
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the fibrillation mechanism of ovalbumin in the presence of mercury at its isoelectric pH†
Manjumol Mathewa,
Charuvila T. Aravindakumarb and
Usha K. Aravind*c
aAdvanced Centre of Environmental Studies and Sustainable Development, Mahatma Gandhi University, Kottayam-686 560, India
bSchool of Environmental Sciences, Inter University Instrumentation Centre, Mahatma Gandhi University, Kottayam-686 560, India
cSchool of Environmental Studies, Cochin University of Science and Technology, Kochi-682022, Kerala, India. E-mail: uka@cusat.ac.in
First published on 24th April 2020
Abstract
The intriguing resemblances of amyloid fibrils and spider silk in protein aggregation diseases have instigated the exploration of identical structural features if any in their oligomeric pathways. The serpin group protein, ovalbumin, on defolding in HgCl2 shares commonness to the micellar pathway of spidroins for their aggregation in response to a pH trigger. The structural feature changes from monomer to worm like fibril with a shift in the primary protein pH to slightly acidic pH (4.5), and then proceeds through a secondary nucleation pathway to ‘hillock’ and ‘hydra’ like protofibrils rich in β-sheet and random coil conformers upon exposure to mercury. The findings are backed by atomic force microscopy, confocal Raman spectroscopy and fluorescence measurements. Unlocking such structural features can favorably assist in the design of therapeutics.
1 Introduction
Nature has an amazing protocol in the maintenance of a good balance of all kind of phenomena in every respect. There are numerous examples to illustrate this protocol. The tropic level is one of the best examples. At each tropic level the number of organisms presented at any point of time, and generated over time, is precisely controlled. A closer look into the cell of each organism also presents a similar phenomenon; a well decided balance with respect to the life span, formation and destruction of proteins. There are many cellular events that generate misfolded proteins which are cleared by chaperons. It is such an intriguing fact to know that in living systems various molecular chaperons interfere in the formation of amyloid state. But when the equilibrium is perturbed, molecular chaperons fails to manage the mechanistic pathway leading to the dissolution of aggregates. This disrupts the routine ubiquitin–proteasome pathway and autophagy.1,2Protein unfolding studies to elucidate its stepwise structural pathways that lead to amyloid fibril formation has been an active research topic for quite some time. Some of the well-studied proteins include human prions, Aβ peptide and α-synuclein fibrils.3–5 The significance lies in the direct association with a broad spectrum of protein aggregation diseases. These protein misfolding diseases can be initiated by a number of environmental triggers, leading to cellular toxic intermediates (β-sheet oligomers). The oligomeric forms of amyloidogenic proteins interact with cell membranes and are known to perturb structural integrity and permeation character.6 The health crisis due to human degenerative diseases led to several investigations to identify the cellular toxic β-sheet rich intermediates of proteins, their mechanistic pathways that lead to fibrillar formation and the subsequent interaction with cellular membranes. The findings indicate that the low molecular weight soluble oligomers and the high molecular weight protofibrils are more toxic than the final insoluble fibrils which form the amyloid plaque.7
Environmental factors will, obviously, have a huge share in protein aggregation. Increased exposure level of endocrine disrupting compounds, traditional contaminants (cadmium, mercury, PAHs, pesticides etc.) and similar toxins pose novel challenge in amyloid fibril formation.8–11 Human health is not an exception due to dermal and dietary exposures. The consumption of aquatic and wildlife species increase the intake level. Among the contaminants, mercury can be considered as a typical model due to its capacity to bio-accumulates and bio-magnify at each tropic levels is well known.12,13 Over the years, the entry of mercuric compounds has only increased at aquatic and terrestrial ecosystems.14 The intake level and hence its presence in various matrices is regulated by EPA. But most of the studies indicate its accumulation in the human body.15,16 Therefore the study of its interaction with any of the cell component is of very relevant.17–19 However, protein is unique in several respects. Most of its functions are related to its structure, conformation, and micropolarity of the environment. Care is also taken to have the right choice of a protein from the serpin family as they are found in the silk gland.20 Serpin group of proteins are interesting candidate from several perspectives.21 The proteins that are responsible for blood coagulation are serpins, antithrombin and antitrypsin.22 Serpinopathy, where protein aggregation leads to many diseases including the widespread liver cirrhosis is presently a hot topic of research.23,24 Many of these diseases share a common end stage that is elongated fibril formation. The functional proteins aggregate and become inactive. In the present study egg white ovalbumin (OVA), a non-inhibitory member of serpin super family, is chosen.
Here we address the perturbation in the form of toxins at slightly acidic pH normally encountered in intracellular regions. The remarkably distinct toxic pathway with features of spider silk (fibrillar) is discussed with its molecular structure and evolution. The structural similarities of the oligomers with spider spinning dope to precursor silk are brought out at molecular level. Atomic force microscopic images and Raman spectroscopic features of prefibrils formed by OVA, on exposure to mercuric chloride (HgCl2) at isoelectric solution pH 4.5 (pI) support the spider dope like conformation. Each added concentration (0–6.25 μM level) represent protofibrils on route to spider silk like amyloids. The amyloidogenic nature and mechanistic pathway is supported by fluorescence (steady-state and life-time) measurements.
2 Materials and methods
2.1 Materials
Ovalbumin was purchased from Sigma-Aldrich and was used without further purification. Protein solutions (0.1 mg ml−1) were prepared in sodium acetate–acetic acid buffer (pH 4.5). Ultra-pure water was used for the preparation of all the solutions and buffers. Other reagents: sodium acetate, acetic acid and mercuric chloride (HgCl2) were procured from Merck-India and glycine from SRL-Mumbai. All other chemicals were of analytical grade and were used as supplied. The pH of the buffer was checked with digital pH meter (model 335 Systronics, Kochi).2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The 295 nm laser diode was used as a light source for the excitation of protein and emission was monitored at respective emission wavelengths. The data was analyzed by using DAS6 software attached with the system.:7 mixture of H2O2/H2SO4 at 80 °C for 20 min. The wafers were then washed several times, dipped in a 1:1:5 mixture of H2O2/NH4OH/water at 70 °C for 15 min, and washed several times. These wafers were kept in deionized water and dried under a gentle stream of nitrogen gas. A fiber coupled DPSS laser at 532 nm (70 mW) was used as an excitation source for Raman spectroscopic studies. The laser line was focused onto the sample using a 100× Zeiss objective. The samples were drop coated on a quartz coverslips and air dried for approximately 20 minutes. All the spectra were averaged over 20 scans with an acquisition time of 20 s. Data was then collected using a multimode fiber (UHTS 300) with 300 mm focal length and grating 1800 g mm−1, and the signal was finally detected using an air-cooled CCD detector. The microscope base is also equipped with an active vibration isolation system, active in the range 0.7–1000 Hz. The baseline corrected and smoothened Raman spectra were plotted in Origin 6 Software. The deconvolution of Raman spectra was done with Origin 6 Software. The acquired spectra and AFM images were processed and analyzed using the Witec Project 4 program.
000. The 295 nm laser diode was used as a light source for the excitation of protein and emission was monitored at respective emission wavelengths. The data was analyzed by using DAS6 software attached with the system.:7 mixture of H2O2/H2SO4 at 80 °C for 20 min. The wafers were then washed several times, dipped in a 1:1:5 mixture of H2O2/NH4OH/water at 70 °C for 15 min, and washed several times. These wafers were kept in deionized water and dried under a gentle stream of nitrogen gas. A fiber coupled DPSS laser at 532 nm (70 mW) was used as an excitation source for Raman spectroscopic studies. The laser line was focused onto the sample using a 100× Zeiss objective. The samples were drop coated on a quartz coverslips and air dried for approximately 20 minutes. All the spectra were averaged over 20 scans with an acquisition time of 20 s. Data was then collected using a multimode fiber (UHTS 300) with 300 mm focal length and grating 1800 g mm−1, and the signal was finally detected using an air-cooled CCD detector. The microscope base is also equipped with an active vibration isolation system, active in the range 0.7–1000 Hz. The baseline corrected and smoothened Raman spectra were plotted in Origin 6 Software. The deconvolution of Raman spectra was done with Origin 6 Software. The acquired spectra and AFM images were processed and analyzed using the Witec Project 4 program.3. Results and discussion
The concentration dependent evolution of topographical features of self-assembled nanoscale structures were recorded using AFM. The oligomeric structural features at the pI are presented in Fig. 1. Prior to the exposure, the predominant morphology observed is worm like fibrils (WL) (Fig. 1A) with height ranging from 4–7 nm (ESI Fig. S1B†) and W = 20–100 nm (Fig. 1B). WL fibrils are considered as the end species, which are incapable of being converted to LS fibrils. WL are distinct in many respects from long straight fibrils (LS). WL is found to exist in many protein aggregation pathways and were considered as off pathway species until recently. Their on pathway, nature to amyloid, is explicitly expressed in β2m as pointed out by Radford.25 WL carries the features of mature fibrils but has identity to toxic oligomers of other proteins especially in sharing a common epitope. Worm like fibrils normally appear from partially unfolded state of protein via non-nucleated pathway.26 These are less ordered compared to LS fibrillar counterparts. Both LS and WL fibrils can bind to the amyloid-specific dye Congo red and Thioflavin-T.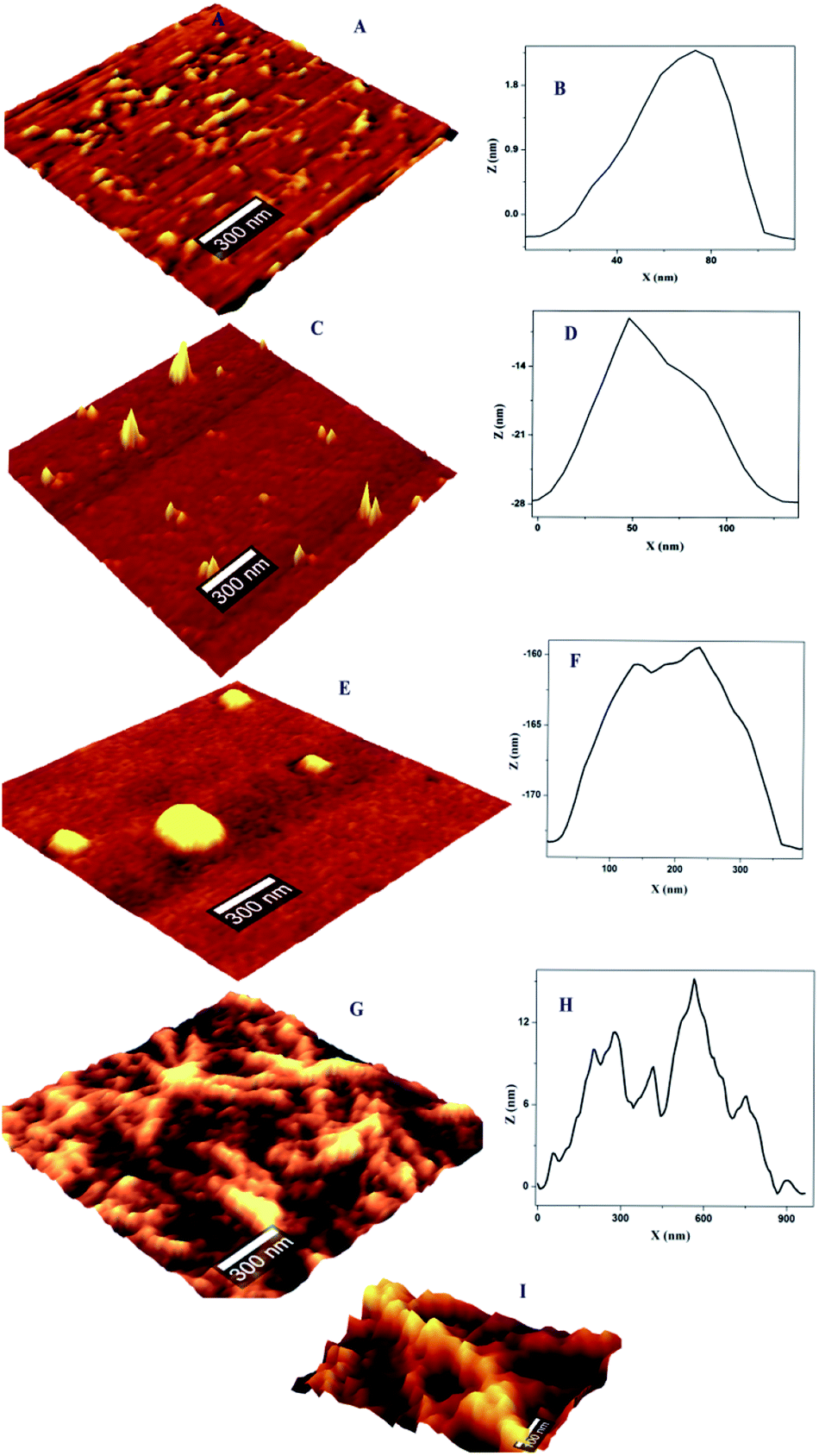 | ||
| Fig. 1 Topographical features of OVA and OVA–HgCl2 complex at pI; (A) AFM images of OVA (pH 4.5) prior to exposure of HgCl2, showing WL fibrils and the corresponding (B) width profile; (C) formation of ‘hillock’ like oligomer after introducing of 1.5 μM HgCl2 and the corresponding (D) width profile (E) globule structure of oligomer with the increment of HgCl2 to 2.5 μM and the corresponding (F) width profile (G) ‘hydra’ like oligomer with the increment of HgCl2 to 6.25 μM and the corresponding (H) width profile (I) magnified view of fibrils of ‘hydra’ like structure. | ||
On the introduction of HgCl2 a new kind of oligomeric feature has appeared (Fig. 1C). It is less populated with a reduction in height and width. Subsequent exposure resulted in globules like nano structure where only few could be seen (Fig. 1E). On further increment of HgCl2 interesting structural feature appeared with proliferation (Fig. 1G). It appears that the third increment motivated the nano-scale assembly in such a way that ‘hillock’ like structural features joined like arms to the globules head with abundance (Fig. 1I). Only the globules like structural feature is familiar in protein fibrillation pathway. But the other structural feature of protofibrils is not encountered in the mechanistic pathway of fibrillation. A series of spectroscopic evaluations were carried out to get more insight into the aggregation pathway. Fluorescence quenching experiments and life time decay curves gave clear cut depictions of aggregation and early growth stages. A plot of log[F0 − F/F] against log[HgCl2] at isoelectric pH is shown in Fig. 2. It is evident from the figure that the double logarithmic plot also shows three distinct regions. The magnitudes of binding constant (Kb), number of binding site (n), and free energy of binding (ΔG0) have been estimated for the first two regions (I–II of Fig. 2) and are summarized in Table 1. Region I of pH 4.5 showed a highly cooperative nature of binding. It has been widely accepted that worm-like fibril formation lead to short or no lag phase with a rapid growth phase.27 This phenomenon too is in concurrence with the present observation. The magnitude of binding constant value (8.72 × 107 M−1) number of binding site (1.3) and free energy of binding (−45.30 kJ mol−1) are found to higher in the Region I. Thus a highly cooperative nature of binding was observed during the initial stage itself. On the other hand, non-cooperative nature of binding is reflected during the Region II where, the binding constant (8.41 × 103 M−1), number of binding site (0.62) and free energy (−22.39 kJ mol−1) values are lower than Region I. Low ‘n’ value indicates that the binding of HgCl2 to the site is only partial. The results indicate that for OVA at pH 4.5, the initial additions has high affinity while at high concentration, HgCl2 involved other sites with lower binding affinity and selectivity. Beyond Region II (Region III), the process is mostly non interactive and the addition of Hg2+ to OVA gets almost saturated.
 | ||
| Fig. 2 Modified Stern–Volmer plots of the OVA–HgCl2 at pI. log[(F0 − F)/F] vs. log[HgCl2]; the insets show a representative plot corresponding to Region II. | ||
| Kq1 (M−1 s−1) | Kq2 (M−1 s−1) | n1 | n2 | Kb1 (M−1) | Kb2 (M−1) |
|---|---|---|---|---|---|
| 1.55 × 1014 | 1.03 × 1014 | 1.3 | 0.62 | 8.72 × 107 | 8.41 × 103 |
The intrinsic fluorescence spectrum of OVA in the presence of a series of concentrations of HgCl2 under pH 4.5 is presented in Fig. 3A. Up to a quencher concentration of 1.5 μM the fluorophore experiences more or less the same environment. But the increase of concentration of HgCl2 to 2.5 μM leads to a spectral shift of 7 nm, more hydrophobic environment indicating probable changes in the secondary structure (onset of molten globule). Further increase in the quencher concentration does not result in further reduction in intensity change and λmax. The variation of fluorescence intensity of OVA with increase in concentration of HgCl2 is shown in Fig. 3B. It is evident from the plot that the addition of HgCl2 at the initial stage (Region I) have prominent effect on the quenching of florescence. The decay parameters too were in favor of early onset of aggregation (ESI Table S1†). The first addition of HgCl2 itself showed a significant reduction in the life time value of OVA. The average life time component decreased by 24.21% (τm 4.13–3.13 ns) on initial additions. The most prominent decrease in life time (3.13–2.14 ns) took place on further increase in 2.5 μM HgCl2 where blue shift in the environment around tryptophan take place. FRET/electron transfer plays the major role here. The protein is more prone to aggregation during this region.28 The clear distinction between Region I and II is missing in the sigmoidal curve and a continuous decrease is observed (ESI Fig. S2†). Such growth stage is associated with secondary nucleation pathway for fibril formation. When the perturbation is brought at a stage which is already seeded, a deeper understanding is expected in the aggregation pathway. Slightly acidic pH values increase the chances of secondary nucleation where the multiplications of aggregates are much faster.29,30 Secondary nucleation promotes proliferation of aggregates when seeded.31 Since the template already exist the energy requirements for aggregation is much low.
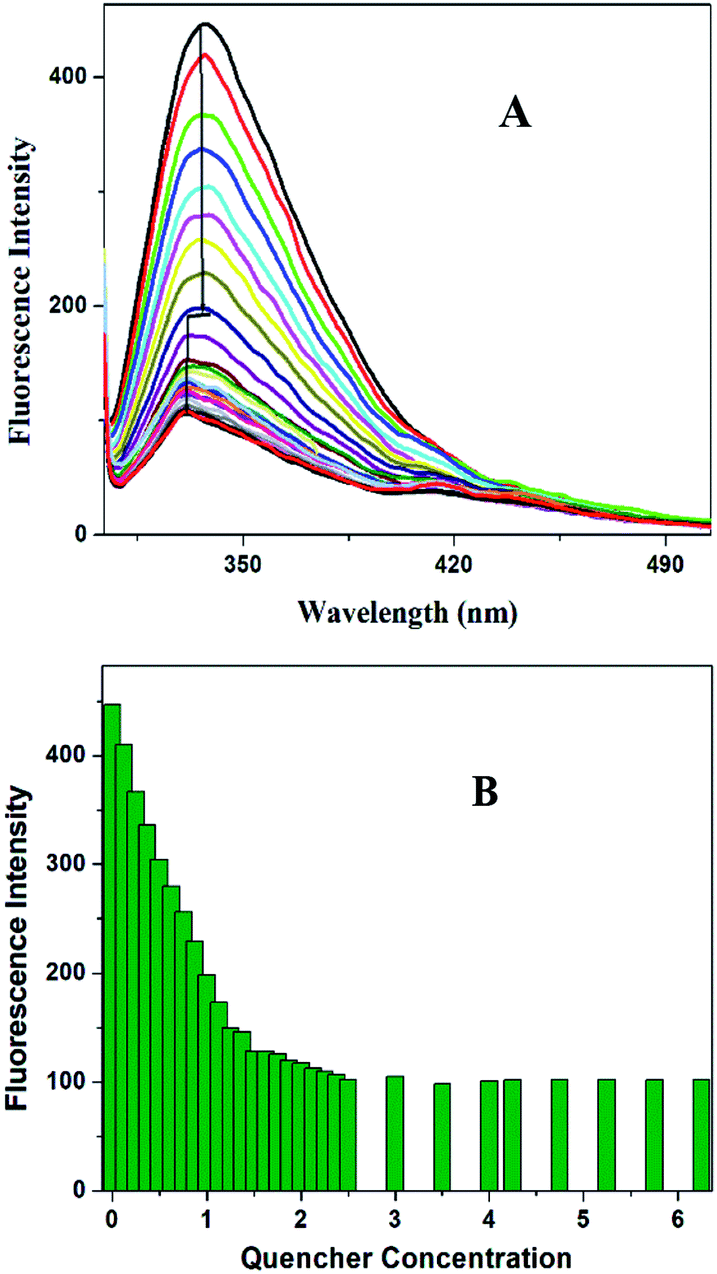 | ||
| Fig. 3 (A) Fluorescence quenching profile of OVA with the addition of HgCl2 and the corresponding shift in the maximum emission peak to more hydrophobic side. (B) Comparison of variation in the fluorescence intensity of OVA against the varying concentrations of HgCl2. | ||
The population distribution pattern from AFM and early stage growth suggest a surface catalyzed secondary nucleation pathway. This point of discussion is supported by our observation in AFM where the height of ‘hilllock’ is reduced to 0.5–1.25 nm (ESI Fig. S1D†). Any perturbation (pH or ionic strength) can depolymerize WL. Here the change in solution environment by the introduction of HgCl2 might have induced particle formation. In depolymerized state, we expect both parent fibrils and monomers with a higher population. But interestingly, population gets reduced with substantial width increase (30–200 nm) (Fig. 1D). These particles (monomers) might have aggregated on parent fibril. Literature supports this argument by a closer analysis of some of the well-established pathway of protein aggregation of sickle hemoglobin,32 Aβ-42 (ref. 31) and amyloid.33 The decrease in the monomer population, and early stage growth phase with a less sharp sigmoidal curve (missing distinct lag phase) point towards the surface-catalyzed secondary nucleation.30,31
In order to have a much closer analysis on the structural variations at the molecular level, Raman spectra were recorded. The peptide backbone analysis is mainly carried out depending on Amide I and III regions. The Raman spectrum in the region of 800–1800 cm−1 of OVA at pI is shown in Fig. 4A. Careful examination of the spectra reveals a broad Amide I band at 1662 cm−1 (Fig. 4B) and this indicates that protein adopt exclusively α-helical conformation. Further increments in HgCl2 (6.25 μM) caused shift in Amide I band (1667 cm−1) with a drop in α-helical content. There was a major rearrangement with hike in β-sheet, random coil conformation and β-turns for ‘hydra’ like structure (Table 2).
 | ||
| Fig. 4 (A) Raman spectra of OVA alone at isoelectric pH and in the presence of different concentration of HgCl2 (800–1800 cm−1), (B) Amide I (1600–1700 cm−1) region of OVA in the presence and absence of different concentration of HgCl2. (C) Deconvoluted spectrum of Amide I region of OVA alone and (D) with the addition of 6.25 μM HgCl2. | ||
| OVA 4.5 | +1.5 μM HgCl2 | +2.5 μM HgCl2 | +6.25 μM HgCl2 | |
|---|---|---|---|---|
| β-Sheet | 20.72 | 20.89 | 23.18 | 48.10 |
| α-Helix | 49.33 | 19.98 | 18.50 | 0.71 |
| Random coils | 8.71 | 19.43 | 29.90 | 33.51 |
| β-turns | 8.81 | 3.31 | 2.66 | 15.98 |
| Antiparallel β | 9.23 | 25.80 | 24.03 | — |
Increase in β-sheet conformation is typical of amyloid pathway of proteins but increase of random coil is seldom seen. A closer comparison of structural features and conformational rearrangement reminds similarities to the spinning dope and spider silk precursors. The deconvoluted Raman bands showed secondary structural components with close similarities to that of spin dope and spider silk where a large increase in the β-sheet content from α-helical rich spin dope is usually observed.34,35 The formation of spider silk has two known pathways, liquid crystalline and micellar. The observed structural features appear to have closer association with micellar pathway. Silk proteins are amazing as it is able to remain in solution state at high concentrations unless an indicative signal is received for fibril formation. The silk spinning is triggered by pH gradient which further proceeds via dimerization, micelle, gel and precursor silk.36 The band position had closeness to dragline silk from the spider Nephila clavipes where the band is initially located at 1660 cm−1 (rich in helices) and got shifted to 1670 cm−1 (rich in β-sheet).37 In the present system, Amide I band is observed at 1662 cm−1 (again rich in helices) that got shifted to 1667 cm−1 (precursor silk) (Fig. 4B). Interestingly both β-sheet and helices are seen in WL as in Nephila clavipes (in the C- and N-terminal regions) for nucleation38 (Table 2). This on addition of HgCl2 proceeds to precursor silk in a similar manner as that of spider silk. The dimensions of ‘hillock’ (30–200 nm) and micelles (100–200 nm) agree with each other. The globules of OVA have a dimension of 80–350 nm which is less than that of spidroin globules (0.8–15 μm). But this is quite dependable on the proportion of OVA and quencher as was seen in the case of fibroin/polyethylene oxide blends.39 ‘Hydra’ like structural feature is comparable to the precursor silk which proceeds to fibrils with respect to time (ESI Fig. S3†).
The Amide III band shows switchability in the conformational transition between β-sheet and random coil. The initially present β-sheet decreases on initial exposure to quencher which further enhances on final exposure (Fig. 5A and ESI Table S2†). There are further similarities in spidroin pathway and that of OVA fibrillation.34 Normally in the pathway of spider silk in the last step, a lot of change in the surroundings of the protein will occur with respect to pH and metal ion concentration. There will be subsequent water loss from the micellar structure to the hydrophobic phase resulting in the formation of crystalline β-sheet conformation. The final fibril consists of crystalline domains embedded in amorphous domains thus giving brittleness and elasticity to the silk. The water loss from the lumen is the essential step in the formation of crystalline structure of spider silk. This has been experimentally proven by Tongyin et al. from the globular structure of silk fibroin by analyzing the Amide III peak.40 Here too we adopted the same method and analyzed the Amide III peak. The increase in intensity of peak at 1233 cm−1 depicts (ESI Fig. S4†) the loss of water which could be the driving force. The change of protein into more hydrophobic environment is further supported by the steady state fluorescence data (Fig. 3A). The amorphous structure in our fibril can be seen from the presence of random coil and β-turns content. Whereas the α-helical content present in the initial step has been almost disappeared during the final phase similar to that observed in the case of Nephila clavipes. The plasticization effect of water in β sheet formation is discussed using NMR by Jelinski et al.41
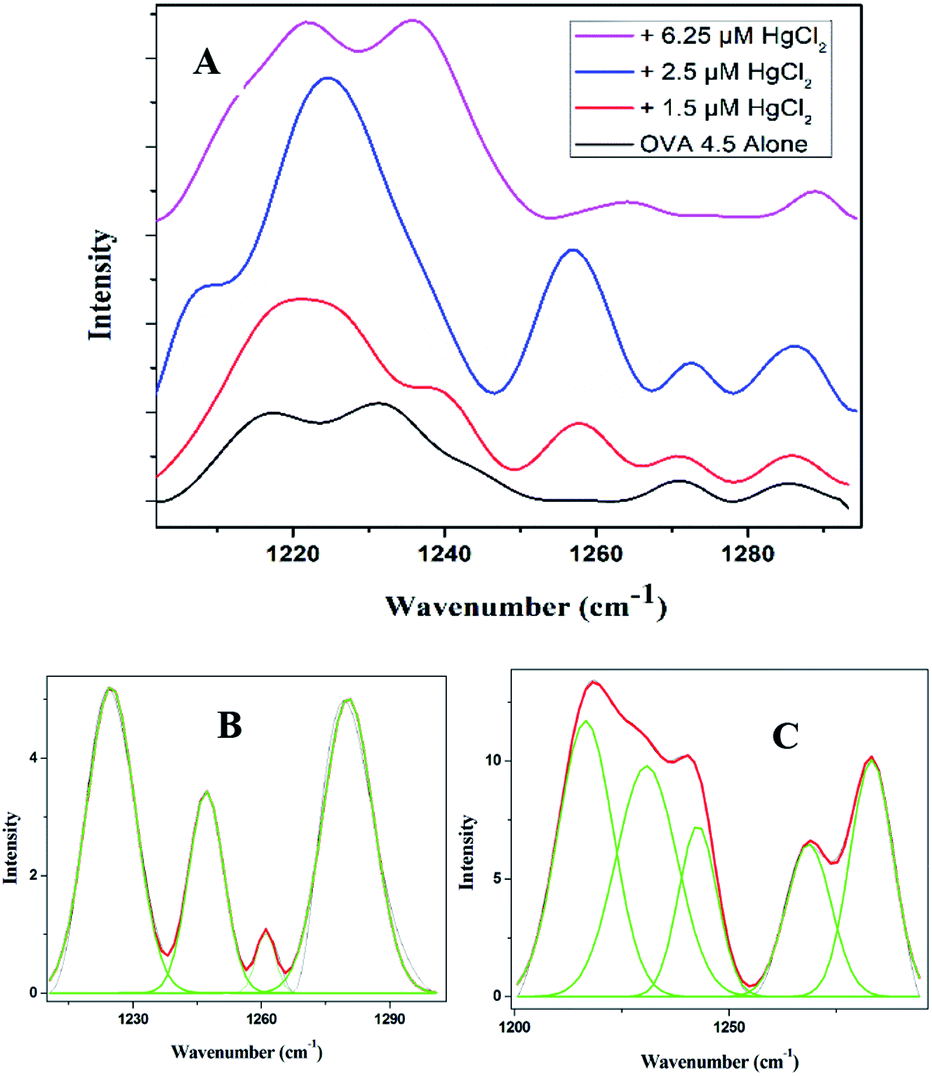 | ||
| Fig. 5 (A) Amide III (1200–1300 cm−1) region of OVA in the presence and absence of different concentration of HgCl2. (B) Deconvolution of the Amide III region of OVA alone and (C) with the addition of 6.25 μM HgCl2. | ||
The knowledge about the shapes of protofibrils and their nature of interaction opens the discussion about membrane penetration. The hydrophobic shifts in the protein environment have a greater influence in the membrane penetration.6 The similarity of the present system to spider dope also reminds the torsional memory of such species. This simply indicates its nature to memorise its structural features even after facing destructive conditions. As and when requires it can restore the original shape and function.36
4. Conclusions
The present study provides complete mechanistic pathway of fibrillation of a typical serpin. The occurrence of conformational transitions (α to β-sheet/antiparallel β-sheet/random coil) is elucidated within these structures using Raman spectroscopy. The study provides evidence for the new protofibril with shape similar to hydra which finally with time proceeds to fibril. The similarity of amyloid in neurodegenerative and systemic diseases to that of the spider silk is experimentally established. The findings and the aggregation stage is well supported by time resolved and steady state fluorescence analysis. This is again a remarkable discussion point where more attention has to be paid in designing the therapeutic agents for the treatment of amyloid diseases. The occurrence of toxic oligomers at the studied pH indicates the possible perturbation of biological clock posed by the increased presence of chemicals in the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge SAIF (DST), MG University, Kottayam for providing the instrumental facilities. Financial support from UGC and DST is also acknowledged.Notes and references
- N. F. Bence, R. M. Sampat and R. R. Kopito, Science, 2001, 292, 1552–1555 CrossRef CAS PubMed.
- A. L. Goldberg, Nature, 2003, 426, 895–899 CrossRef CAS PubMed.
- K. R. Cho, Y. Huang, S. Yu, S. Yin, M. Plomp, S. R. Qiu, R. Lakshminarayanan, J. Moradian-Oldak, M.-S. Sy and J. J. De Yoreo, J. Am. Chem. Soc., 2011, 133, 8586–8593 CrossRef CAS PubMed.
- J. S. Jacobsen, P. Reinhart and M. N. Pangalos, NeuroRx, 2005, 2, 612–626 CrossRef PubMed.
- S. Jain, N. W. Wood and D. G. Healy, Clin. Sci., 2005, 109, 355–364 CrossRef CAS PubMed.
- S. M. Butterfield and H. A. Lashuel, Angew. Chem., Int. Ed., 2010, 49, 5628–5654 CrossRef CAS PubMed.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson and C. Cecchi, Nat. Chem. Biol., 2010, 6, 140 CrossRef CAS PubMed.
- Y. Manivannan, B. Manivannan, T. G. Beach and R. U. Halden, Curr. Alzheimer Res., 2015, 12, 116–146 CrossRef PubMed.
- M. Schlumpf, K. Kypke, M. Wittassek, J. Angerer, H. Mascher, D. Mascher, C. Vökt, M. Birchler and W. Lichtensteiger, Chemosphere, 2010, 81, 1171–1183 CrossRef CAS PubMed.
- Y. Wei, I.-K. Han, M. Hu, M. Shao, J. Zhang and X. Tang, Chemosphere, 2010, 81, 1280–1285 CrossRef CAS PubMed.
- Z. Klimová, J. Hojerová and M. Beránková, Food Chem. Toxicol., 2015, 83, 237–250 CrossRef PubMed.
- M. H. Hazelhoff and A. M. Torres, Chemosphere, 2018, 202, 330–338 CrossRef CAS PubMed.
- C. Y. Chen, C. T. Driscoll, C. A. Eagles-Smith, C. S. Eckley, D. A. Gay, H. Hsu-Kim, S. E. Keane, J. L. Kirk, R. P. Mason, D. Obrist, H. Selin, N. E. Selin and M. R. Thompson, Environ. Sci. Technol., 2018, 52, 9556–9561 CrossRef CAS PubMed.
- C. A. Hartman, J. T. Ackerman and M. P. Herzog, Environ. Sci. Technol., 2019, 53, 5396–5405 CrossRef CAS PubMed.
- M. Rutkowska, G. Bajger-Nowak, D. Kowalewska, S. Bzoma, E. Kalisińska, J. Namieśnik and P. Konieczka, Chemosphere, 2019, 219, 140–147 CrossRef CAS PubMed.
- M. A. Bradley, B. D. Barst and N. Basu, Int. J. Environ. Res. Public Health, 2017, 14, 169 CrossRef PubMed.
- M. Mathew, S. Sreedhanya, P. Manoj, C. T. Aravindakumar and U. K. Aravind, J. Phys. Chem. B, 2014, 118, 3832–3843 CrossRef CAS PubMed.
- I. Chakraborty, U. Saha, D. Mandal, S. Mukherjee, N. Joardar, S. P. Sinha Babu, G. Suresh Kumar and K. Mandal, Phys. Chem. Chem. Phys., 2019, 21, 10726–10737 RSC.
- A. Chaudhary, A. Gupta, S. Khan and C. K. Nandi, Phys. Chem. Chem. Phys., 2014, 16, 20471–20482 RSC.
- P.-C. Guo, Z. Dong, P. Zhao, Y. Zhang, H. He, X. Tan, W. Zhang and Q. Xia, Sci. Rep., 2015, 5, 11863 CrossRef PubMed.
- H. T. Wright, H. X. Qian and R. Huber, J. Mol. Biol., 1990, 213, 513–528 CrossRef CAS PubMed.
- D. van Gent, P. Sharp, K. Morgan and N. Kalsheker, Int. J. Biochem. Cell Biol., 2003, 35, 1536–1547 CrossRef CAS PubMed.
- D. Kaiserman, J. C. Whisstock and P. I. Bird, Expert Rev. Mol. Med., 2006, 8, 1–19 CrossRef PubMed.
- D. Belorgey, P. Hägglöf, S. Karlsson-Li and D. A. Lomas, Prion, 2007, 1, 15–20 CrossRef PubMed.
- W. S. Gosal, I. J. Morten, E. W. Hewitt, D. A. Smith, N. H. Thomson and S. E. Radford, J. Mol. Biol., 2005, 351, 850–864 CrossRef CAS PubMed.
- S. Arya, A. Kumari, V. Dalal, M. Bhattacharya and S. Mukhopadhyay, Phys. Chem. Chem. Phys., 2015, 17, 22862–22871 RSC.
- J. M. D. Kalapothakis, R. J. Morris, J. S. Nossan, K. Eden, S. Covill, S. Tabor, J. Gillam, P. E. Barran, R. J. Allen and C. E. MacPhee, Biophys. J., 2015, 108, 2300–2311 CrossRef CAS PubMed.
- C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 18775–18784 CrossRef CAS PubMed.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. J. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef CAS PubMed.
- T. C. T. Michaels, A. Šarić, J. Habchi, S. Chia, G. Meisl, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Annu. Rev. Phys. Chem., 2018, 69, 273–298 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- F. A. Ferrone, J. Hofrichter and W. A. Eaton, J. Mol. Biol., 1985, 183, 611–631 CrossRef CAS PubMed.
- A. M. Ruschak and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12341–12346 CrossRef CAS PubMed.
- F. Vollrath and D. P. Knight, Nature, 2001, 410, 541–548 CrossRef CAS PubMed.
- M. McGill, G. P. Holland and D. L. Kaplan, Macromol. Rapid Commun., 2019, 40, 1800390 CrossRef PubMed.
- M. Heim, D. Keerl and T. Scheibel, Angew. Chem., Int. Ed., 2009, 48, 3584–3596 CrossRef CAS PubMed.
- T. Lefèvre, J. Leclerc, J.-F. Rioux-Dubé, T. Buffeteau, M.-C. Paquin, M.-E. Rousseau, I. Cloutier, M. Auger, S. M. Gagné, S. Boudreault, C. Cloutier and M. Pézolet, Biomacromolecules, 2007, 8, 2342–2344 CrossRef PubMed.
- T. Lefèvre, S. Boudreault, C. Cloutier and M. Pézolet, Biomacromolecules, 2008, 9, 2399–2407 CrossRef PubMed.
- H.-J. Jin and D. L. Kaplan, Nature, 2003, 424, 1057–1061 CrossRef CAS PubMed.
- S. Zheng, G. Li, W. Yao and T. Yu, Appl. Spectrosc., 1989, 43, 1269–1272 CrossRef CAS.
- D. H. Hijirida, K. G. Do, C. Michal, S. Wong, D. Zax and L. W. Jelinski, Biophys. J., 1996, 71, 3442–3447 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10655c |
| This journal is © The Royal Society of Chemistry 2020 |