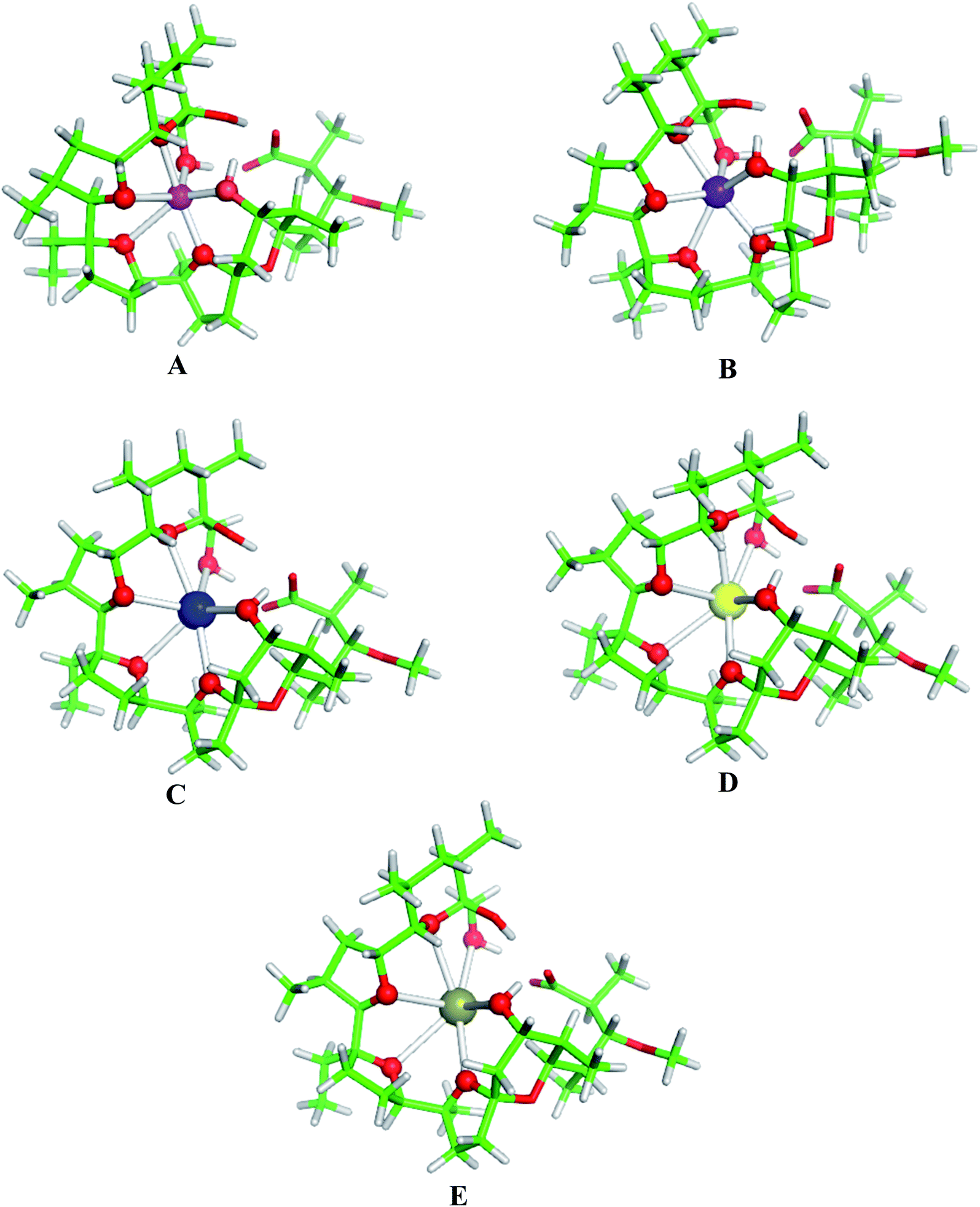 | ||
| Fig. 2 B3LYP/6-31+G(d,p) fully optimized structures of monensinate A anion bound to (A) Li+, (B) Na+, (C) K+, (D) Rb+ and (E) Cs+. Color scheme: C – green, O – red, H – light grey, Li – magenta, Na – purple, K – blue, Rb – yellow, Cs – deep olive. | ||
The outcome of the metal ion competition in solution (reaction (1)) is a balance between electronic and solvation effects. The role of the former can be assessed by following the trends of changes in the gas-phase free energies, ΔG1. As the data in Table 4 indicate, ΔG1 changes in agreement with the affinity of the metal cation for the ligands (measured as the amount of the charge transferred from the ligand to the metal; the greater the charge accepted, the higher the metal affinity; Table 3). It is the greatest for Li+ ions and gradually attenuates in going down the group. Accordingly, ΔG1 increases from the Li+ to Cs+/Na+ substitution. Thus, the electronic effects align the affinity of the metal cations for monensinate A anion as Li+ > Na+ > K+ > Rb+ > Cs+.
| Reaction | ΔGε (kcal mol−1) |
|---|---|
| a In assessing the relative metal affinities, the affinity of the reference metal cation (Na+) is taken as 0, whereas that for the rest of the metal cations follows the trends in ΔGε. | |
| [Li+-solution] + [Mon−Na+] → [Mon−Li+] + [Na+-solution] | ΔG1 = −20.5 |
| ΔG2 = −5.4 | |
| ΔG4 = 0.0 | |
| ΔG32 = 5.7 | |
| ΔG78 = 5.9 | |
| [K+-solution] + [Mon−Na+] → [Mon−K+] + [Na+-solution] | ΔG1 = 27.2 |
| ΔG2 = 17.4 | |
| ΔG4 = 14.0 | |
| ΔG32 = 9.2 | |
| ΔG78 = 9.3 | |
| [Rb+-solution] + [Mon−Na+] → [Mon−Rb+] + [Na+-solution] | ΔG1 = 61.7 |
| ΔG2 = 49.4 | |
| ΔG4 = 44.7 | |
| ΔG32 = 36.8 | |
| ΔG78 = 35.4 | |
| [Cs+-solution] + [Mon−Na+] → [Mon−Cs+] + [Na+-solution] | ΔG1 = 76.2 |
| ΔG2 = 60.7 | |
| ΔG4 = 54.3 | |
| ΔG32 = 45.3 | |
| ΔG78 = 43.6 | |
| [Cu+-solution] + [Mon−Na+] → [Mon−Cu+] + [Na+-solution] | ΔG1 = −31.6 |
| ΔG2 = −2.0 | |
| ΔG4 = 8.7 | |
| ΔG32 = 15.0 | |
| ΔG78 = 14.9 | |
| [Ag+-solution] + [Mon−Na+] → [Mon−Ag+] + [Na+-solution] | ΔG1 = 13.5 |
| ΔG2 = 23.1 | |
| ΔG4 = 26.4 | |
| ΔG32 = 28.7 | |
| ΔG78 = 28.3 | |
| [Au+-solution] + [Mon−Na+] → [Mon−Au+] + [Na+-solution] | ΔG1 = 2.4 |
| ΔG2 = 18.4 | |
| ΔG4 = 24.1 | |
| ΔG32 = 27.3 | |
| ΔG78 = 26.6 | |
The solvation effects, however, affect the affinity order: while it remains the same in low-polarity solvents (with dielectric constant 2–4), it changes in polar solvents (methanol and water). The respective ΔG32 and ΔG78 for Li+/Na+ exchange, due to the high desolvation penalty of Li+ (left-hand side of reaction (1) and Table 2), become positive. The free energies in polar solvents for the rest of the metals decrease but remain on a positive ground. Thus, the affinity series in polar solvents (methanol for example) becomes Na+ > Li+ > K+ > Rb+ > Cs+. This is in good agreement with the experimental results indicating that monensinate A is selective for Na+ in polar solvents among the group IA cations.31,36
Group IB
The physico-chemical properties of the coinage metal cations (Cu+, Ag+ and Au+), as compared to those of the respective alkali metal ions, are strongly influenced by the presence of d orbitals in the outer electron shell, which by hybridizing with the valence s orbitals lower their energy thus reducing the ionic radius and enhancing their charge accepting ability. Indeed, as the data in Table 3 (last two columns) indicate, the charge transfer from the ligands donor groups to the group IB metal cations is greater (meaning higher affinity) than that to the respective alkali metal ions from the same period. Furthermore, the Wiberg indices52, evaluated from the NBO population analysis revealed that the bonds between the group IB metals and ligating oxygen atoms from monensin A are stronger than those of the respective IA metal–oxygen counterparts: the mean Wiberg indices for the Cu+–O, Ag+–O and Au+–O bonds are 0.0578, 0.0601 and 0.0917 a.u., respectively, whereas those for the Li+–O, Na+–O, K+–O, Rb+–O and Cs+–O bonds are 0.0159, 0.0135, 0.0157, 0.0063 and 0.0104 a.u., respectively.The structures of fully optimized complexes of monensin A with Cu+, Ag+ and Au+ are presented in Fig. 3. In all the structures, the metal cation is coordinated to six oxygen atoms.7 The smaller ionic radii of the coinage metal ions are compatible with shorter M+–O distances in monensinate A complexes relative to their alkali metal counterparts (Table 3). The gas-phase M+ → Na+ substitution energies for Cu+, Ag+ and Au+ are much more favorable than those of the respective group IA metals from the same period (K+, Rb+ and Cs+, respectively; Table 4). The affinity order for the entire group I in the gas phase reads Cu+ > Li+ > Na+ > Au+ > Ag+ > K+ > Rb+ > Cs+.
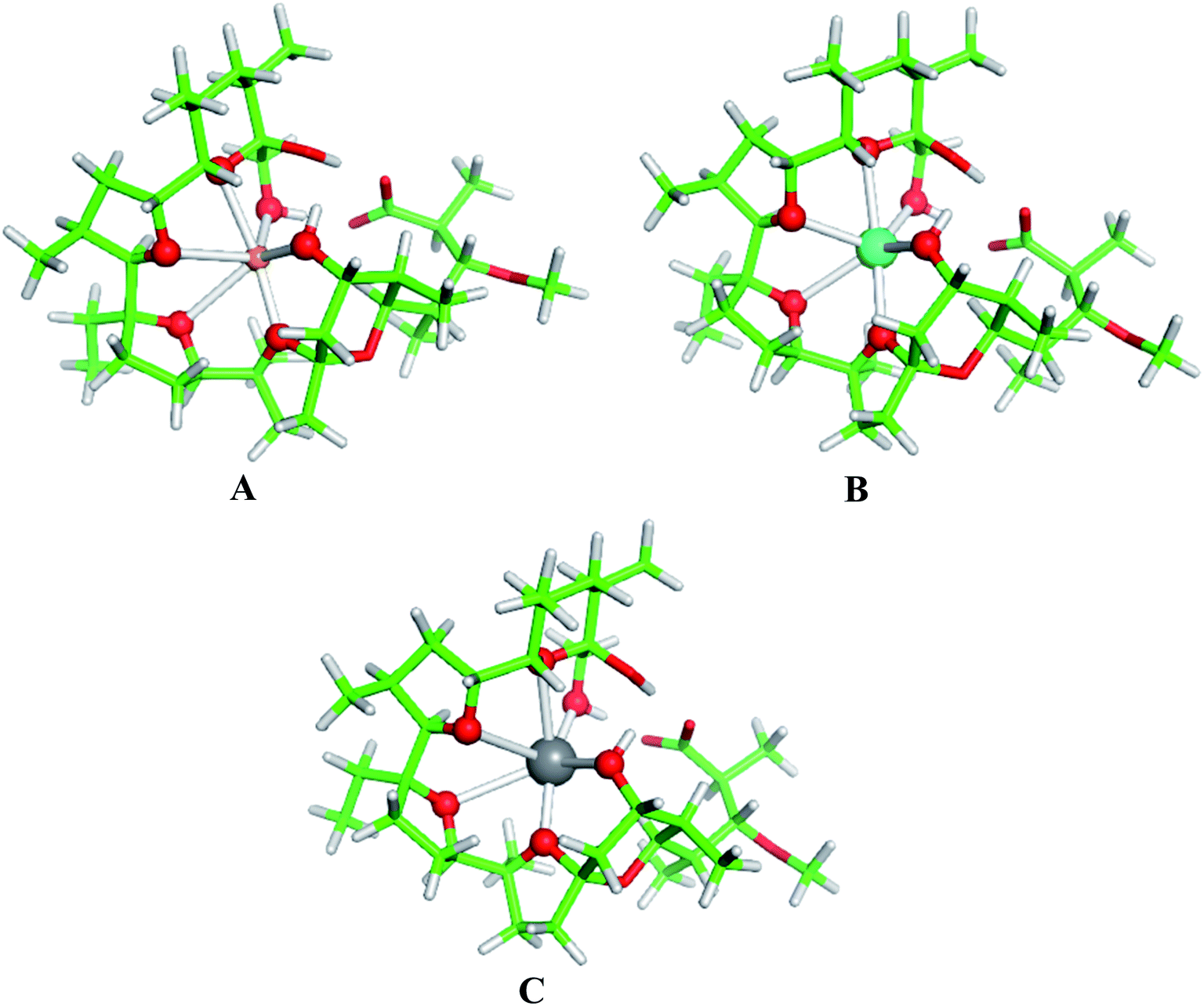 | ||
| Fig. 3 B3LYP/6-31+G(d,p) fully optimized structures of monensinate A bound to (A) Cu+, (B) Ag+ and (C) Au+. Color scheme: C – green, O – red, H – light grey, Cu – orange, Ag – cyan, Au – grey. | ||
In low polarity solvents (for example ε ≈ 2) this order changes in favor of Li+: Li+ > Cu+ > Na+ > K+ > Au+ > Ag+ > Rb+ > Cs+. The sequence changes again in polar solvents (methanol), this time in favor of Na+: Na+ > Li+ > K+ > Cu+ > Au+ > Ag+ > Rb+ > Cs+. Note that the coinage metal ions, although possessing higher affinity to the ligand than the respective alkali metal cations, are relegated to lower positions in the solution affinity scale due to the high desolvation penalty to be paid in condensed phase (reaction (1) and Table 2).
Concluding remarks
The calculations performed reveal the following key determinants of the monovalent metal selectivity in monensinate A anion:- The metal ion radius: smaller size cations, with higher positive charge density, are more competitive than their bulkier counterparts;
- The metal cation charge accepting ability: increasing the metal charge accepting ability, especially for d-elements, which translates into increased affinity toward the surrounding ligands (donor atoms), enhances the metal ion selectivity;
- The dielectric properties of the medium: low-polarity solvents favor the smaller ions possessing high ligand affinity (Li+ and Cu+); in polar solvents, characterized with high dielectric constants, the competitiveness of the medium-size cations, particularly Na+, increases.
The size of the internal cavity appears to be a secondary factor of the metal selectivity as the pore is relatively flexible and adaptable to certain extent to the spatial requirements of the incoming metal cations.
Note that the role of the solvent in governing the metal affinity of monensinate A anion is evaluated for the first time here (to the best of our knowledge). Our results imply that the metal selectivity of monensin A can be manipulated by changing the solvent used: the polyether host selectively binds Na+ in polar solvents (methanol and water) but could become Li+ or Cu+-selective in low-polarity solvents such as alkyl ethers, hydrocarbons and their halogenated derivatives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the support by National Science Fund, Bulgarian Ministry of Education and Science (contract no. KP-06-H29/3).References
- A. Agtarap, J. W. Chamberlin, M. Pinkerton and L. K. Steinrauf, Structure of monensic acid, a new biologically active compound, J. Am. Chem. Soc., 1967, 89, 5737–5739 CrossRef CAS PubMed.
- L. K. Steinrauf, M. Pinkerton and J. W. Chamberlin, The structure of nigericin, Biochem. Biophys. Res. Commun., 1968, 33, 29–31 CrossRef CAS PubMed.
- M. Mitani, T. Yamanishi and Y. Miyazaki, Salinomycin: a new monovalent cation ionophore, Biochem. Biophys. Res. Commun., 1975, 66, 1231–1236 CrossRef CAS PubMed.
- M. Pinkerton and L. K. Stenrauf, Molecular structure of monovalent metal cation complexes of monensin, J. Mol. Biol., 1970, 49, 533–546 CrossRef CAS PubMed.
- W. K. Lutz, F. K. Winkler and J. D. Dunitz, Crystal structure of the antibiotic monensin. Similarities and differences between free acid and metal complex, Helv. Chim. Acta, 1971, 54, 1103–1108 CrossRef CAS PubMed.
- W. L. Duax, G. D. Smith and P. D. Strong, Complexation of metal ions by monensin. Crystal and molecular structure of hydrated and anhydrous crystal forms of sodium monensin, J. Am. Chem. Soc., 1980, 102, 6725–6729 CrossRef CAS.
- D. M. Walba, M. Hermsmeier, R. C. Haltiwanger and J. H. Noordik, Crystal structures of monensin B lithium and silver salts, J. Org. Chem., 1986, 51, 245–247 CrossRef CAS.
- W. Pangborn, W. Duax and D. Langs, The hydrated potassium complex of the ionophore monensin A, J. Am. Chem. Soc., 1987, 109, 2163–2165 CrossRef CAS.
- A. Huczynski, M. Ratajczak-Sitarz, A. Katrusiak and B. Brzezinski, Molecular structure of the 1:1 inclusion complex of monensin A sodium salt with acetonitrile, J. Mol. Struct., 2007, 832, 84–89 CrossRef CAS.
- A. Huczynski, M. Ratajczak-Sitarz, A. Katrusiak and B. Brzezinski, Molecular structure of the 1:1 inclusion complex of monensin A lithium salt with acetonitrile, J. Mol. Struct., 2007, 871, 92–97 CrossRef CAS.
- A. Huczynski, M. Ratajczak-Sitarz, A. Katrusiak and B. Brzezinski, Molecular structure of rubidium six-coordinated dihydrate complex with monensin A, J. Mol. Struct., 2008, 883, 224–229 CrossRef.
- P. Butaye, L. A. Devriese and F. Haesebrouck, Antimicrobial growth promoters used in animal feed: effects of less well known antibiotics on gram-positive bacteria, Clin. Microbiol. Rev., 2003, 16, 175–188 CrossRef CAS PubMed.
- D. A. Kevin II, D. A. F. Meujo and M. T. Hamann, Polyether ionophores: broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites, Expert Opin. Drug Discovery, 2009, 4, 109–146 CrossRef PubMed.
- H. D. Chapman, T. K. Jeffers and R. B. Williams, Forty years of monensin for the control of coccidiosis in poultry, Poult. Sci., 2010, 89, 1788–1801 CrossRef CAS PubMed.
- V. Rajendran, H. S. Ilamathi, S. Dutt, T. S. Lakshminarayana and P. C. Ghosh, Chemotherapeutic potential of monensin as an antimicrobial agent, Curr. Top. Med. Chem., 2018, 18, 1976–1986 CrossRef CAS PubMed.
- A. Huczyński, G. Klejborowska, M. Antoszczak, E. Maj and J. Wietrzyk, Anti-proliferative activity of monensin and its tertiary amide derivatives, Bioorg. Med. Chem. Lett., 2015, 25, 4539–4543 CrossRef PubMed.
- V. Kaushik, J. S. Yakisich, A. Kumar, N. Azad and A. K. V. Iyer, Ionophores: potential use as anticancer drugs and chemosensitizers, Cancers, 2018, 10, 360 CrossRef CAS PubMed.
- D. Carriere, P. Casellas, G. Richer, P. Gros and F. K. Jansen, Endocytosis of an antibody ricin A-chain conjugate (immuno-A-toxin) adsorbed on colloidal gold. Effects of ammonium chloride and monensin, Exp. Cell Res., 1985, 156, 327–340 CAS.
- E. V. Wattenberg, P. L. McNeil, H. Fujiki and M. R. Rosner, Palytoxin down-modulates the epidermal growth factor receptor through a sodium-dependent pathway, J. Biol. Chem., 1989, 264, 213–219 CAS.
- W. T. Shier and D. J. DuBourdieu, Sodium- and calcium-de-endent steps in the mechanism of neonatal rat cardiac myocyte killing by lonophores. I. The sodium-carrying lonophore, monensin, Toxicol. Appl. Pharmacol., 1992, 116, 38–46 CrossRef CAS PubMed.
- N. R. Bury, M. Grosell, A. K. Grover and C. M. Wood, ATP-dependent silver transport across the basolateral membrane of rainbow trout gills, Toxicol. Appl. Pharmacol., 1999, 159, 1–8 CrossRef CAS PubMed.
- C. C. Häse, N. D. Fedorova, M. Y. Galperin and P. A. Dibrov, Sodium ion cycle in bacterial pathogens: evidence from cross-genome comparisons, Microbiol. Mol. Biol. Rev., 2001, 65, 353–370 CrossRef PubMed.
- W. Li, B. Dasgeb, T. Phillips, Y. Li, M. Chen, W. Garner and D. T. Woodley, Wound-Healing Perspectives, Dermatol. Clin., 2005, 23, 181–192 CrossRef CAS PubMed.
- D. Łowicki, A. Huczyński, J. Stefańska and B. Brzezinski, Syntheses, structural and antimicrobial studies of a new N-allylamide of monensin A and its complexes with monovalent metal cations, Tetrahedron, 2009, 65, 7730–7740 CrossRef.
- D. Łowicki, A. Huczyński, J. Stefańska and B. Brzezinski, Structural characterization and antibacterial activity against clinical isolates of Staphylococcus of N-phenylamide of monensin A and its 1:1 complexes with monovalent cations, Eur. J. Med. Chem., 2010, 45, 4050–4057 CrossRef PubMed.
- P. J. Henderson, J. D. McGiven and J. B. Chappell, The action of certain antibiotics on mitochondrial, erythrocyte and artificial phospholipid membranes. The role of induced proton permeability, Biochem. J., 1969, 111, 521–535 CrossRef CAS PubMed.
- R. Ashton and L. K. Stenrauf, Thermodynamic consideration of the ion transporting antibiotics, J. Mol. Biol., 1970, 49, 547–556 CrossRef CAS PubMed.
- W. K. Lutz, P. U. Früh and W. Simon, Microcalorimetric determination of ΔH0, ΔG0 and ΔS0 for the interaction of the carrier antibiotics nigericin and monensin with sodium and potassium ions, Helv. Chim. Acta, 1971, 54, 2767–2770 CrossRef CAS PubMed.
- G. Cornelius, W. Gartner and D. H. Haynes, Cation complexation by valinomycin- and nigericin-type ionophores registered by the fluorescence signal of Tl+, Biochemistry, 1974, 13, 3052–3057 CrossRef CAS PubMed.
- P. G. Gertenbach and A. I. Popov, Solution chemistry of monensin and its alkali metal ion complexes. Potentiometric and spectroscopic studies, J. Am. Chem. Soc., 1975, 97, 4738–4744 CrossRef CAS.
- J. G. Hoogerheide and A. I. Popov, Study of monensin complexes with monovalent metal ions in anhydrous methanol solutions, J. Solution Chem., 1978, 7, 357–372 CrossRef CAS.
- J. G. Hoogerheide and A. I. Popov, A study of metal complexes of a naturally occurring macrocyclic ionophore – monensin, J. Solution Chem., 1979, 8, 83–95 CrossRef CAS.
- B. Cox, N. van Truong, J. Rzeszotarska and H. Schneider, Stability constants of complexes of monensin and lasalocid with alkali-metal and alkaline-earth-metal ions in protic and polar aprotic solvents, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 3275–3284 RSC.
- B. G. Cox, N. van Truong, J. Rzeszotarska and H. Schneider, Rates and equilibria of alkali metal and silver ion complex formation with monensin in ethanol, J. Am. Chem. Soc., 1984, 106, 5965–5969 CrossRef CAS.
- M. Hebrant, Y. Pointud and J. Juillard, Thermodynamics of reaction in heterogeneous systems (water-organic phases) between the ionophore monensin and alkali metal cations, J. Phys. Chem., 1991, 95, 3653–3662 CrossRef CAS.
- M. Mimouni, S. Perrier, Y. Pointud and J. Juillard, Selectivity of bacterial ionophore monensin for monovalent metal cations. Solvent effects in methanol and biphasic water-organic systems, J. Solution Chem., 1993, 22, 769–785 CrossRef CAS.
- S. Filipek, J. Rzeszotarska and M. K. Kalinowski, Polarographic Study of Tl+, Li+, Na+ K+ and Cs+ complexes with monensin anion in dipolar aprotic solvents, Monatsh. Chem., 1994, 125, 801–809 CrossRef CAS.
- Y. Pointud, C. Bernard, S. Touzain, L. Astier, B. Sabatier and J. Juillard, Thermodynamics of complexation of monovalent metal cations by the ionophore monensin free acid in acetonitrile, J. Solution Chem., 1997, 26, 479–495 CrossRef CAS.
- E. Wagner-Czauderna, J. Rzeszotarska, E. Orlowska and M. K. Kalinowski, Complexing ability of monensin anion for alkali metal cations in binary mixtures of dipolar aprotic solvents, Ber. Bunsenges. Phys. Chem., 1997, 101, 1154–1157 CrossRef CAS.
- N. Ben-Tal, D. Sitkoff, S. Bransburg-Zabary, E. Nachliel and M. Gutman, Theoretical calculations of the permeability of monensin cation complexes in model bio-membranes, Biochim. Biophys. Acta, 2000, 1466, 221–233 CrossRef CAS.
- M. W. Forbes, D. A. Volmer, G. J. Francis and D. K. Böhme, A comparison of data analysis methods for determining gas phase stabilities by CID: alkali metal complexes of polyether ionophore antibiotics, J. Am. Soc. Mass Spectrom., 2005, 16, 779–791 CrossRef CAS PubMed.
- S. E. Angelova, V. K. Nikolova and T. M. Dudev, Determinants of the host-guest interactions between α-, β- and γ-cyclodextrins and group IA, IIA and IIIA metal cations: a DFT/PCM study, Phys. Chem. Chem. Phys., 2017, 19, 15129–15136 RSC.
- T. Dudev, D. Cheshmedzhieva and L. Doudeva, Competition between abiogenic Al3+ and native Mg2+, Fe2+ and Zn2+ ions in protein binding sites: implications for aluminum toxicity, J. Mol. Model., 2018, 24, 55 CrossRef PubMed.
- T. Dudev, C. Grauffel and C. Lim, How Pb2+ binds and modulates properties of Ca2+-signaling proteins, Inorg. Chem., 2018, 57, 14798–14809 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Ion selectivity strategies of sodium channel selectivity filters, Acc. Chem. Res., 2014, 47, 3580–3587 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi and G. Scalmani, et al., Gaussian 09, Gaussian Inc., 2009 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- N. Kircheva and T. Dudev, Novel insights into gallium's mechanism of therapeutic action: a DFT/PCM study of the interaction between Ga3+ and ribonucleotide reductase substrates, J. Phys. Chem. B, 2019, 123, 5444–5451 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Determinants of K+ vs Na+ selectivity in potassium channels, J. Am. Chem. Soc., 2009, 131, 8092–8101 CrossRef CAS PubMed.
- H. L. Friedman and C. V. Krishnan, in Water: A Comprehensive Treatise, ed. F. Franks, Plenum Press, New York, 1973, vol. 3, pp. 1–118 Search PubMed.
- R. D. Shannon, Revised effective ionic radii and systematic studies in interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |