
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient donor–acceptor emitter based nonsymmetrical connection for organic emitting diodes with improving exciton utilization†
Jayaraman Jayabharathi *,
Jagathratchagan Anudeebhana,
Venugopal Thanikachalam,
Sekar Sivaraj and
Annadurai Prabhakaran
*,
Jagathratchagan Anudeebhana,
Venugopal Thanikachalam,
Sekar Sivaraj and
Annadurai Prabhakaran
Department of Chemistry, Material Science Lab, Annamalai University, Annamalai Nagar, Tamilnadu-608 002, India. E-mail: jtchalam2005@yahoo.co.in
First published on 23rd January 2020
Abstract
A new strategy developed to construct blue emissive materials having an unsymmetrical connection with identical conjugated phenanthrimidazole groups results in the separation of the frontier orbitals and leads to donor–acceptor (D–A) architecture. The blue device with 2-(naphthalen-1-yl)-1-(4-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (p-PPI)/2-(naphthalen-1-yl)-1-(3-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (m-PPI) emissive layer (λEL – 434/420 nm) shows high efficiencies: current efficiency (ηc) – 5.83/3.56 cd A−1; power efficiency (ηp) – 5.73/3.48 lm W−1; external quantum efficiency (ηex) – 8.98/6.48% at 3.0 V. Their cyano derivatives, p-CNPPI/m-CNPPI (λEL – 422/406 nm) exhibit maximum efficiencies (ηc – 6.28/4.38 cd A−1; ηp – 6.14/4.01 lm W−1; ηex – 9.01/6.72%) at 2.9 V compared to the p-PPI/m-PPI devices. The weak charge transfer in the D–A emitters results in deep blue emission. The anisotropic structural characteristics of p-PPI, p-CNPPI, m-PPI and m-CNPPI induced horizontal dipole orientation in films and enhanced EL efficiency. These bipolar materials with suitable triplet energy can be used as hosts in green as well as red phosphorescent organic light emitting devices (PHOLEDs). The green/red device (λEL – 504/618 nm) with p-CNPPI: Ir(ppy)3/Ir(MDQ)2 (acac) exhibits a maximum L – 8823/38418 cd m−2; ηex – 24.56/17.31%; ηc – 84.30/18.09 cd A −1; ηp – 86.43/21.43 lm W −1with CIE (0.33, 0.61)/(0.65, 0.34).
1 Introduction
Organic donor–acceptor (D–A) conjugated compounds exhibit high maximum efficiency in fluorescent OLEDs,1 however, there is still restriction for commercialization due to a lack of blue (B) emitters.2 Although green and red emitters have been exploited widely, because of their wide band gap (Eg) with unbalanced carrier injection, the fabrication of blue devices is of utmost importance.3–8 Therefore, development of blue emitters with narrow FWHM (full width at half maximum) is an important task in the OLED family.9,10 The aggregation of the emitter induced molecular interaction results in a bathochromic shift with low quantum efficiency,11 however, non-doped blue emitters consisting of restricted intermolecular interactions exhibit a high quantum yield.12 Their potential carrier injection and transportation provides balanced charge recombination and results in enhanced efficiency.13–15 The blue OLEDs of CIEx,y (0.14, 0.08) and (0.15, 0.06) have been the requirement of NTSC (national television system committee) and HDTV (high-definition television), respectively. The pyrene16 and anthracene17 derivatives exhibit high efficiency with poor blue color purity. D–A emitters with twisted configuration was employed to harvest pure blue emission as a result of reduced π-conjugation:18–24 D–A molecules have been designed by connecting electron deficient fragment (benzonitrile, triazine, etc.) with electron-rich fragment (triphenylamine, carbazole, etc.).12,13 However, the blue emitters with D–A configuration is limited because of lower excitation energy and the broad emission spectra is red-shifted with poor colour purity.14 Herein, we report an effective strategy to construct deep blue emitters namely, 2-(naphthalen-1-yl)-1-(4-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (p-PPI), 4-(2-(4-(2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (p-CNPPI), 2-(naphthalen-1-yl)-1-(3-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (m-PPI) and 4-(2-(3-(2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (m-CNPPI) (Fig. S1–S12†) having D–A configuration with unsymmetrical connection between two identical components without typical donor or acceptor characteristics. The electron densities are distributed separately on the two conjugated groups of D–A and exhibit deep blue emission. The non doped blue device with p-PPI, p-CNPPI, m-PPI and m-CNPPI shows high efficiencies and these bipolar materials used as a host for green and red PHOLEDs. The green/red device (λEL – 504/618 nm) with p-CNPPI: Ir(ppy)3/Ir(MDQ)2 (acac) exhibit maximum L – 78823/38418 cd m−2; ηex – 24.56/17.31%; ηc – 84.30/18.09 cd A−1; ηp – 86.43/21.43 lm W−1 with CIE (0.33, 0.61)/(0.65, 0.34). The unsymmetrical connection strategy is used as a tool for developing highly efficient deep blue materials.2 Experimental section
2.1. Measurements
H1 and C13 NMR measurements were recorded on Bruker 400 MHz spectrometer using TMS as internal standard and mass spectra was recorded on Agilent LCMS VL SD. The UV-optical spectra were measured on Lambda 35 PerkinElmer (solution)/Lambda 35 spectrophotometer (RSA-PE-20) (film) instrument. The emission spectra were recorded with PerkinElmer LS55 spectrometer. The quantum yield was measured with fluorescence spectrometer Model-F7100 with integrating sphere. The decomposition temperature (Td) and Tg (glass transition temperature) was measured with PerkinElmer thermal analysis system (10 °C min−1; N2 flow rate – 100 ml min−1) and NETZSCH (DSC-204) (10 °C min−1; N2 atmosphere), respectively. Fluorescence lifetime was estimated by time correlated single-photon counting (TCSPC) method on Horiba Fluorocube-01-NL lifetime system: nano LED is excitation source with TBX-PS is detector; DAS6 software was employed to analyze the decay by reconvolution method. Oxidation potentials of emissive materials were measured from potentiostat electrochemical analyzer (CHI 630A). Ferrocene was used as internal standard with highest occupied molecular orbital (HOMO) of −4.80 eV and 0.1 M tetrabutylammonium perchlorate in CH2Cl2 as supporting electrolyte. The frontier energies were calculated by EHOMO = −(Eox + 4.8 eV) and ELUMO = (Ered − 4.8 eV), respectively.2.2. Computational details
The ground (S0) (DFT)/excited (TD-DFT) characteristics were studied by using Gaussian 09 program25 and multifunctional wavefunction analyzer (Multiwfn).25 The natural transition orbitals (HONTOs & LUNTOs) were analyzed with dominant hole–particle pair contribution and associated transition weights. Multiwfn software was employed for wave function of electron–hole pair from TDM (transition density matrix) by TD-DFT and was plotted in two-dimensional (2D) color-filled map for easily differentiating ground state from excited-state character.
(TD-DFT) characteristics were studied by using Gaussian 09 program25 and multifunctional wavefunction analyzer (Multiwfn).25 The natural transition orbitals (HONTOs & LUNTOs) were analyzed with dominant hole–particle pair contribution and associated transition weights. Multiwfn software was employed for wave function of electron–hole pair from TDM (transition density matrix) by TD-DFT and was plotted in two-dimensional (2D) color-filled map for easily differentiating ground state from excited-state character.
2.3. Device fabrication and measurement
ITO glass (resistance 20 Ω sq−1) were cleaned with acetone, deionized water and isopropanol, dried (120 °C) followed by UV-zone treatment (20 min) and transferred into deposition system. The devices were fabricated by the multiple source beam deposition method in a vacuum at a pressure of 4 × 10−5 mbar. Evaporation rates of 2–4 Å s−1 (organic materials) and 0.1 and 4 Å s−1 for LiF and metal electrodes respectively were applied. The thickness of each deposition layer was monitored with quartz crystal thickness monitor. The EL measurement with CIE coordinates was recorded with USB-650-VIS-NIR spectrometer (Ocean Optics, Inc, USA). The current density–voltage–luminance (J–V–L) characteristics were performed using source meter (Keithley 2450) equipped with LS-110 light intensity meter. The ηex (external quantum efficiency) was determined from luminance, current density and EL spectrum assuming Lambertian distribution.To evaluate the potential application of these compounds as emitting materials, non-doped OLEDs with configuration of ITO/4,4-bis(N-(1-naphthyl)-N-phenylamino)biphenyl (NPB) (60 nm)/p-PPI or p-CNPPI or m-PPI or m-CNPPI (20 nm)/lithium fluoride (LiF) (1 nm)/Al (100 nm) have been fabricated. Green device: ITO/NPB (40 nm)/4,4′,4′′-tri(N-carbazolyl)-triphenylamine (TCTA) (5 nm)/p-PPI (30 nm): 5 wt% Ir(ppy)3 or p-CNPPI (30 nm): 5 wt% Ir(ppy)3 or m-PPI (30 nm): 5 wt% Ir(ppy)3 or m-CNPPI (30 nm): 5 wt% Ir(ppy)3/1,3,5-tris(N-phenylbenzimidazol-2-yl)-benzene (TPBI) (50 nm)/LiF (1 nm)/Al (100 nm) and red device: ITO/NPB (40 nm)/TCTA (5 nm)/p-PPI (30 nm): 8 wt% bis(2-methyldibenzo-[f,h]quinoxaline)acetylacetonate iridium(III) (Ir(MDQ)2 (acac)) or p-CNPPI (30 nm): 8 wt% Ir(MDQ)2 (acac) or m-PPI (30 nm): 8 wt% Ir(MDQ)2 (acac) or m-CNPPI (30 nm): 8 wt% Ir(MDQ)2 (acac)/TPBI(50 nm)/LiF (1 nm)/Al (100 nm) were also fabricated.
3 Results and discussion
The synthetic route for the emissive materials is presented in Scheme S1:† efficient blue emitters namely, 2-(naphthalen-1-yl)-1-(4-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (p-PPI), 4-(2-(4-(2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (p-CNPPI), 2-(naphthalen-1-yl)-1-(3-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)-1H-phenanthro[9,10-d]imidazole (m-PPI) and 4-(2-(3-(2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (m-CNPPI) were synthesized by Suzuki-coupling reaction with appreciable yield and characterized by 1H and 13C NMR and high resolution mass.Geometry optimization of m-PPI, p-PPI, m-CNPPI and p-CNPPI have been performed by DFT/B3LYP/6-31G (d,p) using Gaussian-03 and the optimized geometry is shown in Fig. 1 along with their corresponding molecular orbital distribution. The ground state torsion angles between the bridged phenyl ring and two phenanthrimidazole groups are: N23–C25–C44–C45 (m-PPI-10°; p-PPI-15°; m-CNPPI-6° & p-CNPPI-8°) and N52–C50–C46–C45 (m-PPI-60°; p-PPI-65°; m-CNPPI-46° & p-CNPPI-58°). The dihedral angle about N-side coupling (C24–C25–C42–C44): m-PPI-70°; p-PPI-75°; m-CNPPI-62° & p-CNPPI-66° and C-side coupling C(C36–C53–C57–C59): m-PPI-30°; p-PPI-35°; m-CNPPI-22° & p-CNPPI-26° reveal that only C–H⋯π interaction exist between adjacent molecules. The distorted configuration with weak intermolecular interaction in film can be effectively lowers the quenching (aggregation induced-AIQ) which leads to enhanced efficiency.14 The excited state twist angle is increased relative to ground state twist angle and this may promote the suppression of non-radiation (knr) for the enhancement of photoluminescence efficiency (ηPL).25,26 The potential energy surface of twisted geometry (Fig. S13†) reveal that these materials need small relaxation energy to form excited state excimer corresponding to slightly increased interplanar separation (1.40 Å to 1.42 Å) of phenanthrimidazole moieties from the phenyl linker fragment and only small energy needs to return to equilibrium geometry at ground state. The rigid geometry from initial state to excited state excimer reveals that the minimized non-radiative (knr) energy dissipation contributes to the enhanced emission. Suppression of non-radiative (knr) pathway is likely to be the reason for increasing lifetime which leads to higher photoluminescence efficiency.26,27
 | ||
| Fig. 1 Molecular structure, ground and excited state geometries with dihedral angles. | ||
The HOMO orbital of m-PPI, p-PPI, m-CNPPI and p-CNPPI is localized at phenanthrimidazole with side coupling and linker moiety, whereas the LUMO orbital distributes on another phenanthrimidazole with side coupling (Fig. 2). The HOMO and LUMO orbitals display adequate separation in electron density beneficial for carrier transportation and act as bipolar material.28,29 Moreover, these compounds exhibit reduction and oxidation which reveal that these derivatives possess good charge transport abilities (Fig. 2). The two phenanthrimidazole fragments with twisted D–A architecture were constructed by unsymmetrical connection through para- and meta-linking of bridged phenyl ring. The twisted structure of these molecules promote the frontier orbital distribution which enhanced the excition utilization (ηs) through efficient up conversion of non-radiative triplet to radiative singlet.
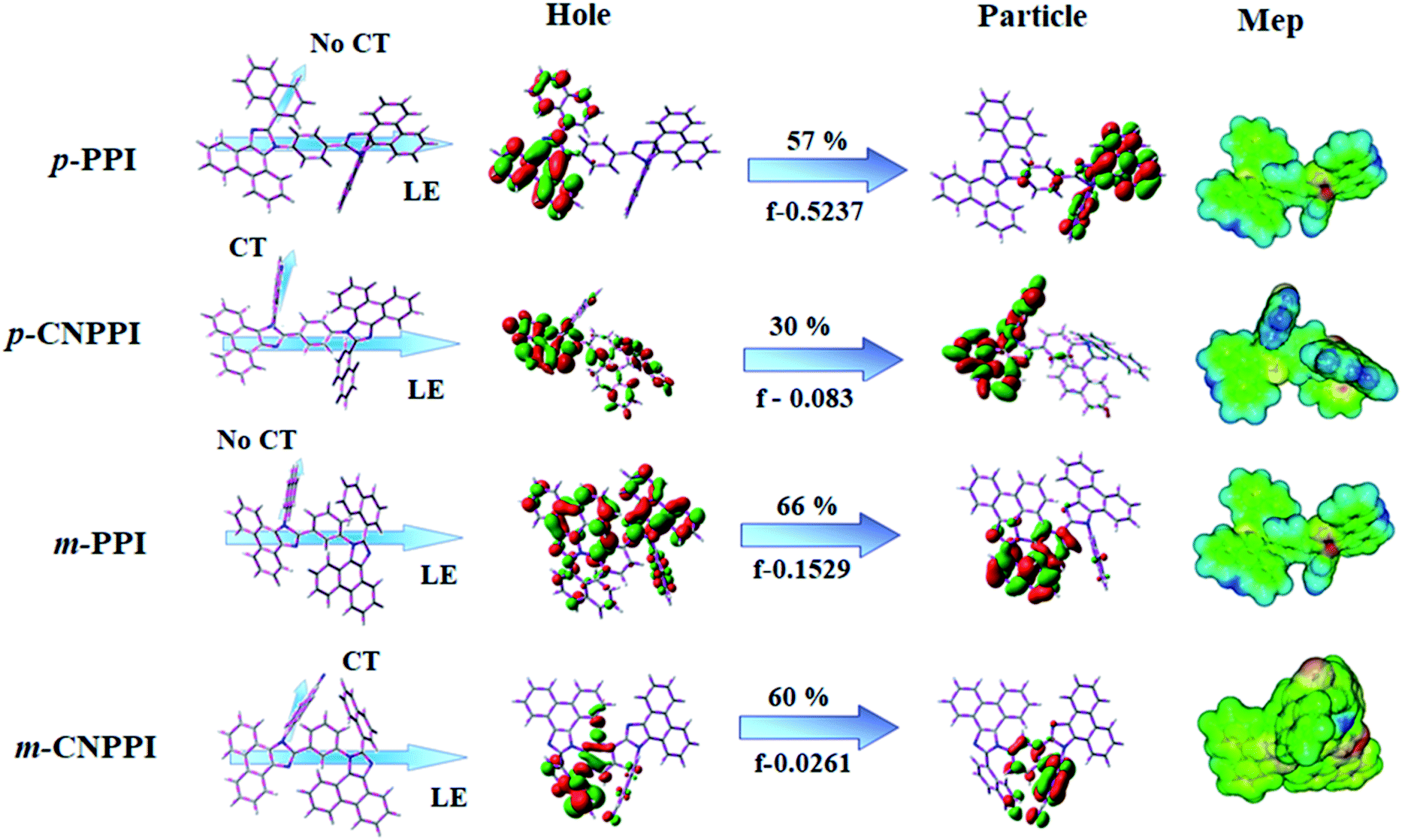 | ||
| Fig. 2 Optimized geometry, HOMO and LUMO and MEP images. | ||
In order to supplement the experimental results, theoretical calculation was made to describe the excited state characteristics of m-PPI, p-PPI, m-CNPPI and p-CNPPI materials (HONTOs & LUNTOs) (Fig. S14–S17 and Tables S1–S16†). The overlap of hole and particle for p-PPI and p-CNPPI is larger than that of m-PPI and m-CNPPI and para coupled derivatives having stronger oscillator strength (f) [0.5237 – p-PPI; 0.083 – p-CNPPI) than meta coupled derivatives (0.1529 m-PPI; 0.0261 – m-CNPPI]. The HONTOs and LUNTOs of m-PPI, p-PPI, m-CNPPI and p-CNPPI show that these materials show weak intramolecular-charge-transfer (ICT) transition derived from the same functional group and the small overlap between HONTOs and LUNTOs will promote the deep blue fluorescence and high photoluminescence quantum yield. The overlap between HOMO and LUMO is extremely small for p-CNPPI and m-CNPPI ensuring small ΔEST and ensures fairish radiative decay from intramolecular CT exciton.28 Moreover, large steric hindrance of these emitters lead to a rigid geometry thus, the amount of charge transfer (CT) is increased due to extent of conjugation. The small inter ring torsion angle of twisted conformation suppress the intermolecular π–π stacking in solid state and thus, fluorescence quenching was suppressed which leads to high ηex in film.29–32
3.1. Thermal and electrochemical properties
The twisted molecular architecture has increased their thermal stability. The excellent thermal stability of these new donor–acceptor compounds was analyzed by high decomposition temperature (Td, 5% weight loss). The high Td (520 °C – p-PPI; 510 °C – m-PPI; 515 °C – m-CNPPI; 528 °C – p-CNPPI) reveals that these compounds would be capable of enduring vacuum thermal sublimation process. Both compounds show high glass transition temperature (Tg: 166 °C – m-PPI; 170 °C – p-PPI; 168 °C – m-CNPPI; 178 °C – p-CNPPI) indicating that introduction of rigid phenanthrimidazole moiety improves greatly their morphological stability (Table 1: Fig. 3). The interaction of substituent at C2- and N1 with phenanthrimidazole core improved the Tg which can induce more condensed molecular packing. The higher Tg implicates that they could form morphologically stable amorphous film upon thermal evaporation which reduces phase separation upon heating and prolonging the lifetime of devices. The thermal morphological stability of thin film of these compounds was examined using atomic-force microscopy measurement at 30 °C and 90 °C for 12 h. The root-mean-square (0.32 nm – m-PPI; 0.28 nm – p-PPI; 0.36 nm – m-CNPPI; 0.31 nm – p-CNPPI) reveal absence of remarkable surface modifications before and after annealing which supports the suitability of these materials for fabrication (Fig. 4).| Parameters | p-PPI | p-CNPPI | m-PPI | m-CNPPI |
|---|---|---|---|---|
| λab (nm) (sol/film) | 285![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 355/288358 355/288358 |
283350/286349 |
281350/284354 |
270339/279342 |
| λem (nm) (sol/film) | 420/367436 |
412/359425 |
409/362422 |
395/360408 |
| Tg/Td (°C) | 170/520 | 178/528 | 166/510 | 168/515 |
| ES/ET/ΔEST (eV) | 3.37/2.51/0.80 | 3.45/2.61/0.84 | 3.42/2.62/0.80 | 3.44/2.66/0.79 |
| ϕ (soln/film) | 0.86/0.80 | 0.89/0.86 | 0.78/0.70 | 0.82/0.73 |
| τ (ns) | 1.86 | 1.91 | 2.01 | 2.58 |
| kr/knr (108 s−1) | 4.6/0.8 | 4.7/0.6 | 3.8/1.1 | 3.2/0.5 |
| HOMO/LUMO (eV) | −5.62/−2.28 | −5.52/−2.23 | −5.59/−2.26 | −5.48/−2.23 |
 | ||
| Fig. 3 (a) Cyclic voltammogram; (b)TGA graph; (c) DSC graph; (d) lifetime decay curve of p-PPI, p-CNPPI, m-PPI and m-CNPPI. | ||
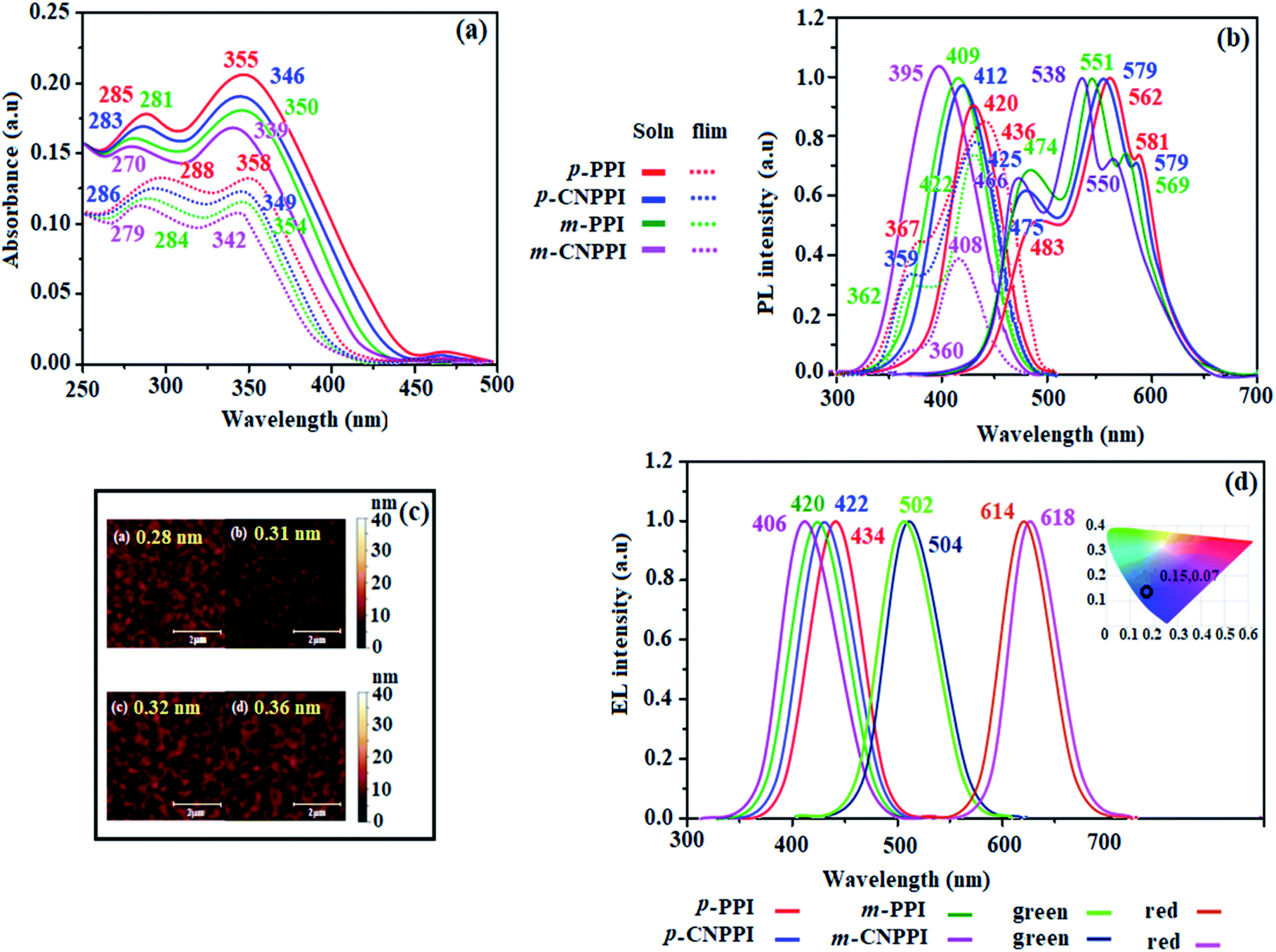 | ||
| Fig. 4 (a) Normalized absorption spectra; (b) normalized emission spectra; (c) AFM images; (d) EL spectra of p-PPI, p-CNPPI, m-PPI and m-CNPPI. | ||
Electrochemical characterization of these materials was made by cyclic voltammetry.33,34 The reduction onset potentials are of −2.54 V – m-PPI; −2.52 V – p-PPI; −2.54 V – m-CNPPI and −2.55 V – p-CNPPI and oxidation onset potentials are of +0.79 V – m-PPI; +0.82 V – p-PPI; +0.68 V – m-CNPPI and +0.72 V – p-CNPPI. Based on redox onset potentials, the frontier orbital energies are calculated as ELUMO: −2.26 eV – m-PPI; −2.28 eV – p-PPI; −2.23 eV – m-CNPPI and −2.25 eV – p-CNPPI and the EHOMO: −5.59 eV – m-PPI; −5.62 eV – p-PPI; −5.48 eV – m-CNPPI and −5.52 eV – p-CNPPI.35 This reflects the more balanced carrier injection properties of these materials (Fig. 3).
The UV and emission spectra of m-PPI, p-PPI, m-CNPPI and p-CNPPI materials in dichloromethane were recorded in solution and in neat film (Fig. 4).36 The strong and sharp absorption (270 nm) is due to π–π* transition originates from phenanthrimidazole ring and the shoulder peak at longer wavelength ranging from 339 to 355 nm is attributed to ICT (intramolecular charge-transfer) from donor to acceptor unit.37 The extinction coefficient of D–A molecules is higher due to increase of conjugation length.38,39 The cyano derivatives m-CNPPI and p-CNPPI show very strong absorption on comparison to parent compounds m-PPI/m-CNPPI: 281/270 nm (emax = 35587.19 cm−1 M−1)/(emax = 37037.04 cm−1 M−1) and p-PPI/p-CNPPI: 285/283 nm (emax = 35087.72 cm−1 M−1)/(emax = 35335.69 cm−1 M−1) due to intramolecular CT from donor to acceptor (Fig. 4). The emissive materials in film state show absorption at 284, 354 nm (m-PPI), 288, 358 nm (p-PPI), 279, 342 nm (m-CNPPI) and 286, 349 nm (p-CNPPI) and the small shift is due to weak π–π* intermolecular stacking.40 The emission of m-PPI, p-PPI, m-CNPPI and p-CNPPI in solution/film show emission at 409/422, 395/436, 420/408 and 412/425 nm, respectively (Fig. 4). The PL spectrum in thin film is slightly red-shifted with respect to solution and the new born emitters exhibit high quantum yield (sol/film): m-PPI (0.78/0.70), p-PPI (0.86/0.80), m-CNPPI (0.82/0.73) and p-CNPPI (0.86/0.80) due to decreased proportion of non-radiative transition (knr).41 The intermolecular interaction in aggregation state is decreasing the quantum yield in film state. However, because of their distorted molecular configuration, the negative effect of aggregation is highly suppressed. Compared to solution, para and or meta-coupling enhanced the steric hindrance forced these molecules to form twisted geometry in film results in less aggregation and lowers the quantum yield in film. The highly rigid geometry reduced the non-radiative exciton which results high quantum yield. The kr/knr (radiative transition rate and non-radiative transition rate) have been calculated from lifetime (τ) and quantum yield (ΦPL) (Table 1). The relative contribution of radiative as well as non-radiative processes in the excited state deactivation was analyzed by kr and knr decay constants: kr = τ/Φ: 3.8 s−1 (m-PPI), 4.6 s−1 (p-PPI), 3.2 s−1 (m-CNPPI) and 4.7 s−1 (p-CNPPI); knr = τ/(1 − Φ): 1.1 s−1 (m-PPI), 0.8 s−1 (p-PPI), 0.5 s−1 (m-CNPPI) and 0.6 s−1 (p-CNPPI) (Fig. 3). These emitters show larger kr and smaller non-radiative rate constant (knr) which leads to enhanced photoluminescence quantum yield (ηPL).
The calculated high triplet energy (ET) of m-PPI (2.62 eV), p-PPI (2.51 eV), m-CNPPI (2.66 eV) and p-CNPPI (2.61 eV) is enough to sensitize the dopants having ET below 2.3 eV and become host materials for green as well as red dopants. The singlet–triplet energy gap (ΔEST) is greater than ≥0.8 eV suggesting that TADF behavior was eliminated.42 The wide vibrational emission peak with small red shift by increasing solvent polarity informed the strong CT of excited state relative to ground state43,44 which reveal that these emitters are different from common D–A molecules with prominent solvation effect.45–48 The red shifted emission is due to twisted conformation which enable for charge transfer from donor to accept or via linker. The intramolecular charge transfer is further confirmed by molecular electrostatic potential (MEP).
The vibrational structured absorption and emission (Fig. S18 and S19†) is because of unclassical rigid D–A architecture.49–54 For conjugated molecules, the para-substitution is more beneficial for CT process than meta-substitution. The m-PPI and m-CNPPI exhibit more blue-shifted emission relative to p-PPI and p-CNPPI. Furthermore, the D–A molecules based on two identical fragments display different emission behavior relative to typical D–A molecules. The small red shift in film compared to solution show suppressed π–π* stacking.55 The emission of p-PPI, m-PPI, p-CNPPI and m-CNPPI gives blue-shift relative to parent PPI which is in controversy to the general observation i.e., extension of π-conjugation leads to red shifted emission.53 In addition to that there is an overlap between UV and PL spectra because of enhanced LE character in these title materials than their parent compounds (Fig. 4).
The overlap between hole and particle of p-PPI, p-CNPPI, m-PPI and m-CNPPI is displayed in Fig. S20–S23,† respectively. The similar hole–electron wave function indicates the efficient hybridization between LE and CT states. The composition of CT and LE can be analyzed by TDM (Fig. S20–S23†). The diagonal part reflects that the LE component localized on main backbone while off-diagonal region represents CT component. The colored map indicates that the left-lower part corresponds to LE and right-lower part attributed to CT of donor to acceptor. The right-high zone is due to LE of donor. In this way, the LE:CT proportion of each structure could be directly calculated according to the calculus of matrix elements.
3.2. Single carrier devices
To understand the carrier transport abilities of p-PPI, m-PPI, p-CNPPI and m-CNPPI, hole-only device (HOD) and electron-only device (EOD) have been fabricated with configuration of ITO/NPB (8 nm)/p-PPI or p-CNPPI or m-PPI or m- CNPPI (40 nm)/NPB (8 nm)/Al (100 nm) (hole-only device): NPB (LUMO: −2.3 eV)31 in the hole-only device can prevent electron injection from Al cathode (Ef – 4.3 eV) to p-PPI or p-CNPPI or m-PPI or m-CNPPI layer31 and ITO/TPBi (8 nm)/p-PPI or p-CNPPI or m-PPI or m-CNPPI (40 nm)/TPBi (8 nm)/LiF (1 nm)/Al (100 nm) (electron-only device): TPBi (HOMO: −6.2 eV)31 in the electron-only device with ITO (Ef – 4.8 eV) as the anode can prevent hole injection were fabricated.32 The current density versus voltage characteristics of hole-only and electron-only devices was displayed in Fig. 5. The hole-only and electron-only devices show the same current density values at same voltage indicating that these compounds are bipolar materials. The electron current density of p-PPI, m-PPI, p-CNPPI and m-CNPPI based device is higher than CBP-based device which reveal that these compounds are bipolar-transporting materials with effectively transporting electrons and or holes than CBP.49–51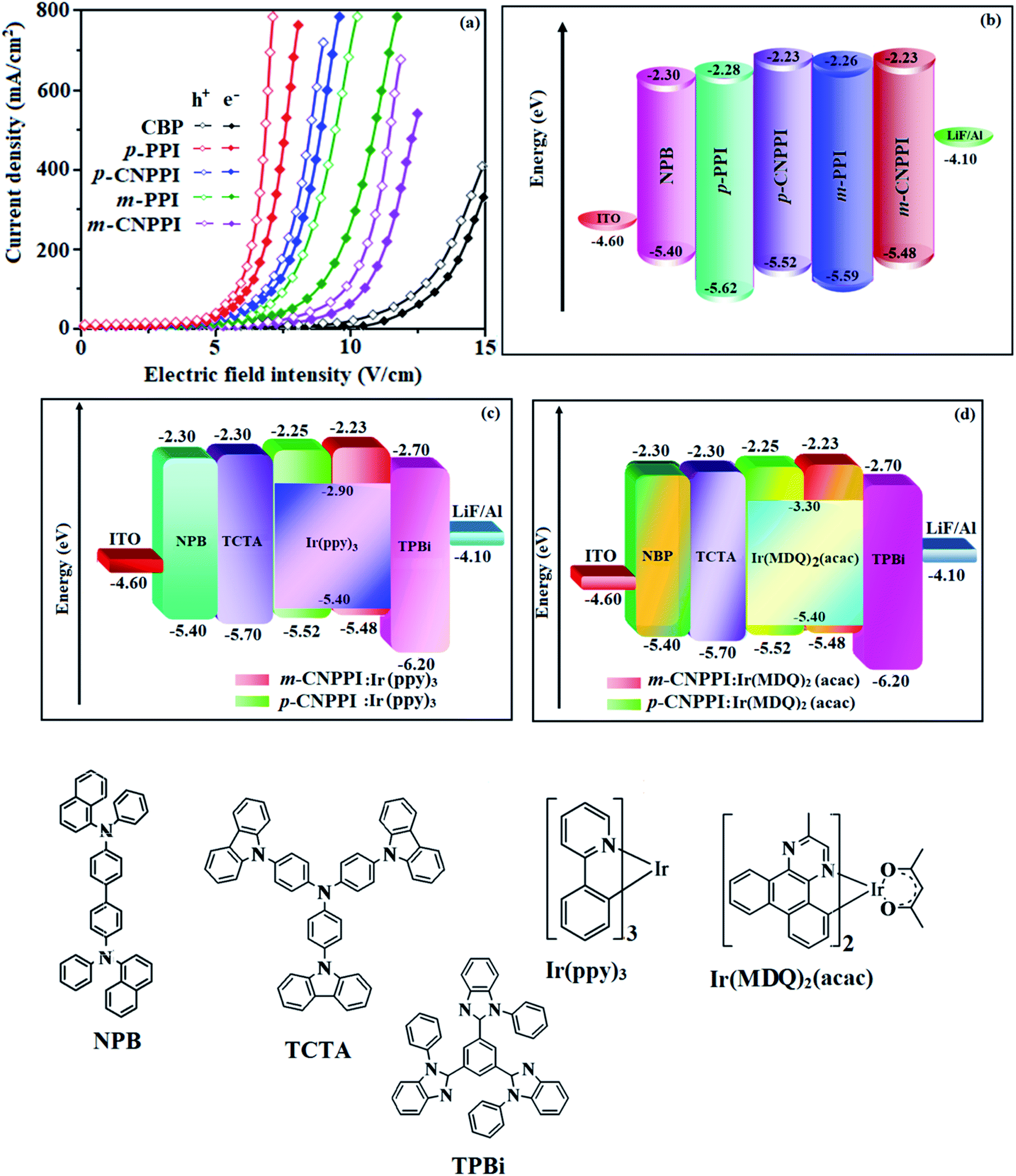 | ||
| Fig. 5 Hole-only and electron only devices (a); energy level diagram of blue (b), green (c) and red (d) devices with molecular structures of functional materials. | ||
However, p-PPI or m-PPI or p-CNPPI or m-CNPPI: Ir(ppy)3 based devices, increase of carrier current is due to direct injection of carrier to HOMO of dopant and hopping transport through p-PPI, m-PPI, p-CNPPI and m-CNPPI sites. The balanced carrier transportation in emissive layer broadening the exciton recombination zone leads to small efficiency roll off.
3.3. Electroluminescent studies
The effective film forming properties of emissive materials are important for device efficiency. The better nanoscale morphology of p-PPI, m-PPI, p-CNPPI and m-CNPPI thin film is attributed to low turn-on voltage (Fig. 4). Energy diagram of the devices are shown in Fig. 5. The TADF materials will show flat decay because of time consuming TADF process for exciton conversion of triplet to singlet, however, the observed single-exponential sharp decay shows that radiative exciton is short-lived without TADF role (Fig. 3). Therefore, exciton utilization efficiency (ηs) in p-PPI, m-PPI, p-CNPPI and m-CNPPI are neither TADF nor TTA mechanism.We have fabricated non-doped blue OLEDs by vacuum evaporation technique with the device structure of ITO/NPB (40 nm)/p-PPI or m-PPI or p-CNPPI or m-CNPPI (20 nm)/TPBi (35 nm)/LiF (0.8 nm)/Al (100 nm). The p-PPI, m-PPI, p-CNPPI and m-CNPPI based device show Commission Internationale de L'Eclairage (CIE) coordinate of (0.15, 0.07), (0.15, 0.06), (0.15, 0.06) and (0.15, 0.06) which is very close to NTSC blue standard. The electroluminescence (EL) spectrum is similar to their PL spectrum which shows both EL and PL originates from the same radiative decay of the singlet exciton. High device performances at low-turn on voltage are extracted from non-doped devices based on p-PPI, m-PPI, p-CNPPI and m-CNPPI (Fig. 4; Table 2). The p-PPI/m-PPI based device (EL – 434/420 nm) shows high efficiencies (ηc – 5.83/3.56 cd A−1; ηp – 5.73/3.48 lm W−1; ηex – 8.98/6.48% at 3.0 V). The cyano derivatives p-CNPPI/m-CNPPI (EL – 422/406 nm) show maximum efficiencies (ηc – 6.28/4.38 cd A−1; ηp −6.14/4.01 lm W−1; ηex – 9.01/6.72% at 2.9 V) than p-PPI/m-PPI based devices (Fig. 6). The high ηex harvested from cyano derivatives based device is due to the co-emission from intercrossed excited state of LE and CT; the isoenergies of singlet (1CT) and triplet (3CT) make 3CT → 1CT transition as spin-allowed transition.52
| Emitters | Host | Von (V) | λEL(nm) | L (cd m−2) | ηc (cd A−1) | ηp (lm W−1) | ηex (%) | CIE (x, y) |
|---|---|---|---|---|---|---|---|---|
| p-PPI | — | 3.0 | 434 | 18845 |
5.83 | 5.73 | 8.98 | 0.15, 0.07 |
| p-CNPPI | — | 2.9 | 422 | 19689 |
6.28 | 6.14 | 9.01 | 0.15, 0.06 |
| m-PPI | — | 3.0 | 420 | 4628 | 3.56 | 3.48 | 6.48 | 0.15, 0.06 |
| m-CNPPI | — | 2.9 | 406 | 10923 |
4.38 | 4.01 | 6.72 | 0.15, 0.06 |
| Green | p-CN | 2.6 | 504 | 78823 |
84.30 | 86.43 | 24.56 | 0.33, 0.61 |
| Green | m-CN | 2.6 | 502 | 90568 |
80.98 | 80.03 | 23.98 | 0.33, 0.61 |
| Red | p-CN | 2.6 | 618 | 38418 |
18.09 | 21.43 | 17.31 | 0.65, 0.34 |
| Red | m-CN | 2.6 | 614 | 34619 |
17.83 | 20.68 | 16.08 | 0.65, 0.34 |
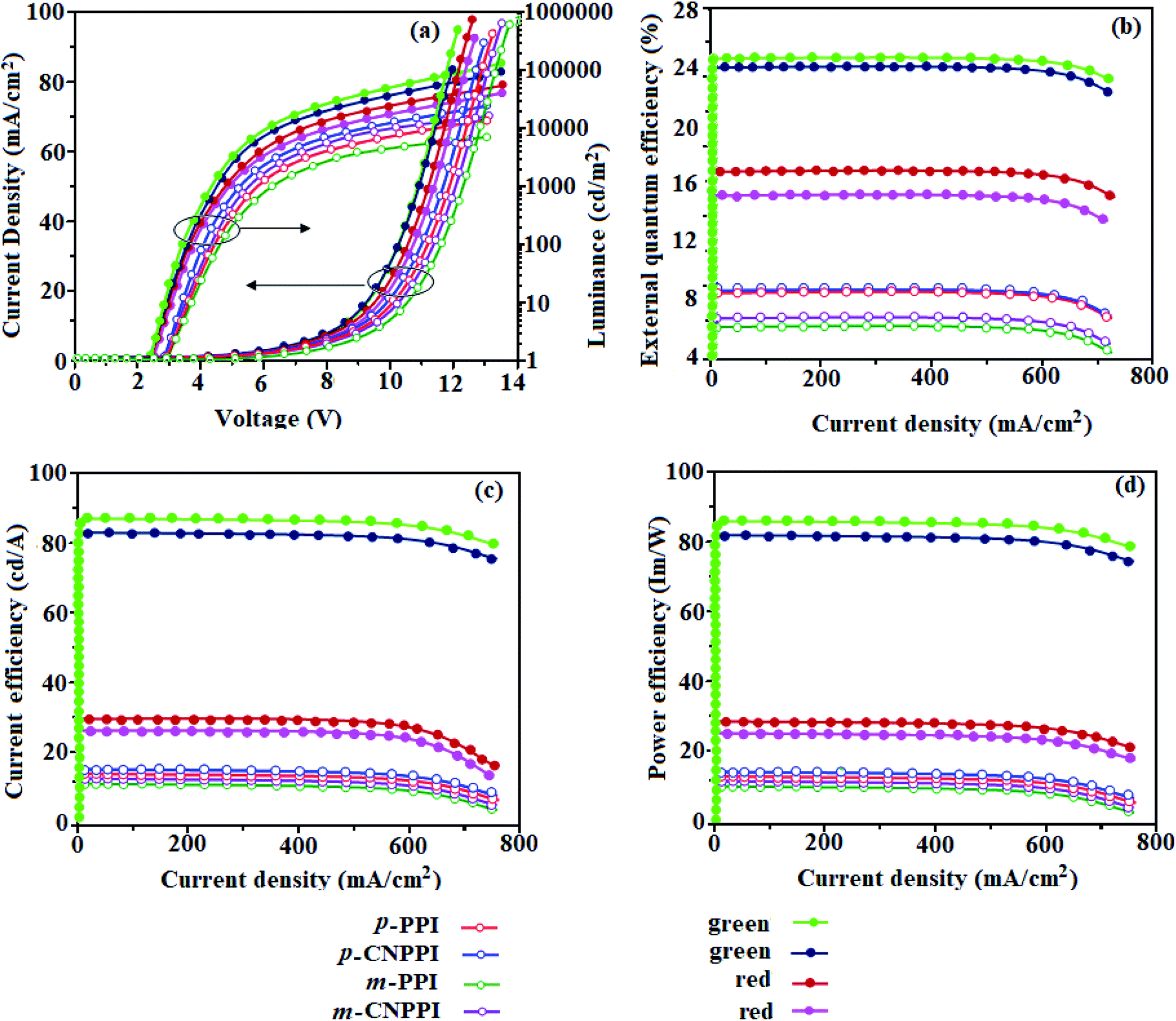 | ||
| Fig. 6 Device efficiencies: (a) luminance/current density – voltage; (b) external quantum efficiency – current density; (c) current efficiency – current density and (d) power efficiency – current density of p-PPI, p-CNPPI, m-PPI and m-CNPPI. | ||
The inevitable vibration splitting in the strongly rigid phenanthrimidazole structure may be enhanced in OLEDs to show large full peak width with red-shifted CIE. These devices have low efficiency roll-off and their ηex can still remain 7.73%, 5.98%, 3.02% and 3.87% at a brightness of 1000 cd m−2 because of the bipolar transport property of these materials. To our knowledge is concern these results are among the best of non-doped blue OLEDs.53 The limitation of internal quantum efficiency (ηIQE) of normal fluorescent emitters is 0.25.54 Therefore, the theoretically calculated (ηex) for p-PPI, m-PPI, p-CNPPI and m-CNPPI based OLED are of 6.25%, 7.14%, 5.81% and 6.85% estimated using quantum yield in film as well as out-coupling efficiency (ηout). The experimental ηex are of p-PPI, m-PPI, p-CNPPI and m-CNPPI based deep blue OLEDs are beyond the theoretical limitation. To understand high EL efficiency mechanism of p-PPI, m-PPI, p-CNPPI and m-CNPPI, detailed characterization were performed. Although the brightness-current density (J) graph in low current density region exhibits linear relationship, no delayed fluorescence component is observed at room as well as low-temperature lifetime measurement (Fig. 3). The triplet–triplet annihilation (TTA) induced enhancement of ηex cannot be ruled out because TTA is complex in OLEDs.55,56 The horizontal-dipole array of anisotropic emitting molecules can remarkably improved the ηout thereby enhancing ηex of OLEDs57–61 (Fig. 7).
 | ||
| Fig. 7 Transition dipole moments of p-PPI, p-CNPPI, m-PPI and m-CNPPI. | ||
The p-PPI, m-PPI, p-CNPPI and m-CNPPI have an anisotropic molecular structure and their transition dipole moment of S1 state calculated by TD-DFT are along the molecular long axis reveal that the exciting dipole adopt horizontal orientation in film (Fig. 7). The film quantum yield of p-PPI, m-PPI, p-CNPPI and m-CNPPI films are of 80.0%, 70.0%, 86.0% and 73.0% respectively, thus the upper limit of ηex calculated from the following equation: ηex = ηout × ηrc × ηγ × ΦPL,62–64, [ηout – light-out-coupling efficiency (20%), ηrc – product of the charge recombination efficiency (100%), ηγ – efficiency of radiative exciton production (25%) and ΦPL – photoluminescence quantum yield]. The ηr calculated for p-PPI (49–56%), m-PPI (37–46%), p-CNPPI (41–52%) and m-CNPPI (37–46%) indicates γ is less than 100% due to very small unbalanced carrier transportation.64 This result could be attributed more balanced charge-transporting properties in the emissive zone achieved by better charge injection provided by hole transport layer. The ηIQE can be calculated from ηEQE ÷ ηout as 31% (p-PPI), 35% (m-PPI), 29% (p-CNPPI) and 34% (m-CNPPI) and maximum ηs of 39% (p-PPI), 51% (m-PPI), 33% (p-CNPPI) and 46% (m-CNPPI) of EL devices is determined using the equation, ηs = ηres × ηPL × ηout ÷ ηEL. The enhanced ηs and ηIQE is due to maintained CT component of D–A compounds. The maximum ηs was breaking the 25% limit (spin statistics): 14.0%, 26.0%, 8.0% and 21.0% of triplet transfer to singlet exciton via RISC (reverse intersystem crossing) to increase the fluorescence emission and the rest undergo non-radiative progress. This provides the possibility of high efficiency blue OLEDs. Hence, the high efficiency of blue OLEDs is attributed to horizontal dipole orientation. Some novel fluorescence OLEDs displayed high ηex due to hot exciton or HLCT (hybridized local and charge-transfer excited state) mechanism.34,35 These processes may exist in p-PPI, m-PPI, p-CNPPI and m-CNPPI based devices to enhance ηex.
Moreover, triplet–triplet annihilation in the non-doped film also makes a possible contribution to high efficiency. In line with this molecular-design strategy, bipolar transport properties and high triplet energy, we report the multifunctional efficient blue OLED materials namely, m-CNPPI and p-CNPPI used as (i) emitters in blue OLEDs and (ii) host for phosphorescent OLEDs (green/red). The m-CNPPI and p-CNPPI was adopted as a host to fabricate PHOLEDs which endowed triplet energy (ET) of these materials exhibit high quantum yield in film (ϕfilm) to ensure that the triplet excited state energy m-CNPPI (2.66 eV) and p-CNPPI (2.61 eV) is high enough to excite phosphorescent dopant (green/red). The green device (λEL – 504 nm) based on p-CNPPI: Ir(ppy)3 exhibit maximum L – 78823 cd m−2; ηex – 24.56%; ηc −84.30 cd A−1; ηp −86.43 lm W−1 with CIE 0.33, 0.61 than m-CNPPI: Ir(ppy)3: L – 90568 cd m−2; ηex – 23.98%; ηc −80.98 cd A−1; ηp −80.03 lm W−1 with CIE (0.33, 0.61). Similarly the red device with p-CNPPI: Ir(MDQ)2 (acac) exhibit maximum L – 38418 cd m−2; ηex – 17.31%; ηc −18.09 cd A−1; ηp −21.43 lm W−1 with CIE (0.65, 0.34). The red device exhibit maximum ηex with low efficiency roll-off.65–69 Moreover, p-CNPPI and m-CNPPI also have a small excited-state dipole moment which can relieve host dopant dipole–dipole interaction explaining the reason for low roll-off efficiency in PHOLEDs adopting them as host materials.41 These findings are greatly helpful to construct low-cost fluorescent materials with subtle molecular modification.
4 Conclusion
We have developed new donor–acceptor compounds, p-PPI, p-CNPPI, m-PPI and m-CNPPI by non-symmetrically connecting π-conjugated building block with bipolar properties and exhibit superior thermal characteristics. The non-doped blue device with p-PPI/m-PPI emissive layer (λEL – 434/420 nm) shows high efficiencies ηc – 5.83/3.56 cd A−1; ηp – 5.73/3.48 lm W−1; ηex – 8.98/6.48% at 3.0 V. Their cyano derivatives, p-CNPPI/m-CNPPI (λEL – 422/406 nm) exhibit maximum efficiencies (ηc – 6.28/4.38 cd A−1; ηp −6.14/4.01 lm W−1; ηex – 9.01/6.72%) at 2.9 V than p-PPI/m-PPI based devices. The anisotropic molecular structural characteristic of p-PPI, p-CNPPI, m-PPI and m-CNPPI induced horizontal dipole orientation in thin film and enhanced the EL efficiency. The well-balanced carrier transportation of these materials relieves efficiency roll-off as well as makes them approving host for PHOLEDs. The green/red device (λEL – 504/618 nm) based on p-CNPPI: Ir(ppy)3/Ir(MDQ)2 (acac) exhibit maximum L – 78823/38418 cd m−2; ηex – 24.56/17.31%; ηc −84.30/18.09 cd A−1; ηp – 86.43/21.43 lm W−1 with CIE (0.33, 0.61)/(0.65, 0.34). The change of connecting mode has great potential for developing new high-performance deep blue EL materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the DST (Department of Science and Technology - EMR/2014/000094, F.No. SR/S1/1C-73/2010, F.No. SR/S1/1C-07/2007), DRDO (Defence Research and Development Organization -213/MAT/10-11), CSIR (Council of Scientific and Industrial Research -No. 01/ (2707)/13EMR-II), UGC (University Grant Commission -6-21/2008, F.No. 30-71/2004(SR)) and DST-Nano Mission (SR/NM/NS-1001/2016).References
- W. J. Li, Y. Y. Pan, R. Xiao, Q. M. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476–8483 CrossRef CAS PubMed.
- A. Islam, Q. Wang, L. Zhang, T. Lei, L. Hong, R. Yang, Z. Liu, R. Peng, L. S. Liao and Z. Ge, Dyes Pigm., 2017, 142, 499–506 CrossRef CAS.
- Y. Park, J. H. Lee, D. H. Jung, S. H. Liu, Y. H. Lin, L. Y. Chen, C. C. Wud and J. Park, J. Mater. Chem., 2010, 20, 5930–5936 RSC.
- S. J. Su, C. Cai and J. J. Kido, Chem. Mater., 2011, 23, 274–284 CrossRef CAS.
- N. Matsumoto, T. Miyazaki, M. Nishiyama and C. Adachi, J. Phys. Chem. C, 2009, 113, 6261–6266 CrossRef CAS.
- M. A. Baldo, O. D. F. Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- G. H. Dobrikov, G. M. Dobrikov and M. Aleksandrova, Cent. Eur. J. Chem., 2011, 9, 1126–1132 CAS.
- C. Xiang, W. Koo and F. So, Light Sci. Appl., 2013, 2, 74–80 CrossRef.
- (a) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC; (b) M. C. Gather, A. Kohnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed; (c) D. M. Kang, J. W. Kang, J. W. Park, S. O. Jung, S. H. Lee, H. D. Park, Y. H. Kim, S. C. Shun, J. J. Kim and S. K. Kwon, Adv. Mater., 2008, 20, 2003–2007 CrossRef CAS; (d) T. J. Park, W. S. Jeon, J. J. Park, S. Y. Kim, Y. K. Lee, J. Jang, J. H. Kwon and R. Pode, Appl. Phys. Lett., 2008, 92, 113308 CrossRef.
- (a) P. I. Shih, C. Y. Chuang, C. H. Chien, E. W. G. Diau and C. F. Shu, Adv. Funct. Mater., 2007, 17, 3141–3146 CrossRef CAS; (b) S. J. Lee, J. S. Park, K. J. Yoon, Y. I. Kim, S. H. Jin, S. K. Kang, Y. S. Gal, S. Kang, J. Y. Lee, J. W. Kang, S. H. Lee, H. D. Park and J. J. Kim, Adv. Funct. Mater., 2008, 18, 3922–3930 CrossRef CAS.
- B. Wei, J. Z. Liu, Y. Zhang, J. H. Zhang, H. N. Peng, H. L. Fan, Y. B. He and X. C. Gao, Adv. Funct. Mater., 2010, 20, 2448–2458 CrossRef CAS.
- J. W. G. Hunt, Z. Q. Jiang, Z. Y. Liu, C. L. Yang, C. Zhong, J. G. Qin, G. Yu and Y. Q. Liu, Adv. Funct. Mater., 2009, 19, 3987–3995 CrossRef.
- Z. Q. Gao, Z. H. Li, P. F. Xia, M. S. Wong, K. W. Cheah and C. H. Chen, Adv. Funct. Mater., 2007, 17, 3194–3199 CrossRef CAS.
- Y. H. Kim, H. C. Jeong, S. H. Kim, K. Yang and S. K. Kwon, Adv. Funct. Mater., 2005, 15, 1799–1805 CrossRef CAS.
- S. Tao, Z. Peng, X. Zhang, P. Wang, C. S. Lee and S. T. Lee, Adv. Funct. Mater., 2005, 15, 1716–1721 CrossRef CAS.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef CAS.
- J. Ye, Z. Chen, M. K. Fung, C. Zheng, X. Ou and X. H. Zhang, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS.
- (a) B. Liu, J. W. Zhao, C. Y. Luo, F. Lu, S. L. Tao and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC; (b) G. Li, J. W. Zhao, D. Zhang, Z. C. Shi, Z. L. Zhu, H. Q. Song, J. J. Zhu, S. L. Tao, F. Lu and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 8787–8794 RSC.
- W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, L. Tang, Q. X. Tong, F. L. Wong and C. S. Lee, Adv. Opt. Mater., 2014, 2, 626–631 CrossRef CAS.
- W. C. Chen, G. F. Wu, Y. Yuan, H. X. Wei, F. L. Wong, Q. X. Tong and C. S. Lee, RSC Adv., 2015, 5, 18067–18074 RSC.
- C. L. Li, J. Wei, J. Han, Z. Li, X. Song, Z. Zhang, J. Zhang and Y. Wang, J. Mater. Chem. C, 2016, 4, 10120–10129 RSC.
- (a) Y. Qian, Q. Wei, G. D. Pozo, M. M. Mróz, L. Lüer, S. Casado, J. C. Gonzalez, Q. Zhang, L. Xie, R. Xia and W. Huang, Adv. Mater., 2014, 26, 2937–2942 CrossRef CAS PubMed; (b) D. He, Y. Yuan, B. Liu, D. Y. Huang, C. Y. Luo, F. Lu, Q. X. Tong and C. S. Lee, Dyes Pigm., 2017, 136, 347–353 CrossRef CAS.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. Kim, S. Lee, K. H. Kim, Y. J. Lee, S. K. Kwon, J. J. Kim and Y. H. Kim, Chem. Commun., 2013, 49, 4664–4666 RSC.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- W. Liu, C. J. Zheng, K. Wang, Z. Chen, D. Y. Chen, F. Li, M. X. Ou, Y. P. Dongand and X. H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 18930–18936 CrossRef CAS PubMed.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
- C. Fan, Y. H. Chen, Z. Q. Jiang, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, J. Mater. Chem., 2010, 20, 3232–3237 RSC.
- J. Jayabharathi, P. Nethaji, V. Thanikachalam and R. Ramya, ACS Omega, 2019, 4, 4553–4570 CrossRef CAS PubMed.
- J. Jayabharathi, R. Ramya, V. Thanikachalam, P. Jeeva and E. Sarojpurani, RSC Adv., 2019, 9, 2048–2966 Search PubMed.
- D. D. Zhang, L. Duan, Y. L. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. D. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS.
- V. Thanikachalam, P. Jeeva and J. Jayabharathi, J. Phys. Org. Chem., 2017, 30, 3695–3708 CrossRef.
- (a) C. C. Lai, M. J. Huang, H. H. Chou, C. Y. Liao, P. Rajamalli and C. H. Cheng, Adv. Funct. Mater., 2015, 25, 5548–5556 CrossRef CAS; (b) C. Wang, J. Zhang, G. Long, N. Aratani, H. Yamada, Y. Zhao and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6292–6296 CrossRef CAS; (c) Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C. J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS.
- (a) W. Z. Yuan, S. M. Chen, J. W. Lam, C. M. Deng, P. Lu, H. M. H. Sung, I. D. Williams, H. S. Kwok, Y. M. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 11216 RSC; (b) X. J. Xu, S. Y. Chen, G. Yu, C. A. Di, H. You, D. G. Ma and Y. Q. Liu, Adv. Mater., 2007, 19, 1281 CrossRef CAS.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 2834 CrossRef.
- W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, J. Ye, M. Chen, F. Lu, Q. X. Tong, F. L. Wong and C. S. Lee, Org. Electron., 2015, 17, 159 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171 CrossRef CAS PubMed.
- (a) Y. P. Li, F. Li, H. Y. Zhang, Z. Q. Xie, W. J. Xie, H. Xu, B. Li, F. Z. Shen, L. Ye, M. Hanif, D. G. Ma and Y. G. Ma, Chem. Commun., 2007, 3, 231–233 RSC; (b) Y. P. Li, F. Z. Shen, H. Wang, F. He, Z. Q. Xie, H. Y. Zhang, Z. M. Wang, L. L. Liu, F. Li, M. Hanif, L. Ye and Y. G. Ma, Chem. Mater., 2008, 20, 7312–7318 CrossRef CAS.
- X. Y. Cai and S. J. Su, Adv. Funct. Mater., 2018, 28, 1802558–1802590 CrossRef.
- X. Du, G. Li, J. Zhao, S. Tao, C. Zheng, H. Lin, Q. Tong and X. Zhang, Adv. Opt. Mater., 2017, 5, 1700498–1700508 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef.
- D. D. Zhang, M. H. Cai, Y. G. Zhang, D. Q. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef.
- M. Y. Lai, C. H. Chen, W. S. Huang, J. T. Lin, T. H. Ke, L. Y. Chen, M. H. Tsai and C. C. Wu, Angew. Chem., Int. Ed., 2008, 47, 581–585 CrossRef CAS PubMed.
- S. T. Zhang, L. Yao, Q. M. Peng, W. J. Li, Y. Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. M. Wang, P. Lu, F. Li, S. J. Su, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- Y. Liu, L. S. Cui, M. F. Xu, X. B. Shi, X. B. Zhou, Z. K. Wang, Z. Q. Jiang and L. S. Liao, J. Mater. Chem. C, 2014, 2, 2488–2495 RSC.
- Y. H. Lou, M. F. Xu, L. Zhang, Z. K. Wang, S. Naka, H. Okada and L. S. Liao, Org. Electron., 2013, 14, 2698–2704 CrossRef CAS.
- Z. Wang, Y. Lou, S. Naka and H. Okada, Appl. Phys. Lett., 2011, 98, 043302–043305 CrossRef.
- J. Liu, Z. Zeng, X. Cao, G. Lu, L. H. Wang, Q. L. Fan, W. Huang and H. Zhang, Small, 2012, 8, 3517–3522 CrossRef CAS.
- W. C. Chen, Z. L. Zhu and C. S. Lee, Adv. Opt. Mater., 2018, 6, 1800258–1800300 CrossRef.
- M. Pope and C. E. Swenberg, Electronic Processes in Organic Crystals and Polymers, Oxford University Press, New York, 2nd edn, 1999, p. 1351, ISBN: 9780195129632 Search PubMed.
- D. Y. Kondakov, J. Soc. Inf. Disp., 2009, 17, 137–144 CrossRef CAS.
- N. A. Kukhta, T. Matulaitis, D. Volyniuk, K. Ivaniuk, P. Turyk, P. Stakhira, J. V. Grazulevicius and A. P. Monkman, J. Phys. Chem. Lett., 2017, 8, 6199–6205 CrossRef CAS PubMed.
- M. H. Lu and J. C. Sturm, J. Appl. Phys., 2002, 91, 595–604 CrossRef CAS.
- W. Brutting, J. Frischeisen, T. D. Schmidt, B. J. Scholz and C. Mayr, Phys. Status Solidi A, 2013, 210, 44–65 CrossRef.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- M. Liu, R. Komatsu, X. Y. Cai, K. Hotta, S. Sato, K. K. Liu, D. C. Chen, Y. Kato, H. Sasabe, S. Ohisa, Y. Suzuri, D. Yokoyama, S. J. Su and J. Kido, Chem. Mater., 2017, 29, 8630–8636 CrossRef CAS.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961–1704968 CrossRef PubMed.
- J. Jayabharathi, P. Sujatha, V. Thanikachalam and P. Nethaji, RSC Adv., 2018, 8, 20007–20015 RSC.
- (a) S. Tokito, T. Tsutsui and Y. Taga, J. Appl. Phys., 1999, 86, 2407–2411 CrossRef CAS; (b) S. H. Cho, J. R. Oh, H. K. Park, H. K. Kim, Y. H. Lee, J. G. Lee and Y. R. Do, Opt. Express, 2010, 18, 1099–1104 CrossRef CAS PubMed.
- A. O. Eseola, O. Adepitan, H. Gorls and W. Plass, J. Chem., 2012, 36, 891–899 CAS.
- J. Jayabharathi, G. Goperundevi and S. Panimozhi, ACS Omega, 2019, 4, 15030–15042 CrossRef CAS.
- X. Du, G. Li, J. Zhao, S. Tao, C. Zheng, H. Lin, Q. Tong and X. Zhang, Adv. Opt. Mater., 2017, 5, 1700498–1700508 CrossRef.
- Y. Yuan, J. X. Chen, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, F. L. Wong, Z. Q. Guo, J. Ye, Z. Chen, X. H. Zhang and C. S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef CAS.
- B. Wang, X. Lv, J. Tan, Q. Zhang, Z. Huang, W. Yi and L. Wang, J. Mater. Chem. C, 2016, 4, 8473–8482 RSC.
- G. Li, D. Zhu, T. Peng, Y. Liu, Y. Wang and M.-R. Bryce, Adv. Funct. Mater., 2014, 24, 7420–7426 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10513a |
| This journal is © The Royal Society of Chemistry 2020 |