DOI:
10.1039/C9RA10760F
(Paper)
RSC Adv., 2020,
10, 9431-9437
Enantioselective hydrosilylation of unsaturated carbon–heteroatom bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N, CO) catalyzed by [Ru–S] complexes: a theoretical study†
N, CO) catalyzed by [Ru–S] complexes: a theoretical study†
Received
20th December 2019
, Accepted 14th February 2020
First published on 4th March 2020
Introduction
Nowadays, the demand for enantiomerically pure compounds is increasing gradually in the fields of biochemistry and pharmaceutical chemistry.1 Chiral alcohols and amines are common organic compounds as important building blocks for organic synthesis.2 The asymmetric reduction of unsaturated compounds, such as ketones and imines, remains one of the fundamental approaches to synthesize biologically active agents and pharmaceutical products in the laboratory and industry.3,4 At the same time, silanes have been developed as an alternative to hydrogenation reagents because the H–Si bond is easier to be heterolytically splitted by catalysts than H–H bonds in organic synthesis.5 Furthermore, hydrosilylation reactions play a crucial role across industrial fields due to the easily handled, low cost and non-toxic characteristics of silicon reagents,6 together with the mild reaction conditions required and various applications. The silyl group is also retained as a protecting group to facilitate further functionalization of the reaction products.7,8 Currently, the asymmetric hydrosilylation of prochiral ketones and imines has been investigated by using transition metal catalysts, such as Rh,9 Ir,10 Co,11 Fe,12 Ru13a,c,d and so on.
In order to make progress in asymmetric catalytic hydrosilylation of ketones and imines, low reaction activity, narrow substrate range, harsh reaction conditions and difficult recovery of catalysts need to be overcome, great progress and important breakthroughs need to be made in the challenging hydrosilylation of ketones and imines.13 Interestingly, in the hydrosilylation of ketones, rhodium catalysts can reach more than 90% ee for a broad spectrum of substrates despite a reaction temperature of about −60 °C.14 Cobalt(III) complexes have also been used as catalysts for this reaction but require temperatures of 60 °C and long reaction times to give the final products.15 In 2019, Koga and coworkers explored the mechanism of hydrosilylation of imines by using iron catalysts.16a An iron three membered ring intermediate was suggested.16a Hasegawa's group investigated the Rh-catalyzed hydrosilylation of acetone or ethylene with tertiary silane to examine three existing mechanisms, the Chalk–Harrod (CH) mechanism, the modified Chalk–Harrod (mCH) mechanism, and the outer-sphere mechanism.16b They also proposed another two reaction pathways, the double hydride (DH) mechanism and the alternative Chalk–Harrod (aCH) mechanism.16b It was found that the entire hydrosilylation reaction takes place only at the center of the reaction, without the participation of the ligand. Single active site catalysts can be used for the molecular catalysts, but also for periodic surface catalysis. The 2D materials are a platform to design efficient single active site catalysts for various reactions.17
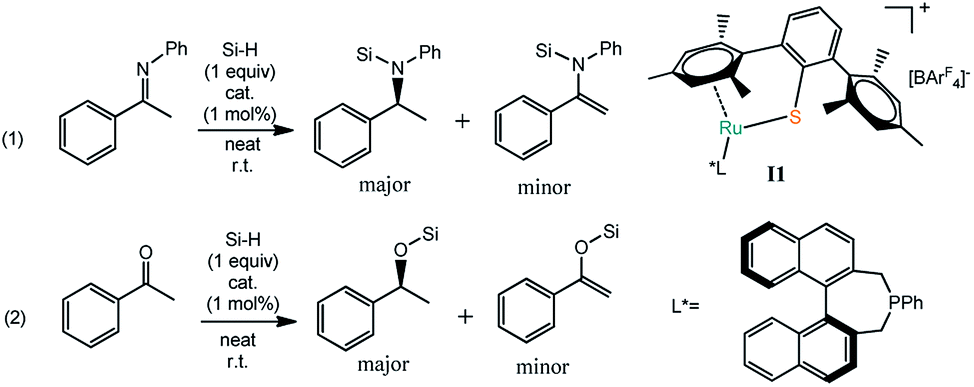
Moreover, coordinatively unsaturated ruthenium(II) thiolate complexes with bulky ligands reported by Tatsumi and Oestreich have been used in the cooperative activation of H–H bonds,18 Si–H bonds,19 B–H bonds20 and Al–H bonds.21 Based on these works, the Oestreich group changed the phosphine ligand to a chiral monodentate phosphine ligand to achieve enantioselective hydrosilylation of enolizable imines and ketones, which provided the corresponding chiral products at low catalyst loadings (as low as 1 mol%) at room temperature and with no additives.22 Differently, in the Ru–S catalyzed hydrosilylation reaction, it is assumed that the transition metal Ru shows Lewis acidity while the thiolate ligand works as the Lewis base to split X–H (X = B, Al, Si) bonds. That is to say, the [Ru–S] complex was performed bifunctional catalytic activation. Although (S)-silyl amines and (S)-silyl ethers were observed as the major products, but the ee value is not very high. Intrigued by these experimental results, we are interested in studying the mechanism of Ru–S cooperatively catalyzed enantionselective hydrosilylation of imines and ketones, in order to confirm the role of basic thiolate ligand in these reactions and investigate the methods to improve the ee value.
Computational details
Gaussian 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 was used for all calculations in this work. Geometric structures optimization and frequency calculation were conducted by M062X24 functional in gas phase at T = 298.15 K and 1 atm pressure. The LANL2DZ basis set was used for Ru atom25 with the polarization functions added for Ru (f = 1.235).26 The all-electron basis set 6-31G27 was used in describing the atoms (C, H, O, N, P, S). There is no imaginary frequency for all the intermediates while only-one imaginary frequency is in transition state (TS) structures. In addition, the intrinsic reaction coordinate (IRC) calculation28 which correctly bridged the intermediates further demonstrate the credibility of transition state structures. Based on the gas-phase optimized geometries, solvation effect of toluene was incorporated with the SMD29 model at the level of B3LYP-D330 while LANL2DZ basis set for Ru atom and 6-311++G** basis set for all other main group atoms. We used simplified Si reagent PhSiH3 rather than PhMeSiH2 to avoid the overestimation of the steric effect from reactant. All 3D molecular structures were generated by using the CYLview (Version) program.31 The numbers shown in energy profiles are solvent corrected Gibbs free energies based on SMD model.
23 was used for all calculations in this work. Geometric structures optimization and frequency calculation were conducted by M062X24 functional in gas phase at T = 298.15 K and 1 atm pressure. The LANL2DZ basis set was used for Ru atom25 with the polarization functions added for Ru (f = 1.235).26 The all-electron basis set 6-31G27 was used in describing the atoms (C, H, O, N, P, S). There is no imaginary frequency for all the intermediates while only-one imaginary frequency is in transition state (TS) structures. In addition, the intrinsic reaction coordinate (IRC) calculation28 which correctly bridged the intermediates further demonstrate the credibility of transition state structures. Based on the gas-phase optimized geometries, solvation effect of toluene was incorporated with the SMD29 model at the level of B3LYP-D330 while LANL2DZ basis set for Ru atom and 6-311++G** basis set for all other main group atoms. We used simplified Si reagent PhSiH3 rather than PhMeSiH2 to avoid the overestimation of the steric effect from reactant. All 3D molecular structures were generated by using the CYLview (Version) program.31 The numbers shown in energy profiles are solvent corrected Gibbs free energies based on SMD model.
Results and discussion
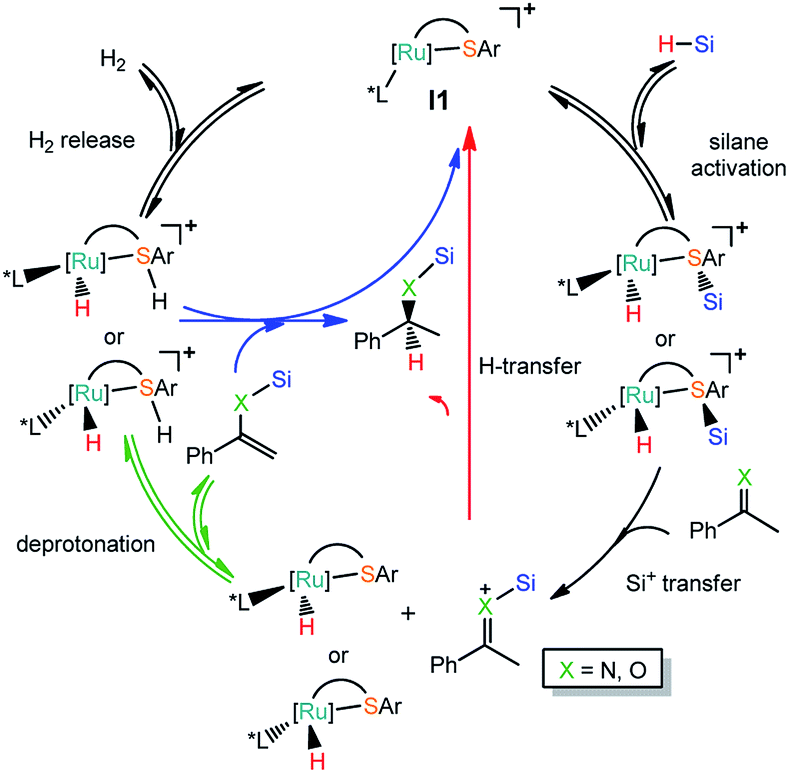
Based on the experimental observation, we studied the reaction mechanism of the Ru–S catalyzed hydrosilylation of imines and ketones in order to understand better on the dual role of Ru–S catalyst and how the enantioselectivity is controlled by using bulky ligand on Ru–S catalyst.22 Since imines and ketones can react with silanes to give hydrosilylation and dehydrogenative silylation products as shown in Scheme 1. Reactions of imines and ketones with silanes promoted by catalyst I1 can have reaction pathways: (1) the cleavage of Si–H bond by Ru–S bond; (2) silyl group transfer from S to X (X = N, O); (3) hydrogen transfer from Ru to carbon of imine or ketone or proton transfer from imine or ketone to sulfur ligand; (4) [Ru–S] complex regeneration through H2 release or hydrogenation reaction of alkenyl silane (Scheme 2). The detailed mechanism of these pathways will be studied and discussed and the origin of the competition between hydrosilylation and dehydrogenative silylation reactions is addressed. Unless specified, all the figures of the potential energy profiles are presented in the relative Gibbs free energies in solution (kcal mol−1). The relative Gibbs free energies and electronic energies in gas-phase are shown in Table S1 (see ESI† for details). The relative Gibbs free energies and relative electronic energies in gas-phase are similar in cases when the number of the reactant and product molecules is equal, for example, one-to-one or two-to-two transformations. However, this differs significantly for one-to-two or two-to-one transformations because of the entropic contribution.
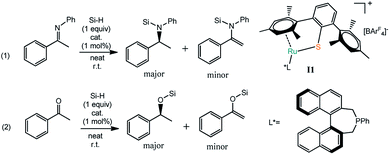 |
| | Scheme 1 Hydrosilylation or dehydrogenative silylation of imine and ketone catalyzed by [Ru–S] complex (left) and the structure of [Ru–S] catalyst I1 and L* (right). | |
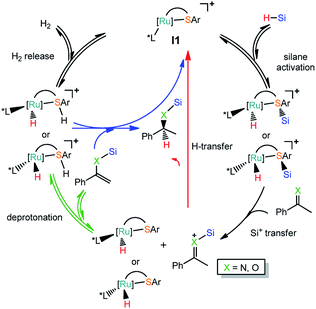 |
| | Scheme 2 Reactions pathways for Ru thiolate complex catalyzed hydrosilylation (red line) or dehydrogenative silylation of imine and ketone (green line). | |
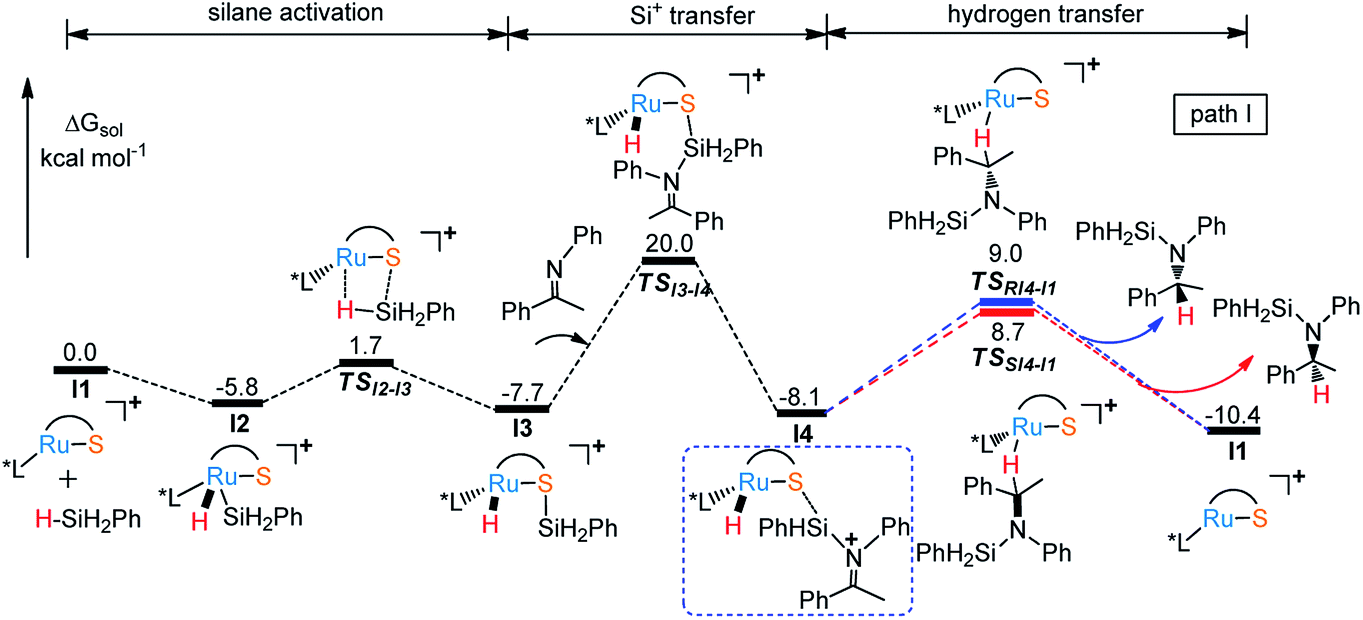
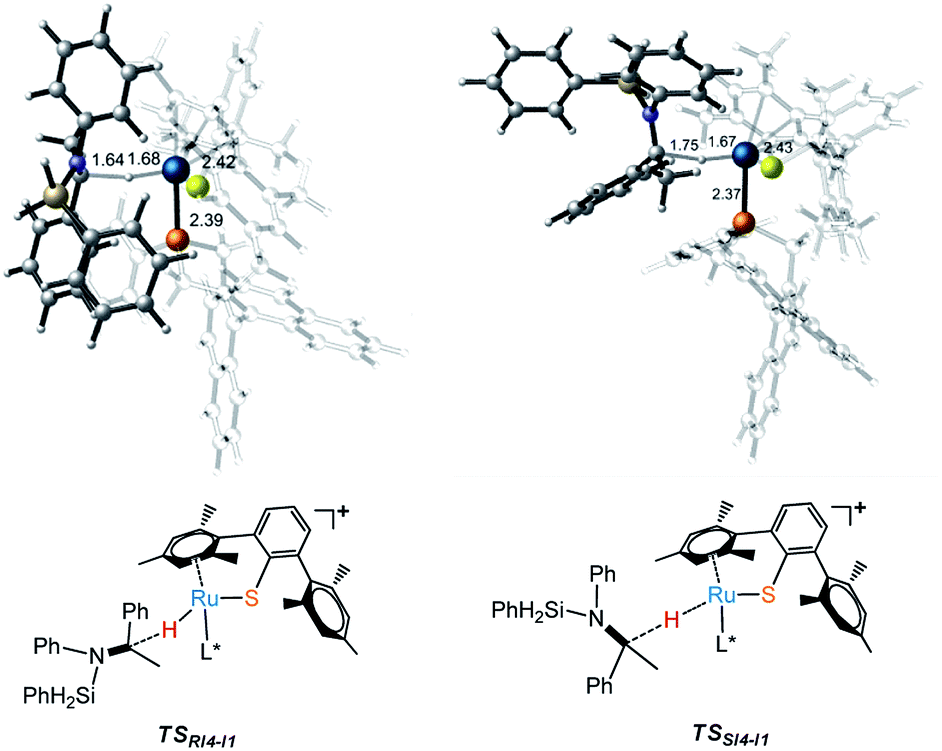
Free energy profile for I1 catalyzed hydrosilylation of imine is shown in Fig. 1. The catalytic reaction is triggered by the coordination of silane on Ru center to form intermediate I2. Then, H–Si bond is splitted heterolytically by Ru–S bond, which undergoes silane activation process via a silicon-stabilized quadrilateral transition state TSI2-I3 (ΔG‡ = 7.5 kcal mol−1) to give a silicon electrophile and a metal hydride due to the Lewis acidic Ru center and basic thiolate ligand. Next, the silyl group migrates from S to N of imine through transition state TSI3-I4 with a free energy barrier of 27.7 kcal mol−1, which is the rate determining step of the whole reaction. After that, the hydride on Ru transfers to silyl imide cation with enantioselectivity. At last, R- and S-silyl amines are generated by the transition states TSRI4-I1 and TSSI4-I1 with the energy barriers 17.1 kcal mol−1 and 16.8 kcal mol−1, respectively. Therefore, S-silyl amine is the main product in the whole reaction with 24.79% ee by calculation. Taking a close look at the structures of transition states TSRI4-I1 and TSSI4-I1, we found that the repulsion between amine group and phosphine ligand is larger in TSRI4-I1 than that between methyl group and phosphine ligand in TSSI4-I1, leading to lower energy of TSSI4-I1 (Fig. 2).
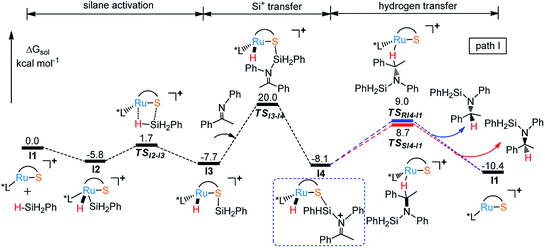 |
| | Fig. 1 Free energy profile for I1 catalyzed hydrosilylation of imine by route 1. | |
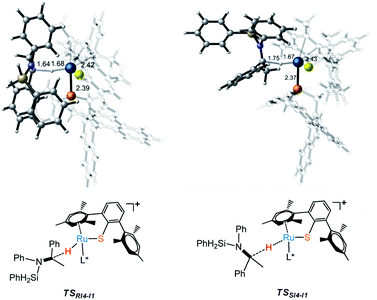 |
| | Fig. 2 Transition states (TSs) for the formation of R- and S- alkyl silyl amines. | |
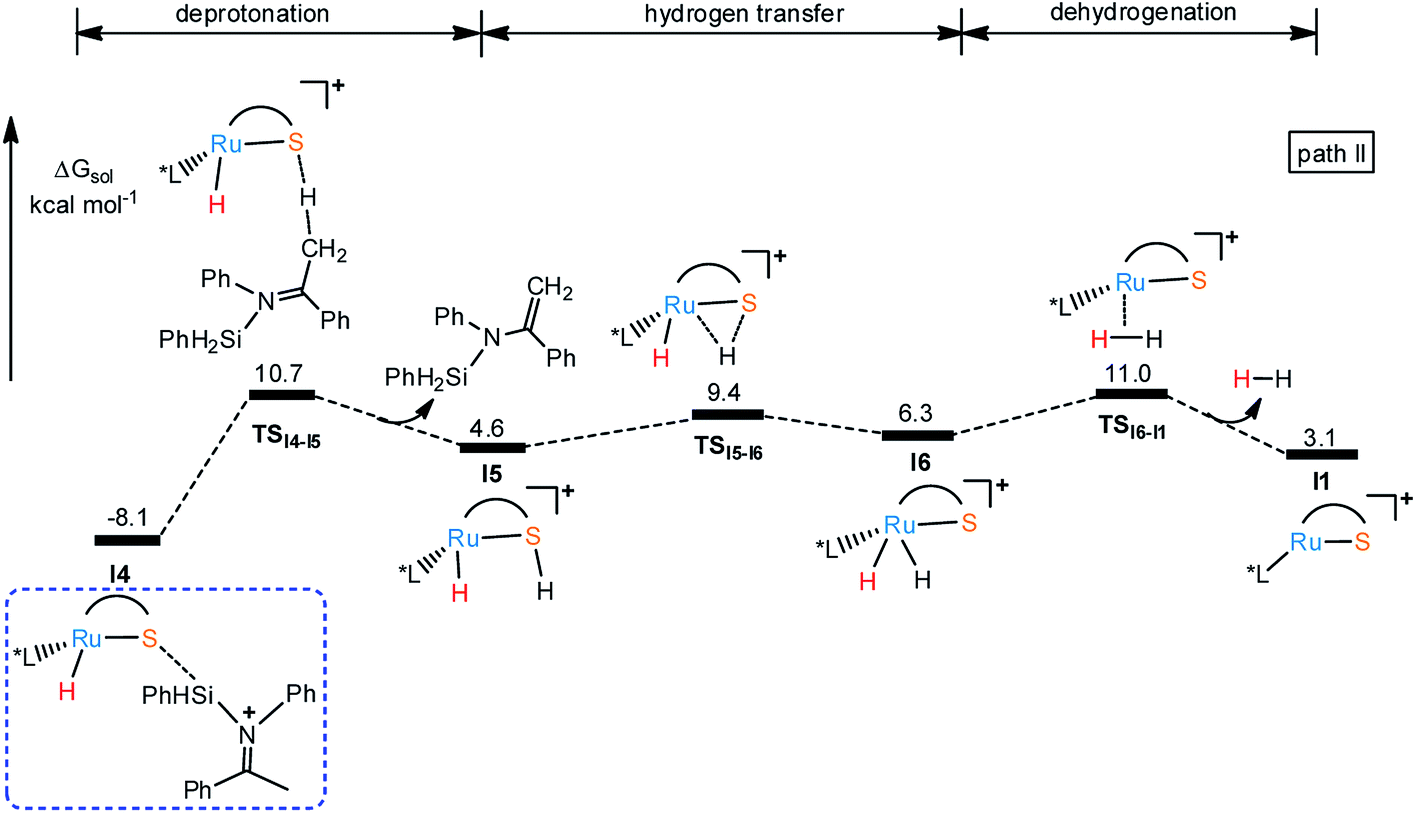
Considering the basicity of thiolate ligand, a possible reaction pathway is the proton transfer from silyl imide cation to S in I4 through transition state TSI4-I5 (18.8 kcal mol−1 energy barrier) to release silyl enamine and generate intermediate I5 as shown in Fig. 3. And hydrogen elimination takes place through transition state TSI5-I6 (ΔG‡ = 17.5 kcal mol−1) to give intermediate I6. At last, dehydrogenation happens to regenerate I1 via the TSI6-I1 with the barrier of 19.1 kcal mol−1, finishing the catalytic cycle. Although the reaction barriers for all these processes are low, the whole reaction is endothermic, about 3.1 kcal mol−1, which makes the pathway in Fig. 4 thermodynamically unfavorable. In summary, the main product of this reaction can be silyl amine instead of silyl enamine.
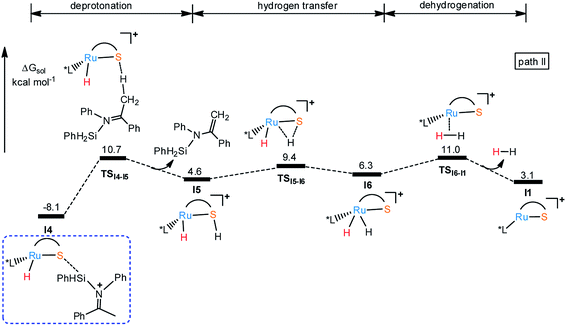 |
| | Fig. 3 Free energy profile for I1 catalyzed dehydrogenative silylation of imine. | |
 |
| | Fig. 4 Free energy profile for I1 catalyzed hydrosilylation of ketone. | |
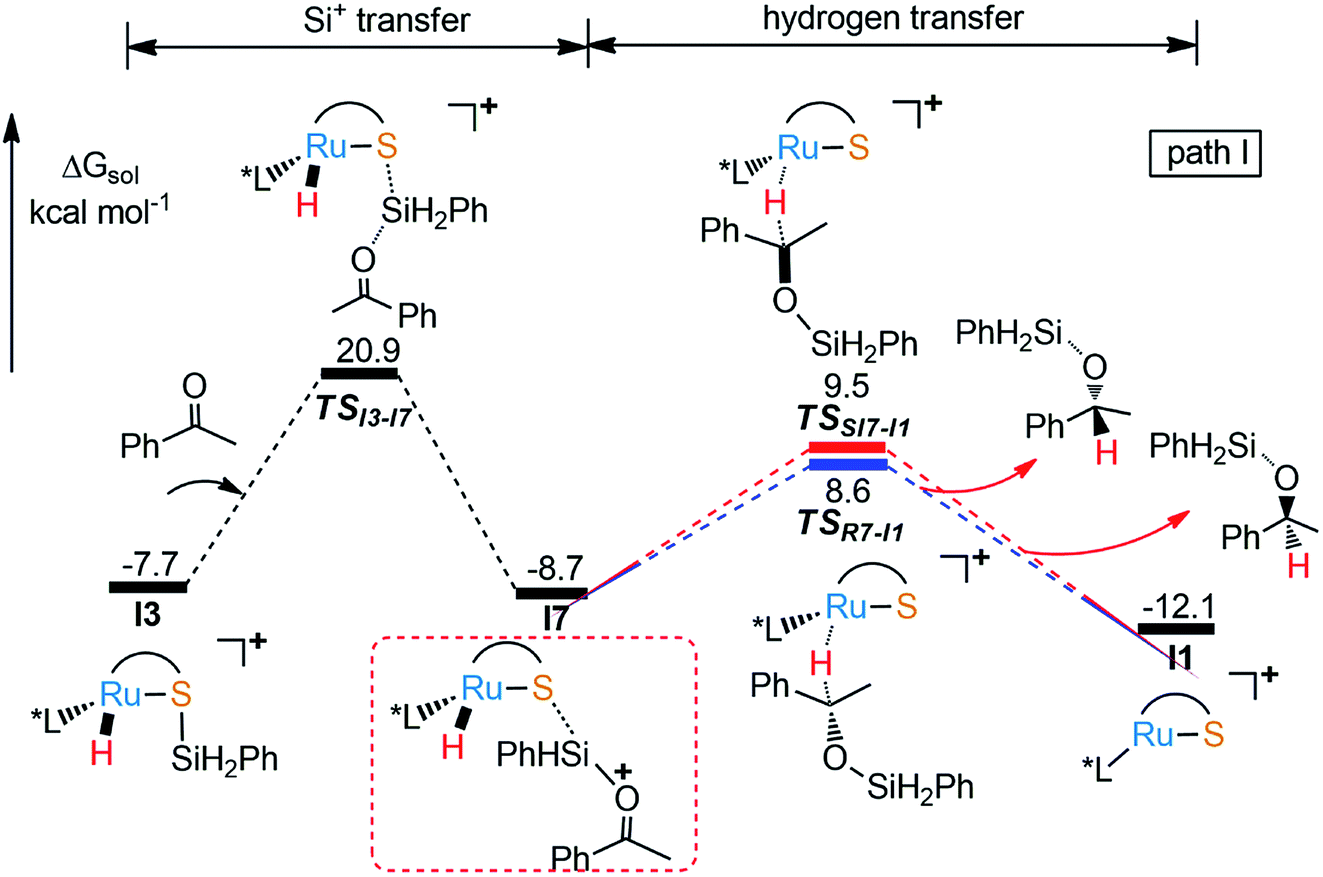
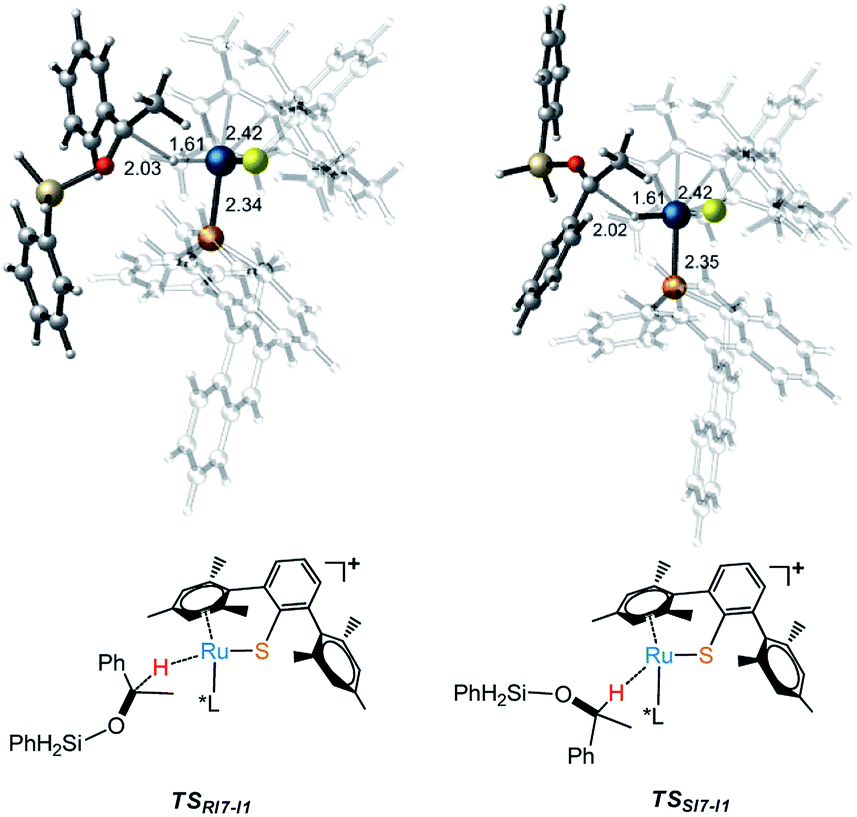
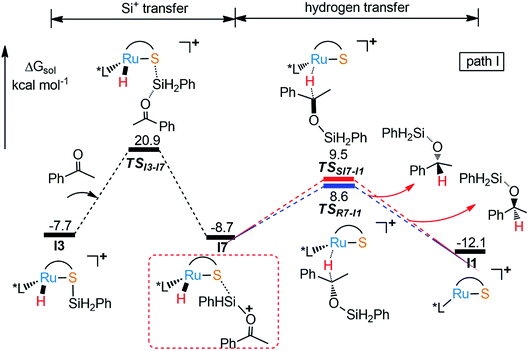
As part of the ongoing effort to explore the reduction of aryl ketone. Similar to the calculation of Ru–S catalyzed reaction of imine with silane, we also study the reaction mechanism of ketone with PhSiH3. As shown in Fig. 4, when I3 reacts with ketone, silyl group migrates from I3 to O of keto to give I7 via a very high energy barrier (TSI3-I7, ΔG‡ = 28.6 kcal mol−1), which is also the rate determining step of the whole reaction. After that, the hydride transfer from Ru to carbonyl carbon to give R- and S-silyl ether through transition states TSRI7-I1 and TSSI7-I1 with the energy barriers 17.3 kcal mol−1 and 18.2 kcal mol−1, respectively. Here, the calculation results show that the formation of R- and S-silyl ether have similar reaction barriers resulting of moderate enantioselectivity of products, about 64.08% ee of R-silyl ether. Taking a close look at the structures of transition states TSRI7-I1 and TSSI7-I1, we found that the repulsion between silyl ether group and phosphine ligand in TSRI7-I1 and the repulsion between phenyl group and phosphine ligand in TSSI7-I1 are all not obvious, leading to similar energy of TSRI7-I1 and TSSI7-I1 (Fig. 5).
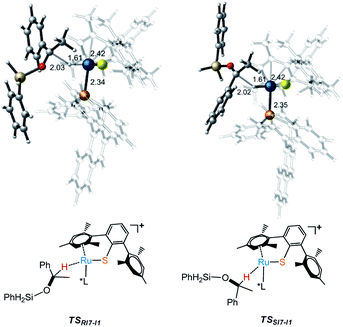 |
| | Fig. 5 Transition states (TSs) for the formation of R- and S- alkyl silyl ethers. | |
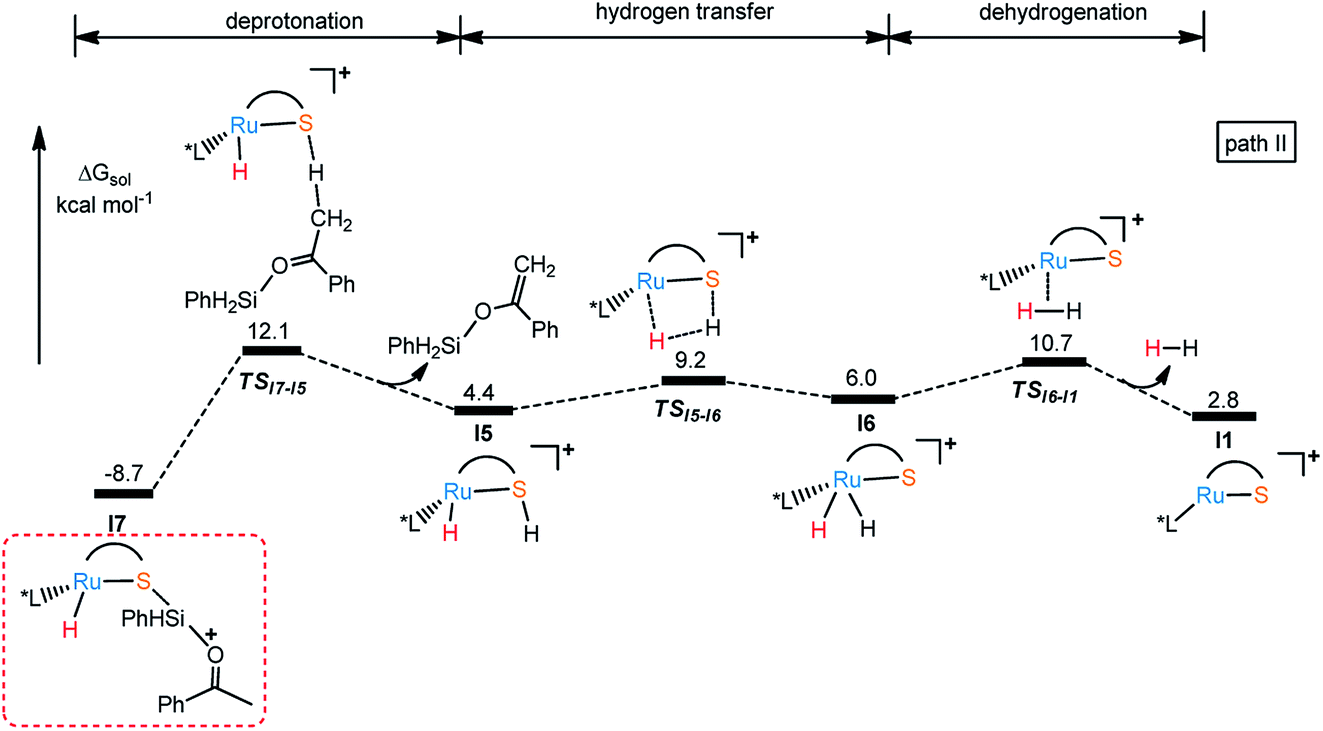
In the same way, there is another reaction pathway from intermediate I7 when considering the Lewis basicity of sulfur ligand. As shown in Fig. 6, hydrogen transfer from methyl group to sulfur occurs because of the electronegativity of thiolate ligand resulting in the formation of I5 and release the alkenyl silyl ether. I6 is generated through transition state TSI5-I6 with the energy barrier 17.9 kcal mol−1 by transferring hydrogen from S to Ru. At last, reductive elimination occurs to regenerate I1 and release dihydrogen via TSI6-I1 with the energy barrier 19.4 kcal mol−1. Similar to dehydrogenative silylation of imine, the reaction pathway in Fig. 4 is endothermic, which makes the dehydrogenative silylation of ketone thermodynamically unfavorable. Therefore, the main product of this Ru–S cooperatively catalyzed reaction is silyl ether instead of silyl-ketene.
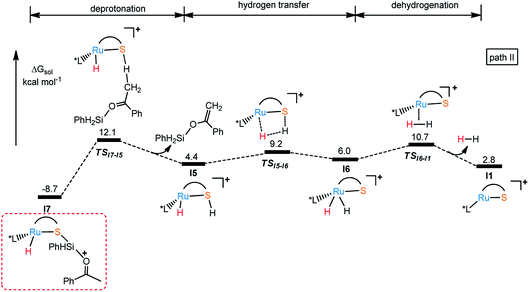 |
| | Fig. 6 Free energy profile for I1 catalyzed dehydrogenative silylation of ketone. | |
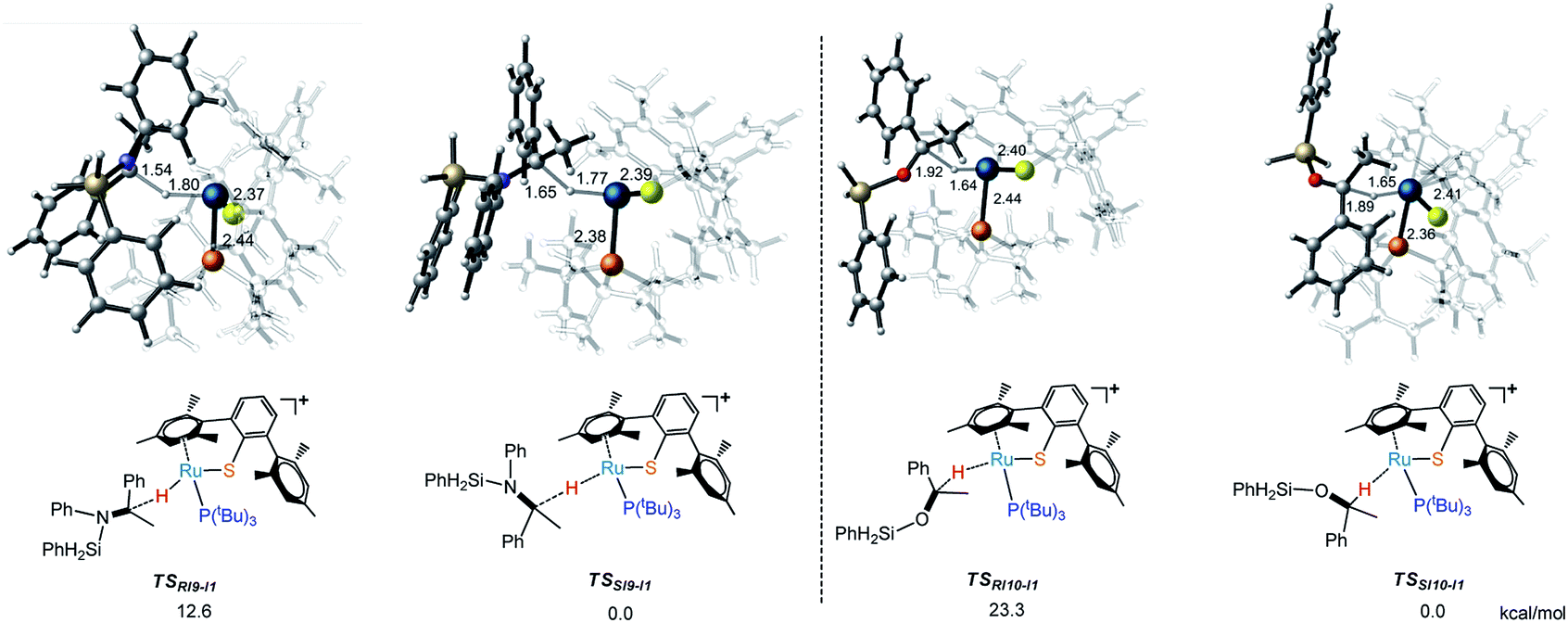
When the phosphine ligand is changed from L* to P(iso-butanyl)3, the energy difference between R- and S- transition states (TSs) became larger and enantioselectivity is enhanced a lot (Fig. 7). Comparing the structural character of transition states TSRI9-I1, TSSI9-I1, TSRI10-I1 and TSSI10-I1, the repulsion between silyl group and phosphine ligand in TSRI9-I1 and TSRI10-I1 is much larger than the repulsion between phenyl group and phosphine ligand in TSSI9-I1 and TSSI10-I1, which explains the ee value predicted from model ligand P(iso-butanyl)3. The larger repulsion makes the transition state more distorted, which destabilizes the TSRI9-I1 and TSRI10-I1,32 while for catalysts with smaller repulsion in TS, the attractive, dynamic, and cooperative non-covalent interactions can make other R- and S- transition states have similar flexibility and stability.32d
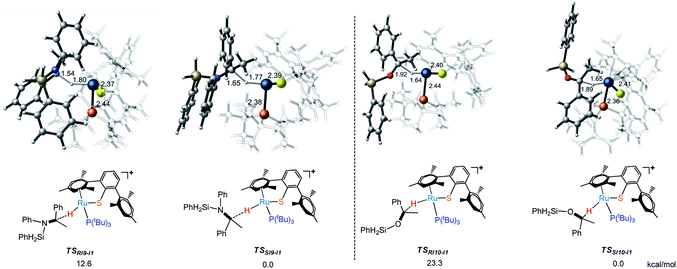 |
| | Fig. 7 Transition states (TSs) for the formation of R- and S- alkyl silyl amine and ether catalyzed by [RuS]P(tBu)+. | |
Conclusion
In summary, we set out to elucidate the mechanism of the [Ru–S] complex as catalysts for hydrosilylation of ketones and imines and explain the chemo- and enantio-selectivity involved in these reactions. In the catalytic process, the [Ru–S] complex is assumed to play a decisive role by heterolytically splitting the Si–H bond to form a sulfur-stabilized ruthenium hydride intermediate and a Si electrophile that can attract electron rich N and O atoms to form a cation. Although both hydrosilylation and dehydrogenative silylation processes afterward have barriers less than 30 kcal mol−1, the dehydrogenative silylation is reversible, making this pathway unfavourable. At the same time, the regioselectivity of these reactions is not high according to our calculations, we can predict that a bulky phosphine ligand such as P(iso-butanyl)3 can enhance the enantioselectivity significantly.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from the National Natural Science Foundation of China (No. 21573102) and Department of Education of Guang-dong Province are appreciated.
Notes and references
-
(a) M. P. Schneider and U. Goergens, Tetrahedron: Asymmetry, 1992, 3, 525–528 CrossRef CAS;
(b) R. K. Pandey, R. A. Fernandes and P. Kumar, Tetrahedron Lett., 2002, 43, 4425–4426 CrossRef CAS;
(c) X. Xu, R. Fu, J. Chen, S. Chen and X. Bai, Bioorg. Med. Chem. Lett., 2007, 17, 101–104 CrossRef CAS;
(d) D. J. Wardrop and J. Fritz, Org. Lett., 2006, 8, 3659–3662 CrossRef CAS PubMed;
(e) J. D. Katz, J. P. Jewell, D. J. Guerin, J. Lim, C. J. Dinsmore, S. V. Deshmukh, B. S. Pan, C. G. Marshall, W. Lu and M. D. Altman, J. Med. Chem., 2011, 54, 4092–4108 CrossRef CAS PubMed.
-
(a) G. Zanoni, A. Porta, Q. D. Toma, F. Castronovo and G. Vidari, J. Org. Chem., 2003, 68, 6437–6439 CrossRef CAS PubMed;
(b) P. Hoyos, J. V. Sinisterra, F. Molinari and A. R. Alcántara, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS;
(c) B. M. Trost, I. Fleming and C. H. Heathcock, Comprehensive Organic Synthesis, Pergamon, Oxford, 1991, vol. 2, pp. 1–53 Search PubMed;
(d) T. Mukaiyama, Tetrahedron, 1999, 55, 8609–8670 CrossRef CAS;
(e) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS;
(f) D. A. Evans, A. M. Ratz, B. E. Huff and G. S. Sheppard, J. Am. Chem. Soc., 1995, 117, 3448–3467 CrossRef CAS;
(g) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506–7525 CrossRef CAS;
(h) I. Paterson and M. M. Mansuri, Tetrahedron, 1985, 41, 3569–3624 CrossRef CAS;
(i) S. Masamune, G. S. Bates and J. W. Corcoran, Angew. Chem., Int. Ed., 1977, 16, 585–607 CrossRef CAS.
-
(a) P. J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72–76 CrossRef CAS;
(b) X. Chen, J. Z. Fong, J. Xu, C. Mou, Y. Lu, S. Yang, B. A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7212–7215 CrossRef CAS PubMed;
(c) C. Chen, H. Wang, Z. Zhang, S. Jin, S. Wen, J. Ji, L. W. Chung, X. Q. Dong and X. Zhang, Chem. Sci., 2016, 7, 6669–6673 RSC;
(d) S. F. Zhu, Y. Cai, H. X. Mao, J. H. Xie and Q. L. Zhou, Nat. Chem., 2010, 2, 546–551 CrossRef CAS;
(e) M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS;
(f) L. Sutin, S. Anderson, L. Bergquist, V. M. Castro, E. Danielsson, S. James, M. Henriksson, L. Johansson, C. Kaiser, K. Flyren and M. Williams, Bioorg. Med. Chem. Lett., 2007, 17, 4837–4840 CrossRef CAS PubMed.
-
(a) S. Lou and C. Fu, J. Am. Chem. Soc., 2010, 132, 1264–1266 CrossRef CAS PubMed;
(b) P. M. Lundin, J. Esquivias and C. Fu, Angew. Chem., Int. Ed., 2009, 121, 160–162 CrossRef;
(c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed.
-
(a) J. M. Brunel, Int. J. Hydrogen Energy, 2010, 35, 3401–3405 CrossRef CAS;
(b) J. M. Brunel, Int. J. Hydrogen Energy, 2017, 42, 23004–23009 CrossRef CAS.
- F. Meinardi, S. Ehrenberg, L. Dhamo, F. Carulli, M. Mauri, F. Bruni, R. Simonutti, U. Kortshagen and S. Brovelli, Nat. Photon., 2017, 11, 177–185 CrossRef CAS.
-
(a) M. C. Pirrung, L. Fallon, J. Zhu and Y. R. Lee, J. Am. Chem. Soc., 2001, 123, 3638–3643 CrossRef CAS PubMed;
(b) M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef CAS PubMed.
-
(a) T. Murai, T. Sakane and S. Kato, J. Org. Chem., 1990, 55, 449–453 CrossRef CAS;
(b) A. C. Fernandes and C. C. Romao, Tetrahedron Lett., 2005, 46, 8881–8883 CrossRef CAS;
(c) A. C. Fernandes and C. C. Romao, J. Mol. Catal. A: Chem., 2007, 272, 60–63 CrossRef CAS;
(d) R. G. Noronha, A. C. Fernandes and C. C. Romao, Tetrahedron Lett., 2009, 50, 1407–1410 CrossRef;
(e) R. G. Noronha, C. C. Romao and A. C. Fernandes, J. Org. Chem., 2009, 74, 6960–6964 CrossRef;
(f) R. G. Noronha, C. C. Romao and A. C. Fernandes, Catal. Commun., 2011, 12, 337–340 CrossRef;
(g) G. Du and P. E. Fanwick, Inorg. Chim. Acta, 2008, 361, 3184–3192 CrossRef CAS;
(h) K. A. Nolin, R. W. Ahn, Y. Kobayashi, J. J. Kennedy-Smith and F. D. Toste, Chem.–Eur. J., 2010, 16, 9555–9562 CrossRef CAS PubMed;
(i) G. I. Nikonov and D. V. Gutsulyak, Angew. Chem., Int. Ed., 2010, 49, 7553–7556 CrossRef PubMed.
-
(a) K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613–621 CrossRef CAS;
(b) I. Ojima, M. Kogure and M. Kumagai, J. Org. Chem., 1977, 42, 1671–1679 CrossRef CAS;
(c) M. Sawamura, R. Kuwano and Y. Ito, Angew. Chem., Int. Ed., 1994, 33, 111–113 CrossRef;
(d) D. Enders, H. Gielen and K. Breuer, Tetrahedron: Asymmetry, 1997, 8, 3571–3574 CrossRef CAS;
(e) K. Kromm, P. L. Osburn and L. Gladysz, Organometallics, 2002, 21, 4275–4280 CrossRef CAS;
(f) D. A. Evans, E. M. Forrest, J. S. Tedrow and K. R. Campos, J. Am. Chem. Soc., 2003, 125, 3534–3543 CrossRef CAS PubMed;
(g) W. L. Duan, M. Shi and G. B. Rong, Chem. Commun., 2003, 35, 2916–2917 RSC;
(h) C. G. Arena and R. J. Pattacini, J. Mol. Catal. A: Chem., 2004, 222, 47–52 CrossRef CAS;
(i) V. Cesar, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2005, 11, 2862–2873 CrossRef CAS PubMed;
(j) T. Imamoto, T. Itoh, Y. Yamanoi, R. Narui and K. Yoshida, Tetrahedron: Asymmetry, 2006, 17, 560–565 CrossRef CAS;
(k) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609–1613 CrossRef CAS PubMed;
(l) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Chem.–Eur. J., 2010, 15, 11515–11529 CrossRef PubMed;
(m) N. Schneider, M. Kruck, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2009, 4, 493–500 CrossRef.
-
(a) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS;
(b) R. S. Tanke and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 7984–7989 CrossRef CAS;
(c) S. Doherty, J. G. Knight, T. H. Scanlan, M. R. J. Elsegood and W. J. Clegg, Organomet. Chem., 2002, 650, 231–248 CrossRef CAS;
(d) M. Martín, E. Sola, O. Torres, P. Pablo and L. A. Oro, Organometallics, 2003, 22, 5406–5417 CrossRef;
(e) S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2002, 21, 4648–4661 CrossRef CAS;
(f) L. D. Field, B. A. Messerle and S. L. Wren, Organometallics, 2003, 22, 4393–4395 CrossRef CAS.
-
(a) H. Brunner, R. Becker and G. Rielp, Organometallics, 1984, 3, 1354–1359 CrossRef CAS;
(b) L. H. Gade, V. Cesar and S. Bellemin-Laponnaz, Angew. Chem., Int. Ed., 2004, 43, 1014–1017 CrossRef CAS PubMed;
(c) L. M. Newman, J. M. J. Williams, R. McCague and G. A. Potter, Tetrahedron: Asymmetry, 1996, 7, 1597–1598 CrossRef CAS;
(d) T. Langer, J. Janssen and G. Helmchen, Tetrahedron: Asymmetry, 1996, 7, 1599–1602 CrossRef CAS;
(e) B. Tao and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 3892–3894 CrossRef CAS.
-
(a) X. Sun, L. Zhou, W. Li and X. Zhang, J. Org. Chem., 2008, 73, 1143–1146 CrossRef CAS;
(b) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed;
(c) C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS;
(d) Y. Deng, C. V. Karunaratne, E. Csatary, D. L. Tierney, K. Wheeler and H. Wang, J. Org. Chem., 2015, 80, 7984–7993 CrossRef CAS PubMed;
(e) H. Teng, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16506–16509 CrossRef CAS PubMed;
(f) F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Sumner, G. P. McGovern and M. Findlater, Organometallics, 2015, 34, 5051–5056 CrossRef CAS;
(g) A. Kišić, M. Stephan and B. Mohara, Adv. Synth. Catal., 2015, 357, 2540–2546 CrossRef;
(h) B. Tao and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 3892–3894 CrossRef CAS;
(i) I. Takei, Y. Nishibayashi, Y. Ishii, Y. Mizobe, S. Uemura and M. Hidai, Chem. Commun., 2001, 2360–2361 RSC;
(j) S. Hosokawa, J. I. Ito and H. Nishiyama, Organometallics, 2010, 29, 5773–5775 CrossRef CAS.
-
(a) M. L. Huw, C. V. Davies, H. Tore and W. H. Darrin, J. Am. Chem. Soc., 2003, 125, 6462–6468 CrossRef PubMed;
(b) J. W. Patrick, Acc. Chem. Res., 2003, 36, 739–749 CrossRef PubMed;
(c) Y. Nishibayashi, I. Takei, S. Uemura and M. Hidai, Organometallics, 1998, 17, 3420–3422 CrossRef CAS;
(d) B. Li, J. Sortais, C. Darcel and P. H. Dixneuf, ChemSusChem, 2012, 5, 396–399 CrossRef CAS PubMed;
(e) C. Song, C. Ma, Y. Ma, W. Feng, S. Ma, Q. Chai and M. B. Andrus, Tetrahedron Lett., 2005, 46, 3241–3244 CrossRef CAS.
- V. César, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2004, 43, 1014–1017 CrossRef.
- H. Zhou, H. Sun, S. Zhang and X. Li, Organometallics, 2015, 34, 1479–1486 CrossRef CAS.
-
(a) A. A. Dahy and N. Koga, J. Comput. Chem., 2019, 40, 62–71 CrossRef CAS PubMed;
(b) L. Zhao, N. Nakatani and Y. Sunada, et al., J. Org. Chem., 2019, 84, 8552–8561 CrossRef CAS PubMed.
-
(a) B. Song, Y. Zhou, H.-M. Yang, J.-H. Liao, L.-M. Yang, X.-B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 141, 3630–3640 CrossRef CAS;
(b) J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC;
(c) J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC;
(d) J.-H. Liu, L.-M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC;
(e) J.-H. Liu, L.-M. Yang and E. Ganz, Energy Environ. Mater., 2019, 2, 193–200 CrossRef CAS;
(f) J. H. Liu, L. M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS;
(g) L. Xu, L.-M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed;
(h) L.-M. Yang, V. Bacic, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS PubMed.
-
(a) Y. Ohki, Y. Takikawa, H. Sadohara, C. Kesenheimer, B. Engendahl, E. Kapatina and K. Tatsumi, Chem.–Asian J., 2008, 3, 1625–1635 CrossRef CAS PubMed;
(b) A. Lefranc, Z. W. Qu, S. Grimme and M. Oestreich, Chem.–Eur. J., 2016, 22, 10009–10016 CrossRef CAS PubMed.
-
(a) T. Stahl, K. Müther, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed;
(b) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS.
- T. Stahl, K. Müther, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS.
- F. Forster, T. T. Metsanen, E. Irran, P. Hrobarik and M. Oestreich, J. Am. Chem. Soc., 2017, 139, 16334–16342 CrossRef CAS PubMed.
- S. Bahr and M. Oestreich, Organometallics, 2017, 36, 935–943 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed;
(b) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS PubMed;
(c) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194120 CrossRef PubMed;
(d) S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS;
(c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
-
(a) A. W. Ehlers, M. Biihme, S. Dapprich, A. Gobbi, A. Hijllwarth, V. Jonas, K. F. Kiihler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS;
(b) A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. J. Mantzaris, Chem. Phys., 1988, 89, 2193–2218 CAS.
- C. Gonzalez and H. B. Gonzalez, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
-
(a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154123 CrossRef PubMed;
(b) S. Grimme, S. Ehrlich and L. J. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
-
(a) D. G. Truhlar, B. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS;
(b) J. Lucas Bao and D. G. Truhlar, Chem. Soc. Rev., 2017, 46, 7548–7596 RSC;
(c) L. Pavlovic, J. Vaitla, A. Bayer and K. H. Hopmann, Organometallics, 2018, 37, 941–948 CrossRef CAS;
(d) J. M. Crawford and M. S. Sigman, Synthesis, 2019, 1021–1036 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational methods, alternative energy profiles, tables of calculated energies and computed cartesian coordinates, full ref. 23. See DOI: 10.1039/c9ra10760f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Li Dang
* and
Li Dang![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/O
C/O