
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase molybdenum-99 separation from uranium dioxide by fluoride volatility using nitrogen trifluoride
Bruce K. McNamara *a,
Matthew J. O'Haraa,
Richard A. Clarka,
Samuel S. Morrisona,
Chuck Z. Soderquista and
Randall D. Scheeleb
*a,
Matthew J. O'Haraa,
Richard A. Clarka,
Samuel S. Morrisona,
Chuck Z. Soderquista and
Randall D. Scheeleb
aPacific Northwest National Laboratory, PO Box 902, Battelle Blvd, Richland, Wa 99352, USA. E-mail: bruce.mcnamara@pnnl.gov
bFlibe Energy, Richland, Wa 99352, USA
First published on 21st January 2020
Abstract
Production of the important 99mTc medical isotope parent, molybdenum-99 (99Mo), via the fissioning of high- and low-enriched uranium (HEU/LEU) targets followed by target dissolution in acid and solution-phase purification of 99Mo is time-consuming, generates quantities of corrosive radioactive waste, and can result in the release of an array of radionuclides to the atmosphere. An alternative 99Mo purification method has been devised that has the potential to alleviate many of these issues. Herein, we demonstrate the feasibility of a rapid Mo/Tc gas-phase separation from UO2. The results indicate that volatile [99Mo]Mo can be captured downstream of the reacted solid mixture on a column bed (trap) of alumina; the majority of the captured [99Mo]Mo can be subsequently eluted from the alumina trap with a few milliliters of water. >1.0 × 105 single pass decontamination of U and the collected [99Mo]Mo product is demonstrated. This simple thermo-fluorination technique has the potential to provide a rapid methodology for routine 99Mo production.
1. Introduction
Technetium-99m (99mTc, t1/2 = 6.01 h) is the most widely used diagnostic radionuclide world-wide. It is dispensed at radiopharmacies and hospitals via 99Mo/99mTc generators.1 The 99Mo (t1/2 = 65.98 h, ∼6.1% fission yield) parent is produced via the fissioning of highly enriched uranium (HEU) targets. The 99Mo is chemically purified from the target material and other fission and activation products by processing of the acid-dissolved HEU targets.2 Processing of the dissolved HEU targets requires several days, produces corrosive liquid waste streams, and an enriched uranium waste stream. Further issues related to chemical processing for planned conversions of HEU target materials to LEU targets have been discussed by Vandergrift and other researchers.2,3In this article, we discuss the volatility profiles and separation protocol for the 99Mo/99mTc couple from uranium using nitrogen trifluoride. In most of the proposed fluorination methods such as the FLUOREX process,4 or those described earlier by workers in the Czech Republic,5 the former Soviet Union,6 or the US,7–9 fluorine gas is used to rapidly form volatile UF6 from an irradiated matrix, generally UO2 or U metal10 A large set of volatile fluoride products such as PuF6, NpF6, IFx (for x = 3, 5, 7), TeF6, TcF6, etc., can be binned cryogenically, or can be sorbed onto solid traps that have specific capture affinities for the volatile products.10,11 Regardless of the exact process used, fluorination requires the use of rigorously closed reactor and trapping systems. These systems are thus more suited to complete trapping of volatile fission products than the liquid digestion processes currently in use.
The usefulness of NF3 for volatility separations is related to its slightly lower thermal reactivity compared to more potent fluorinating reagents. The lower reactivity of NF3 allows for volatility of a reduced set of fission products, in particular, Mo and Tc, without volatilization of U, Np and Pu. The basis for the separations is the formation of thermally stable, nonvolatile UO2F2 produced as the first product in the fluorination of UO2 (eqn (1)), or of UF4 in the case of the U metal fluorination (eqn (2)):
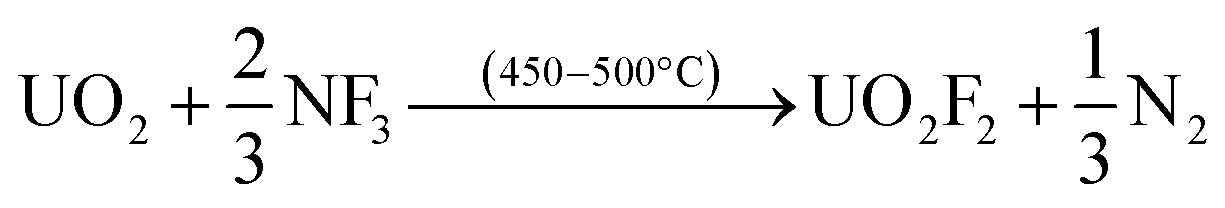 | (1) |
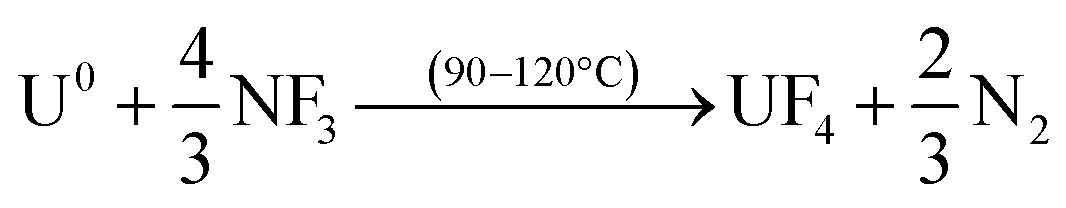 | (2) |
The fluorinated solid matrix formed per eqn (1) or (2) can be further reacted with NF3 to extract volatile fluorides without formation of gaseous UF6. The onset temperature for the conversion of UO2F2 to UF6 is usually near 500 °C, but can be stalled nearly completely by lowering the NF3 concentration to 1 or 5% in Ar.12 Similarly, in a metal target the conversion of UF6 from UF4 can be considerably slowed using reduced temperature or lower NF3 concentrations.13 This feature of the reactions allows for gaseous leaching of the solid U-bearing sample for the time required to volatilize lower boiling point components (such as 99Mo/99mTc) that are generally shown to be rapidly separated at or below 400 °C. Separation and recovery of volatile MoF6/TcF6 from other fission products has been demonstrated by use of selective sorbents, such as solid MgF2.14
High temperature oxidation of irradiated U has been widely cited as being effective at removal of gaseous fission products such as Xe and Kr.15 This is more rapidly and completely realized by the lattice disruption of the U solid, as induced by fluorination (eqn (2) and (3)). Fluorination using NF3 will volatilize Nb, Mo, Sb, Tc, Te16,17 and I from a solid matrix at or below ∼400 °C. Ru will be released near 500 °C.18 Rhodium, Pd18 and Pu17 do not form volatile fluorides using NF3 as the fluorination reagent. Americium,19 the lanthanides, and the Group I and II elements do not form volatile fluorides using any fluorinating reagent.20
After down-stream capture of the volatile fission products has been performed, uranium can be recovered as gaseous UF6 per eqn (3) or (4), leaving the lanthanides, Pu, Am, and the other non-volatiles in the reactor furnace.
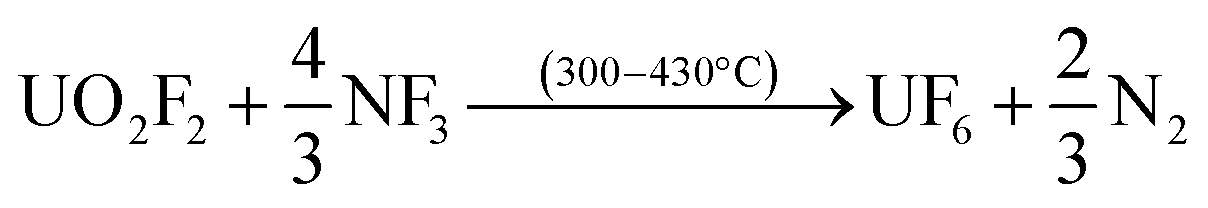 | (3) |
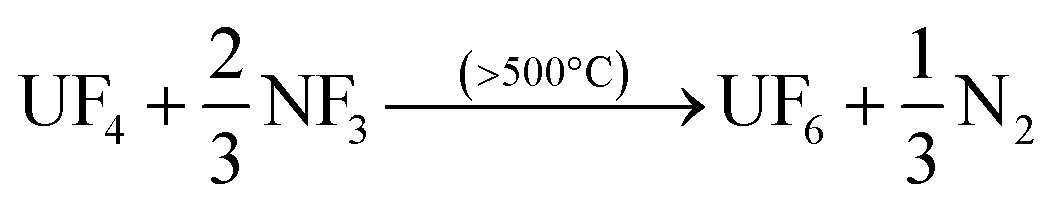 | (4) |
2. Experimental
2.1 Reagents and materials
Mo metal, MoO2 and MoO3 were purchased from Alfa Aesar (Haverville, MA). Deionized water from a Barnstead E-Pure (Thermo Fisher, Waltham, MA) water purification system was 18.0 MΩ cm. Activated alumina spheres (0.125 inch dia.) were purchased from Delta Adsorbents (Roselle, IL). The sample and reference pans used in the TGA/DT and thermo-fluorination reactor were pressed in-house from 99.999% nickel, as 0.254 mm thick sheet purchased from EPSI Metals (Ashland, OR) and were preconditioned by treatment with NF3 up to 610 °C. Monel screens (400 mesh) were purchased from Cleveland Wire Cloth & Mfg. Co (Cleveland, OH).Technetium-99 dioxide (99TcO2) was freshly prepared by thermal decomposition of NH4TcO4 (ref. 21) from house stocks at PNNL. Technetium-99 metal was prepared by heating 99TcO2 in a thermo-gravimetric furnace in a gas stream of 4% H2/Ar at 600 °C. The resulting 99Tc metal was a silver granular material. The metal was used in fluorination experiments immediately after each preparation.
For the mixed [99Mo]MoO2/UO2 experiment, sodium molybdate and sodium borohydride were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. No-carrier-added (NCA) 99Mo/99mTc solution in physiological saline solution was used as received from a commercial medical isotope supplier. A depleted uranium dioxide powder source from AREVA (Richland, WA) was used for the [99Mo]Mo/UO2 experiment.
2.2 [99Mo]MoO2/UO2 sample preparation
A homogeneous mixture of fine UO2 and [99Mo]MoO2 crystals, in a ∼7.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio, was prepared in a nickel pan. The sample was prepared via the following steps, with Table 1 summarizing the reagent inputs: sodium molybdate (Na2MoO4) salt (2.35 mg) was added to a microcentrifuge tube (“tube 1”). A 0.76 mL aliquot of NCA 99Mo (79.9 ± 2.3 kBq, equivalent to 4.50 ± 0.13 pg) was added to the tube. The Na2MoO4 salts were allowed to completely dissolve and equilibrate in the tube, thus creating a homogeneous mixture of [99Mo]MoO42− ions. In a separate microcentrifuge tube (“tube 2”), NaBH4 salts (12.43 mg) were added and 0.2 mL H2O was used to dissolve the salts. The resulting NaBH4 solution was added to tube 1 and the solutions were mixed thoroughly.
:1 mole ratio, was prepared in a nickel pan. The sample was prepared via the following steps, with Table 1 summarizing the reagent inputs: sodium molybdate (Na2MoO4) salt (2.35 mg) was added to a microcentrifuge tube (“tube 1”). A 0.76 mL aliquot of NCA 99Mo (79.9 ± 2.3 kBq, equivalent to 4.50 ± 0.13 pg) was added to the tube. The Na2MoO4 salts were allowed to completely dissolve and equilibrate in the tube, thus creating a homogeneous mixture of [99Mo]MoO42− ions. In a separate microcentrifuge tube (“tube 2”), NaBH4 salts (12.43 mg) were added and 0.2 mL H2O was used to dissolve the salts. The resulting NaBH4 solution was added to tube 1 and the solutions were mixed thoroughly.
| Reagent | Reagent mass, mg | Mass ratio, reagent: Na2MoO4 | Moles reagent | Mole ratio, reagent: [99Mo]MoO2 |
|---|---|---|---|---|
| a Dissolved salts spiked to 34.0 ± 1.0 kBq 99Mo/mg Na2MoO4.b Equivalent to moles [99Mo]MoO2 reaction product. | ||||
| Na2MoO4 | 2.35a | — | 1.14 × 10−5 b | — |
| NaBH4 | 12.43 | 5.29 | 3.29 × 10−4 | 28.8 |
| UO2 | 23.05 | 9.81 | 8.54 × 10−5 | 7.48 |
Precipitates of [99Mo]MoO2 began to form quickly in the presence of the reducing agent. After several hours, it was determined that the Mo(VI) → Mo(IV) reduction was complete. Tube 1 was centrifuged at ∼8000 rpm using Sorvall MC 12V centrifuge (Dupont, Newtown, CT). Next, the supernate was removed. Water (1 mL) was added to tube 1, and the [99Mo]MoO2 crystals were re-suspended. Depleted uranium dioxide powder (23.05 mg) was added to the tube and the [99Mo]MoO2/UO2 mixture was thoroughly mixed by sonication. Tube 1 was again centrifuged and the supernate discarded. The mixture was re-suspended in ∼250 μL water, and 50 μL aliquots of the suspension were added to a Ni sample pan that had been placed under an infrared heat lamp. The solid suspension was quantitatively added to the pan in successive ∼50 μL aliquots as the liquid in the pan was evaporated. Once thoroughly dried, the Ni pan containing the mixture of [99Mo]MoO2 and UO2 was transferred to a thermo-fluorination apparatus for gas-phase [99Mo]Mo separation from UO2.
2.3 Thermo-fluorination apparatus
Thermogravimetric (TG) and differential thermal (DTA) screening data for the reaction of NF3 on samples of UO2, Mo and Tc metal, MoO2 and TcO2, and MoO3 (Fig. 1B) was acquired using a modified Seiko TG/DTA 320.12 The gases used for thermoanalytical experiments were 99.995% purity NF3 from Advanced Specialty Gases (Reno, NV) and 99.9995% ultra-high purity (UHP) Ar from OXARC (Pasco, WA). The same instrument was used in the reduction of 99TcO2 to 99Tc metal, wherein a stream of 4% H2 (99.99%) (OXARC) in Ar was used at 600 °C for 1 h.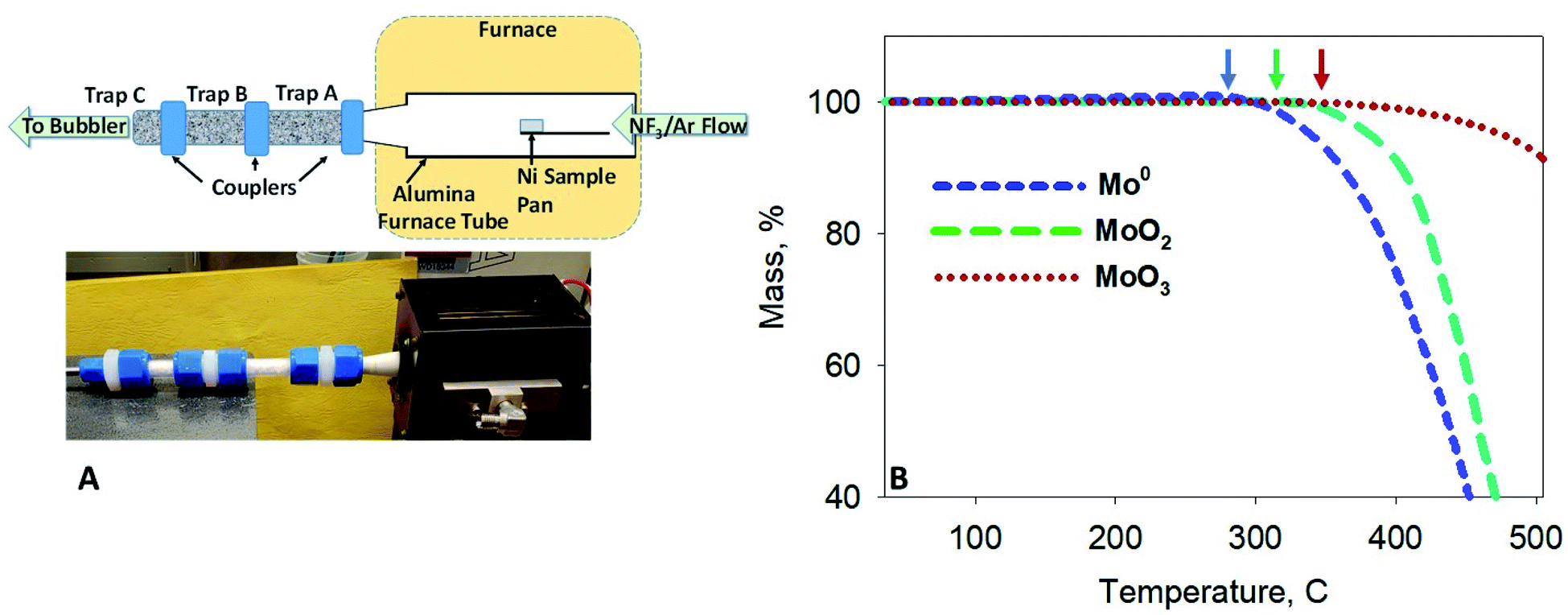 | ||
| Fig. 1 (A) Schematic and image of the fluorination apparatus. Two packed alumina traps (Traps A and B) and a quartz wool (Trap C) were coupled to the furnace tube outlet. Effluent gases were scrubbed through a bubbler trap prior to release. (B) Thermogravimetric scans of Mo metal, MoO2, and MoO3 powders exposed to a 5% NF3/Ar gas mixture. Arrows indicate the onset of volatility below 350 °C. | ||
Modification of the TG/DTA system included conditions for adequate gas mixing and improvements for corrosion resistance. NF3/UHP Ar gas mixtures were premixed in 4 linear feet of SS tubing (0.25 inch OD) prior to their entry into the furnace chamber of the TG apparatus. The premixed gas was routed through the analytical microbalance chamber by a 1/16 inch OD nickel tube to an area about 2.54 cm from the sample and reference pans. This distance reduced buoyancy motion of the sample and reference arms as the dense gas mixture was released from the nickel tube and also allowed for some laminar flow of the gas mixture along the direction of the sample. A larger UHP Ar flow was passed though the analytical balance and sensitive electronic components to protect them from a backflow of hot NF3 and other reaction product gases. Three flow meters were used to adjust the NF3/Ar concentration to a total gas flow rate of 200 mL min−1. The platinum thermocouples inside the balance beams were plated with nickel and the plating was covered in ceramic. The coatings help to reduce hot NF3 corrosion of the thermocouples for extended reaction screening of Mo, Tc and U samples, below 550 °C. The coatings were supplied by RT Instruments (Woodland, CA).
2.4 Fluorination protocol for [99Mo]MoO2/UO2 samples
The thermogravimetric apparatus described above was used to react the homogeneous [99Mo]MoO2/UO2 mixture with NF3. Two sequential activated alumina traps were attached to the 0.75 inch output of the TG alumina furnace tube as shown in Fig. 1A. The traps were made of 0.75 inch o.d. Teflon® PFA tubing (McMaster-Carr, Chicago, IL) assembled with Swagelok (Solon, OH) Teflon® PFA tube unions. The activated alumina spheres were contained in the tubing with the use of Monel screens placed within the tube unions; the spheres were slightly crushed to produce trap packing media that allowed unimpeded flow of gases. Behind the rear trap, a quartz wool plug was placed after the rear Monel screen. From there, a Teflon tube routed effluent gases through a 125 mL Erlenmeyer flask configured as a water bubbler.At the end of a thermo-fluorination experiment, the traps and the furnace tube were disassembled, and each component was washed using a series of solvent washes as is described in detail below. The residual components in the nickel sample pan were fully analyzed by dissolution of the entire sample pan in nitric acid. The distribution of 99Mo was evaluated by gamma counting, and that of the U was evaluated by inductively coupled plasma-mass spectrometry (ICP-MS).
2.5 Radiometric measurements
1. HPGe: reference standards were prepared by spiking known volumes of 99Mo-bearing solutions (in secular equilibrium with 99mTc) into 2.0 mL of 0.1 M HCl in 20 mL glass scintillation vials. These samples were analyzed using several high purity germanium (HPGe) gamma detectors (Ortec, Oak Ridge, TN) that had been energy and efficiency calibrated for this geometry using NIST traceable standards. Gamma spectra were evaluated using Genie 2000 Gamma Acquisition and Analysis software (v. 3.4.1) (Canberra, Meriden, CT). The mean 99Mo activity obtained in the reference standards using the HPGe detector analysis was used to establish the various detection efficiencies (Ed) for 99Mo-bearing samples of non-standardized geometries using NaI(Tl) scintillation detectors (described below).2. Auto-gamma counter: aqueous samples were prepared as 2.0 mL aliquots in 12 × 74 mm test tubes for counting on a Wizard 1470 (PerkinElmer, Meriden, CT) automatic gamma counter containing a well-type NaI(Tl) scintillation detector. The detector was configured with a counting protocol specific to the 99mTc gamma emission region of interest (corresponding to 140.57 keV (89 ± 4% intensity)). Samples were not analyzed until secular equilibrium between 99Mo and 99mTc was attained (sample analyses were performed ∼24 h after each experiment was conducted). The Ed for the Wizard 1470 was determined by comparing the count rate of a 2.0 mL aliquot of 99Mo/99mTc solution in the test tube vs. the 2.0 mL aliquot activity determined by the calibrated HPGe detector.
3. Benchtop NaI(Tl) detector/scaler: non-aqueous samples (e.g., sample pan, trap components, furnace tube) were counted using a Ludlum 2200 scaler/rate meter coupled to a 2′′ dia. NaI(Tl) scintillation detector (Sweetwater, TX). Sample observation distance was maximized to ≥15 cm to minimize geometry effects. At a given sample/detector distance, Ed was determined by comparing the NaI(Tl) detector count rate with that of the HPGe-analyzed standard as described above. Again, samples were not analyzed until secular equilibrium between 99Mo and 99mTc was attained.
2.6 Mass spectrometric measurements
After complete decay of 99Mo, dilutions of the dissolved Ni sample pan and trap leachates were prepared in 2% Optima grade HNO3. Quantification of U in the diluted solutions was performed by an Agilent 7700X (Ventura, CA) ICP-MS. Sample solutions were delivered to the mass spectrometer with a fluoride-resistant polyfluoroalkoxy alkane sample intake and nebulizer (Glass Expansion, Pocasset, MA). A ten-point calibration curve was prepared by gravimetric dilutions from a NIST traceable 1000 ppm single element U standard obtained from High Purity Standards (Charleston, SC). The calibration curve had a regression coefficient of 0.9999.3. Results and discussion
3.1 Evaluation of Mo, Tc, and UO2 volatility by fluorination
Fluoride volatility of Tc was likewise evaluated under the same conditions. Fig. 2A shows the evolution of 99TcF6 from 99Tc metal, which initiates at ∼180 °C, and that from 99TcO2, which initiates at or below ∼250 °C isothermal in Fig. 2B. The volatile reaction products are analogous to the Mo complexes discussed above. In Fig. 1B and 2A and B, the volatile species of Mo and Tc were removed from the reaction system by the Ar gas purge as demonstrated by the steep downward slopes of the TG scans. The Mo and Tc volatility profiles were found to be quite similar, and the complete removal of Te, Ru, Nb, Sb, and several other elements have been shown previously to follow suit.17,18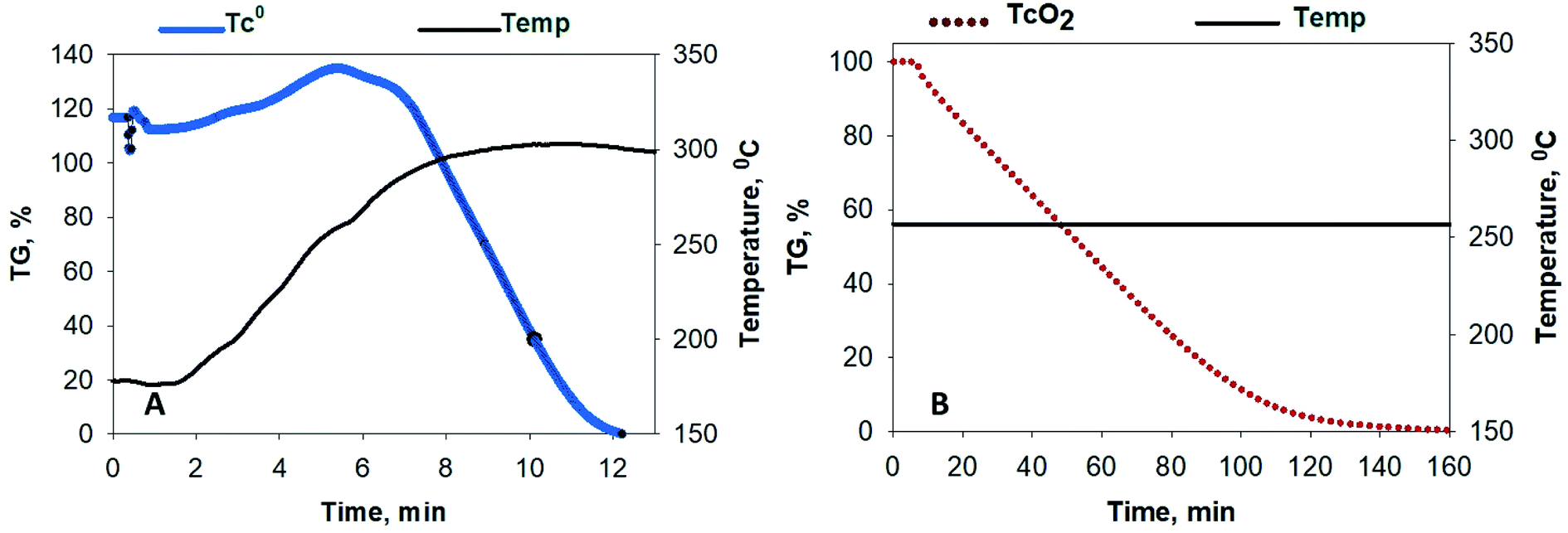 | ||
| Fig. 2 Conversion of 99Tc metal and 99TcO2 to their volatile fluorides (99TcF6) by exposure to 5% NF3 (in Ar). (A) Evolution of 99Tc metal, which initiates at ∼180 °C, and (B) 99TcO2, which initiates at or below ∼250 °C. | ||
The behavior of UO2 with exposure to NF3 provides a stark contrast to that observed with Mo and Tc species, as shown in Fig. 3. Fluorination of UO2 is quite unique to this oxide of U and has been described previously by members of this research team.12 Using the same 5% NF3/Ar mixture employed for Mo- and Tc-bearing materials, UO2 was converted to non-volatile UO2F2 once the temperature approached 420 °C, after which a plateau region was sustained for several hours with the proper NF3 exposure conditions before significant production of gaseous UF6 occurred. The thermogravimetric evaluations with gas streams of heated 5% NF3/Ar indicate that gas-phase separations of Mo (metal and MoO2) and Tc (metal and TcO2) from UO2 is feasible.
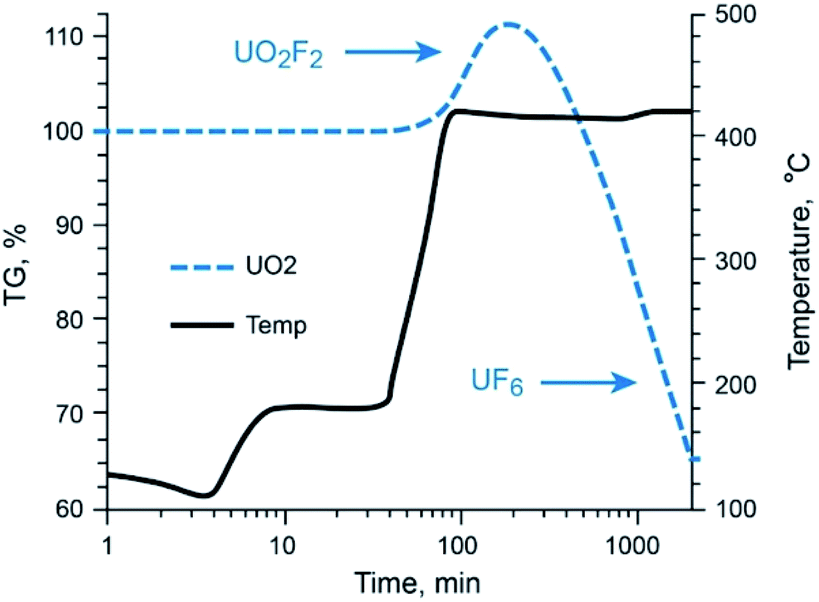 | ||
| Fig. 3 Thermo-fluorination conversion of UO2 to UO2F2 to UF6(g) with 5% NF3. UO2F2 to UF6(g) conversion occurs at temperatures well above that required for volatile MoF6/99TcF6 formation. | ||
Gaseous fluorides of these transition metals can be generated at temperatures below the conversion temperature of UO2 to UF6 (via UO2F2 formation). This permits NF3 leaching of a fissioned UO2 solid with no UF6 attendant in the gaseous Mo (Tc) phase.
3.2 Gas-phase separation of [99Mo]MoO2 from UO2
Given the preceding thermogravimetric results for metal and metal oxide constituents and UO2, a gas-phase separation of 99Mo (as MoO2) from UO2 was evaluated. A sample was prepared in a nickel sample pan that consisted of a homogeneous mixture of fine UO2 (23 mg) and MoO2 crystals (1.5 mg); NaBH4 was initially used to reduce an aqueous solution of Na2[99Mo]MoO4 to form a composite isotope solid of [99Mo]MoO2 via eqn (5).22| Na2MoO4 + NaBH4 + 2H2O → NaBO2 + MoO2 + 2NaOH + 3H2 | (5) |
For this experiment, the outlet of the modified TG furnace tube was connected to tandem traps (A and B) that were packed with activated alumina. A third trap (C) was packed with a compact bundle of quartz wool (Fig. 1A). A fluorination experiment was performed with a 5% NF3/Ar mixture, and the furnace temperature held at ∼400 °C for 2 h. At the end of the experiment, the trapping system components were disconnected from the furnace tube, and each of the three traps was disconnected from each other. Next, each component in Fig. 1A was leached using a series of solvent washes. These washes included that of the furnace tube and each of the three traps. The nickel sample pan (and salt residues) was completely dissolved in nitric acid. The water in the bubbler trap was acidified and evaporated to near dryness. Each component and wash solution was analyzed by gamma counting (99Mo/99mTc) and ICP-MS (U).
Analysis of the distribution of [99Mo]Mo and U revealed an excellent separation of the fission product from the simulated fissioned source material. The Mo was almost completely removed from the sample pan, with only 4% remaining (Table 2). Approximately 10% was deposited on the walls of the furnace tube, and 71% was captured in Trap A. Less than 1% of Mo was measured in Traps B, C, and the bubbler. In total, 86% of the Mo was accounted for in the assays of the trapping components. Of the Mo captured in Trap A, ∼70% was removed with a 5 mL H2O rinse (representing ∼50% of the total Mo pan deposit), and an additional 21% was recovered in two sequential washes with NaOH (Table 3). Within the three Trap A aqueous washes, ∼65% of the pan-deposited Mo was recovered.
| Treatment | Reagent | Volume, mL | Trap A recoverya, % | Trap B recoveryb, % |
|---|---|---|---|---|
| a Total recovered 99Mo activity fraction = 71.38% (from Table 2).b Total recovered 99Mo activity fraction = 0.42% (from Table 2).c Elutes 2 and 3 were combined into single vessel.d Al2O3 in traps emptied into vessel followed by hot leaching with NaOH; leachate assayed for 99Mo activity. | ||||
| Elute 1 | H2O | 5 | 69.8 | 55.0 |
| Elute 2 | 4 M NaOH | 5 | 17.0 | 7.6c |
| Elute 3 | 4 M NaOH | 5 | 4.0 | — |
| Al2O3 leach | 4 M NaOH, Δd | 5 | 5.7 | 23.5 |
| Al2O3 residue | — | — | 3.4 | 13.9 |
Radiometric counting of the trapping components immediately after disassembly (before 99Mo/99mTc secular equilibrium was attained) provided qualitative indication that 99mTc was transported efficiently out of the pan and was successfully deposited primarily in the furnace tube and Trap A. Unfortunately, quantitative determination of the 99mTc depositions were not possible with the use of the NaI(Tl) scintillation detector/scaler. However, an HPGe detector scan of the post-reacted Ni sample pan indicated that 99mTc was successfully volatilized and transported out of the pan, thereby corroborating the observed volatilization profile shown in Fig. 2.
In sharp contrast, the U remained in a non-volatile state; 95.3 ± 3.4% of the original U deposit remained in the nickel sample pan, and ∼0.027% was found in the furnace tube (0.024%) and the three traps (0.003%, Table 2). Based on the mass of U measured in the combined trap leaches, the U decontamination factor in the Trap A [99Mo]Mo product was >1.0 × 105. Total U recovery in all fractions was found to be 95.5 ± 3.2%, a value that was within the analytical uncertainty of the experiment.
4. Conclusions
We show that exposure of a homogeneous mixture of [99Mo]Mo/UO2 to 5% NF3/Ar mixture at ∼400 °C for ∼2 h results in a rapid, high yield extraction of [99Mo]Mo from U.Of the ∼86% of 99Mo activity accounted for in the various furnace/trap components, ∼71% of the 99Mo activity was deposited in the first alumina trap. A simple 5 mL water wash of the trap's alumina bed resulted in ∼70% of the trapped 99Mo activity removal, which represented ∼50% of the total 99Mo activity originally deposited in the nickel pan. Technetium-99m was likewise transported and collected on the alumina trap with the separated 99Mo product, although quantitative distribution was not possible in this first test. The results indicate that the gas-phase [99Mo]Mo product was largely devoid of U contamination.
Aqueous processing releases I, Te, Xe and Kr potentially at every step of processing of irradiated targets. Acid dissolution, in particular promotes, volatile behavior in several elements as Tc, and Ru. While fluoride volatility must release these species as well, we believe that the front-end processing of irradiated uranium targets by volatility-based separations is better suited by its rigorous closed engineering to sequester radionuclide populations than the digest and back end, clean-up approach historically and currently used by most nuclear-related enterprises. Fluoride volatility separations of 99Mo from uranium, so described, has a sound chemical basis. Its practical implementation for radiopharmaceutical scale processing still requires elucidation of transport and capture technologies that are optimized for high efficiency retention of isotopes of pharmaceutical interest.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed at Pacific Northwest National Laboratory. The Pacific Northwest National Laboratory is operated for the U.S. Department of Energy by Battelle under contract DE-AC06-67RLO 1830.References
- R. M. Ball, Characteristics of nuclear reactors used for the production of molybdenum-99, Production Technologies for Molybdenum-99 and Technetium-99m, IAEA-TECDOC-1065, International Atomic Energy Agency, Vienna, 1999, pp. 5–17 Search PubMed.
- G. F. Vandegrift and J. L. Snelgrove, et al., Converting targets and processes for fission-product molybdenum-99 from high- to low-enriched uranium, Production Technologies for Molybdenum-99 and Technetium-99m, IAEA-TECDOC-1065, International Atomic Energy Agency, Vienna, 1999, pp. 25–74 Search PubMed.
- G. F. Vandegrift, C. Conner, G. L. Hofman, R. A. Leonard, A. Mutalib, J. Sedlet, D. E. Walker, T. C. Wiencek and J. L. Snelgrove, Modification of Targets and Processes for Conversion of 99Mo Production from High- to Low-Enriched Uranium, Ind. Eng. Chem. Res., 2000, 39(9), 3140–3145 CrossRef CAS.
- H. Kobayashi and O. Amano, et al., FLUOREX reprocessing system for the thermal reactors cycle and future thermal/fast reactors (coexistence) cycle, Prog. Nucl. Energy, 2005, 47(1), 380–388 CrossRef CAS.
- J. Uhlir, An experience on dry nuclear fuel reprocessing in the Czech Republic, in 5th International Information Exchange Meeting on Actinide and Fission Product Partitioning and Transmutation, Czech Republic, 1999 Search PubMed.
- V. K. Ezhov, Commercial rectification facility for deep purification of sublimate uranium hexafluoride, At. Energ., 2007, 103(5), 890–894 CrossRef CAS.
- A. A. Jonke, Reprocessing of Nuclear Reactor Fuels by Processes Based on Volatilization Fractional Distillation, and Selective Adsorption, At. Energy Rev., 1965, 3, 3–60 CAS.
- A. A. Jonke, N. M. Levitz and M. J. Steindler, The potential of the fluoride volatility process for fast breeder reactor fuels, in Symposium on Reprocessing of Nuclear Fuels, The Metallurgical Society and Ames Laboratory, Ames, IA, 1969 Search PubMed.
- N. M. Levitz, A Conceptual Design Study of a Fluoride-Volatility Plant for Reprocessing LMFBR Fuels. ANL-7583, Argonne National Laboratory, Argonne, IL, 1969, p. 295 Search PubMed.
- W. H. Carr., L. J. King, F. G. Kitts, W. T. McDuffee and F. W. Miles, Molten-Salt Fluoride Volatility Pilot Plant: Recovery of Enriched Uranium from Aluminum-Clad Fuel Elements, ORNL-4574, Oak Ridge National Laboratory, Oak Ridge, TN, 1971, p. 82 Search PubMed.
- W. L. Carter and M. E. Whatley, Fuel and Blanket Processing Development for Molten Salt Breeder Reactors, ORNL-TM-1852, Oak Ridge National Laboratory, Oak Ridge, TN, 1967, p. 52 Search PubMed.
- B. K. McNamara, M. J. O'Hara, A. M. Casella, J. C. Carter, R. S. Addelman and P. J. MacFarlan, Uniform deposition of uranium hexafluoride (UF6): Standardized mass deposits and controlled isotopic ratios using a thermal fluorination method, Talanta, 2016, 154, 219–227 CrossRef CAS PubMed.
- B. K. McNamara, R. D. Scheele, A. M. Casella, A. E. Kozelisky and D. Neiner, Fluorination of Uranium Metal to UF3 and UF4 by Nitrogen Trifluoride: Evidence for Elusive UF2, Mater. Res. Soc. Symp. Proc., 2010, 1264, 1264-Z06-03, DOI:10.1557/PROC-1264-Z06-03.
- D. Watanabe, A. Sasahira, K. Hoshino and F. Kawawamura, Adsorption of Molybdenum Hexafluoride on Magnesium Difluoride for Uranium Purification in FLUOREX Reprocessing, J. Nucl. Sci. Technol., 2011, 48(12), 1413–1419 CrossRef CAS.
- R. Jubin, Used Fuel Reprocessing, in Nuclear Fuel Cycle Course, Consortium for Risk Evaluation with Stakeholder Participation (CRESP): Fuel Cycle and Isotopes Division, Oak Ridge National Laboratory, 2011 Search PubMed.
- R. A. Clark, B. K. McNamara, C. J. Barinaga, J. M. Peterson, N. Govind, A. Andersen, D. G. Abrecht, J. M. Schwantes and N. E. Ballou, Electron ionization mass spectrum of tellurium hexafluoride, Inorg. Chem., 2015, 54(10), 4821–4826 CrossRef CAS PubMed.
- R. D. Scheele, B. K. McNamara, A. M. Casella and A. E. Kozelisky, On the use of thermal NF3 as the fluorination and oxidation agent in treatment of used nuclear fuels, J. Nucl. Mater., 2012, 424(1–3), 224–236 CrossRef CAS.
- B. K. McNamara, E. C. Buck, C. Z. Soderquist, F. N. Smith, E. J. Mausolf and R. D. Scheele, Separation of metallic residues from the dissolution of a high-burnup BWR fuel using nitrogen trifluoride, J. Fluorine Chem., 2014, 162, 1–8 CrossRef CAS.
- T. R. Mills and L. W. Reese, Separation of plutonium and americium by low-temperature fluorination, J. Alloys Compd., 1994, 213, 360–362 CrossRef.
- D. Brown, Halides of the Lanthanides and Actinides, in Halides of the Transition Elements, Wiley, New York, 1968 Search PubMed.
- P. P. Chinenov, S. I. Rovnyi, V. V. Ershov and V. A. Mezentsev, Production of technetium preparations of high radiochemical purity, At. Energ., 1996, 81(1), 467–470 CrossRef.
- C. F. Tsang and A. Manthiram, Synthesis of lower-valent molybdenum oxides in aqueous solutions by reducing Na2MoO4 with NaBH4, J. Mater. Chem., 1997, 7(6), 1003–1006 RSC.
| This journal is © The Royal Society of Chemistry 2020 |