
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhytochemistry by design: a case study of the chemical composition of Ocotea guianensis optimized extracts focused on untargeted metabolomics analysis†
Ananda da Silva Antonio a,
Ana Tayná Chaves Aguiara,
Gustavo Ramalho Cardoso dos Santosb,
Henrique Marcelo Gualberto Pereirab,
Valdir Florêncio da Veiga-Junior*ac and
Larissa Silveira Moreira Wiedemanna
a,
Ana Tayná Chaves Aguiara,
Gustavo Ramalho Cardoso dos Santosb,
Henrique Marcelo Gualberto Pereirab,
Valdir Florêncio da Veiga-Junior*ac and
Larissa Silveira Moreira Wiedemanna
aChemistry Department, Institute of Exact Sciences, Federal University of Amazonas, Avenida Rodrigo Octávio, 6200, Coroado, CEP: 69.077-000, Manaus, AM, Brazil. E-mail: valdir.veiga@gmail.com
bFederal University of Rio de Janeiro, Chemistry Institute, Brazilian Doping Control Laboratory (LBCD – LADETEC/IQ – UFRJ), Avenida Horácio Macedo, 1281 – Pólo de Química – Cidade Universitária, Ilha do Fundão, CEP: 21941-598, Rio de Janeiro, RJ, Brazil
cChemical Engineering Section, Military Institute of Engineering, Praça General Tibúrcio, 80, Praia Vermelha, Urca, CEP: 22.290-270, Rio de Janeiro, RJ, Brazil
First published on 21st January 2020
Abstract
Untargeted metabolomics aim to provide a global chemical fingerprint of biological matrices. This research field can be used in phytochemical screenings for bioactive species or in the identification of species. Despite its importance in providing a global chemical profile, little research has focused on the optimization of the extraction methods, as each type of matrix requires a specific procedure. Therefore, we propose to evaluate the effect of different extraction features in an ultrasound-assisted extraction for the untargeted metabolomic study of an Ocotea species, a genus of great economical interest but little chemical exploitation. Method optimization was performed in a full factorial 2232 design, evaluating the solvent composition, extraction temperature, sample particle size and sample![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :solvent ratio effects in the metabolomic response. The effect of these parameters on the quality of the untargeted metabolomic profiles was studied by analysis of the extraction yield as well as the chromatographic and spectrometric profiles. Most substances identified were glycosylated flavonoids and aporphinic alkaloids. The application of 70% ethanol enhanced the extraction of several specialized metabolites. Statistical analysis of extraction yield and chemical profiles indicates that high temperatures and low proportion between sample and extracting solvent reduce the quality and modify the chemical profile, both qualitatively and quantitatively. The use of 70% ethanol as the extracting solvent, 1:12 sample:solvent ratio, 40 °C as the extraction temperature and particle size of 0.595 mm were the optimized conditions to produce a comprehensive chemical profile for Ocotea guianensis.
:solvent ratio effects in the metabolomic response. The effect of these parameters on the quality of the untargeted metabolomic profiles was studied by analysis of the extraction yield as well as the chromatographic and spectrometric profiles. Most substances identified were glycosylated flavonoids and aporphinic alkaloids. The application of 70% ethanol enhanced the extraction of several specialized metabolites. Statistical analysis of extraction yield and chemical profiles indicates that high temperatures and low proportion between sample and extracting solvent reduce the quality and modify the chemical profile, both qualitatively and quantitatively. The use of 70% ethanol as the extracting solvent, 1:12 sample:solvent ratio, 40 °C as the extraction temperature and particle size of 0.595 mm were the optimized conditions to produce a comprehensive chemical profile for Ocotea guianensis.
Introduction
Untargeted metabolomics is currently defined as a research field focused on monitoring and providing a global chemical profile of biological matrices regarding their specialized metabolites composition.1,2 The major goal is to provide as much chemical information about the sample as possible, without focusing on pre-determined chemical class, thus generating a holistic chemical fingerprint of the sample.1–3 The global chemical profiles provided by untargeted metabolomics studies have been efficiently used in the research of natural products for the exploitation of botanic species,4,5 determination of new quality control features for bioproducts,6,7 discovery of unusual chemical markers,8,9 species identification by fingerprint comparison10,11 and comprehension of botanic response to endogenous and exogenous modification.12,13 One of the major advantages of untargeted metabolomics in the phytochemical screening of plants is the agility with which a huge set of chemical data is produced, especially when compared to previously applied strategies, typically involving the use of time-consuming methods of metabolite isolation.1,14,15Technological development of high performance analytical methods, such as liquid chromatography and mass spectrometry, has greatly advanced this research field. The enhancement of the resolution and sensitiveness of those methods are crucial in the production of global chemical profiles in botanic samples, as they can biosynthesize more than 50000 different metabolites.1,2,15 In addition to the importance of the instrumental method in untargeted metabolomics, other steps of its common workflow2,3 are just as crucial but usually neglected, such as sample obtention/extraction.1–3 This step is the second in the untargeted metabolomics workflow and the first which will influence the composition of the extract. The application of a specific solvent, as well as the selection of the extraction method and its operational features, plays an important role on which metabolites will compose the final sample fingerprint.3,16
Despite its importance, extraction method development in untargeted metabolomics is poorly explored. The lack of studies regarding extraction optimization is due to by the complexity and diversity of sample types. Each biological matrix has distinct physical and chemical characteristics, driving some authors to assert that extraction methods should be developed specifically to each study case.2,3,15–18 As a standard method to obtain the chemical profile of angiosperms does not exist, we proposed to evaluate the influence of extraction parameters within an ultrasound-assisted extraction (UAE) in the untargeted metabolomic study of an angiosperm. The species chosen for this study belongs to a renowned bioactive genus but poorly chemically described, the Ocotea, from the Lauraceae botanical family.
Results and Discussion
Effects in the extraction yield
The extraction yield (Y%) was the first evaluated factor. In natural products metabolomics, this response variable plays an important role regarding specimens preservation, as it can reduces the sampling amount necessary for research development. As it is expected that the studied specimen will be accessible for further sampling, it is recommended that as little stress as possible be caused to the plant. A significant level of stress, caused by the removal of large quantities of leaves and branches can cause specimen death or significant modifications to its metabolism, preventing the reproduction or continuity of the study. Thus, a high extraction yield can enable the execution of several analytical methods with minimal stress to the specimens as smaller amounts of raw material are required.During extraction method assessment the Y% achieved ranged from 1.97 to 10.76% with relative standard deviations (RSD) lower than 10% between the technical triplicates (Table 1). The RSD values highlight the repeatability of the extraction procedure. As the operational characteristics of the extraction method can significantly affect the procedure performance, they must be specifically evaluated before metabolomics studies. The highest Y% obtained (10.76%) also reveals that the ultrasound-assisted extraction is a promising method for the phytochemical analysis of angiosperms. Our optimal Y% results present similar values to those obtained by previous studies with Lauraceae, which applied a 5 day times exhaustive maceration.19–21 Therefore, we demonstrate a substantial decrease in the extraction time from a time scale in days to just 30 minutes.
| Sample code | Factors | Y% (n = 3) | RSD (%) | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| T (°C) | PS (mm) | SSP (g:mL) |
% of ethanol | |||
| a T = extraction temperature, PS = sample particle size, SSP = sample to solvent proportion, Y% = extraction yield, RSD = Relative standard deviation. | ||||||
| 1A-2A-3C-4A | 40 | 0.595 | 1:12 |
70 | 9.96 | 8.74 |
| 1A-2B-3C-4A | 40 | 0.841 | 1:12 |
70 | 9.76 | 5.61 |
| 1A-2C-3C-4A | 40 | 2.000 | 1:12 |
70 | 7.03 | 6.04 |
| 1A-2A-3A-4A | 40 | 0.595 | 1:8 |
70 | 7.39 | 6.34 |
| 1A-2A-3C-4C | 40 | 0.595 | 1:12 |
99.8 | 5.75 | 2.10 |
| 1B-2A-3C-4A | 50 | 0.595 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :12 :12 |
70 | 10.76 | 8.88 |
| 1B-2B-3C-4A | 50 | 0.841 | 1:12 |
70 | 9.25 | 2.05 |
| 1B-2C-3C-4A | 50 | 2.000 | 1:12 |
70 | 5.87 | 4.09 |
| 1B-2A-3A-4A | 50 | 0.595 | 1:8 |
70 | 7.08 | 2.85 |
| 1B-2A-3C-4C | 50 | 0.595 | 1:12 |
99.8 | 6.53 | 6.26 |
| 1C-2A-3C-4A | 60 | 0.595 | 1:12 |
70 | 9.29 | 6.75 |
| 1C-2B-3C-4A | 60 | 0.841 | 1:12 |
70 | 8.43 | 2.99 |
| 1C-2C-3C-4A | 60 | 2.000 | 1:12 |
70 | 6.39 | 5.46 |
| 1C-2A-3A-4A | 60 | 0.595 | 1:8 |
70 | 7.84 | 5.08 |
| 1C-2A-3C-4C | 60 | 0.595 | 1:12 |
99.8 | 6.23 | 8.60 |
Each evaluated parameter presented a distinct effect on the extraction yield as they are related to different chemical and physicochemical phenomena involved in sonochemistry. The analysis of the main effects of each operational feature on the Y% (Fig. 1) highlighted the composition of the extracting solvent as the most influential factor on its response, whilst the temperature was the only factor which did not cause significant modifications in Y% values. The significant influence of the extraction solvent is mainly related to its polarity. As Ocotea is currently known as a rich source of aporphine alkaloids, lignoids and flavonoids, decreases in solvent polarity can reduce the interaction and solubility of those medium polarity chemical classes with the solvent, reducing extraction efficacy.22–24 Additionally, the TLC results (Fig. 2) also corroborates bibliographic survey of Ocotea, as the stain pattern indicates the presence of substances related to alkaloids, anthraquinone, flavonoids and phenolic substances. Regarding the NP-PEG reagent (natural products/polyethylene glycol reagent) revelation (Fig. 2A), colors suggest the presence of quercetin, kaempferol and luteolin derivates.25
 | ||
| Fig. 1 Pareto diagram of the factors main effect (effect) on the extraction yield (%). Confidence level of the analysis was 95.0% (p = 0.05). | ||
 | ||
| Fig. 2 Example of the TLC chemical profile obtained during optimization procedures, where 1 = sample extracted at 50 °C, 0.595 mm particle size, 1:12 sample to solvent ratio and 70% ethanol; 2 = sample extracted at 50 °C, 0.595 mm particle size, 1:12 sample to solvent ratio and 99.8% ethanol. (A) NP-PEG reactive, (B) UV 254 nm, (C) UV 366 nm, (D) sulfuric vanillin, (E) ferric chloride. | ||
Solvent influence was observable qualitatively by preliminary tests using thin layer chromatography (TLC) (Fig. 2). In this analysis, samples extracted with 99.8% ethanol or at 60 °C presented the same profile, with fewer observed stains when compared to samples extracted with 70% ethanol at milder temperatures. The distinction in chemical profile of the samples was mainly observed between lower and medium polarity compounds (Fig. 2B, D and E). Untargeted metabolomics studies are intimately related to the quality of the chemical fingerprint produced. Although most phytochemical screening of Ocotea use 99.8% ethanol or do not inform its grade,26–29 our results indicate the relevance of evaluating this parameter prior to sample extraction in order to provide a reliable, representative and high quality chemical profile for untargeted metabolomics.
The particle size was the second most influential factor in the Y% response. As particle sizes reduce, the surface area of the sample increases enhancing the contact with the solvent and consequently maximizing the mass transference30,31 and Y% value (Table 1). Regarding the ratio between sample and solvent, the increase of Y% (Table 1) occurred by a large concentration gradient formed when the highest ratios were applied.32 A large concentration gradient induces the diffusion of metabolites through the solvent, enhancing the mass transference and consequently increasing the Y%.33 In addition, as the volume of the extracting solvent increases, its extraction efficacy is also improved as it will enable the solubilization of a larger concentration of metabolites without being saturated. This is an important issue that should be accessed by several techniques such as exhaustive maceration and Soxhlet method.30,31
To evaluate the qualitative effect of each parameter on the extract composition, a multivariate analysis of chromatographic data was performed. TLC evaluation by HCA and PCA (Fig. 3) demonstrates that though temperature does not significantly influence Y%, it qualitatively affects the chemical profile, causing clustering formation in distant groups. Samples were separated within 3 groups, regarding the solvent type and extraction temperature (For the sample nomenclature in figure please refer to subsection botanic sample in the Experimental Section, Table 3). The blue group was mainly composed of samples obtained in milder temperatures with 70% ethanol, while the red and yellow groups represent those obtained at 60 °C and 99.8% ethanol. Samples extracted by different conditions of particle size and ratio between sample and solvent did not form specific groups, demonstrating that they do not qualitatively affect the TLC profile.
 | ||
| Fig. 3 Multivariate analysis of TLC results by (A) principal component analysis and (B) hierarchical clustering analysis. Sample nomenclature in accordance with Table 3: extraction temperature (1) at 40 °C (A), 50 °C (B) or 60 °C (C); particle size (2) at 0.595 mm (A) or 0.841 mm (B); sample to solvent ratio (3) at 1:8 (A) or 1:12 (B); and % of ethanol in the extraction solvent (4) at 70% (A) or 99.8% (C). | ||
Although the temperature did not significantly affect the Y% value, there were substantial qualitative modifications on the chemical profile, as can be observed by the formation of a group representing the extractions performed at 60 °C. This data may indicate a thermal degradation of metabolites as TLC revelators react with specific chemical classes and therefore their degraded products usually are not detected.25 As most of the conventional extraction methods applied in Lauraceae phytochemical studies involve heating procedures,34–36 our results emphasize that the reckless application of relatively high temperatures (above 50 °C) can degrade the sample without affecting the Y%. The formation of 3 clusters even by the usage of a low resolution method, such as TLC, to evaluate the chemical profile highlights the importance of method development for untargeted metabolomics, even in well-known botanic groups such as Lauraceae.23,37 Multivariate analysis emphasizes the significant influence of the evaluated operational parameters on the chemical profile composition and quality.
Extraction effects in the HPLC-DAD chemical profile
HPLC-DAD profiles (Fig. 4A) presented significant modifications qualitatively (quantity of peaks) and quantitatively (peaks area) due to modification in the extracting solvent and temperature, corroborating our previous results by TLC. Significant modifications can be observed in the retention times ranging from 18 to 30 minutes (Fig. 4). The use of milder temperatures and 70% ethanol promoted an increase in chromatogram total area, corroborating TLC conclusions regarding solvent efficacy and thermal degradation of metabolites. As untargeted metabolomics quantitative approaches use relative concentration data (e.g. peak area or total height),2,3 decreases in this characteristics due to thermal degradation or low extraction efficacy, can affect the holistic view and biological interpretation of the botanic sample. Therefore, a matrix rich in medium to high polarity metabolites could have the representatives of their chemical profile significantly altered by excessive temperature. Notwithstanding, these parameters must be carefully optimized, since extraction temperature can enhance cavitation process and influence mass transference in UAE.31 Multivariate analysis was initially performed using all 57 peaks as variables in order to determine which one of them was more significant to the analysis. This procedure was performed by the analysis of loading plot and established 20 significant peaks to use as variables in HCA and PCA (Fig. 4B). Both chemometric analyses clustered the samples in 3 groups (Fig. 4C and D), mainly correlated to the percentage of ethanol and sample-solvent ratio. These separation patterns are consistent to the factor effects in Y% (Fig. 1), in which the temperature was the least influential parameter in the response-variable, thus, being unable to form a specific cluster characterized by samples of distinct extraction temperature. | ||
| Fig. 4 HPLC-DAD data analysis (A) comparison of samples extracted under different conditions; (B) peaks with highest loading values; (C) dendrogram; (D) score plot of PCA; (E) PCA analysis of green and blue groups. Sample nomenclature in accordance with Table 3: extraction temperature (1) at 40 °C (A), 50 °C (B) or 60 °C (C); particle size (2) at 0.595 mm (A) or 0.841 mm (B); sample to solvent ratio (3) at 1:8 (A) or 1:12 (B); and % of ethanol in the extraction solvent (4) at 70% (A) or 99.8% (C). | ||
In this analysis, the distinction among the clusters is related to a decrease in the ratio between sample and solvent. The chemometric analysis emphasizes the red group as the most different cluster of samples, presenting the largest Euclidian distance in comparison to the other groups (Fig. 4C and D). The distinction of the red group (composed of samples extracted within a proportion of 1:8) is related to an efficacy reduction in the mass transference process. This is caused by low volume of extracting solvent, which reduces metabolite extraction and modifies the qualitative and quantitative features of the chemical profile. Though it is important, the proportion between sample and extracting solvent is not usually provided in most phytochemical studies, even in those whose data is used in metabolomics approaches.35,36,38
The PCA of chromatographic data from groups blue and green (Fig. 4D) qualitatively distinguish samples by the solvent composition and extraction temperature (Fig. 4E). Loadings analysis evidences that the clustering process was caused by presence/absence of chromatographic peaks in the time ranging from 18 to 30 min. and decreases in the peaks area of the signals between 12 to 17 min. The cyan group, mainly composed of samples under extraction conditions with milder temperatures and 70% ethanol, and cluster A (Fig. 4C and E) grouped the extraction conditions with greater number of peaks and highest peak area. An increase in water content of the extracting solvent can enhance the sample hydration increasing its permeability in the matrix and solubilization of specialized metabolites in sonochemical methods. While this phenomena enhances extraction efficacy of cyan group, the usage of 60 °C during the extraction can cause thermal degradation and reduction of the cavitation process, reducing the method efficacy and creating the samples distinction observed in chemometric analysis.33
Evaluation of MS fingerprint
The evaluation of mass spectrometry chemical profile was performed using the full scan spectra of the samples by direct injection. The evaluation of loading values of negative and positive modes demonstrated that only peaks within the range of 100 to 600 m/z present a significant effect during multivariate analysis (Fig. 5). Among the 214 signals detected on negative-ion mode in this range, 16 were significantly relevant in PCA grouping as their absolute and relative abundances were drastically modified in function of the different extraction conditions. In positive mode, only 35 m/z signals were detected in the same m/z range, with only 8 significantly relevant peaks for group formation. As positive-ionization was less sensitive to modifications in the extraction method conditions, generating fewer results regarding the studied extraction parameters, further multivariate analysis was run only with data from negative mode.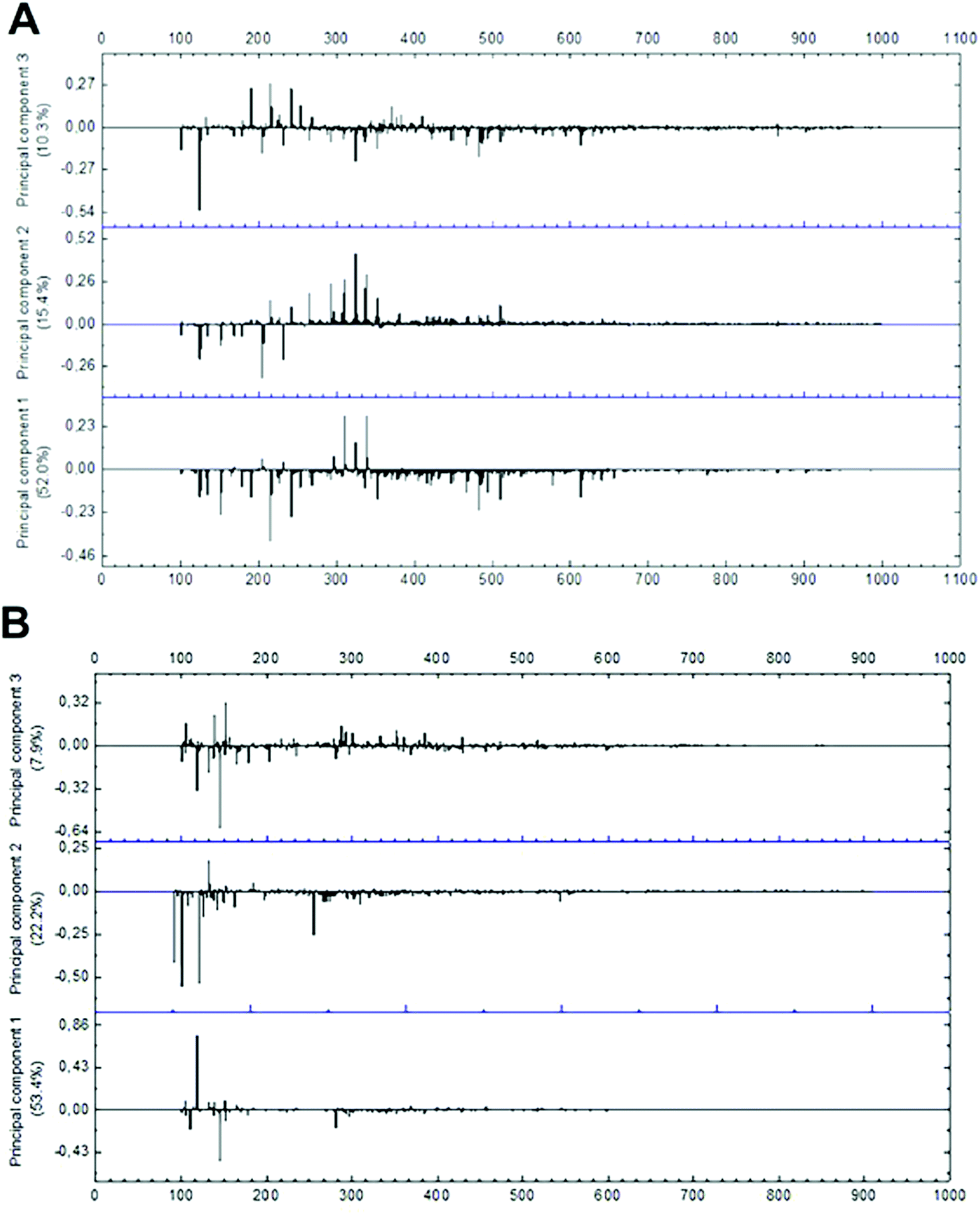 | ||
| Fig. 5 Loading plot of mass spectra data in (A) negative-ion mode and (B) positive-ion mode from m/z 100 to 1000. | ||
Multivariate analysis of MS data performed with the 16 more significant signals (Fig. 6B and C) formed 3 clusters mainly related to the difference in the solvent type and particle size applied in the extraction. Cluster A was composed only of samples extracted with 70% ethanol, while clusters B and C represent those extracted with 99.8% ethanol. Additionally, cluster C primarily represents samples extracted with higher particle sizes.
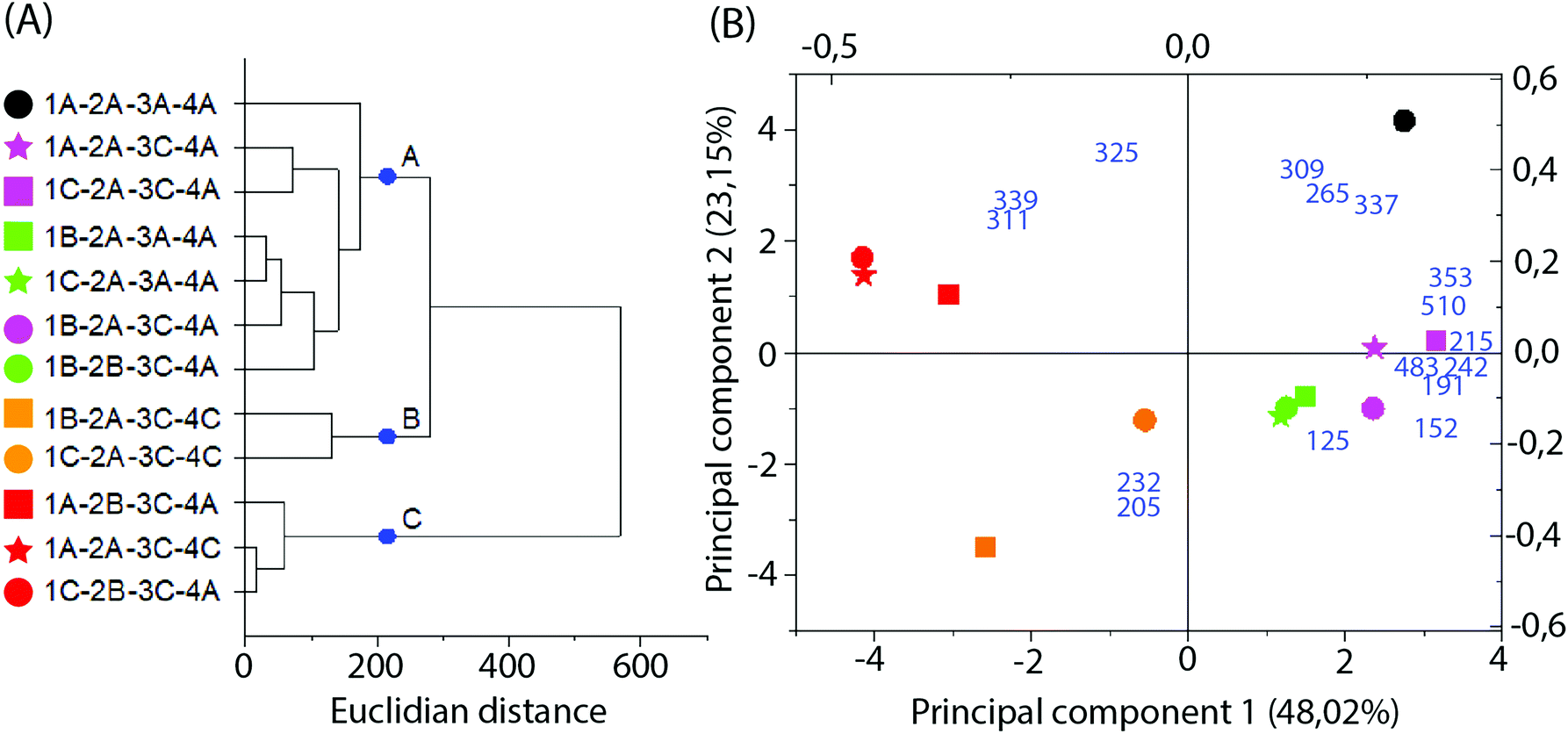 | ||
| Fig. 6 Multivariate analysis of MS data in the negative-ion mode. (A) Dendrogram; (B) PCA biplot, with loadings as blue numbers. Sample nomenclature in accordance with Table 3: extraction temperature (1) at 40 °C (A), 50 °C (B) or 60 °C (C); particle size (2) at 0.595 mm (A) or 0.841 mm (B); sample to solvent ratio (3) at 1:8 (A) or 1:12 (B); and % of ethanol in the extraction solvent (4) at 70% (A) or 99.8% (C). | ||
Loadings analysis indicates that the red group (Cluster C) is characterized by high relative intensities in peaks 311, 325 and 339 m/z and low values of all other signals. In the other groups those peak signals present small to intermediary intensities (10 to 80%) (Fig. 7). This indicates that the red group experimental conditions did not favor the solubility of most of the specialized metabolites or presented high specificity to a small set of compounds. Both occasions are undesirable in untargeted metabolomics as its goal is to obtain as much data as possible.
 | ||
| Fig. 7 ESI full scan in the negative mode of samples extracted with (A) 70% ethanol and (B) 99.8% ethanol. | ||
Considering the extraction yield, the qualitative and quantitative information on the chemical profile and the modifications in the chromatographic and spectrometric profiles, evidenced by PCA and HCA, the extraction conditions using 70% ethanol, extraction temperature of 40 °C, particle size of 0.584 mm and proportion between sample and solvent of 1:12 (g:mL) (sample 1A-2A-3C-4A, Table 1) presented the optimized holistic view of the sample (Fig. S1†). With these conditions, the sample presented a greater quality in the chemical fingerprint produced by both, chromatographic and spectrometric methods. Although these conditions presented the second greatest extraction yield, T-test of this response variable shows that it is statistically similar to the condition where the largest Y% value was achieved (sample 1B-2A-3C-4A, Table 1). Briefly, the use of 70% ethanol at milder temperatures (40 °C) and low particle size enhances the quality of extraction of Ocotea secondary metabolites for untargeted approaches.
The chemical exploitation of Lauraceae species, as it is currently performed in most research does not have a standard procedure. In many cases information regarding particle size and the sample to solvent ratio are not even mentioned.21,39–41 In chemosystematics and phytochemical screening for bioactive compounds several researchers vary the type of extraction solvent and extraction temperature.26–29,41 This prevents any reliable comparison among studies, as our results demonstrated that these parameters can significantly change the metabolites of the sample chemical profile. Therefore, our results emphasize that the further development of phytochemistry depends on a standardization of analytical methods for each purpose, including the extraction methods.
Phytochemical screening of Ocotea guianensis
O. guianensis is the species-type used to define the Ocotea genus, however few publications describing its chemical composition are available, and most of these are focused on its essential oil,23,24,37,42,43 even though it was the first species taxonomically described for the genus. In order to provide a preliminary characterization of the phytochemicals present in O. guianensis a MS2 data-dependent analysis of the sample was performed with 1A-2A-3C-4A (which represents the optimized extraction conditions) in an UHPLC-HRMN. With this analysis it was possible to suggestively identify 16 metabolites within the sample (Table 2) based on their fragmentation pattern (Table 2 and ESI†). This analysis demonstrates that the method extracted mainly glycosylated flavonoids and aporphinic alkaloids, such as kaempferol-3-O-xyloside and ocoteine (Fig. 8). The identified compounds are commonly found within Ocotea, as already described in several species (Table 2 and Fig. 8).23,37,44| Observed m/z | Retention time (min) | Observed MS2 data | Elemental composition | Error (ppm) | Negative-ion mode | |
|---|---|---|---|---|---|---|
| Possible identity | Previous reports within Ocotea | |||||
| a m/z signals contained in PCA analysis of the full scan data. | ||||||
| 353.0887a | 3.01 | 191.0552, 179.0340, 173.0449, 135.0439 | C16H17O9 | 5.6643 | Chlorogenic acid | O. preciosa51 |
| 483.1295a | 4.56 | 341.0662, 289.0717, 245.0816, 205.0500, 203.0704, 109.0282 | — | — | Catechin glycosylated derivate | — |
| 191.0551a | 4.11 | 173.0446, 127.0389, 111.0439, 93.0332 | C7H11O6 | 0.5234 | Quinic acid | O. foetens40 |
| 289.0714 | 3.23 | 245.0816, 125.0229a, 137.0230, 203.0708, 205.0500a | C15H13O6 | 2.4215 | Catechin | O. notata35 |
| 301.0355 | 5.49 | 271.0248, 255.0297, 178.9976, 151.0025 | C15H9O7 | 3.9862 | Quercetin | O. notata35 |
| 285.0401 | 6.00 | 255.0300, 229.0500 | C15H9O6 | 2.4558 | Kaempferol | O. acutifolia52 |
| 417.0827 | 5.64 | 284.0325, 255.0294, 227.0337 | C20H17O10 | 2.6373 | Kaempferol-3-O-pentoside | — |
| 447.0934 | 5.82 | 314.0432, 301.0344, 271.0251, 179.9977, 151.0028 | — | — | Isorhamnetin glycosylated derivate | O. lancifolia53 |
| Observed m/z | Retention time (min) | Observed MS2 data | Elemental composition | Error (ppm) | Error (ppm) | |
|---|---|---|---|---|---|---|
| 342.1697 | 4.28/4.56 | 311.1273, 296.1039, 279.1013, 264.0777, 248.0828 | C20H24NO4 | −0.8767 | Corydine/predicentrine | O. vellosiana54 |
| 330.1697 | 4.07 | 299.1275, 192.1017, 175.0753, 137.0597 | C19H24NO4 | −0.9086 | Reticuline | O. caudata38 |
| 370.3310 | 8.49 | 309.2783, 268.2632 | C21H25NO5 | 448.7189 | Ocoteine | O. acutifolia52 |
| 286.1436 | 3.83 | 269.1170, 237.0908, 178.0856, 175.0752, 143.0491, 107.0494 | C17H21NO3 | −0.6989 | Coclaurine | O. lancifolia55 |
| 328.1537 | 3.91/4.27 | 297.1117, 282.0882, 237.0907, 265.0855 | C19H22NO4 | −1.8284 | Corytuberine/isoboldine | O. lancifolia55 |
| 314.1745 | 3.90 | 297.1115, 283.0941, 265.0855, 237.0904 | C18H20NO4 | 113.9623 | Norisoboldine | O. lancifolia55 |
 | ||
| Fig. 8 Molecular structure of the specialized metabolites identified within O. guianensis. | ||
The identification of phenolic compounds and alkaloids within the sample corroborates the clustering pattern observed in the chemometric analyses. Previous studies with other botanic matrices evidences that the optimal extraction conditions for phenolic compounds are related to the use of moderate temperatures (up to 37 °C) and aqueous solvent concentrations varying from 50 to 70%.45,46 Since Ocotea is known as one of the most difficult genus to identify species by taxonomic features,1,47 the application of untargeted metabolomics can bring further light in the development of identification methods through chemical profile. Based on the identified compounds (Table 2), O. guianensis presented a certain level of chemical similarity with 5 different morphological groups and 2 phylogenetic clades.48,49 Regarding Ocotea phylogenetics, it is worth mentioning that O. guianensis currently is not classified as an Old World species but as a dioecious one, in a not well supported clade (Ocotea s.str., Endlicheria, and Rhodostemonodaphne clade),49 underlining the doubt of this classification model. One of the phylogenetic groups which present species with chemical compounds also identified in our study is the Old-World clade. The metabolomic similarity of O. guianensis with O. foetens and O. macrophylla (Old World species) could indicate its possible ancestralism and evolutionary importance, since metabolomic composition is a response of genetic expression.
Unlike morphological and phylogenetic systems of Ocotea, which presents more than 10 classificatory groups, the current chemosystematic model of the genus proposes to separate the species in only two groups, according to the presence or absence of alkaloids and lignans based on the compounds which had been isolated in each species.44,50 Nevertheless, our results demonstrate that this strategy is very trick, as only light changes in the extraction procedures can significantly affect the metabolites that will be extracted as much as their concentration. At this point it becomes clear that the usage of isolated compounds by different extraction methods, as it has been done during the Ocotea chemical exploitation throughout history, produces equivocal conclusions regarding genus chemosystematics. Therefore, this model, as proposed to date, should not be considered reliable, bringing an urgency to develop Ocotea chemosystematics by a single and specific method for this purpose.
As our data demonstrated that O. guianensis present a certain level of chemical similarity with species from other systematic groups within the genus, it is possible to estimate that further chemosystematic evaluation including a larger set of species may produce new sets of classificatory groups within Ocotea, aiding its species identification. In addition, our results also demonstrated the importance of untargeted metabolomics in this approach as the minor compounds within the sample were also responsible to distinguish the extraction methods. This fact emphasizes that untargeted metabolomics can be readily applied even in the differentiation of closely related species, becoming a reliable tool in the study of unresolved genera, such as Ocotea itself.
Experimental
Botanic sample
Ocotea guianensis leaves were collected at the Amazonas Federal University (Manaus, Amazonas – Brazil, S03°05′395′′, W059°57′958′′) from a single specimen. Sampling was performed at different heights of the tree canopy to produce a composite sample. A voucher sample was deposited under the number 11417 at the Amazonas Federal University Herbarium (Rodrigo Octavio 6200 Avenue, Coroado I–Manaus Federal University Campus, South Sector, ICB Block I – Ground floor, room 9. Manaus, Amazonas 69080-900 – Brazil). The composite sample was air dried (25 °C), divided in three portions and ground (Willey Mill SP-32, SPLabor) into specific particle sizes (Table 3).
| Number | Factors | Levels | ||
|---|---|---|---|---|
| A | B | C | ||
| 1 | Temperature of extraction (°C) | 40 | 50 | 60 |
| 2 | Particle size (mm) | 0.595 | 0.841 | 2.000 |
| 3 | Sample: extracting solvent proportion (g:mL) |
1:8 |
— | 1:12 |
| 4 | % of ethanol in the extraction solvent | 70 | — | 99.8 |
Sample coding were assigned in accordance with the factor's identification numbers (first column in Table 3) followed by the upper letter which represents the level factor applied during sample extraction procedure. Thus, each sample was identified by a sequence of four Arabic numbers and upper letters.
Method evaluation
The extraction method was optimized by a full factorial design with mixed levels 2232 (Table 3).56 Ethanol was chosen as the extracting solvent due to its usage in phytochemical screenings of the Lauraceae family. The factors themselves and their levels were chosen based on bibliographic survey of Lauraceae, considering the most used extraction characteristics and the absence of details in Experimental Section.21,57–59 All optimization extractions (Table 3) were performed as technical triplicates in an ultrasonic bath (Unique Ultrasonic Cleaner) operating at 40 kHz for 30 minutes. Crude extracts were evaporated to dryness under vacuum before further analysis. The response variables used to establish the optimal experimental condition for the untargeted metabolomics approach were the extraction yield (Y%) and the quality of the chemical profile obtained by chromatographic and spectrometric methods as stated by Klein-Júnior et al. (2016).16The extraction yield was calculated by gravimetric determination, considering the total weight of the crude extract and the initial weight of botanical material used for the extraction. This parameter was used to establish the main effect (or just effect) of each factor (Table 3) during optimization.56 The main effects of the factors in Y% were evaluated by the confounding of the variables with 3 levels in new factors of 2 levels.56 The confounding turns the asymmetrical design 2232 in a symmetric 26, enabling an easier assessment of the main effects.
Chromatographic and mass spectrometric analysis
The crude ethanolic extracts obtained in each experiment of UAE optimization (Table 3) were fractionated by liquid–liquid partition. Extracts were diluted in methanol:water (4:1) and partitioned in three individual fractions. The applied solvent followed an increasing polarity, using in sequence hexane, ethyl acetate and methanol. The medium polarity fraction (ethyl acetate fraction) was used to obtain the chemical profile of O. guianensis applied in the UAE optimization and untargeted metabolomic evaluation of the species. This fraction was selected since most chemotaxonomic evaluation of Lauraceae emphasizes the importance of medium polarity substances as chemical markers and prominent bioactive fraction.23,43,44,50,60,61
Ethyl acetate fraction was analyzed by thin layer chromatography (TLC) and high performance liquid chromatography (HPLC). The TLC was performed on a pre-activated silica gel HPTLC analytical plate (Merck Alugram® Xtra SIL G/UV254) developed with a mobile phase composed of chloroform:ethyl acetate:methanol:formic acid (5:3:2:0.5). TLC visualization was achieved by the irradiation with UV light at 254 nm and 366 nm, vanillin-sulfuric acid, NP/PEG reagent (natural products/polyethylene glycol reagent) and 5% w/v of ferric chloride, as they are commonly applied on the visualization of phenolic compounds (such as flavonoids), alkaloids and lignoids.25
For the optimization study, HPLC-DAD (Table S1†) and full scan MS (Table S2†) analysis by direct injection were applied to determine the optimal extraction condition regarding the quality of the chemical profile provided by those methods (Fig. S1†). HPLC-DAD analysis was performed on a 250 × 4.6 mm Shim-Pack ODS(H) Shimadzu® 5.0 μm C18 column in a Dionex Ultimate 3000 (Thermo Scientific, Bremen, Germany) system equipped with an autosampler and a diode array detector (DAD). Column and injection system were kept at 40 °C while injection volume and flow rate were set as 20 μL and 1.0 mL min−1. Samples were injected at 1 mg mL−1 in methanol. The elution was performed as a gradient of 0.5% formic acid (Solvent A) and acetonitrile (Solvent B), from 5 to 70% of Solvent B with a rate of 2% min−1. DAD detector was set to register from 200 to 700 nm. Chromatography profile evaluation by HPLC-DAD was performed at 300 nm, where 70% of its peaks had a resolution equal or higher than 1.5.62 The quality of HPLC-DAD data was evaluated in function of chromatographic resolution, total quantity of peaks and individual and total peak area.16
Mass spectrometric (MS) analysis in full scan mode was performed by direct injection on a TSQ Quantum Access (Thermo Scientific, Bremen, Germany), with triple quadrupole and electrospray ionization (ESI) source with the ionization being monitored in positive and negative-ion modes, in the range of 100 to 1000 Da. Full scan spectra acquisition was set as an average of 10 scans obtained with an isolation window of 4.0 and resolution of 75000 FHWM. ESI operational conditions was set as follow: spray voltage of 3.0 kV (negative mode) and 4.5 kV (positive mode); sheath gas flow of 35 (arb); sweep gas flow of 0.0 arb; auxiliary gas flow of 15 (arb); capillary temperature at 350 °C; source temperature at 250 °C; collision energy of 30 eV; and nebulizing gas flow (N2) of 2.5 L min−1. The quality of chemical profile obtained by MS was evaluated considering the total number of peaks and their relative intensity.
The identification of substances present in O. guianensis were performed by High Resolution Mass Spectrometry (HRMS) in a Q-Exactive (Thermofisher Scientific, Bremen, Germany) high resolution spectrometer equipped with an ESI source (Table S3†). The fragmentation experiments (MS2) by HRMS were acquired in data depend acquisition (DDA) mode in both negative and positive ionization modes. The m/z signal was monitored from 100 to 900 Da. All HRMS experiments were performed with a sheath gas flow rate of 60 arb, auxiliary gas flow rate at 20 arb, spray voltage of 3.9 kV in positive ionization mode and 2.9 kV in negative ionization mode, capillary temperature at 380 °C, resolution of 70000 FHWM, isolation window for MS2 experiments at 4.0 m/z, collision energy of 30 eV and mass error of 5.0 ppm. DDA MS2 acquisition algorithm applied was “TopN” set to 5 precursors fragmentation scan.63 MS and MS2 accurate mass measure were performed with Xcalibur software option “Lock mass” set as “best” for equipment calibration with the most intense ions of polydimethylcyclosiloxane. Suggestive identification was performed by comparison of the MS2 data with Lauraceae literature and metabolomics databases, such as the MassBank of North America (MoNA – http://mona.fiehnlab.ucdavis.edu/), the Human Metabolome Database (HMDB – http://www.hmdb.ca/) and PubChem (https://pubchem.ncbi.nlm.nih.gov/). It is worth mentioning that UHPLC-HRMS was only applied after extraction method optimization.
Data analysis
The data acquired by HPLC-DAD were processed in the software Chromeleon 7.2.1 (Thermo Fisher Scientific) for the baseline correction, peak detection and integration. TLC record and retention factor calculation were performed by the software winCATS 1.4.9 (CAMAG, Switzerland). MS data analysis was processed in the software XCalibur 3.0.1 (Thermo Fisher Scientific).Chemometric analysis was performed on OriginPro 2017, including one-way ANOVA test, hierarchical clustering analysis (HCA) by Ward's method with Euclidian distance and principal component analysis (PCA), with confidence levels of 95% (α = 0.05). Multivariate analysis of chromatographic and spectroscopic data considered each observed peak as a discrete variable. In HPLC-DAD data, absorbances (mAU) intensity were used as variable responses, without normalization to preserve the quantitative differentiation among samples. In MS data, the response was normalized with supremum norm in function of the m/z peak with higher intensity, converting absolute in relative abundance of each peak (from 0 to 100%).
Conclusions
It was demonstrated that operational features of the extraction method can produce significant qualitative and quantitative modification in the generation of a chemical profile for untargeted metabolomic. The composition of the extracting solvent was the factor which most affected the results, both qualitatively and quantitatively. Meanwhile, extraction temperature presents its major effects in the qualitative composition of the fingerprint, which can be attributed to the thermal degradation of phenolic compounds. Briefly, multivariate data analysis enables the evaluation of modifications in the quality of the O. guianensis fingerprint during method optimization. In addition, several minor compounds were relevant in the differentiation of samples, emphasizing the reliability of untargeted methods in the systematic study of similar species within unresolved genera. A comprehensive chemical characterization of this species was achieved applying the optimized method which applied 70% ethanol as extracting solvent, 1:12 ratio between sample and solvent, 40 °C as the extraction temperature and a particle size of 0.595 mm. This first chemical description, regarding O. guianensis ethanolic extract, indicates the presence of glycosylated flavonoids and aporphinic alkaloids and possible chemical similarity with Old World Ocotea species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors wish to thank Amazonas Federal University Herbarium and Multidisciplinary Analytical Center. We acknowledge the contribution of Dr Alberto Vicentini (INPA) and the support of the Coordination for the Improvement of Higher Education Personnel (CAPES) and the National Council for Scientific and Technological Development (CNPq).References
- G. A. B. Canuto, J. L. da Costa, P. L. R. da Cruz, A. R. L. de Souza, A. T. Faccio, A. Klassen, K. T. Rodrigues and M. F. M. Tavares, Quim. Nova, 2018, 41, 75–91 CAS
.
- O. Begol, H. G. Gika, G. A. Theodoridis and I. D. Wilson, in Metabolomic Profiling: Methods in Molecular Biology, ed. G.Theodoridis, H.Gika and I.Wilson, Humana Press, New York, 1st edn, 2018, vol. 1738 Search PubMed
- S. Naz, M. Vallejo, A. García and C. Barbas, J. Chromatogr. A, 2014, 1353, 99–105 CrossRef CAS PubMed
- B. C. Aranha, J. F. Hoffmann, R. L. Barbieri, C. V. Rombaldi and F. C. Chaves, Phytochem. Anal., 2017, 28, 439–447 CrossRef CAS PubMed
- E. Cadahía, F. De Simón, I. Aranda, M. Sanz, D. Sánchez-gómez and E. Pinto, Phytochem. Anal., 2015, 26, 171–182 CrossRef PubMed
- Y. Liu, G. Sun, J. Luan, J. Ling, J. Zhang and F. Yang, RSC Adv., 2015, 6, 366–375 RSC
- F. Van Der Kooy, F. Maltese, H. C. Young, K. K. Hye and R. Verpoorte, Planta Med., 2009, 75, 763–775 CrossRef CAS PubMed
- F. Souard, C. Delporte, P. Sto, E. A. Thévenot, N. Noret, B. Dauvergne, J. Kau, P. Van Antwerpen and C. Stévigny, Food Chem., 2018, 245, 603–612 CrossRef CAS PubMed
- Y. Wahyuni, A. R. Ballester, Y. Tikunov, R. C. H. de Vos, K. T. B. Pelgrom, A. Maharijaya, E. Sudarmonowati, R. J. Bino and A. G. Bovy, Metabolomics, 2013, 9, 130–144 CrossRef CAS PubMed
- M. Lv, Y. Tian, Z. Zhang, J. Liang, F. Xu and J. Sun, RSC Adv., 2015, 5, 15700–15708 RSC
- L. N. Zhang, L. Wang, Z. Q. Shi, P. Li and H. J. Li, RSC Adv., 2018, 8, 9074–9082 RSC
- W. Chen, G. Zhang, W. Chen, Q. Zhong and H. Chen, RSC Adv., 2018, 8, 31396–31405 RSC
- N. Villa-Ruano, R. Velásquez-Valle, L. G. Zepeda-Vallejo, N. Pérez-Hernández, M. Velázquez-Ponce, V. M. Arcos-Adame and E. Becerra-Martínez, Food Res. Int., 2018, 106, 870–877 CrossRef CAS PubMed
- A. C. Schrimpe-rutledge, S. G. Codreanu, S. D. Sherrod and J. A. Mclean, J. Am. Soc. Mass Spectrom., 2016, 27, 1897–1905 CrossRef CAS PubMed
- H. Gika, C. Virgiliou, G. Theodoridis, R. S. Plumb and I. D. Wilson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1117, 136–147 CrossRef CAS PubMed
- L. C. Klein-Júnior, J. Viaene, J. Salton, M. Koetz, A. L. Gasper, A. T. Henriques and Y. Vander Heyden, J. Chromatogr. A, 2016, 1463, 60–70 CrossRef PubMed
- M. L. Doran, N. Mykytczuk, A. Bieniek, A. Methé and T. J. S. Merritt, Metabolomics, 2017, 13, 1–10 CrossRef CAS
- R. t'Kindt, L. De Veylder, M. Storme, D. Deforce and J. Van Bocxlaer, J.
Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 871, 37–43 CrossRef
- S. Zschocke, S. E. Drewes, K. Paulus, R. Bauer and J. Van Staden, J. Ethnopharmacol., 2000, 71, 219–230 CrossRef CAS
- A. L. Batista, N. C. Yoshida, F. R. Garcez and W. S. Garcez, Biochem. Syst. Ecol., 2015, 61, 229–231 CrossRef CAS
- L. T. Albarracin, W. A. Delgado, L. E. Cuca and M. C. Ávila, Biochem. Syst. Ecol., 2017, 74, 60–62 CrossRef CAS
- D. P. Xu, J. Zheng, Y. Zhou, Y. Li, S. Li and H. Bin Li, Food Chem., 2017, 217, 552–559 CrossRef CAS PubMed
- D. L. Custódio and V. F. da Veiga Junior, RSC Adv., 2014, 4, 21864 RSC
- S. Maria, W. Zanin, A. Luísa and L. Lordello, Quim. Nova, 2007, 30, 92–98 CrossRef
- M. Waksmundzka-Hajnos, J. Sherma and T. Kowalska, Thin layer chromatography in phytochemistry, CRC Press, Boca Raton, 2008 Search PubMed
- E. D. C. Barrera, L. E. C. Suárez, C. Sua, E. David and C. Barrera, Biochem. Syst. Ecol., 2009, 37, 522–524 CrossRef
- C. C. Andrei, R. Braz-Filho and O. R. Gottliebt, Phytochemistry, 1988, 27, 3992–3993 CrossRef CAS
- W. S. Garcez, F. R. Garcez, L. M. G. E. Da Silva and A. A. Shimabukuro, J. Braz. Chem. Soc., 2005, 16, 1382–1386 CrossRef CAS
- E. D. Coy-Barrera, L. E. Cuca-Suárez and M. Sefkow, Phytochemistry, 2009, 70, 1309–1314 CrossRef CAS PubMed
- J. Azmir, I. S. M. Zaidul, M. M. Rahman, K. M. Sharif, A. Mohamed, F. Sahena, M. H. A. Jahurul, K. Ghafoor, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2013, 117, 426–436 CrossRef CAS
- B. K. Tiwari, TrAC, Trends Anal. Chem., 2015, 71, 100–109 CrossRef CAS
- J. P. Maran and B. Priya, Carbohydr. Polym., 2015, 115, 732–738 CrossRef CAS
- F. Chemat, N. Rombaut, A. G. Sicaire, A. Meullemiestre, A. S. Fabiano-Tixier and M. Abert-Vian, Ultrason. Sonochem., 2017, 34, 540–560 CrossRef CAS PubMed
- S. M. W. Zanin, O. G. Miguel, D. P. Montrucchio, C. K. Costa, J. B. Lagos and A. L. L. Lordello, Quim. Nova, 2011, 34, 743–747 CAS
- R. Garrett, M. T. V Romanos, M. Ricardo, M. G. Santos, L. Rocha and A. Jorge, Rev. Bras. Farmacogn., 2012, 22, 306–313 CrossRef
- D. T. da Silva, R. Herrera, B. F. Batista, B. M. Heinzmann and J. Labidi, Int. Biodeterior. Biodegrad., 2017, 117, 158–170 CrossRef CAS
- M. M. R. Silva Teles, A. A. Vieira Pinheiro, C. Da Silva Dias, J. Fechine Tavares, J. M. Barbosa Filho and E. V. Leitão Da Cunha, The Alkaloids: Chemistry and Biology, 2019, vol. 82, pp. 147–304 Search PubMed
- E. Gil Archila and L. E. Cuca Suárez, Nat. Prod. Res., 2018, 32, 195–201 CrossRef CAS PubMed
- D. C. Gontijo, G. C. Brandão, P. C. Gontijo, A. B. de Oliveira, M. A. N. Diaz, L. G. Fietto and J. P. V. Leite, Food Chem., 2017, 230, 618–626 CrossRef CAS
- E. J. Llorent-Martínez, V. Spínola and P. C. Castilho, Ind. Crops Prod., 2017, 107, 1–12 CrossRef
- X. Liu, J. Yang, J. Fu, T. G. Xie, P. C. Jiang, Z. H. Jiang and G. Y. Zhu, Biochem. Syst. Ecol., 2018, 81, 45–48 CrossRef CAS
- N. F. Roque, Z. S. Ferreira, O. R. Gottliebt, R. L. Stephens and E. Wenkert, Rev. Latinoam. Quim., 1978, 9, 25–27 CAS
- O. R. Gottliebt, Phytochemistry, 1972, 11, 1537–1570 CrossRef
- W. M. N. H. W. Salleh and F. Ahmad, J. Appl. Pharm. Sci., 2017, 7, 204–218 CAS
- M. Irakli, P. Chatzopoulou and L. Ekateriniadou, Ind. Crops Prod., 2018, 124, 382–388 CrossRef CAS
- L. Wang, N. Boussetta, N. Lebovka and E. Vorobiev, Int. J. Food Sci. Technol., 2018, 53, 2104–2109 CrossRef CAS
- H. van der Werff and A. Vicentini, Novon, 2000, 10, 264–297 CrossRef
- J. G. Rohwer, Mitt. Inst. Allg. Bot. Hamburg, 1986, 20, 1–279 Search PubMed
- A. S. Chanderbali, H. van der Werff and S. S. Renner, Ann. Mo. Bot. Gard., 2001, 88, 104–134 CrossRef
- O. R. Gottlieb and K. Kubitzki, Biochem. Syst. Ecol., 1981, 9, 5–12 CrossRef CAS
- A. D. Meinhart, F. M. Damin, L. Caldeirão, T. F. F. da Silveira, J. T. Filho and H. T. Godoy, Food Res. Int., 2017, 99, 522–530 CrossRef CAS PubMed
- F. R. Garcez, A. F. G. Da Silva, W. S. Garcez, G. Linck, M. D. F. C. Matos, E. C. S. Santos and L. M. M. Queiroz, Planta Med., 2011, 77, 383–387 CrossRef CAS PubMed
- D. T. da Silva, R. Herrera, B. F. Batista, B. M. Heinzmann and J. Labidi, Int. Biodeterior. Biodegrad., 2017, 117, 158–170 CrossRef CAS
- W. S. Garcez, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1995, 39, 815–816 CrossRef CAS
- A. Fournet, M. E. Ferreira, A. Rojas de Arias, I. Guy, H. Guinaudeau and H. Heinzen, Fitoterapia, 2007, 78, 382–384 CrossRef CAS PubMed
- D. C. Montgomery, Design and Analysis of Experiments, Jonh Wiley & Sons, New York, 5th edn, 2001 Search PubMed
- M. J. De Camargo, M. Lazaro, D. Miranda, C. M. Kagamida, E. D. Rodrigues, F. Rodrigues, W. Silva, S. Of and L. Six, Quim. Nova, 2013, 36, 1008–1013 CrossRef
- L. E. Cuca, P. Leon and E. D. Coy, Chem. Nat. Compd., 2009, 45, 179–181 CrossRef CAS
- C. C. Andrei, R. Braz-Filho and O. R. Gottlieb, Phytochemistry, 1988, 27, 3992–3993 CrossRef CAS
- S. S. Grecco, H. Lorenzi, A. G. Tempone and J. H. G. Lago, Tetrahedron: Asymmetry, 2016, 27, 793–810 CrossRef CAS
- W. M. N. H. W. Salleh and F. Ahmad, Marmara Pharm. J., 2016, 20, 390 CrossRef CAS
- A. Kruve, R. Rebane, K. Kipper, M. L. Oldekop, H. Evard, K. Herodes, P. Ravio and I. Leito, Anal. Chim. Acta, 2015, 870, 29–44 CrossRef CAS PubMed
- S. Kreimer, M. E. Belov, W. F. Danielson, L. I. Levittsky, M. V. Gorshkov, B. L. Karger and A. R. Ivanov, J. Proteome Res., 2017, 176, 139–148 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10436d |
| This journal is © The Royal Society of Chemistry 2020 |