
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of a natural PTP1B inhibitor from Rhodomela confervoides on the amelioration of fatty acid-induced insulin resistance in hepatocytes and hyperglycaemia in STZ-induced diabetic rats
Shuju Guo abc,
Lijun Wangabc,
Dong Chend and
Bo Jiang*abc
abc,
Lijun Wangabc,
Dong Chend and
Bo Jiang*abc
aKey Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, 7 Nanhai Road, Qingdao, 266071, China. E-mail: jiangbo@qdio.ac.cn
bCenter for Ocean Mega-Science, Chinese Academy of Sciences, 7 Nanhai Road, Qingdao, 266071, P. R. China
cLaboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
dKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Yushan Road, Qingdao, 266003, P. R. China
First published on 21st January 2020
Abstract
PTP1B is a key negative regulator of insulin signaling transduction, and the inhibition of PTP1B has emerged as a potential therapeutic strategy to treat T2DM. 3,4-Dibromo-5-(2-bromo-6-(ethoxymethyl)-3,4-dihydroxybenzyl)benzene-1,2-diol (BPN), a natural bromophenol isolated from marine red alga Rhodomela confervoides, was found to inhibit PTP1B activity in our previous study. Herein, we identified that BPN functioned as a competitive PTP1B inhibitor and enhanced phosphorylation of IRβ, IRS-1 and Akt in palmitate acid-induced insulin-resistant HepG2 cells. Moreover, 2-deoxyglucose uptake technology-based characterization demonstrated that BPN could stimulate glucose uptake in HepG2 cells. Furthermore, the effects of BPN against oxidative stress were investigated and showed that BPN attenuated oxidative stress by attenuating ROS generation. Finally, long-term oral administration of BPN at dose of 20 mg kg−1 significantly reduced blood glucose levels in streptozotocin-induced diabetic mice and no visible toxic effects were observed. Our work is thus expected to provide a natural uncharged PTP1B inhibitor that could be used as a potential lead compound for further research.
1. Introduction
Diabetes mellitus is a polygenic disease, which can be divided into type 1 diabetes and type 2 diabetes. Type 2 diabetes accounts for 90% of the disease. In type 2 diabetes, insulin resistance is a major patho-physiological abnormality, which means a decreased sensitivity to insulin stimulation.1 The molecular mechanism of insulin resistance is closely related to the insulin signaling pathway.2Protein tyrosine phosphatases (PTPs) are enzymes that catalyze protein tyrosine dephosphorylation. In humans, PTPs can function either as negative or positive modulators in various signal transduction pathways.3 As expected, several PTPs are found to regulate the insulin signaling pathway, such as protein tyrosine phosphatases 1B (PTP1B) and src homology 2-containing PTPase (SH-PTP2).4 PTP1B was the first PTP to be isolated in a homogeneous form and sequenced.5,6 PTP1B can associate with and dephosphorylate activated insulin receptor (IR) or insulin receptor substrates (IRS), and thus act as a negative regulator of insulin signaling.7–11 Moreover, mice that lack PTP1B display enhanced sensitivity to insulin and prolonged insulin receptor autophosphorylation.12 Therefore, inhibition of PTP1B could increase insulin sensitivity by negative regulation of the insulin signaling pathway.
At present, a large number of PTP1B inhibitors have been found. But the limited cell membrane permeability and selectivity failed to support them to progress beyond pre-clinical stage.13–17 Hence, there is an urgent need to find new lead compounds with PTP1B inhibitory activity. As a part of our continuous search for lead compounds for PTP1B inhibition from natural marine sources, we investigated the natural bromophenol 3,4-dibromo-5-(2-bromo-6-(ethoxymethyl)-3,4-dihydroxybenzyl)benzene-1,2-diol (BPN) antidiabetic action in vitro and in vivo.
2. Materials and methods
2.1. Reagents
BPN were isolated and characterized from the marine red alga Rhodomela confervoides in our laboratory as described previously.18 The compound used in the experiments was >95% purity as established by HPLC analysis. Palmitic acid (PA), insulin (INS), fatty acid-free BSA, 2,2-diphenyl-l-picrylhydrazyl (DPPH), streptozotocin (STZ) and MTT were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2-NBDG was obtained from Invitrogen (Carlsbad, CA, USA). Ni-NTA agarose resin was purchased from Qiagen (Hildon, Germany). Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum (FBS) and penicillin-streptomycin were purchased from Gibco (Grand Island, NY, USA). IRS1 antibody, insulin receptor β (L55B10) mouse mAb, anti-phospho-IRβ (Tyr1146) rabbit IgG, phospho-Akt (Ser473) (D9E) XP rabbit mAb were obtained from Cell Signaling Technology (Danvers, MA, USA). p-IRS-1 Antibody (Tyr632)-R was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). GLUT1 antibody, anti-β-actin mouse monoclonal IgG and all secondary antibodies were obtained from Proteintech Group (Chicago, IL, USA). PVDF membrane for western blot analysis was purchased from Millipore (Bedford, MA, USA). The BCA Protein Assay Kit and Pierce™ ECL Western Blotting Substrate were purchased from Thermo Scientific (Waltham, MA, USA). ROS assay kit was obtained from Beyotime Biotechnology (Beyotime, Shanghai, China).2.2. Recombinant human PTP1B1-321 purification
Recombinant PTP1B1-321 was purified by the following procedures. The expression plasmid pET28-a(+)-PTP1B1-321 was constructed in our lab and then transformed into BL21(DE3). Protein expression was induced with 0.5 mM IPTG at 37 °C for 3 h. The PTP1B1-321 protein was purified using Ni-NTA agarose resin (Qiagen) and finally dialyzed into the stock solution (20 mM Tris, pH 7.5, 150 mM KCl, 1 mM DTT, and 50% glycerol).2.3. PTP1B enzymatic assay and inhibition kinetics
The PTP1B enzyme reaction was carried out under the conditions described previously.19 The effect of BPN on the PTP1B-catalyzed pNPP hydrolysis was determined at 37 °C and pH 7.5 in a 100 μL reaction system in a 96-well plate. Each reaction contained 1 μL compound in DMSO and 99 μL assay buffer (10 mM Tris–HCl, 25 mM NaCl, 1 mM EDTA and 2 mM DTT) containing pNPP and 60 nM PTP1B. The PTP1B-catalyzed reaction was started by addition of the PTP1B and terminated by the addition of 1 N NaOH after 30 min. The amount of product p-nitrophenol was determined from the absorbance at 405 nm by a 96-well microplate spectrophotometer. In the kinetic analysis, five different concentrations of pNPP (0.5, 1.0, 2.0, 4.0 and 8.0 mM) were used as PTP1B substrate in the presence of BPN (4, 8 and 16 μM). The inhibition mode for the inhibitor was determined from the Lineweaver–Burk plot. The inhibition constant Ki was determined by plotting Km(app) versus various concentrations of BPN.2.4. Surface plasmon resonance (SPR) assay
The binding affinity of BPN towards PTP1B was assayed using the SPR-based Biacore T200 instrument. hPTP1B1-321 in 10 mM sodium acetate buffer (pH 5.0) was immobilized onto CM5 sensor chips using standard amine coupling method to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 response units. Phosphate-buffered saline (PBS) containing 0.05% P20 was used as running buffer. Briefly, using a flow rate of 10 μL min−1, the surface of flow cell was activated for 7 min using a 1:1 mixture of 100 mM EDC and 100 mM NHS, and subsequently hPTP1B1-321 was injected for 7 min over the surface until the desired immobilization level was reached. Excess reactive carboxy methyl groups on the surface were blocked by a 7 min injection of 1 M ethanolamine (pH 8.5). Flow cells used for reference was activate and blocked as described above, but remained uncoupled. Binding was expressed as relative response units (RU), which was defined as the response obtained from the flow cell containing the immobilized hPTP1B1-321 minus the response obtained from the reference flow cell.
000 response units. Phosphate-buffered saline (PBS) containing 0.05% P20 was used as running buffer. Briefly, using a flow rate of 10 μL min−1, the surface of flow cell was activated for 7 min using a 1:1 mixture of 100 mM EDC and 100 mM NHS, and subsequently hPTP1B1-321 was injected for 7 min over the surface until the desired immobilization level was reached. Excess reactive carboxy methyl groups on the surface were blocked by a 7 min injection of 1 M ethanolamine (pH 8.5). Flow cells used for reference was activate and blocked as described above, but remained uncoupled. Binding was expressed as relative response units (RU), which was defined as the response obtained from the flow cell containing the immobilized hPTP1B1-321 minus the response obtained from the reference flow cell.
2.5. Preparation of BSA-conjugated palmitate complexes
PA solutions were prepared as described previously19 with minor modifications. Briefly, 100 mM PA were dissolved in 0.1 mol L−1 NaOH by heating at 70 °C while 5% BSA solution was prepared by dissolving fatty acid free-BSA in ddH2O by heating at 55 °C. Once BSA and FFA solutions were completely dissolved, PA solutions were diluted 25-times into the BSA solution and thoroughly mixed by pipetting. 4 mM PA/BSA conjugates were subsequently incubated at 55 °C for 15 min and cooled to room temperature. Control BSA was prepared by adding the same amount of 0.1 mol L−1 NaOH into 5% BSA solution. All preparations were filtered, aliquoted and stored at −20 °C.2.6. Cell culture
HepG2 cells were obtained from Qingdao Medical University Affiliated Hospital (Qingdao, Shandong, China). Cells were cultured in DMEM supplemented with 100 unit per mL penicillin, 100 μg mL−1 streptomycin, and 10% FBS at 37 °C in a humidified atmosphere composed of 5% CO2. Briefly, HepG2 cells were plated in 6-well plates (5 × 105 cells per well). After overnight incubation, confluent cells were pre-incubated for 1 h with BPN. Then compound-treated cells were exposed to 0.4 mM PA for 24 h and subsequently starved in FBS-free DMEM containing compound and PA for 6 h followed by stimulation with 100 nM insulin for 30 min.2.7. Cell viability test
To determine cell viability, cells (5 × 103 cells per well) were seeded in sterile 96-well culture plates and treated with BPN for 24 h. Cell viability was evaluated in each well via the addition of 10 μL MTT (5 mg mL−1 in PBS) and cells were incubated for 4 h at 37 °C prior to the reading. For analysis, the cell-free supernatants were removed completely from each well, and 150 μL of DMSO was added. The absorbance densities of the wells were measured using a spectrophotometric multi-well microplate reader (BioTek, Winooski, VT, USA) at the wavelength of 490 nm.2.8. Western blot analysis
Cells were lysed with ice-cold RIPA buffer (Solaibo, Beijing, China) containing freshly added protease inhibitor PMSF and phosphatase inhibitors (Beyotime, Shanghai, China). Protein concentration was determined using BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). 20 μg of protein per lane were separated on SDS-polyacrylamide gels and transferred to PVDF membrane (Millipore, Billerica, MA, USA). Protein detection was performed with anti-IRS1, anti-p-IRS-1 (Tyr632), anti-IR, anti-p-IRβ (Tyr1146), anti-AKT1/2/3, anti-p-Akt (Ser473), anti-β-actin and HRP conjugated secondary antibodies. Immunocomplexes were detected with Pierce™ ECL Western Blotting Substrate (Thermo Scientific, Waltham, MA, USA). Protein band densities were analyzed using the ImageJ software.2.9. Glucose uptake assay
Cellular glucose uptake was measured by using a fluorescent 2-deoxyglucose analog, 2-[N-(7-nitrobenz-2-oxa-1,3-diaxol-4-yl) amino]-2-deoxyglucose (2-NBDG) as a sensitive probe according to the published procedure20 with some modification. Briefly, HepG2 cells were seeded in a black clear-bottomed 96-well culture plate (Corning, NY, USA), 1 × 104 cells per well. After overnight incubation, the cells were pre-incubated with compound for 1 h before 0.4 mM PA treatment. After 24 h, cells were starved in FBS-free low-glucose DMEM containing compound and PA for 6 h before stimulated with 100 nM insulin for 30 min. 2-NBDG (100 μM) was added for 30 min after rinsing cells with FBS-free glucose-free DMEM. Finally, cells were washed with PBS for 3 times and 2-NBDG uptake was measured by the increase in fluorescence intensity at the excitation wavelength of 465 nm and at the emission wavelength of 540 nm using a fluorescence microplate reader (BioTek, Winooski, VT, USA). At last a MTT assay was performed to adjust the errors generated from cell number difference. The level of 2-NBDG uptake was expressed as a percentage of relative fluorescence intensity of control.2.10. Indirect immunofluorescence
HepG2 cells were plated on glass coverslips in 24-well culture plate (5 × 104 cells per well) and treated as Cell culture part presented. Then cells were first fixed with 4% PFA for 15 min at room temperature. Then the samples were incubated for 5 min with PBS containing 0.5% Triton X-100 to permeabilize the cells. Cells were then incubated in the diluted GLUT1 antibody (Proteintech, Chicago, IL, USA) for 1 h at room temperature in a humidified chamber after blocked with 5% BSA/5% NGS for 30 min. Before imaging, samples were incubated with secondary antibody (anti-rabbit FITC) for 1 h at room temperature in the dark. After DAPI staining and mounting, images were captured with a fluorescence microscope (Zeiss, Jena, Germany).2.11. Assay for DPPH radical scavenging activity
The scavenging activity of DPPH was determined according to the method described previously.21 Briefly, a series of concentrations of BPN were added to the ethanolic DPPH solution. After a 30 min incubation period in dark, absorbance was read at 519 nm. Data were expressed as percentage of inhibition and was calculated according to the following formula:| Percentage of inhibition (%) = [OD control − OD sample]/OD control × 100%, |
2.12. Determination of intracellular ROS
Intracellular ROS level in HepG2 cells was measured by using 2′,7′-dichlorofluorescin diacetate (DCFH-DA) as fluorescent probes. Cells grown in 6-well plates were treated as indicated and collected with 0.1% trypsin. Rosup (50 μg mL−1) was used as positive control. Harvested cells were adjusted to a concentration of 1 × 106 cells per mL in FBS-free DMEM. For probe loading, cells were incubated with the DCFH-DA (10 μM) for 20 min at 37 °C and then washed 3 times with FBS-free DMEM. ROS level in individual living cells was analyzed by BD FACSAria II™ flow cytometry (Becton–Dickinson, San Diego, CA).2.13. Anti-hyperglycemic experiments in STZ-induced diabetes rats
20 Wistar rats (10 males and 10 females) weighing 260–280 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). After one week of acclimation, the rats were fed on the high fat diet for 6 weeks to establish diet-induced obesity. The obesity rats were subjected to 24 h fast prior to induction of diabetes, then freshly prepared STZ in citrate buffer (0.1 mol L−1, pH 4.5) was administered intraperitoneally at a single dose of 30 mg kg−1 body weight. The rats with blood glucose ≥16.7 mM after one week were considered diabetic and were used in this study. The rats were randomly assigned to two groups: a model group and a medicated group. The medicated group received 20 mg kg−1 body weight of BPN by oral gavage and the control group was administered 0.5% CMC-Na solution. All animal experiments were performed in accordance with the guidelines established by Institute of Oceanology committees for care and use of laboratory animals.All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Institute of Oceanology, Chinese Academy of Sciences (HAIFAJIZI-2013-3, approval date: 2013-12-09) and approved by the Animal Ethics Committee of Institute of Oceanology, Chinese Academy of Sciences.
2.14. Acute toxicity study
20 healthy ICR mice (10 males and 10 females) weighing 18–22 g were housed in rooms where the temperature was 24 ± 2 °C with a relative humidity 60–80%, and in 12 h light–dark cycle. Prior to each experiment, mice were fasted for 12 h and allowed free access to water. BPN suspended in 0.5% CMC-Na solution and administered by oral gavage with 5000 mg kg−1. The mice were observed for general behavior changes, toxicity and mortality continuously for 14 days.2.15. Statistical analysis
All data were presented as means ± SD values. Differences between the mean values were assessed using one-way analysis of variance (ANOVA). For all the analyses, P < 0.05 was considered significant. Statistical analyses were performed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA).3. Results
3.1. BPN acts as a competitive PTP1B inhibitor with moderate affinity with PTP1B
To elucidate the inhibition mode of BPN (Fig. 1A), the enzyme kinetic was conducted by the Lineweaver–Burk method with various concentrations of pNPP (0.5, 1, 2, 4, 8 mM). As shown in Fig. 1B, the plots were intersected at y-axis, and the Km values increased in dose-dependent manner without changing the Vmax value, indicating that BPN is a competitive PTP1B inhibitor. The Ki value of BPN calculated by plotting apparent Km against different concentrations of BPN was 0.54 μM (Fig. 1C). To further determine the direct binding affinity of BPN, SPR technology was used to monitor its association and dissociation rates. After PTP1B was immobilized on the CM5 chip, serial concentrations (0.39–50 μM) of BPN were injected automatically over the flow cell. As shown in Fig. 1D, BPN bound to PTP1B in a concentration-dependent manner, KD value was 3.3 μM.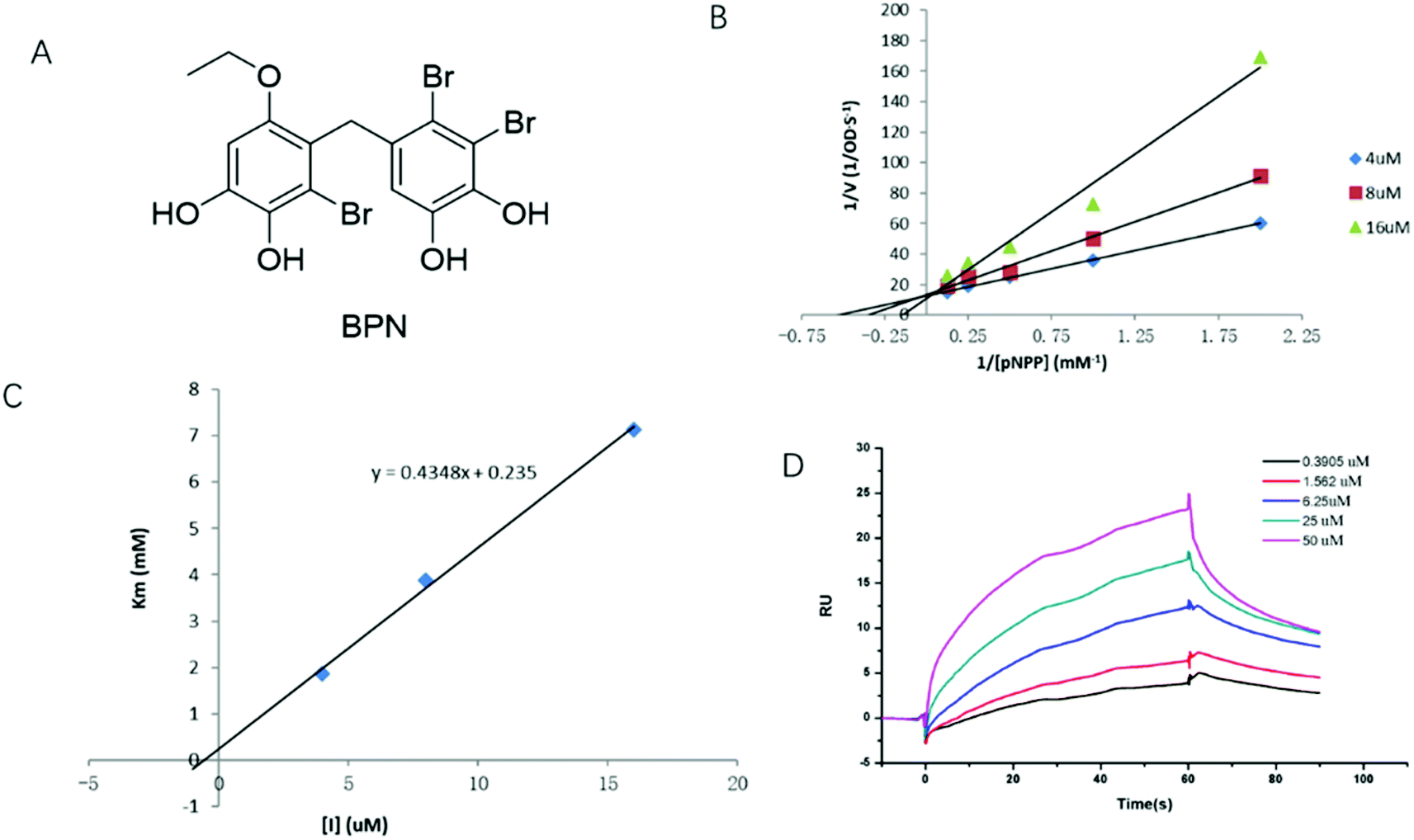 | ||
| Fig. 1 BPN is a competitive PTP1B inhibitor. (A) Chemical structure of BPN. (B) The Lineweaver–Burk plot for inhibition of PTP1B enzyme reaction by BPN. The experiment was performed at 37 °C and pH 7.5. BPN concentrations were 4 (◆), 8 (■) and 16 μM (▲), respectively. (C) Ki determination for BPN. The plot of apparent Km versus the concentrations of BPN. BPN concentrations were 4 (◆), 8 (■) and 16 μM (▲), respectively. (D) SPR dose–response curves of BPN. The Ka, Kd and KD values of BPN were 4.238 × 103 Ms−1, 1.406 × 10−2 s−1, 3.318 × 10−6 M, respectively. KD values of BPN was obtained by fitting the data sets to 1:1 binding model using Biacore T200 Evaluation Software. | ||
3.2. Cytotoxicity of BPN on HepG2 cells
We first examined the cytotoxicity on HepG2 cells treated with various concentrations of BPN, and cell viability was measured by MTT assay. As shown in Fig. 2, up to 1 μM treatment with BPN for 24 h did not reduce the survival of HepG2 cells. Accordingly, further in vitro studies on the anti-insulin resistance activity of BPN were conducted in the range of 0.1–1 μM.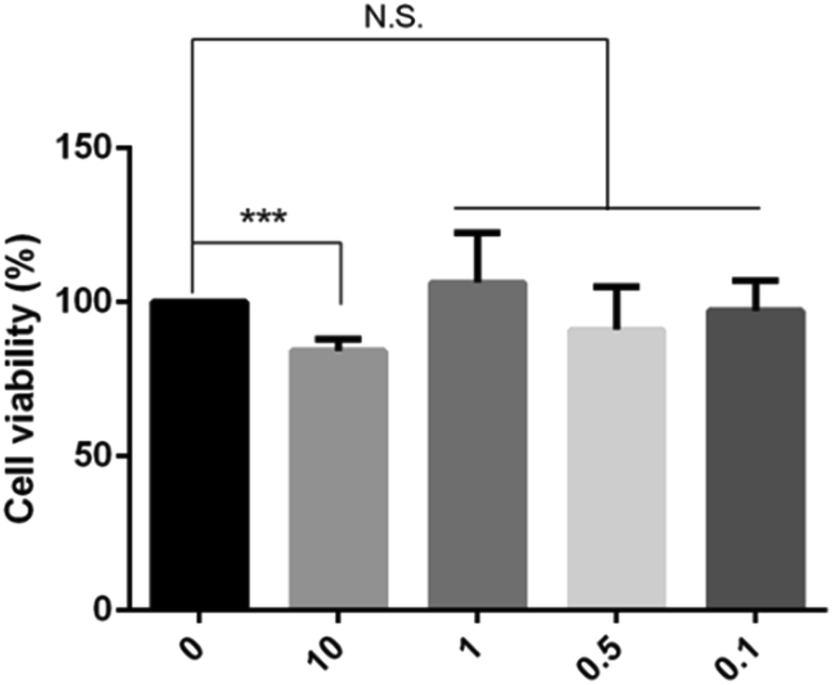 | ||
| Fig. 2 Cytotoxicity of BPN on HepG2 cells. Cells were treated with the indicated concentrations of BPN (0.1–10 μM). Cell viability was evaluated with the MTT Assay. The data shown in the graphs are the mean ± SD values (n = 4). ***P < 0.001 versus the control. N.S., non-significant. | ||
3.3. HPN improves PA-induced insulin resistance in HepG2 cells by activating insulin signaling
The ability of BPN to reverse impaired insulin signaling was investigated in PA-induced insulin resistant HepG2 cells. Cells were treated with 0.25, 0.5 and 1.0 μM BPN and 0.25 mM PA for 16 h, and then 100 nM insulin was added into the cells. Under control condition, insulin stimulated an obvious increase in phosphorylation of IRβ at Tyr1146 and IRS-1 phosphorylation at Tyr632. Incubation with PA caused significant decrease in insulin stimulated IRβ at Tyr1146 and IRS-1 phosphorylation at Tyr632 as expected. As shown in Fig. 3, treatment of BPN caused a significant increase in IRβ and IRS-1 phosphorylation. We also evaluated effects of BPN on phosphorylation level of Akt at Ser473, a key downstream effector of the insulin-signaling cascade. Likewise, a decrease in expression of phosphorylated Akt levels can be observed after PA stimulation. Presence of BPN at 0.25 μM remarkably increased phosphorylated Akt levels, which were comparable to the positive control (insulin-stimulated) group (Fig. 3). The results above demonstrated that BPN was able to recover PA-induced insulin resistance in HepG2 cells.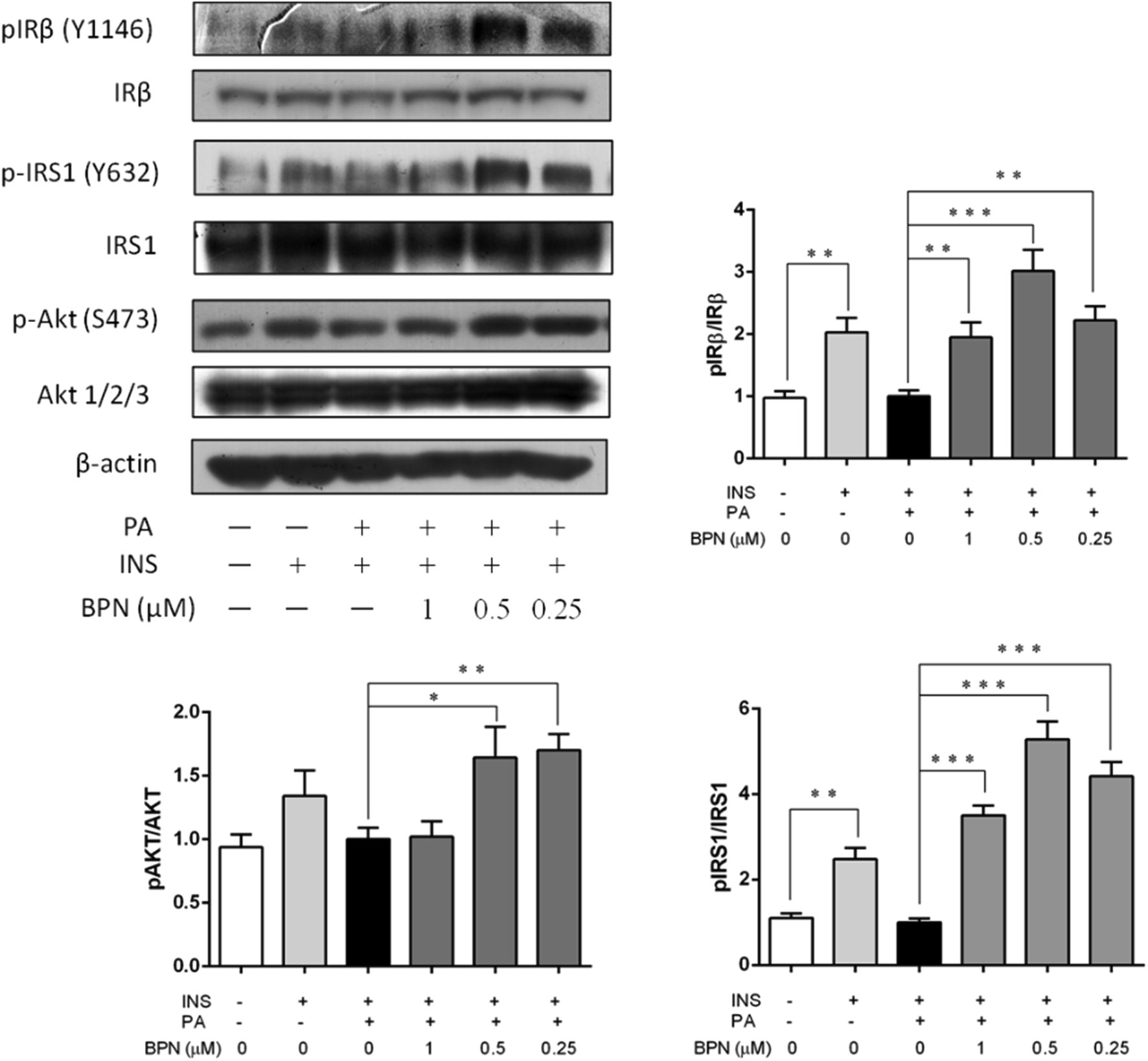 | ||
| Fig. 3 Effects of BPN on restoration of PA-induced insulin signaling. BPN-treated HepG2 cells were incubated with PA for 16 h prior to stimulation with 100 nM insulin for 30 min. Immunoblotting assay was used to determine the phosphorylated and non-phosphorylated protein levels of IRβ, IRS1 and Akt. β-Actin was used as loading control. *P < 0.05, **P < 0.01, ***P < 0.001 versus PA-treated group. | ||
3.4. BPN promotes glucose uptake through GLUT1 translocation in HepG2 cells
To investigate the effect of BPN to increase insulin-stimulated glucose uptake, 2-NBDG, a fluorescent D-glucose analog, was used to monitor glucose uptake in PA-treated HepG2 cells. As shown in Fig. 4A, BPN at concentration of 0.2 μM significantly enhanced the insulin-stimulated uptake of 2-NBDG in insulin-resistant HepG2 cells compared with the control. We next examined whether BPN induced stimulation of insulin signaling promote glucose uptake via the translocation of GLUT1 to the plasma membrane. As shown in Fig. 4B, treatment with BPN significantly increased the GLUT1 levels in the plasma membrane. The above findings demonstrate that BPN improved glucose uptake in PA-treated HepG2 cells.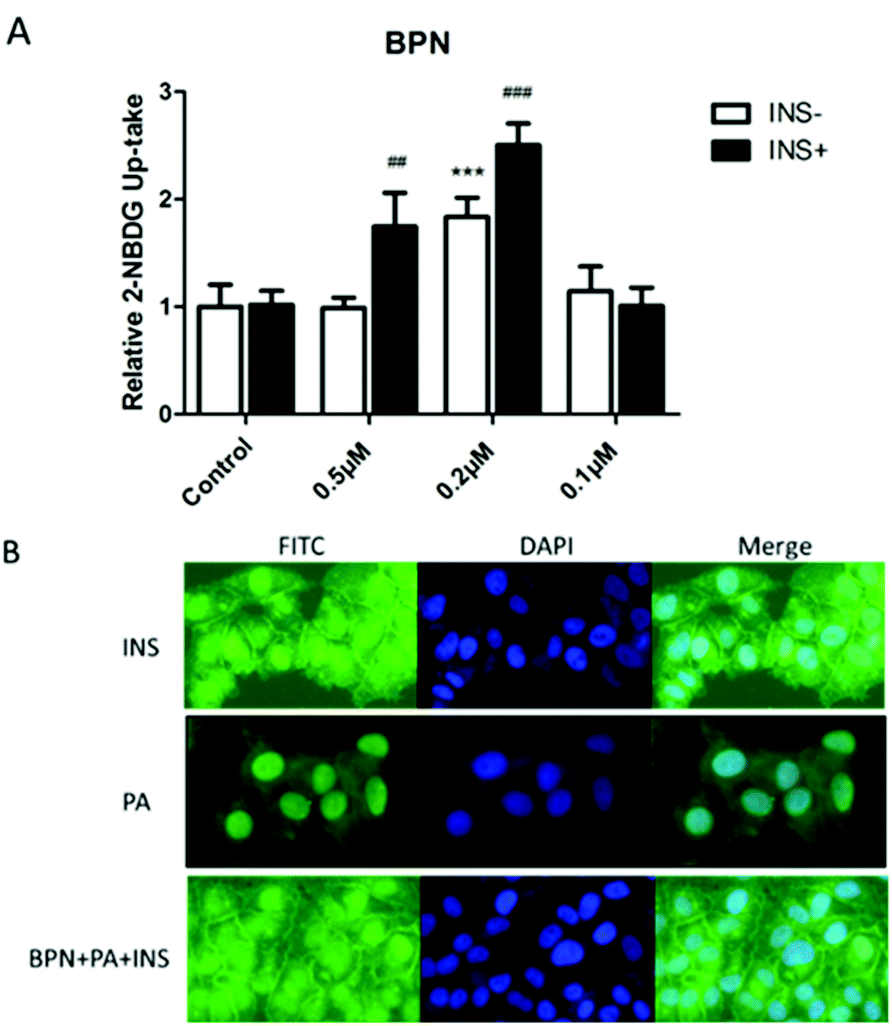 | ||
| Fig. 4 BPN improves glucose uptake ability of HepG2 cells. (A) Microplate fluorimeter measurement of 2-NBDG uptake in PA-treated HepG2 cells. 1 × 104 cells per well were seeded in a black clear-bottomed 96-well culture plate. After overnight incubation, the cells were pre-incubated with BPN for 45 min before PA (0.25 mM) treatment. After 16 h, cells were cultured in a serum-free low glucose culture medium for 3 h before the addition of 2-NBDG. ##P < 0.01, ###P < 0.001, ***P < 0.001 versus PA-treated group. (B) Fluorescent microscopic imaging of GLUT1 expression in cell membrane. Cells were treated with indicated concentrations of BPN for 45 min, and incubated for another 16 h with 0.25 mM PA. Then, 100 nM insulin was added. Immunofluorescence assay was described in the Materials and methods section. | ||
3.5. Antioxidant activities of BPN
BPN belongs to a kind of polyphenols, which are considered to display antioxidant and radical scavenging activities. Therefore, we used DPPH radical scavenging assay to evaluate the antioxidant activities of BPN. As shown in Fig. 5A, BPN exhibited DPPH radical scavenging activity in a concentration-dependent manner. Given that ROS production closely associated with fatty acid-induced insulin resistance, the effect of BPN on PA-induced ROS production in HepG2 cells were also investigated. As shown in Fig. 5B, treatment with BPN at 0.5 μM effectively decreased PA-induced ROS generation (P < 0.01).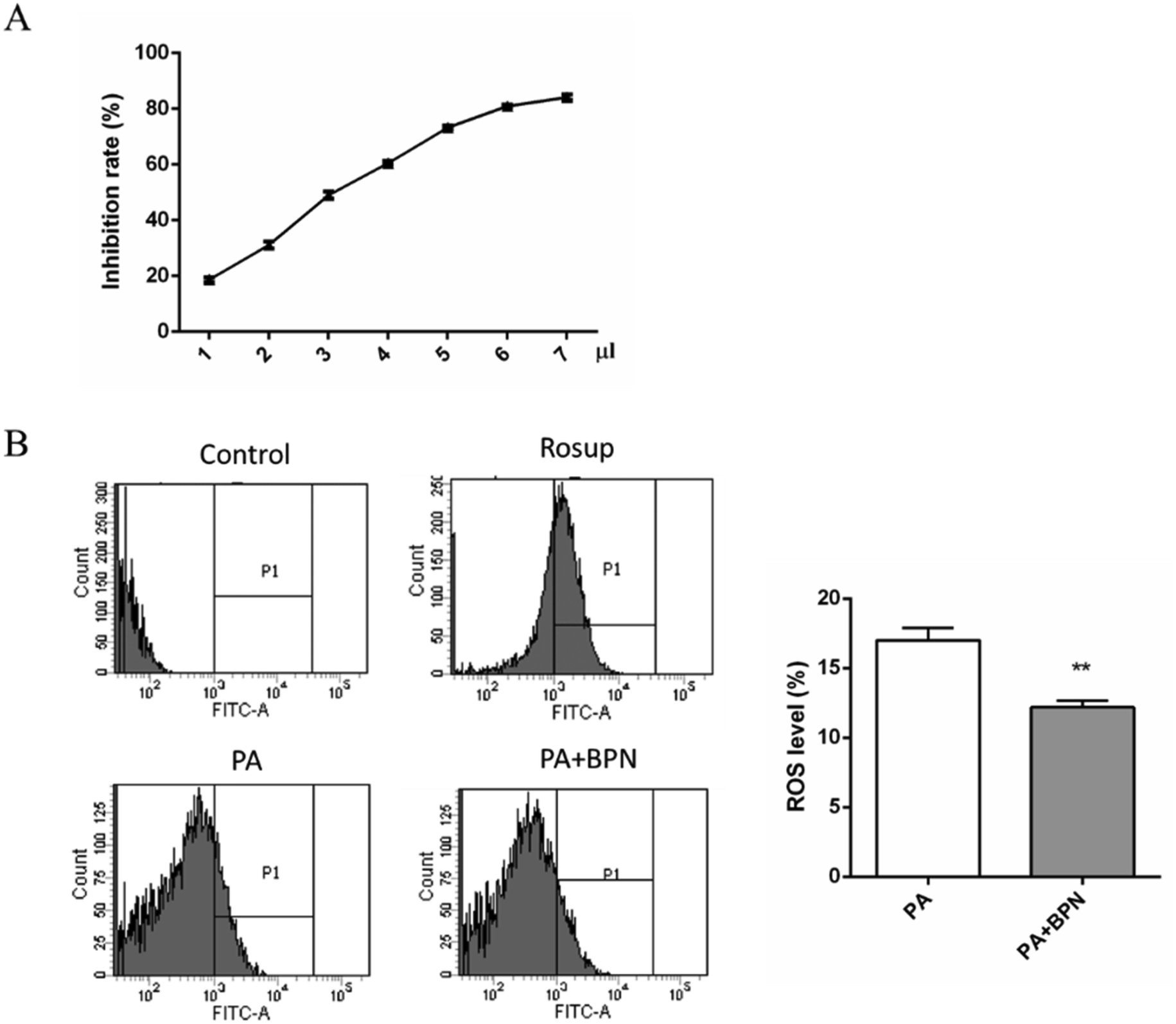 | ||
| Fig. 5 Antioxidant activities of BPN. (A) DPPH-scavenging activity of BPN. Various concentrations of BPN were incubated with DPPH at 25 °C for 30 min, absorbance was read at 519 nm. (B) Flow cytometry assay was used to determine the percentage of PA-induced ROS generation in HepG2 cells. Cells were treated with PA for 16 h in absence or presence of BPN. After incubation, cells were washed with PBS, loaded with probe DCFH-DA and ROS generation was evaluated. **P < 0.01 relative to control. | ||
3.6. BPN decreases blood glucose levels in STZ-induced diabetes rats
BPN at dose of 20 mg kg−1 was administrated orally to STZ-induced diabetic rats once daily for 21 days. Blood glucose was determined at every 7 days. As shown in Fig. 6, BPN treated group exhibited an obvious reduction in blood glucose level (from 25.7 ± 4.4 mM to 15.6 ± 4.0 mM) since the first 7 day time point (P < 0.05). After the treatment for 21 days, the level of fasting blood glucose in the BPN treated group (11.4 ± 3.8 mM) was significantly lower than that in the model group (25.2 ± 4.1 mM) (P < 0.01).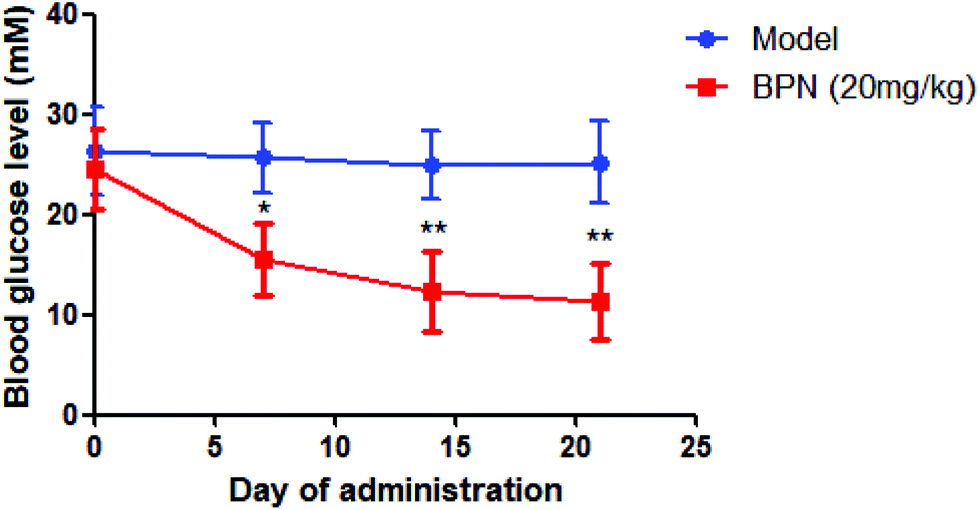 | ||
| Fig. 6 Hypoglycemic effects of BPN in STZ-induced diabetic rats. STZ-induced diabetic rats were administered vehicle alone or 20 mg per kg per day of BPN for 21 days. Data are expressed as mean ± SD (n = 10). *P < 0.05, **P < 0.01 versus model. | ||
3.7. BPN shows low toxicity in ICR mice
BPN was further evaluated for in vivo acute toxicity in ICR mice. As shown in Table 1, the mice were administered by BPN at dose of 5000 mg kg−1 and did not induce any signs of acute toxicity or mortality. The mice had glossy fur, moved with ease and demonstrated normal food intake, urination and defecation during the 14 day observation period. Under limited stomach capacity of mice, therefore, maximum tolerable dose of BPN was decided as 5000 mg kg−1.| Dose (mg kg−1) | Mortality | |
|---|---|---|
| 5000 | Male | 0/10 |
| Female | 0/10 | |
4. Discussions
PTP1B has been considered as a valid therapeutic target for the treatment of type 2 diabetes and obesity. For efficient inhibition of PTP1B, the inhibitors mainly contain negatively-charged non-hydrolyzable pTyr mimetic, which will competitively occupy the catalytic site of PTP1B. However, the low cell permeability and low bioavailability of these inhibitors have limited their application for the development of effective drugs. The naturally derived bromophenol BPN is an uncharged PTP1B inhibitor with IC50 0.84 μM. In the present studies, the results of enzyme kinetics studies revealed that BPN showed competitive inhibition against PTP1B, indicating that BPN can bind to the active site of PTP1B. To eliminate the false positive caused by enzymatic reaction, we used surface plasmon resonance (SPR), which is a well-established screening technique to detect biomolecular interaction,22 to study the binding affinity of BPN with PTP1B. The results showed that the affinity of BPN binding to PTP1B is 3.3 μM, indicating that BPN was a direct PTP1B inhibitor.Insulin resistance and pancreatic β-cell dysfunction are the common causes of type 2 diabetic mellitus.23 In an insulin resistance state, a defective insulin action increases gluconeogenesis in the liver, declines glycogen synthesis and allows enhancement of hepatic glucose output, which contributes to the development of T2DM.24 PTP1B has been implicated as a negative regulator of insulin signaling pathway in vitro.25 It catalyzes dephosphorylation of tyrosine residues in IRβ subunit and IRS-1, which in turn suppresses insulin signaling.26 Early studies have demonstrated that overexpression of PTP1B inhibits phosphorylation of IR and IRS-1 leading to insulin resistance.27,28 As a result, IRβ and IRS-1 have been identified as two major substrates of PTP1B.
Studies have shown that increased plasma fatty acid levels lead to accelerated lipid oxidation in liver and hepatic insulin resistance by inhibiting the insulin signal transduction.29,30 Thereinto PA is the most liable to insulin resistance due to it is an endogenous precursor for the biosynthesis of ceramide and glycosphingolipid. In the present study, HepG2 cells were treated with 0.25 mM PA for 16 h to establish insulin resistance state, which was confirmed by impaired tyrosine phosphorylation of IRβ and IRS-1, and serine phosphorylation of Akt. Consequently, our study revealed that BPN treatment promotes the phosphorylation of IRβ, IRS-1 and Akt in PA-induced HepG2 cells. The glucose transporter GLUT1 has an essential role for constant uptake of glucose in hepatic cells. In the present study, we demonstrated that BPN increased insulin-stimulated 2-NBDG uptake and promoted the translocation of GLUT1 from the cytoplasm to the cell membrane, indicating the treatment of HepG2 cells with BPN greatly facilitates cellular glucose uptake through GLUT1 translocation.
It is well-known that oxidative stress contributes to the progression of various diseases, including T2DM.31 Recent studies have demonstrated that ROS affect insulin signaling.32,33 Insulin-mediated stimulation leads to the activation of intracellular signaling cascade involving the IR, IRS and Akt.34 Given that polyphenol display antioxidant and radical scavenging activities,35,36 we hypothesized that BPN, as a kind of bromophenol, may counteract oxidative stress. To verify our hypothesis, the antiradical activity and ROS levels in HepG2 cells of BPN were investigated. In the present study, as expected, we observed that BPN exhibited DPPH radical scavenging activity in a concentration-dependent manner, and attenuated oxidative stress by decreasing ROS levels. Hence, our results provided evidence the effects of BPN on amelioration of insulin resistance may be attributed to its antioxidant activity and attenuation of ROS generation in HepG2 cells.
With the in vitro data in hands, we employed STZ-induced diabetic rats to assess the hypoglycemic effect of BPN in vivo. Administration of BPN at dose of 20 mg kg−1 exerted an significant reduction of blood glucose levels compared with model group during a 21 day period. The results of acute toxicity assays also suggested that BPN showed extremely low toxicity in oral administration. According to these observations, we found that BPN was a potential lead compound with hypoglycemic potency and safety. Given that, however, there was single dose of BPN used in the assessment, additional research are needed to assess the potency of BPN treatment on blood chemical parameters such as triglyceride, cholesterol, and free fatty acids at various dosages in different diabetic animal models.
5. Conclusion
Despite the intensive efforts and progress in the past few years, many synthetic compounds that showed excellent in vitro inhibitory effects on PTP1B have been limited by their poor cell permeability and oral bioavailability. In the present study, a naturally derived bromophenol BPN has been investigated as a PTP1B inhibitor both in vitro and in vivo. As a competitive PTP1B inhibitor, BPN not only increased phosphorylation levels of IRβ, IRS-1 and Akt, but also promoted glucose uptake in insulin resistant HepG2 cells. In addition, BPN exhibited potent antioxidant activity as well as reduced ROS levels in HepG2 cells. Finally, long-term oral administration of BPN at 20 mg kg−1 significantly reduced blood glucose levels in STZ-induced diabetic mice and no visible toxic effects were observed. Taken together, our work has demonstrated that the natural uncharged PTP1B inhibitor may be useful as potential lead compound for further research.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
This study was supported by grants from the Key Research and Development Project of Shandong province (2018GSF118208 and 2018GSF118223), and NSFC-Shandong Joint Fund (U1706213).References
- L. Chen, D. J. Magliano and P. Z. Zimmet, Nat. Rev. Endocrinol., 2012, 8, 228–236 CrossRef CAS PubMed.
- R. Catanzaro, A. Lorenzetti, F. Allegri, H. Yadav, U. Solimene, A. K. Kumaraju, E. Minelli, C. Tomella, A. Polimeni and F. Marotta, Acta Bio Med. Atenei Parmensis, 2012, 83, 95–102 CAS.
- A. Alonso, J. Sasin, N. Bottini, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon and T. Mustelin, Cell, 2004, 117, 699–711 CrossRef CAS PubMed.
- B. J. Goldstein, F. Ahmad, W. Ding, P. M. Li and W. R. Zhang, Mol. Cell. Biochem., 1998, 182, 91–99 CrossRef CAS PubMed.
- A. B. Comeau, D. A. Critton, R. Page and C. T. Seto, J. Med. Chem., 2010, 53, 6768–6772 CrossRef CAS PubMed.
- L. F. Zeng, J. Xu, Y. He, R. He, L. Wu, A. M. Gunawan and Z. Y. Zhang, ChemMedChem, 2013, 8, 904–908 CrossRef CAS PubMed.
- W. J. Ji, X. L. Chen, J. Lv, M. Wang, S. T. Ren, B. X. Yuan, B. Wang and L. N. Chen, Int. J. Endocrinol., 2014, 312452 Search PubMed.
- M. F. Hsu and T. C. Meng, J. Biol. Chem., 2010, 285, 7919–7928 CrossRef CAS PubMed.
- E. Panzhinskiy, Y. Hua, B. Culver, J. Ren and S. Nair, Diabetologia, 2013, 56, 598–607 CrossRef CAS PubMed.
- B. J. Goldstein, A. Bittner-Kowalczyk, M. F. White and M. Harbeck, J. Biol. Chem., 2000, 275, 4283–4289 CrossRef CAS PubMed.
- M. R. Calera, G. Vallega and P. F. Pilch, J. Biol. Chem., 2000, 275, 6308–6312 CrossRef CAS PubMed.
- M. Elchebly, P. Payette, E. Michaliszyn, W. Cromlish, S. Collins, A. L. Loy, D. Normandin, A. Cheng, J. Himms-Hagen, C. C. Chan, C. Ramachandran, M. J. Gresser, M. L. Tremblay and B. P. Kennedy, Science, 1999, 283, 1544–1548 CrossRef CAS PubMed.
- I. G. Boutselis, X. Yu, Z.-Y. Zhang and R. F. Borch, J. Med. Chem., 2007, 50, 856–864 CrossRef CAS PubMed.
- D. P. Wilson, Z.-K. Wan, W.-X. Xu, S. J. Kirincich, B. C. Follows, D. Joseph-McCarthy, K. Foreman, A. Moretto, J. Wu, M. Zhu, E. Binnun, Y.-L. Zhang, M. Tam, D. V. Erbe, J. Tobin, X. Xu, L. Leung, A. Shilling, S. Y. Tam, T. S. Mansour and J. Lee, J. Med. Chem., 2007, 50, 4681–4698 CrossRef CAS PubMed.
- M. A. T. Blaskovich, Curr. Med. Chem., 2009, 16, 2095–2176 CrossRef CAS PubMed.
- A. P. Combs, J. Med. Chem., 2010, 53, 2333–2344 CrossRef CAS PubMed.
- K. A. Lantz, S. G. E. Hart, S. L. Planey, M. F. Roitman, I. A. Ruiz-White, H. R. Wolfe and M. P. McLane, Obesity, 2010, 18, 1516–1523 CrossRef CAS PubMed.
- X. Fan, N. J. Xu and J. G. Shi, J. Nat. Prod., 2003, 66, 455–458 CrossRef CAS PubMed.
- J. Luo, N. Wu, B. Jiang, L. Wang, S. Wang, X. Li, B. Wang, C. Wang and D. Shi, Mar. Drugs, 2015, 13, 4452–4469 CrossRef CAS PubMed.
- D. W. Jung, H. H. Ha, X. Zheng, Y. T. Chang and D. R. Williams, Mol. BioSyst., 2011, 7, 346–358 RSC.
- Z. Liu, G. Li, C. Long, J. Xu, J. Cen and X. Yang, Food Chem., 2018, 253, 5–12 CrossRef CAS PubMed.
- L. X. Liao, X. M. Song, L. C. Wang, H. N. Lv, J. F. Chen, D. Liu, G. Fu, M. B. Zhao, Y. Jiang, K. W. Zeng and P. F. Tu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5986–E5994 CrossRef CAS PubMed.
- R. A. DeFronzo, R. C. Bonadonna and E. Ferrannini, Diabetes Care, 1992, 15, 318–368 CrossRef CAS PubMed.
- L. Agius, Best Pract. Res., Clin. Endocrinol. Metab., 2007, 21, 587–605 CrossRef CAS PubMed.
- T. O. Johnson, J. Ermolieff and M. R. Jirousek, Nat. Rev. Drug Discovery, 2002, 1, 696–709 CrossRef CAS PubMed.
- J. C. Byon, A. B. Kusari and J. Kusari, Mol. Cell. Biochem., 1998, 182, 101–108 CrossRef CAS PubMed.
- K. A. Kenner, E. Anyanwu, J. M. Olefsky and J. Kusari, J. Biol. Chem., 1996, 271, 19810–19816 CrossRef CAS PubMed.
- J. M. Zabolotny, F. G. Haj, Y. B. Kim, H. J. Kim, G. I. Shulman, J. K. Kim, B. G. Neel and B. B. Kahn, J. Biol. Chem., 2004, 279, 24844–24851 CrossRef CAS PubMed.
- P. Kovacs and M. Stumvoll, Best Pract. Res., Clin. Endocrinol. Metab., 2005, 19, 625–635 CrossRef CAS PubMed.
- G. Boden and G. I. Shulman, Eur. J. Clin. Invest., 2002, 32(suppl. 3), 14–23 CrossRef CAS PubMed.
- J. W. Baynes and S. R. Thorpe, Diabetes, 1999, 48, 1–9 CrossRef CAS PubMed.
- Z. Cheng, Y. Tseng and M. F. White, Trends Endocrinol. Metab., 2010, 21, 589–598 CrossRef CAS PubMed.
- K. Loh, H. Deng, A. Fukushima, X. Cai, B. Boivin, S. Galic, C. Bruce, B. J. Shields, B. Skiba, L. M. Ooms, N. Stepto, B. Wu, C. A. Mitchell, N. K. Tonks, M. J. Watt, M. A. Febbraio, P. J. Crack, S. Andrikopoulos and T. Tiganis, Cell Metab., 2009, 10, 260–272 CrossRef CAS PubMed.
- D. R. Alessi and P. Cohen, Curr. Opin. Genet. Dev., 1998, 8, 55–62 CrossRef CAS PubMed.
- H. T. Balaydin, I. Gulcin, A. Menzek, S. Goksu and E. Sahin, J. Enzyme Inhib. Med. Chem., 2010, 25, 685–695 CrossRef CAS PubMed.
- Y. Cetinkaya, H. Gocer, A. Menzek and I. Gulcin, Arch. Pharm., 2012, 345, 323–334 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |