DOI:
10.1039/C9RA10542E
(Paper)
RSC Adv., 2020,
10, 3479-3486
Metal ion-induced separation of valuable organic acids from a depolymerized mixture of lignite without using organic solvents†
Received
11th November 2019
, Accepted 3rd January 2020
First published on 21st January 2020
Abstract
Due to the low utilization efficiency of lignite as a primary energy source, the valuable and clean use of lignite becomes important. Oxidative depolymerization of lignite into valuable organic acids (VOAs) has been identified to be feasible, but the difficulty in separating VOAs from the complex lignite depolymerized mixture (LDM) limits the potential application of this route. In this study, based on the coordination interactions between metal ions and carboxylate groups in VOAs, the metal ion-induced separation of VOAs from the LDM was proposed. The results proved that most of the studied metal ions (Mn+) could selectively form M–VOA precipitates with the VOAs in LDM and transferred the VOAs from the water phase into the solid precipitates. Then, the intermediate M–VOAs could be dissolved in diluted NaOH solution to release the VOAs, with Mn+ being transformed into M(OH)n. The separation yield and selectivity could be tuned facilely by various metal ions at different dosages, pH, and temperatures. The process could be fulfilled under near-room temperature in water without the use of organic solvents. Due to its efficiency, tunable selectivity, and green nature, the proposed separation strategy may find potential applications in the valuable and clean use of lignite sources.
Introduction
Lignite, as typical low rank coal, is an important carbon resource on earth. However, the utilization efficiency of lignite as a primary energy source is low because it has several specific properties, such as high content of water and volatiles and low calorific value.1 To improve the utilization efficiency, upgradation of lignite by thermal processes such as production of high-quality chars by pyrolysis or conversion to syngas by gasification is currently adopted.2 These strategies could increase the utilization efficiency remarkably compared to the traditional combustion, but they still suffer from some problems.1,3 One issue is the high energy-input for both pyrolysis and gasification. Another issue is that the abundant valuable structure units or components in lignite are often destroyed and/or even converted into pollutants under the critical reaction conditions, leading to the waste of carbon resources to some extent. Therefore, how to fulfill the green and valuable utilization of lignite, particularly based on their unique structures and properties, is still a challenge.3,4
Obtaining valuable chemicals from lignite is attractive because lignite is rich in valuable structural units, such as oxygen-containing groups, aromatic rings, and/or N, S-containing heterocyclic structures.5 Compared to the traditional thermal processes, a more rational choice is to maintain these structures as much as possible during the utilization process and convert them into valuable products.6 Lignite depolymerization provides a potential route to convert lignite into valuable chemicals under much milder conditions compared to pyrolysis and gasification.1 Various processes have been developed to depolymerize lignite.5a,7 Valuable organic acids (VOAs) could be obtained from lignite by oxidative depolymerization, including benzene polycarboxylic acids and small-molecule fatty acids.5a,7c However, the depolymerized products are rather complex mixtures, and the separation of the valuable products from the mixtures is a bottleneck of this route. Several attempts have been reported to separate VOAs from the complex lignite depolymerized mixture (LDM) or their simulated systems. Extraction by traditional organic solvents or ionic liquids was commonly used to separate VOAs from the LDM.6,8 Ion-pair solvent extraction by quaternary ammonium salts was applied to separate VOAs from both the simulated benzene polycarboxylic acids and the real coal-depolymerized mixture.9 Quaternary ammonium salts could also be used to separate the isomers of benzene polycarboxylic acids in the simulated mixture by forming deep eutectic solvents.10 Chromatographic separation was used after alkylation of LDM to obtain different fractions, but each fraction was still rather complex.11 These reported strategies could fulfill the efficient separation of LDM to some extent, but two main shortcomings still existed. On one hand, organic solvents and/or complex pretreatments were required, which could not only increase the separation cost but also bring potential pollution and safety risks. On the other hand, it is not facile to tune the extraction yield and selectivity of the separation process. Overall, although lignite depolymerization has shown attractive potential, the separation process severely limits the development of this route. It is still highly desirable to develop new and efficient separation strategies to promote the valuable utilization of lignite.
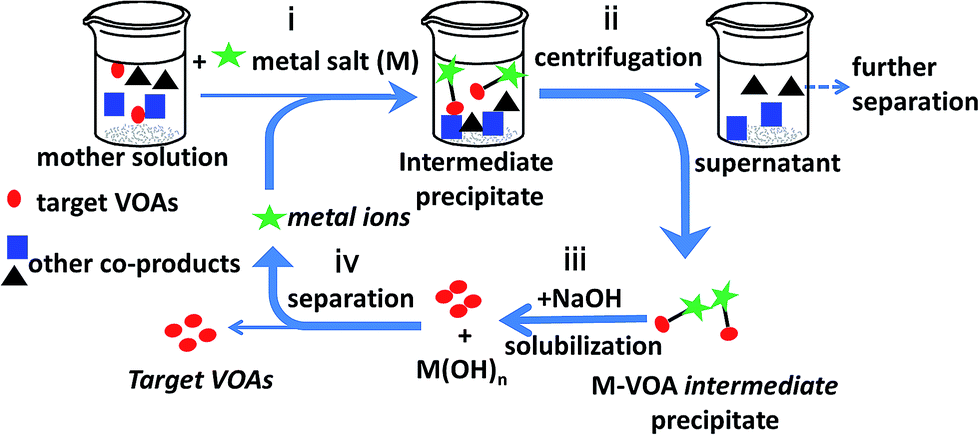
The coordination of carboxylate groups with metal ions is one of the basic properties of organic acids in the field of chemistry. In this study, we applied the interaction between metal ions and carboxylate groups to the separation of organic acids from the depolymerized mixture of lignite. Metal ion-induced separation was proposed to separate VOAs from the real LDM. As shown in Scheme 1, the basic principle is using the selective coordinating interaction between metal ions and the carboxyl groups in VOAs to extract VOAs out of LDM solution. The approach could be accomplished mainly by four steps: (i) adding metal salts (Mn+) into LDM water solution to form M–VOA intermediate precipitates, (ii) separating the intermediate precipitate by centrifugation, (iii) solubilizing the intermediate precipitate using NaOH solution to release the target VOAs into water with Mn+ being converted into M(OH)n precipitates, and (iv) removing M(OH)n precipitates by centrifugation to obtain the target VOA solution. The extraction yield and selectivity of different VOAs in LDM could be simply tuned by varying the types and dosages of metal ions as well as pH and temperature. The advantages of this approach include (i) near room temperature (commonly 50 °C) operation, (ii) no use of organic solvents (totally in water), and (iii) facile tunability of the separation process. The proposed approach may enrich the separation approaches of LDM and promote the green and valuable utilization of lignite resources.
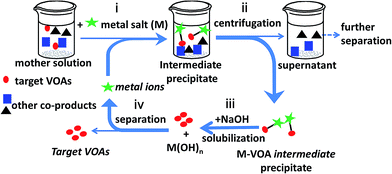 |
| | Scheme 1 Illustration of the metal-induced separation of VOAs from LDM. | |
Experimental
Materials
All the benzene polycarboxylic acids and small molecule fatty acids (analytical reagent) were purchased from J&K Scientific Ltd. The metal salts (analytical reagent) used in this study were supplied by Beijing Innochem Technology Co. Ltd. The reagents were directly used as received without further treatment.
Preparation of the simulated solution
The concentration of each organic acid in the simulated mixture was controlled at 0.001 mol L−1. The calculated amounts of each organic acid was weighed and added into the same flask. To promote the solubilization of organic acids, dilute NaOH solution (0.035 mol L−1) was used as the solvent and the mixture was stirred under 60 °C until a homogeneous solution was formed. Finally, deionized water was added to a total volume of 200 mL. The homogeneous solution was used as mother liquid. The pH of the mother solution was tuned using dilute HCl solution. The pH was detected using a pH meter (PB-10 from Sartorius Scientific Instruments, Beijing, Co., Ltd).
Oxidative depolymerization of lignite
The raw lignite sample was obtained from the Shengli coalfield in Inner Mongolia, China. The raw lignite sample was ground to fine powder with the average sizes being ca. 38–75 μm for the subsequent depolymerization. In this study, lignite was depolymerized by alkali-oxygen oxidation according to a reported method.3,12 The depolymerization of lignite was conducted in a 150 mL high-pressure batch reactor made of Hastelloy alloy (HC276) from Haian Petroleum Scientific Research Co. Ltd, Jiangsu, China. The liner in the autoclave is made of Teflon and graphite, which could sustain the temperature of the alkali-oxygen oxidation. Typically, 9.0 g NaOH was dissolved into 60 mL distilled water and then, 3.0 g coal sample and NaOH solution were mixed in the autoclave. Next, oxygen was charged into the autoclave with typical pressure of 5.0 MPa, and the reaction mixture was magnetically stirred and reacted at 240 °C for 1 h. After the reaction, the autoclave was cooled down under air naturally. The slurry was separated by filtration, and pH of the filtrate was adjusted to neutral with dilute hydrochloric acid. Finally, the neutralized filtrate was evaporated through a rotatory evaporator and then dried at 80 °C under vacuum for 12 h. The alkali-oxygen oxidation products (AOOPs) were ground and solubilized in deionized water for the subsequent separation.
HPLC and HPLC-MS analysis
The mother solution and the samples after separation were analyzed by HPLC (SHIMADZU LC-20AT). A binary gradient elution procedure was used for HPLC analysis. The mobile phase was acetonitrile and 0.1% (volume fraction) phosphoric acid aqueous solution, and the stationary phase was C18 bonded by silica gel (Shim-pack GIST C18, 5 μm). A UV detector at 235 nm was used to quantify the products. The mobile phase flow rate was 0.8 mL min−1, and the column temperature was 45 °C. The gradient elution procedure was as follows: first, the volume ratio of acetonitrile to phosphoric acid aqueous was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95; the ratio was increased to 15:85 linearly over 10 min and then maintained for 38 min; finally, the ratio was decreased to 5:95 over 2 min. HPLC analysis of the real lignite depolymerized mixture (LDM) was the same as above. HPLC-MS analysis was conducted on Agilent 1290/6460 Triple Quad LC/MS.
:95; the ratio was increased to 15:85 linearly over 10 min and then maintained for 38 min; finally, the ratio was decreased to 5:95 over 2 min. HPLC analysis of the real lignite depolymerized mixture (LDM) was the same as above. HPLC-MS analysis was conducted on Agilent 1290/6460 Triple Quad LC/MS.
Results and discussion
Separation of the simulated system by different metal ions
Due to the complexity of the real LDM, this study was started with the separation of the simulated system to check the feasibility of the proposed approach. A mixture containing different organic acids was constructed to mimic the real LDM solution. Typical VOAs were chosen based on the commonly reported composition of LDM.12 The structures and names of the VOAs in the simulated system are given in Fig. 1. The HPLC profile of the simulated system is given in Fig. S1.† The detailed separation process is described in the ESI (Section S1.2†). Various metal ions were attempted as transfer molecules, including the common transition metals (Fe3+, Co2+, Cu2+, and Mn2+), rare earth metals (La3+, Ce3+, Pr3+, Nd3+, and Yb3+), an alkaline-earth metal (Ca2+), and a main group metal (Al3+). The separation process is illustrated in Fig. S2 and S3.† Herein, taking FeCl3 as an example, the separation process is shown in Scheme 2. The intermediate precipitate (Fe–VOAs) was formed once FeCl3 was added into the simulated solution. The slurry was centrifugated to obtain the supernatant A containing the residual VOAs and the Fe–VOA precipitate. The precipitate was washed with deionized water to remove the physically adsorbed VOAs. The obtained precipitate was solubilized using 0.5 mol L−1 NaOH solution (5 mL) to give the supernatant B containing target VOAs separated out of the simulated mixture. Both the residual VOAs (supernatant A) and VOAs separated from the mother solution (supernatant B) were analyzed by HPLC (Fig. 2). After the above separation, the obtained supernatant A could also be further separated by a similar process. In this study, emphasis was put on the first separation process (shown in Scheme 2). The results showed that Fe3+ could combine and separate most of the VOAs, but the extraction yields for different VOAs were different, with BA having the lowest extraction yield (14.4%). The separation results using other metal ions are given in Fig. S4–S11.† Based on their performances in separating different VOAs, the studied metal ions could generally be classified into four groups under the studied separation conditions (Table 1). Interestingly, some metal ions such as Ca2+ and Mn2+ could selectively combine BHA with much weaker or even no combination with other VOAs (Fig. S10 and S11†). These results indicated that the separation selectivity could be tuned by simply choosing suitable metal ions.
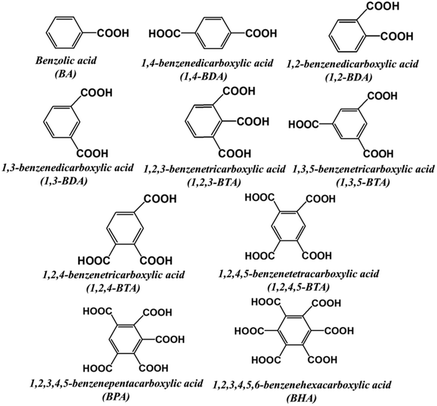 |
| | Fig. 1 Structures and abbreviations of the organic acids in the simulated solution. | |
 |
| | Scheme 2 Typical separation process of the simulated system using FeCl3·6H2O. Separation conditions: the mother solution, 5 mL, pH 6; FeCl3·6H2O, 0.2 g; temperature, 50 °C. | |
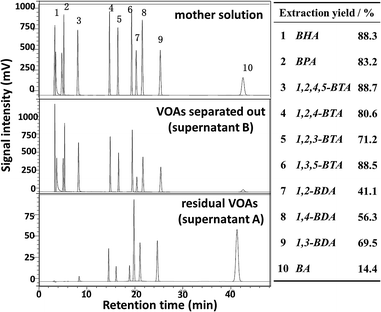 |
| | Fig. 2 Separation results of the simulated mixture using FeCl3·6H2O. The inserted table gave the extraction yields of each organic acid. Separation conditions: the mother solution 5 mL; the dosage of FeCl3·6H2O 0.2 g; 50 °C; 2 h. | |
Table 1 Classification of the studied metal ions based on their performances of separating VOAs from the simulated systema
| Group |
Metal ions (Mn+) |
VOAs combined with Mn+ |
| Separation conditions: the mother solution: 5 mL; the dosage of the metal ions: 0.1 g; 50 °C; pH of the mother solution: 6; 2 h. |
| 1 |
Fe3+, Cu2+ |
Mainly: BHA, BPA, 1,2,4,5-BTA, 1,2,4-BTA, 1,2,3-BTA, 1,3,5-BTA, 1,3 BDA, 1,4-BDA |
| Much less: 1,2-BDA, BA |
| 2 |
La3+, Ce3+, Pr3+, Nd3+, Yb3+ |
Mainly: BHA, BPA, 1,3,5-BTA |
| Much less: 1,2,4,5-BTA, 1,2,4-BTA, 1,2,3-BTA, 1,3-BDA |
| Nearly no: 1,2-BDA, 1,4-BDA, BA |
| 3 |
Ca2+, Mn2+ |
Mainly: BHA |
| Much less: BPA |
| Nearly no: 1,2,4,5-BTA, 1,2,4-BTA, 1,2,3-BTA, 1,3,5-BTA, 1,3 BDA, 1,4-BDA, 1,2-BDA, BA |
| 4 |
Al3+, Co2+ |
No precipitate formed under the present conditions |
In addition to the effect of different metal ions, the effects of other parameters on the separation results were also studied, including the dosages of metal ions (Table 2, Fig. S12†), temperature (Table 3), and the pH values of the mother solution (Fig. 3, Table S1†). The results showed that the dosage of Cu2+ could affect the extraction yields of VOAs to different extents, but had no significant influences on the separation selectivity (Table 2, Fig. S12†). Temperature had no significant effects on both extraction yield and separation selectivity for Cu2+ (Table 3). Interestingly, decreasing the pH value of the mother solution could remarkably increase the selectivity of Cu2+ to 1,3,5-BTA (Fig. 3, Table S1†).
Table 2 Effects of the dosage of Cu(CH3COO)2·H2O on the separation results of the simulated system: comparison of the extraction yields
| Dosage of Cu(CH3COO)2·H2O (g) |
Extraction yield/% |
| 0.1 |
0.2 |
0.4 |
| 1 |
BHA |
63.3 |
77.8 |
40.7 |
| 2 |
BPA |
58.5 |
73.3 |
33.8 |
| 3 |
1,2,4,5-BTA |
41.1 |
55.9 |
15.4 |
| 4 |
1,2,4-BTA |
35.1 |
46.2 |
17.6 |
| 5 |
1,2,3-BTA |
30.9 |
39.0 |
10.5 |
| 6 |
1,3,5-BTA |
70.7 |
80.4 |
58.5 |
| 7 |
1,2-BDA |
2.0 |
4.8 |
0.6 |
| 8 |
1,4-BDA |
56.9 |
92.7 |
62.0 |
| 9 |
1,3-BDA |
18.5 |
24.7 |
10.8 |
| 10 |
BA |
2.7 |
4.9 |
3.3 |
Table 3 Cu(CH3COO)2·H2O as a transfer molecule: effects of separation temperature on the extraction yield for the simulated system. Separation conditions: mother solution, 5 mL; Cu(CH3COO)2·H2O, 0.1 g; 2 h
| Temperature (°C) |
Extraction yield/% |
| 25 |
50 |
| 1 |
BHA |
68.3 |
63.3 |
| 2 |
BPA |
58.2 |
58.5 |
| 3 |
1,2,4,5-BTA |
31.1 |
41.1 |
| 4 |
1,2,4-BTA |
25.7 |
35.1 |
| 5 |
1,2,3-BTA |
20.0 |
30.9 |
| 6 |
1,3,5-BTA |
67.6 |
70.7 |
| 7 |
1,2-BDA |
1.2 |
2.0 |
| 8 |
1,4-BDA |
69.2 |
56.9 |
| 9 |
1,3-BDA |
13.8 |
18.5 |
| 10 |
BA |
1.6 |
2.7 |
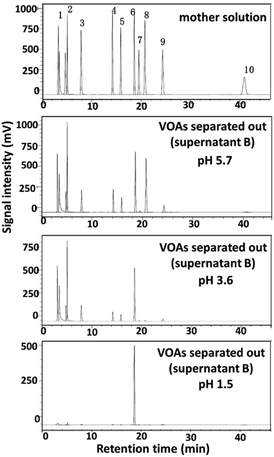 |
| | Fig. 3 Effects of the pH values of the mother solution on the separation results of the simulated mixture using Cu(CH3COO)2·H2O. Separation conditions: mother solution, 5 mL; Cu(CH3COO)2·H2O 0.2 g; 50 °C; 2 h. | |
Overall, the above results showed that the proposed strategy was indeed feasible for the separation of the simulated system, and it was probable to tune the extraction yield and selectivity by changing the type of metal ions and varying the dosages of the ions and pH of the mother solution.
Separation of the real lignite depolymerized mixture by different metal ions
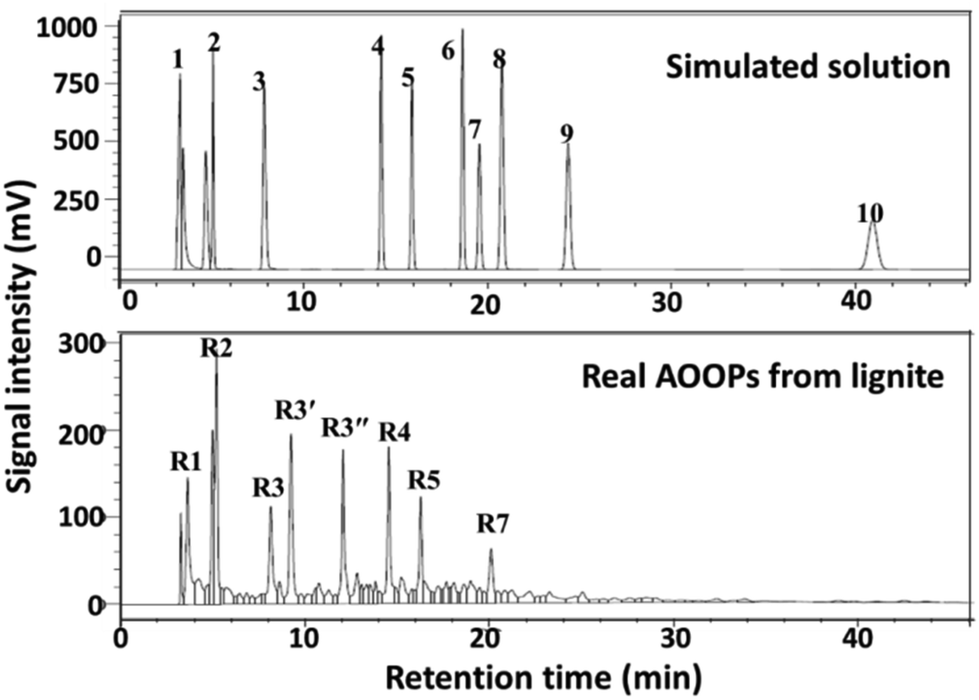
Encouraged by the above results from the simulating system, the proposed strategy was further applied into the real mixture system, i.e., alkali-oxygen oxidation products (AOOPs) from lignite. The structures of the depolymerized products were qualitatively and quantitatively analysed by HPLC-MS and HPLC. The HPLC profile and the composition of AOOPs are given in Fig. 4 and Table 4. The metal ions studied in the simulated system were attempted for the real AOOPs. The separation results are given in Fig. S13–S23.† For the real AOOP system, most of the studied metal ions gave similar separation behavior to the simulated system under similar conditions, but some of them performed differently. For example, using Al3+, no precipitate was formed in the simulated system, while it could separate most of the VOAs in AOOPs (Fig. S14†). Cu2+ gave higher selectivity to OA in AOOPs than in the simulated system (Fig. S15†). Co2+ could form an intermediate precipitate in AOOPs, but no VOAs were detected in the intermediate (Fig. S23†). These differences might be due to the more complex composition of AOOPs than the simulated system. Based on the separation performances for AOOPs under similar conditions, the studied metal ions could be classified into five groups (Table S2†).
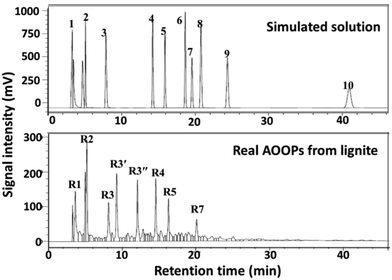 |
| | Fig. 4 HPLC profile of the real AOOPs from lignite and comparison with the simulated solution. | |
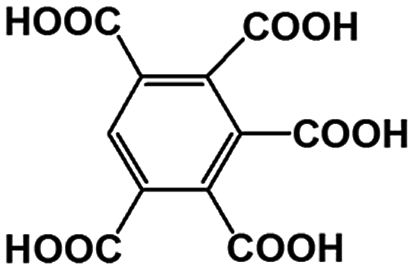
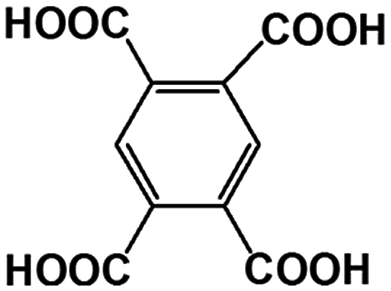
Table 4 The main and known components in the real AOOPs from lignite and concentration
| Peak number |
Abbreviation |
Structure |
Volume content (wt%) |
| R1 |
OA |
HOOC–COOH |
10.8 |
| R2 |
BPA |
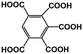 |
1.3 |
| R3 |
1,2,4,5-BTA |
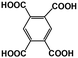 |
0.4 |
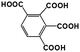
| R3′, R3′′ |
1,2,3,4-BTA |
 |
1.2 |
| 1,2,3,5-BTA |
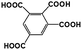 |
| R4 |
1,2,4-BTA |
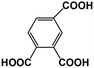 |
0.4 |
| R5 |
1,2,3-BTA |
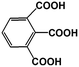 |
0.5 |
| R7 |
1,2-BDA |
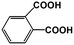 |
0.3 |
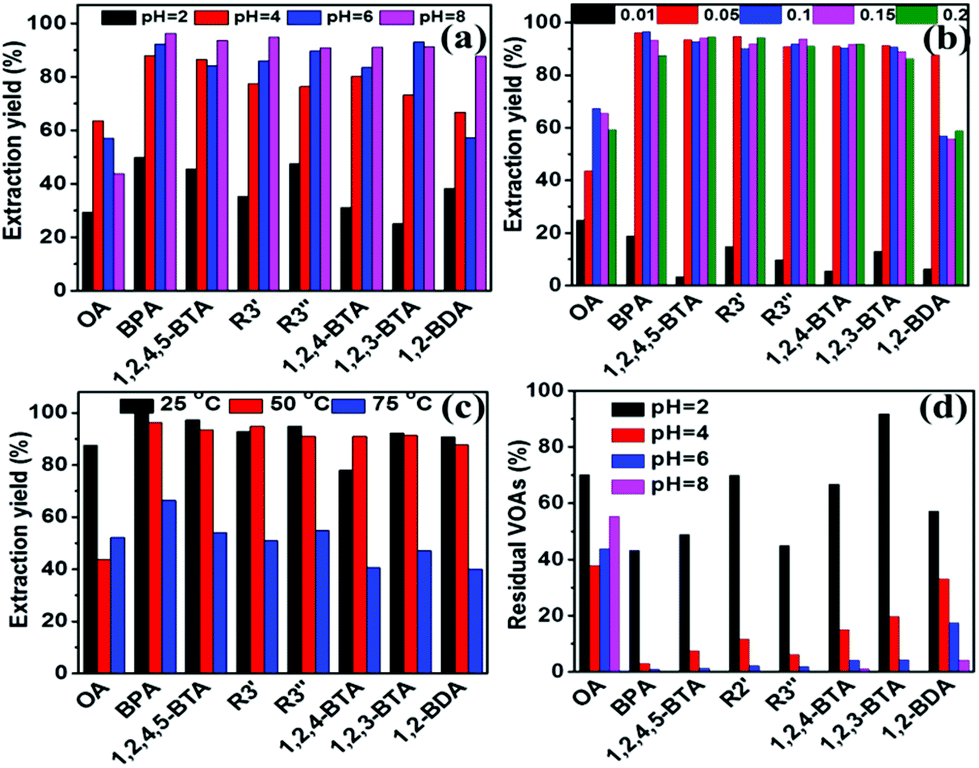
Taking typical Fe3+ and Nd3+ as examples, the effects of separation conditions on the separation results for AOOPs were investigated in detail, including pH of the mother solution, dosages of the metal ions, and the separation temperatures. For Fe3+, increasing the pH of the mother solution and the dosage of Fe3+ could significantly increase the extraction yield of almost all the detected VOAs, while increasing the temperature had a negative effect on the extraction yield (Fig. 5a–c). Interestingly, tuning the pH of the mother solution could significantly change the composition of the residual solution (supernatant A), as shown in Fig. 5d. Under pH 8, the detected VOA in the residual solution was mainly oxalic acid (OA). For Nd3+, pH of the mother solution and dosages of Nd3+ could affect both the extraction yield and the selectivity of VOAs, while temperature had no significant effects on them (Fig. S24†). Increasing the dosage of Nd3+ could decrease the extraction yield of OA but increase the extraction yield of 1,2-BDA. Under pH 2, Nd3+ could nearly selectively separate OA out of AOOPs. The tuning role of pH on the separation selectivity for Fe3+ and Nd3+ could be clearly observed via the HPLC profiles under different pH values (Fig. 6). By controlling the pH of the mother solution, it was feasible to tune the composition of the supernatant A (for Fe3+, Fig. 6A) or the supernatant B (for Nd3+, Fig. 6B). The above results showed that both the extraction yield and selectivity could be tuned by controlling the separation conditions.
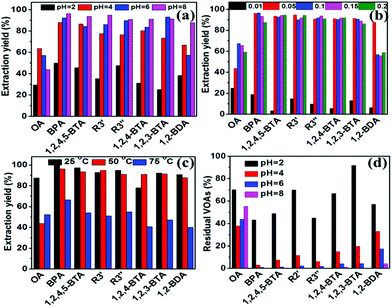 |
| | Fig. 5 Effects of pH (a), dosages (gram of FeCl3·6H2O) (b), and temperature (c) on the composition of the solution separated form AOOPs (supernatant B in Scheme 2). (d) The effect of pH on the composition of the residual solution (supernatant A in Scheme 2). Separation conditions: AOOP solution, 5 mL. | |
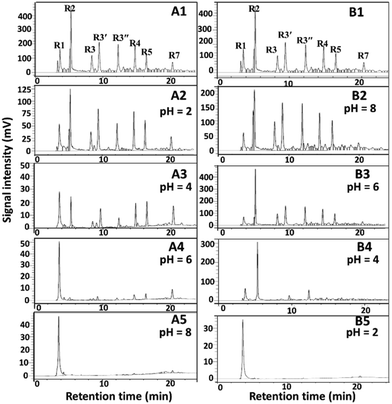 |
| | Fig. 6 Effects of pH on the separation results of the real AOOP mother solution using FeCl3·6H2O (A) and Nd(NO3)3·6H2O (B). A1 and B1 represent HPLC profiles of the real AOOP mother solution. A2–A5 represent HPLC profiles of the residual solution (supernatant A in Scheme 2) after separation by FeCl3·6H2O under different pH values. B2–B5 represent HPLC profiles of the solution (supernatant B in Scheme 2) after separation by Nd(NO3)3·6H2O at different pH values. Separation conditions: AOOPs solution, 5 mL; dosages of metal salts, 0.05 g; temperature, 50 °C; 2 h. R1, OA; R2, BPA; R3, 1,2,4,5-BTA; R3′ and R3′′ represent 1,2,3,4-BTA and 1,2,3,5-BTA, but the retention time was not distinguished due to the highly similar structures; R4, 1,2,4-BTA; R5, 1,2,3-BTA; R7, 1,2-BDA. | |
Based on the experimental data of the simulated system and the real AOOP system, we postulated that the different separation behavior of different metal ions might be related to the valence state and coordination ability with carboxylate groups. Furthermore, the separation results were also related to the composition of the mixtures, the structures of the organic acids, and the separation conditions (particularly pH).
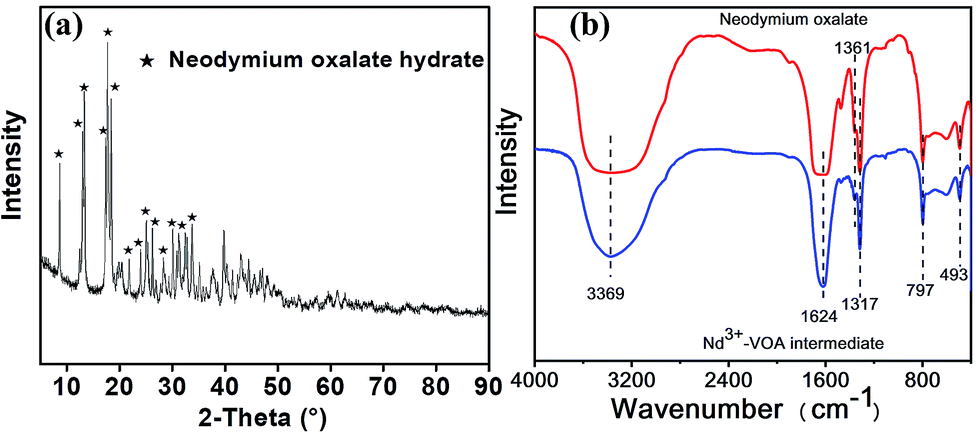
To give an insight into the structures of the separation intermediate precipitate, the structures of the Nd–VOA intermediate were characterized as an example (Fig. 7). The XRD pattern shows typical diffraction peaks of neodymium oxalate hydrate (Fig. 7a). The FTIR spectrum of the Nd–VOA intermediate is shown in Fig. 7b and compared with that of neodymium oxalate hydrate. The absorption bands at about 1624 cm−1 could be assigned to the asymmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and the peaks at 1361 cm−1 and 1317 cm−1 correspond to symmetric stretching of C–O.13 The sharp peak at 797 cm−1 could be ascribed to the presence of metal–oxygen bonds (M–O) and the in-plane bending motion of O–CO.14 The absorption at 493 cm−1 was due to the ring deformation and the bending mode of O–CO.14 The broad band centered at about 3369 cm−1 could be attributed to the anti-symmetric OH stretching in the adsorbed water.13,14 XRD and FTIR data indicated that the VOA in the Nd–VOA intermediate was mainly oxalic acid, which was in agreement with the HPLC results. These results proved that Nd3+ could indeed combine with the carboxyl groups in VOAs (such as oxalic acid) and separated them from the complex mixture.
O, and the peaks at 1361 cm−1 and 1317 cm−1 correspond to symmetric stretching of C–O.13 The sharp peak at 797 cm−1 could be ascribed to the presence of metal–oxygen bonds (M–O) and the in-plane bending motion of O–CO.14 The absorption at 493 cm−1 was due to the ring deformation and the bending mode of O–CO.14 The broad band centered at about 3369 cm−1 could be attributed to the anti-symmetric OH stretching in the adsorbed water.13,14 XRD and FTIR data indicated that the VOA in the Nd–VOA intermediate was mainly oxalic acid, which was in agreement with the HPLC results. These results proved that Nd3+ could indeed combine with the carboxyl groups in VOAs (such as oxalic acid) and separated them from the complex mixture.
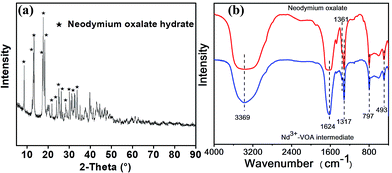 |
| | Fig. 7 XRD pattern (a) and FTIR spectra (b) of the Nd3+–VOA intermediate precipitate. The intermediate was obtained under the following conditions: AOOP solution, 5 mL; pH, 2; 50 °C; 2 h; and Nd(NO3)·6H2O dosage, 0.05 g. | |
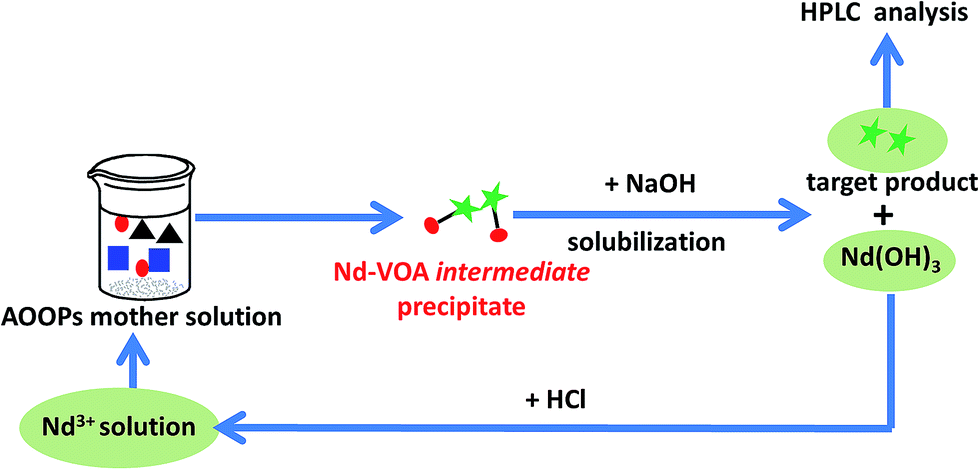
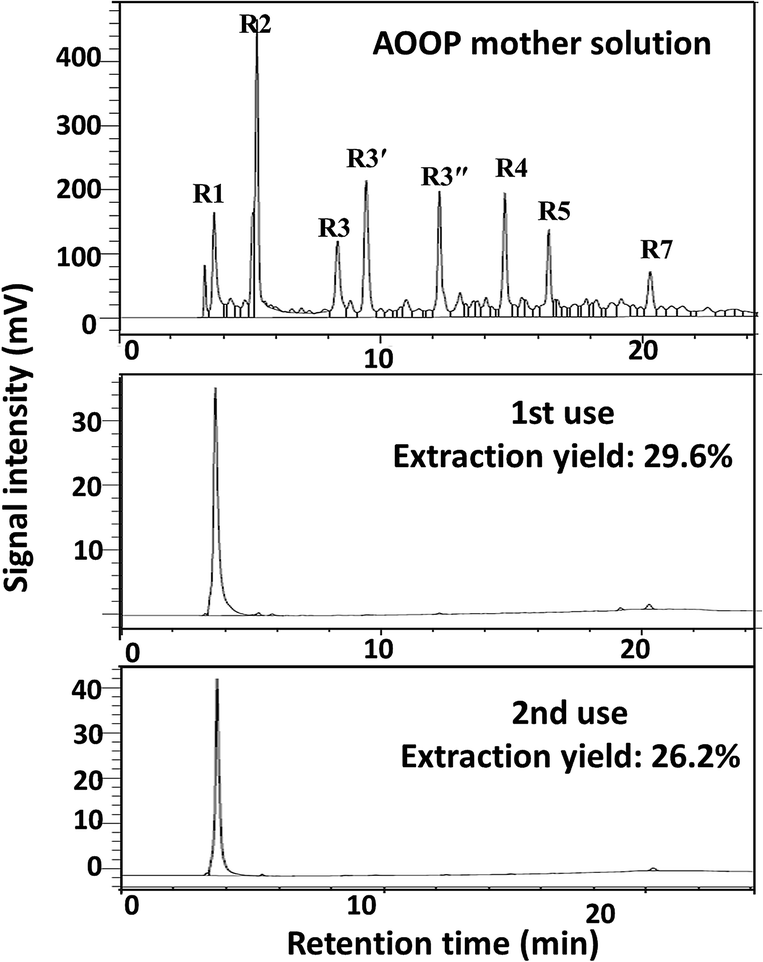
Recycle and reuse of the metal ions
Finally, the possibility of recycling the metal ions based on the design in Scheme 1 was attempted. The regeneration and reuse of Nd3+ was conducted as an example. The recycling process and the separation results in two sequential uses are shown in Scheme 3 and Fig. 8. The results showed that Nd3+ could be simply regenerated by solubilization in HCl solution, and the extraction yield did not decrease significantly during at least two subsequent uses. This result indicated that it was probable to fulfill the recycling of metal ions.
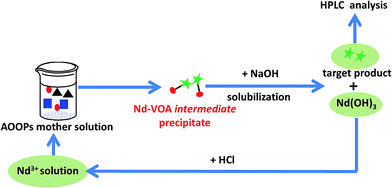 |
| | Scheme 3 The recycling process of Nd3+ during separation of the real AOOPs mixture. | |
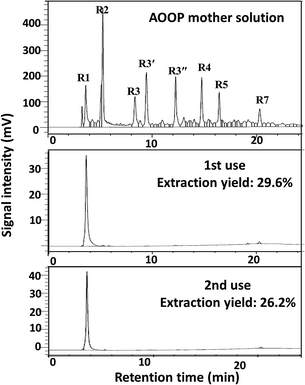 |
| | Fig. 8 The separation results in two sequential uses of Nd3+. Separation conditions: 5 mL AOOPs, pH = 2, 0.05 g Nd(NO3)3·6H2O (initial dosages), 50 °C, 2 h. The extraction yield given in the figure was for peak R1 (oxalic acid). | |
Conclusions
In this study, metal ion-induced separation of VOAs from a lignite depolymerized mixture was proposed. This approach was identified to be efficient in the separation of real lignite depolymerized mixtures. It was also demonstrated that the extraction yield and selectivity could be tuned simply by changing the type and dosages of metal ions, as well as varying the pH and temperature. The approach could be fulfilled under near-room temperature totally in water without the use of organic solvents. With these advantages, this approach may shed light on obtaining valuable chemicals from lignite and promoting the green and valuable utilization of lignite resources.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (21968021, 21606134, 21676149, 21563022, 21868021), CAS “Light of West China” Program, the Innovative and Entrepreneurial Talents Grassland Talents Engineering of Inner Mongolia, Postgraduate Research and Innovation Project in Inner Mongolia Autonomous Region (B20191130Z), and the Incentive Fund for the Scientific and Technology Innovation Program of Inner Mongolia.
Notes and references
- Z. K. Li, X. Y. Wei, H. L. Yan, Y. G. Wang, J. Kong and Z. M. Zong, Energy Fuels, 2015, 29, 6869 CrossRef CAS.
-
(a) Q. Zhou, T. Zou, M. Zhong, Y. M. Zhang, R. C. Wu, S. Q. Gao and G. W. Xu, Fuel Process. Technol., 2013, 116, 35 CrossRef CAS;
(b) A. Bosoaga, N. Panoiu, L. Mihaescu, R. Backreedy, L. Ma, M. Pourkashanian and A. Williams, Fuel, 2006, 85, 1591 CrossRef CAS;
(c) J. Tanner, S. Bhattacharya, M. Bläsing and M. Müller, AIChE J., 2016, 62, 2101 CrossRef CAS;
(d) O. Okuma, Fuel, 2000, 79, 355 CrossRef CAS;
(e) N. Li, Y. Li, H. C. Zhou, Y. Liu, Y. M. Song, K. D. Zhi, R. X. He, K. L. Yang and Q. S. Liu, Fuel, 2017, 203, 817 CrossRef CAS.
- Y. F. Sha, Z. H. Xiao, H. C. Zhou, K. L. Yang, Y. M. Song, N. Li, R. X. He, K. D. Zhi and Q. S. Liu, Green Chem., 2017, 19, 4829 RSC.
- D. K. Sharma and H. Dhawan, Ind. Eng. Chem. Res., 2018, 57, 8361 CrossRef CAS.
-
(a) F. Yang, Y. C. Hou, M. G. Niu, W. Z. Wu and Z. Y. Liu, Fuel, 2017, 202, 129 CrossRef CAS;
(b) F. J. Liu, X. Y. Wei, Y. G. Wang, P. Li, Z. K. Li and Z. M. Zong, RSC Adv., 2015, 5, 7125 RSC;
(c) X. Y. Wei, X. H. Wang and Z. M. Zong, Energy Fuels, 2009, 23, 4848 CrossRef CAS;
(d) M. J. Ding, Z. M. Zong, Y. Zong, X. D. OuYang, Y. G. Huang, L. Zhou, F. Wang, J. P. Cao and X. Y. Wei, Energy Fuels, 2008, 22, 2419 CrossRef CAS.
- F. J. Liu, X. Y. Wei, M. H. Fan and Z. M. Zong, Appl. Energy, 2016, 170, 415 CrossRef CAS.
-
(a) Y. G. Huang, Z. M. Zong, Z. S. Yao, Y. X. Zheng, J. Mou, G. F. Liu, J. P. Cao, M. J. Ding, K. Y. Cai, F. Wang, W. Zhao, Z. L. Xia, L. Wu and X. Y. Wei, Energy Fuels, 2008, 22, 1799 CrossRef CAS;
(b) W. H. Wang, Y. C. Hou, W. Z. Wu and M. G. Niu, Fuel Process. Technol., 2013, 112, 7 CrossRef CAS;
(c) F. Yang, Y. C. Hou, M. G. Niu, W. Z. Wu, D. Y. Sun, Q. Wang and Z. Y. Liu, Ind. Eng. Chem. Res., 2015, 54, 12254 CrossRef CAS.
- Y. G. Wang, X. Y. Wei, H. L. Yan, F. J. Liu, P. Li and Z. M. Zong, Fuel Process. Technol., 2014, 125, 182 CrossRef CAS.
-
(a) K. Kawamura, H. Nagano and A. Okuwaki, Sep. Sci. Technol., 2005, 40, 2761 CrossRef CAS;
(b) K. Kawamura, A. Okuwaki, T. V. Verheyen and G. J. Perry, Sep. Sci. Technol., 2006, 41, 723 CrossRef CAS;
(c) K. Kawamura, A. Okuwaki, T. V. Verheyen and G. J. Perry, Sep. Sci. Technol., 2007, 41, 379 CrossRef;
(d) K. Kawamura, K. Takahashi and A. Okuwaki, Sep. Sci. Technol., 2006, 41, 2795 CrossRef CAS.
- Y. C. Hou, J. Li, S. H. Ren, M. G. Niu and W. Z. Wu, J. Phys. Chem. B, 2014, 118, 13646 CrossRef CAS PubMed.
- J. Arul Leo Bastin, N. Krishnamurthy, D. Madhavan, P. Vallinayagam, M. Palanichamy and A. Chellamani, Solid Fuel Chem., 2017, 51, 256 CrossRef CAS.
- W. H. Wang, Y. C. Hou, W. Z. Wu, M. G. Niu and W. N. Liu, Ind. Eng. Chem. Res., 2012, 51, 14994 CrossRef CAS.
- J. L. Song, L. Q. Wu, B. W. Zhou, H. C. Zhou, H. L. Fan, Y. Y. Yang, Q. L. Meng and B. X. Han, Green Chem., 2015, 17, 1626 RSC.
- B. Want, J. Cryst. Growth, 2011, 335, 90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10542e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Xiaomin Li,
Yanpeng Ban,
Na Li,
Huacong Zhou
,
Xiaomin Li,
Yanpeng Ban,
Na Li,
Huacong Zhou