
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrediction of the [4 + 2]- and [5 + 4]-cycloaddition reactions in zig-zag carbon nanotubes via an ambimodal transition state: density functional theory calculations†
Akanksha Ashok Sangolkar and
Ravinder Pawar *
*
Department of Chemistry, National Institute of Technology (NIT) Warangal, Telangana-506004, India. E-mail: ravinder_pawar@nitw.ac.in
First published on 17th March 2020
Abstract
A unique type of chemical reaction known as an ambimodal reaction has drawn tremendous attention owing to its intriguing feature of forming multiple (two or more) products from the same (single) transition state. In contrast to conventional reactions, bifurcation of the potential energy surface takes place in ambimodal reactions. Density functional theory (DFT) based calculations were performed to probe the Diels–Alder (DA) cycloaddition reactions of various carbon nanotubes (CNTs) with 1,3-butadiene. The present investigation reveals the possibility of ambimodal transition state formation on a potential energy surface (PES) corresponding to an unusual [5 + 4]-cycloadduct along with the conventional [4 + 2]-cycloadduct. The ground state of the [5 + 4]-cycloadduct obtained from butadiene and the H-terminated CNTs is a triplet (3T) state, but on the other hand the [4 + 2]-cycloadduct is a singlet (1S) state. The [5 + 4]-adduct is energetically more stable in comparison with the [4 + 2]-adduct. The possibility of the formation of the [5 + 4]-adduct is validated using frontier molecular orbitals. The length of the nanotube significantly influences the overall kinetics and thermodynamics of the reaction.
Introduction
The utmost attention has been devoted to modelling and understanding new reactions and their mechanisms in chemistry. A single transition state in the chemical reaction that yields two or more dissimilar products via potential energy surface (PES) bifurcation is known as an ambimodal reaction.1–4 While researching the dimerization of cyclopentadiene, Caramella and co-workers proposed a “bispericyclic” (ambimodal pericyclic) transition state.5,6 As mentioned, multiple products can be obtained from the single transition state, the conventional transition state theory plays a small part in probing the reaction kinetics. Therefore, numerous attempts have been made to understand the dynamics of ambimodal reactions.7,8Factors that are proposed to play a decisive role in determining the ratio of various products in ambimodal reactions are: (i) the shape of the PES; (ii) the geometry of the transition structure; and (iii) the dynamic matching.4,9–12 Singleton and co-workers and Houk and co-workers have contributed significantly to the understanding of various factors that influence the ratio of products obtained in ambimodal reactions.4,9,10,13–17 They have also carried out studies to find the ambimodal transition structures (TSs) in various reactions. Ambimodal transition states have also been proposed and explored in the biosynthesis of spinosyn A and heronamide A.15,16 Recently, Burns et al. reported [4 + 3] and [5 + 2] products in the cycloaddition of butadiene and oxidopyrylium ylides via a novel ambimodal transition structure (TS).18,19 Furthermore, Datta and co-workers reported PES bifurcation in different contexts.20,21
Single-walled carbon nanotubes (CNTs) are conceptually generated by enfolding of the graphene sheets. Numerous experimental and theoretical studies have been carried out on CNTs to understand their physical, electronic, and optical properties, as well as the chemical reactivity.22–33 Furthermore, the functionalization of various CNTs and fullerenes was considered to be an enormously important process to separate and tune the properties of these materials. Various chemical reactions have been reported in the context of the successful functionalization of different CNTs and fullerenes.22,34–39 The Diels–Alder (DA) cycloaddition is recognized as a plausible and convenient chemical approach for functionalization, in this context few reports are known.22,40–45 A similar strategy has been used in the present investigation.
The DA cycloaddition of a single walled carbon nanotube (SWCNT) with ester functionalized terminals and o-quinodimethane was carried out under microwave irradiation.46 The results obtained from scanning force microscopy (SFM) and Raman spectral analysis reveal the side-wall functionalization via a cycloaddition reaction. Furthermore, the high-density pentagon–heptagon defects in the nanotube lattice were highlighted as being the preferred zone for functionalization.46 Elsewhere, the functionalization of the single wall CNTs via the DA reaction of a fluorinated single walled carbon nanotube was reported. The presence of electron withdrawing substituents enhances the rate of cycloaddition by providing ‘‘activated’’ C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds on the sidewall.47 Doris and co-workers carried out the functionalization of single walled carbon nanotubes (SWCNTs) using tandem high-pressure/Cr(CO)6 activation.48 They investigated the synergistic effect of pressure and chromium on the DA reaction and found that the nanotube is activated by the combined effect of high pressure and Cr(CO)6.48 The theoretical report by Lu et al. demonstrated the energetics of the DA reaction with the aid of an ONIOM two layer method.49 The report highlights that the stability of the TS is attained mainly owing to the aromaticity.49 Both the experimental and theoretical reports described herein focussed on the formation of only the [4 + 2]-cycloadduct. However, the possibility of formation of other products is unavoidable in the majority of chemical reactions. During our investigation of the DA reaction of the H-terminated CNTs of finite length with butadiene, we noticed an unusual critical point structure on the PES. The unusual point structures are ambimodal transition states which require further exploration.
C bonds on the sidewall.47 Doris and co-workers carried out the functionalization of single walled carbon nanotubes (SWCNTs) using tandem high-pressure/Cr(CO)6 activation.48 They investigated the synergistic effect of pressure and chromium on the DA reaction and found that the nanotube is activated by the combined effect of high pressure and Cr(CO)6.48 The theoretical report by Lu et al. demonstrated the energetics of the DA reaction with the aid of an ONIOM two layer method.49 The report highlights that the stability of the TS is attained mainly owing to the aromaticity.49 Both the experimental and theoretical reports described herein focussed on the formation of only the [4 + 2]-cycloadduct. However, the possibility of formation of other products is unavoidable in the majority of chemical reactions. During our investigation of the DA reaction of the H-terminated CNTs of finite length with butadiene, we noticed an unusual critical point structure on the PES. The unusual point structures are ambimodal transition states which require further exploration.
Therefore, in the present investigation, a systematic attempt has been made to understand the energetics of the DA reaction of various nanotubes at the H-terminating corner. The plausibility of formation of an extremely unusual [5 + 4]-cycloadduct along with the conventional [4 + 2]-cycloadduct in various nanotubes have been scrutinized. The role of the nanotube length in the cycloaddition reaction has also been addressed. The effect of solvent on the cycloaddition reaction has also been investigated.
Computational details
The geometries of various pristine, TSs, and functionalized H-terminated CNTs were fully optimized without any geometrical/symmetrical constraints using density functional theory (DFT)-based Becke's three parameter hybrid exchange functional and the Lee–Yang–Parr correlation functional (B3LYP) method.50,51 To gain an insight into the effect of the inclusion of polarization and to diffuse the basis set functions, the calculations were performed using 6-31G* and the same basis set in combination with 6-311++G** (the fragments considered using 6-31G* and 6-311++G** basis sets are shown in Scheme 1).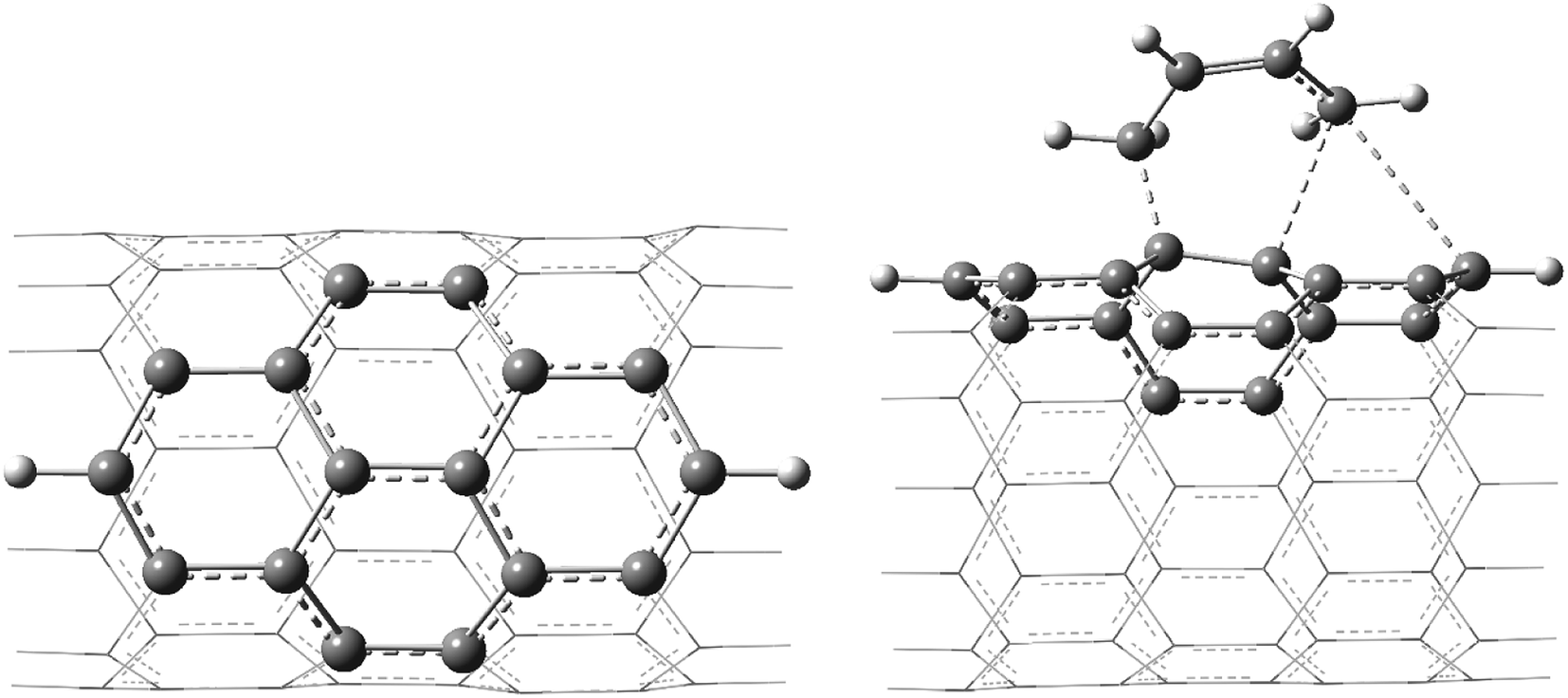 | ||
| Scheme 1 The atoms shown in the ball-stick model were considered using the 6-311++G** basis set and the atoms shown in the wire model were considered using the 6-31G* basis set. | ||
In order to assess the role of inclusion of the dispersion interaction, all of the geometries were re-optimized using the B3LYP method along with inclusion of the empirical dispersion correction (D3) as suggested by Grimme et al., employing both the basis sets, that is 6-311++G**:6-31G*.52 The nature of the critical point structures were characterized as local minima and as first order saddle points by the frequency calculations at the same level of theory. The TSs were confirmed by the existence of a characteristic single imaginary frequency with displacement vectors in the direction of bond formation (breaking). Intrinsic reaction coordinate (IRC) calculations were performed to ensure that the saddle points found were true TSs connecting the reactants and the products. The energetics of the various reactions were calculated at a temperature of 298.15 K and 1 atm of pressure in the gaseous phase.
Continuum solvation model density (SMD) calculations were carried out to probe the effect of solvation on the reaction. In order to understand the role of the polarity of the solvents in the present investigation, toluene (ε = 2.3741) and acetonitrile (ε = 35.688) were utilized. Systematic comparison of the gas-phase DA cycloaddition reaction in the same non-polar and polar reaction medium were carried out. All of the calculations were performed using the Gaussian 09 suite of programs.53
Results and discussion
Various mechanisms were proposed to probe the DA reactions of the small molecule viz. non-polar mechanisms (synchronical mechanism, biradicaloidal one step-mechanism, stepwise biradical mechanism, and polar mechanisms (one step-two stage mechanism, stepwise zwitterionic mechanism)). In this context, Jasinski and co-workers have made significant contributions to analysing the different small molecules.54–59The DA cycloaddition reaction of various H-terminated CNTs with 1,3-butadiene were chosen as the model reactions. Both the moieties considered in the present work are neutral. Therefore, all of the reactions are considered to follow the concerted reaction mechanism path. A plethora of reports on the DA reaction of CNTs revealed that the CNTs conceivably act as model dienophiles in DA cycloaddition reactions.30,40–43 Therefore, in the present investigation the nanotube models were considered as dienophiles. The terminal carbon atoms of CNT, which are connected to the H atoms (H-terminal), are possibly displaced from their equilibrium position owing to the reduced strain. Therefore, the reactivity of the carbon atoms which are linked to the H atoms (terminal) is greater when compared to that of any other C atoms in the CNTs. The reactive nature of the terminal carbon atom enables such units to engage as a dienophile in the [5 + 4]-cycloaddition reaction. Scheme 2 illustrates the different DA cyclic adduct formation routes through path-1 and path-2 via the ambimodal transition state.
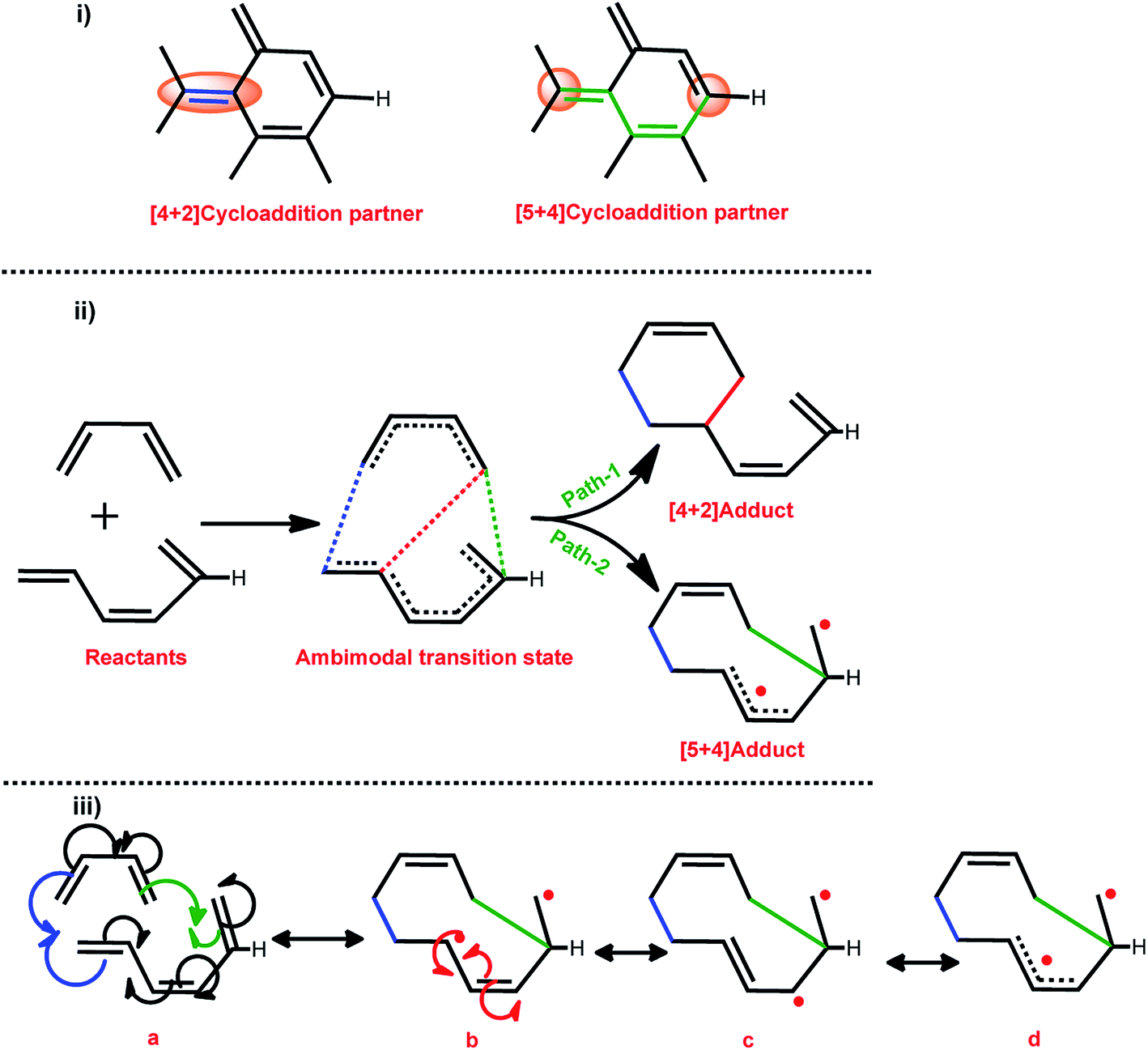 | ||
| Scheme 2 (i) The dual cycloaddition reactivity modes of a H-terminated CNT, and (ii) the possible [4 + 2] and [5 + 4] pathways of the reaction with butadiene. (iii) An arrow pushing diagram of the formation of the [5 + 4]-cycloadduct via biradical formation. | ||
Qualitatively, ambimodal TS formation in cycloaddition reactions indicates that both the reaction paths are kinetically indistinguishable. The red dot in the [5 + 4] adduct represents the free electron and the single electron resonates throughout the nanotube structure. Thus, the possibility of the formation of frustrated biradical species in the reaction is probable. The carbon nanotube model dienophile consists of an extended π-electron conjugation, thus the newly formed biradical may be stabilized via the formation of a resonance structure (as a triplet ground state) and/or followed by flipping of the spin of one of these electrons (as a singlet ground state). Thus, the electron spin inversion is also of considerable importance in the process of product formation.
The thermodynamic quantities obtained for the reaction determines the ratio of the products along with various other factors viz. the shape of the PES and the geometrical features of the TS, and the distribution of the momenta at the TS. A similar approach was selected to understand the cycloaddition reaction of various CNTs with 1,3-butadiene.
Geometry
The optimized geometries of the reactant, TS, and the products obtained in the DA reaction (8,0)CNT with 1,3-butadiene are shown in Fig. 1 and are labelled with the important bond distances. Geometries (optimized coordinates) of all other CNTs, corresponding TSs and products are given in the ESI.†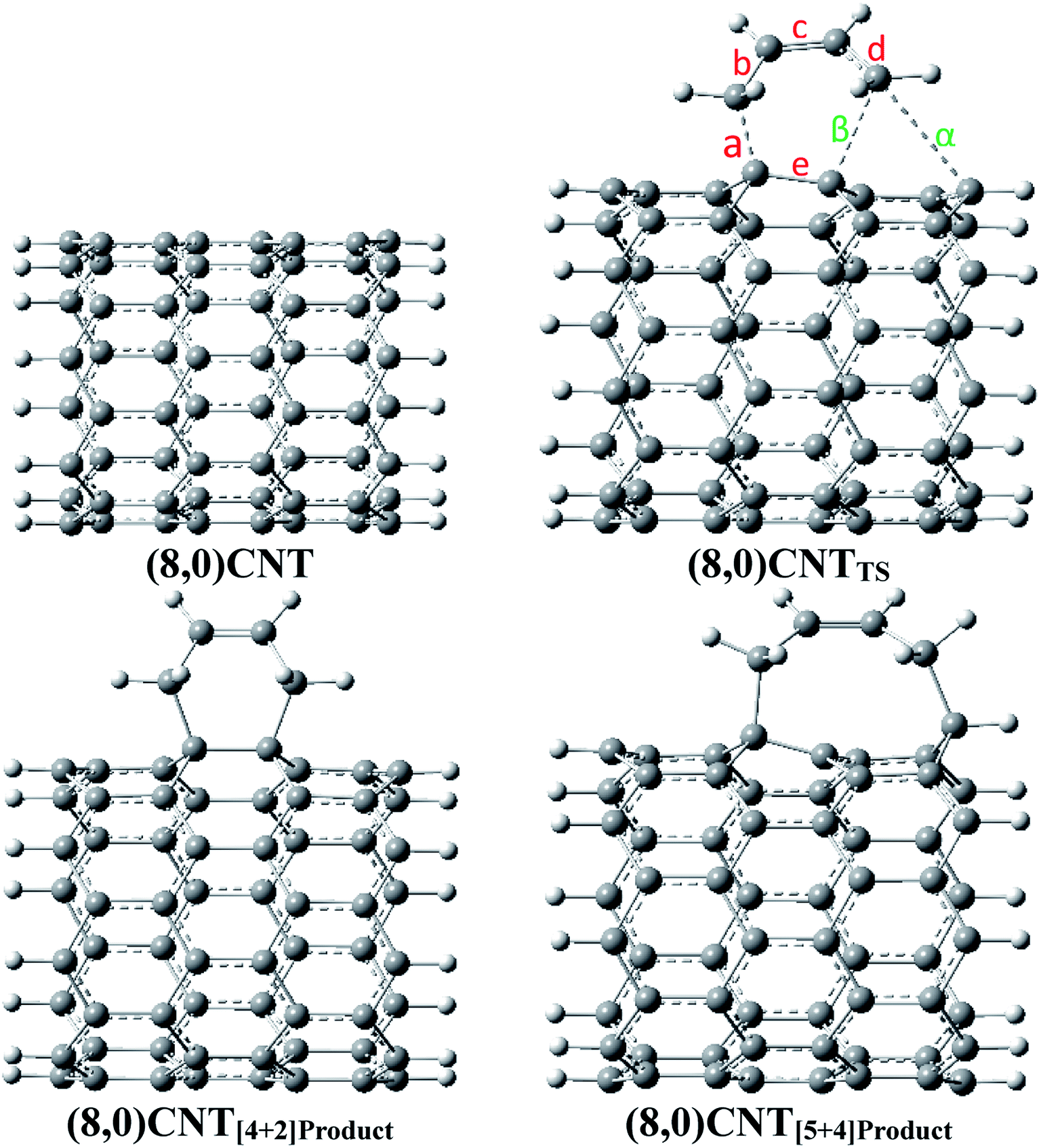 | ||
| Fig. 1 Optimized geometries of the reactant, ambimodal transition structure and products (a, b, c, d, e, α, and β are the most important bond distances in the active site) with a tube length of 9.25 Å; calculations were carried out using the B3LYP-D3/6-311++G**:6-31G* level of theory (colour key: grey = carbon and white = hydrogen atoms). | ||
As observed from the previous investigations, the geometrical parameters of TS play a decisive role in the formation of different products in various ambimodal reactions, in the present investigation geometrical data analysis was carried out and the important results are described herein. Important geometrical parameters of various TSs, [4 + 2]-cycloadduct, and [5 + 4]-cycloadduct are reported in Table 1. Important bond angles and dihedral angles along with the index lx–y, as defined by Jasinski et al.,59,60 are given in Tables S1 and S2 in the ESI.†
| a | b | c | d | e | α | β | |
|---|---|---|---|---|---|---|---|
| a All of the parameters are in Å and a value given in parenthesis represents the same bond in the reactant. | |||||||
| (5,0)CNT-TS | 1.58 | 1.49 | 1.35 | 1.45 | 1.51 (1.40) | 2.53 | 2.35 |
| (6,0)CNT-TS | 1.60 | 1.48 | 1.36 | 1.43 | 1.52 (1.44) | 2.82 | 2.38 |
| (7,0)CNT-TS | 1.61 | 1.47 | 1.36 | 1.42 | 1.52 (1.43) | 2.99 | 2.35 |
| (8,0)CNT-TS | 1.64 | 1.48 | 1.37 | 1.41 | 1.53 (1.44) | 3.01 | 2.49 |
| (5,0)CNT[5+4]Product | 1.59 | 1.52 | 1.35 | 1.53 | 1.47 | 1.60 | |
| (6,0)CNT[5+4]Product | 1.60 | 1.52 | 1.35 | 1.52 | 1.48 | 1.61 | |
| (7,0)CNT[5+4]Product | 1.60 | 1.52 | 1.35 | 1.52 | 1.48 | 1.60 | |
| (8,0)CNT[5+4]Product | 1.60 | 1.52 | 1.35 | 1.52 | 1.48 | 1.60 | |
| (5,0)CNT[4+2]Product | 1.56 | 1.50 | 1.33 | 1.50 | 1.59 | 1.56 | |
| (6,0)CNT[4+2]Product | 1.57 | 1.50 | 1.33 | 1.50 | 1.60 | 1.57 | |
| (7,0)CNT[4+2]Product | 1.57 | 1.50 | 1.33 | 1.50 | 1.60 | 1.57 | |
| (8,0)CNT[4+2]Product | 1.57 | 1.50 | 1.33 | 1.50 | 1.60 | 1.57 | |
All of the TSs contain three partially formed bonds (viz. bonds indicated by the labels a, β, and α in Fig. 1) which indicate that these TSs are possibly ambimodal, but the β and α bond distances are significantly larger when compared with the bond distance designated by label a. As the diameter of the CNT increases, the value of the α bond distance increases enormously in the TSs. Typically, the calculated α bond length in the different TSs range from 2.53 to 3.01 Å. The same value for the β bond ranges from 2.35 to 2.49 Å. The discrimination in the increment of the newly formed bonds may be attributed to the reduced flexibility of the terminal carbon atom. As the tube diameter increases, the ability of the carbon atoms to attain planarity increases, thus the flexibility of the same atom decreases. Close analysis of the newly formed bonds in the various TSs clearly indicate that the formation of all of these TSs is considerably asynchronous in nature.
Herein, the Csp3–Csp3 bond distance is reported which is significantly larger in comparison with the conventional Csp3–Csp3 bond distance. Close analysis of the geometrical parameters of the various adducts reveal that the α value in the [5 + 4]-cycloadduct is marginally larger when compared with the conventional C–C single bond distance (i.e. 1.54 Å). Typically, the calculated α values for the (5,0), (6,0), (7,0), and (8,0)CNT adduct range from 1.60 to 1.61 Å. Similar distances for the C–C bonds were found in the case of the diamondoid dimers (and other highly crowded alkanes).61 In this context Schreiner and co-workers have shown that the molecular systems have an elongated Csp3–Csp3 bond in which the dispersion interaction plays a major role in attaining the stability of the system.61 Therefore, comparative analysis of the C–C bond distances in the functionalized CNTs and the diamondoid dimer indicates that the stability of the [5 + 4]-cycloadduct is governed by the dispersion interaction and is an important factor to consider in the calculation of the geometry and energetics. Nevertheless, in the present case the longer Csp3–Csp3 bond may be attributed to the large strain, allowing it to form a nine membered ring structure involving five C atoms from the CNT.
To gain insight into the role of the length of the CNTs, in this investigation various length CNTs of (5,0) chirality were considered. Typically, the selected model H-terminated CNT lengths were 9.25, 11.34, 13.49, and 15.63 Å, respectively. Optimized geometries of various ambimodal TSs involved in the DA cycloaddition reaction of the (5,0) CNTs with different lengths are shown in Fig. S5.† It should be noted from the geometries of various TSs that the increment in the length of the CNT marginally alters the geometry of the active site.
Energetics
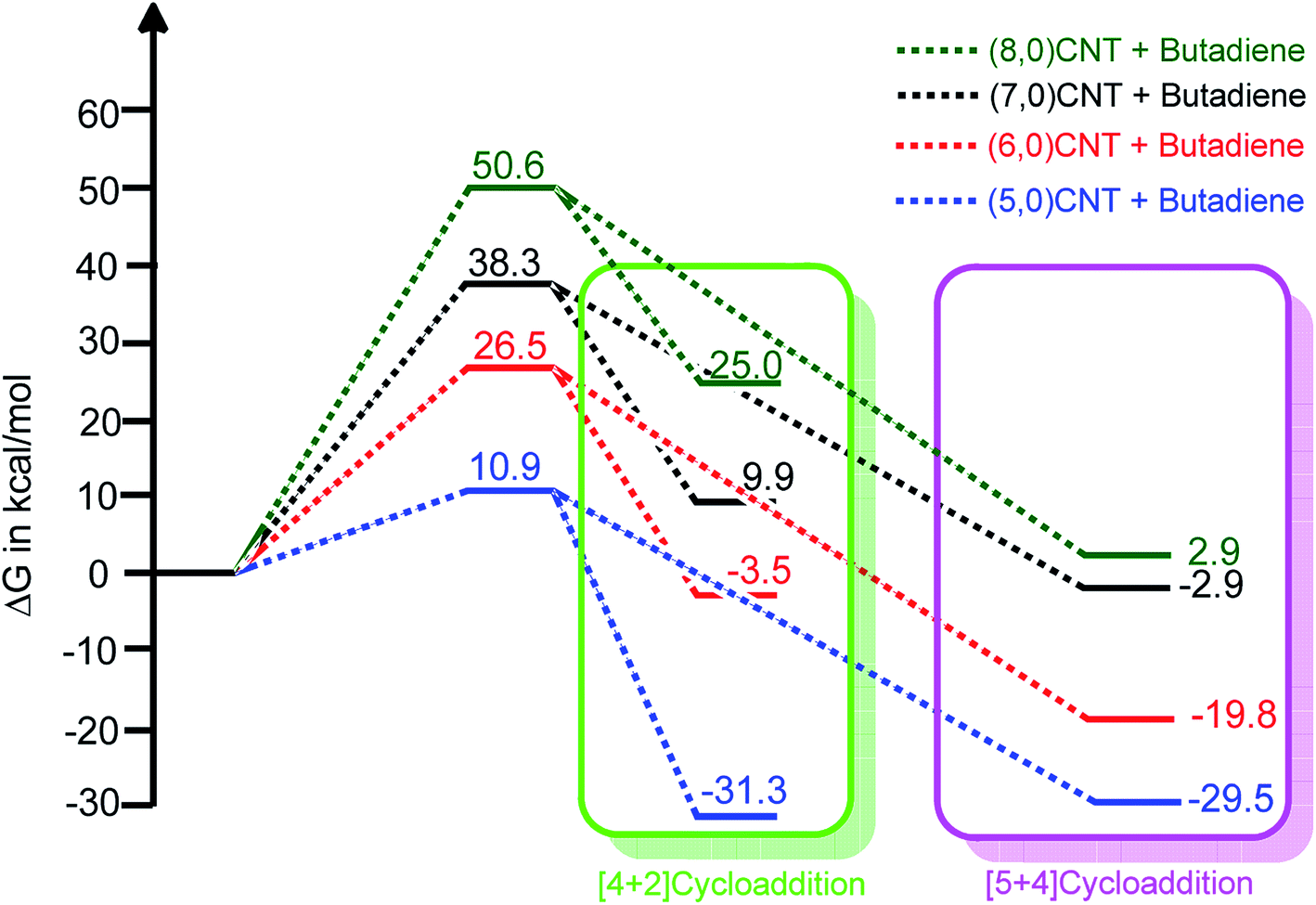 | ||
| Fig. 2 The relative free energy profiles of the [4 + 2] and [5 + 4]-cycloaddition reactions of various carbon nanotubes with 1,3-butadiene (using B3LYP-D3/6-311++G**:6-31G*). | ||
The ambimodal reactions of various CNTs show the same energy barrier for the formation of both products. It can also be noted that the calculated energy barrier of the cycloaddition reaction on different CNTs increased upon the increase in the tube diameter and the results are in agreement with a previous report on single wall (SW) transformation in various nanotubes.62 Furthermore, the role of the charge transfer (polar nature) of these cycloaddition reactions was investigated using the global electron density transfer (GEDT) index (Domingo et al.).63,64 The calculated GEDT values obtained from the B3LYP/6-31G* level of calculation for the TSs involved in the (5,0), (6,0), (7,0), and (8,0) cycloaddition reactions are 0.008, 0.012, 0.034, and 0.036(e), respectively. Interestingly, it was found that the GEDT increases linearly as the energy barrier height increases (r2 = 0.81). Therefore, the charge transfer in TS is a prominent factor in the kinetics of the DA reactions.
Fig. 2 illustrates that the adduct obtained from the [5 + 4]-cycloaddition is energetically more stable when compared with the corresponding [4 + 2] counterpart. The same adduct stability trend is observed for the cycloaddition reactions using (6,0)CNT to (8,0)CNT. The adduct obtained in the case of the same length (5,0)CNT, for the [4 + 2]-cycloaddition is energetically more favourable when compared with the [5 + 4]-cycloaddition. As the tube diameter increases, the thermodynamic stability of the adduct decreases and some of these adducts are thermodynamically less stable when compared to that of the reactant. Thus, it is worth noting that although the kinetics of formation of both products are the same, the thermodynamic stability of these adducts enables the discrimination.
To evaluate the path adopted to convert the [5 + 4]- and [4 + 2]-cyclic products from the ambimodal TSs, the intrinsic reaction coordinates (IRCs) were investigated. Relative energies of various geometries along with the reaction coordinates are depicted in Fig. 3. Total energies along with the IRCs are plotted in Fig. S3† for all of the TSs. As the systems under scrutiny are substantially larger and computationally demanding, the IRC calculations were carried out using the B3LYP-D3/6-31G* level of calculations.
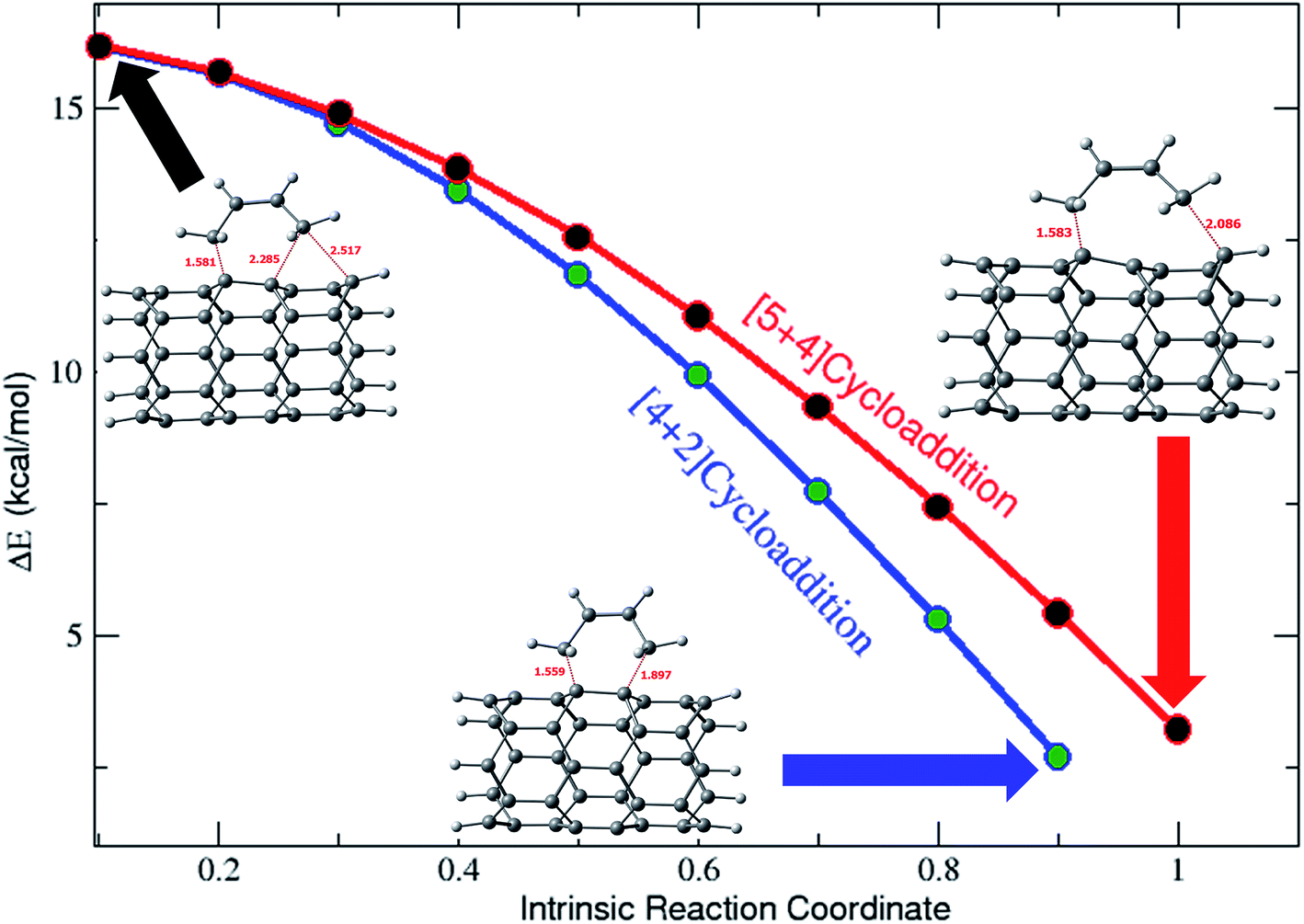 | ||
| Fig. 3 Relative energies plotted along with the IRC (calculated using B3LYP-D3/6-31G*). | ||
Fig. 3 clearly illustrates the formation of a [5 + 4]-cyclic adduct in addition to that of the [4 + 2]-cyclic adduct. It is also interesting to note that the ambimodal TSs are more like the [4 + 2] type. Moreover, the same trend is seen for all other TSs. The calculated PES contour map, along with the distance variation is shown in Fig. S4 in the ESI.†
It can be observed from the above described results that the product obtained from the [5 + 4]-cycloaddition reaction is energetically more stable when compared with that of the corresponding [4 + 2]-cyclic adduct counterpart. Furthermore, the possibility of formation of a triplet state intermediate and products along with the singlet is also ambiguous, thus the inclusion of an unrestricted calculation was found to be more interesting. Therefore, the geometries of the various point structures involved in the cycloaddition reaction of (6,0)CNT with butadiene were re-optimized using the UB3LYP-D3 method employing the 6-31+G*:3-21G* basis set. The energetics obtained using the above mentioned method are given and described in the ESI† (biradical formation). The results clearly indicate that the [5 + 4]-cycloadduct is biradical with a triplet ground state.
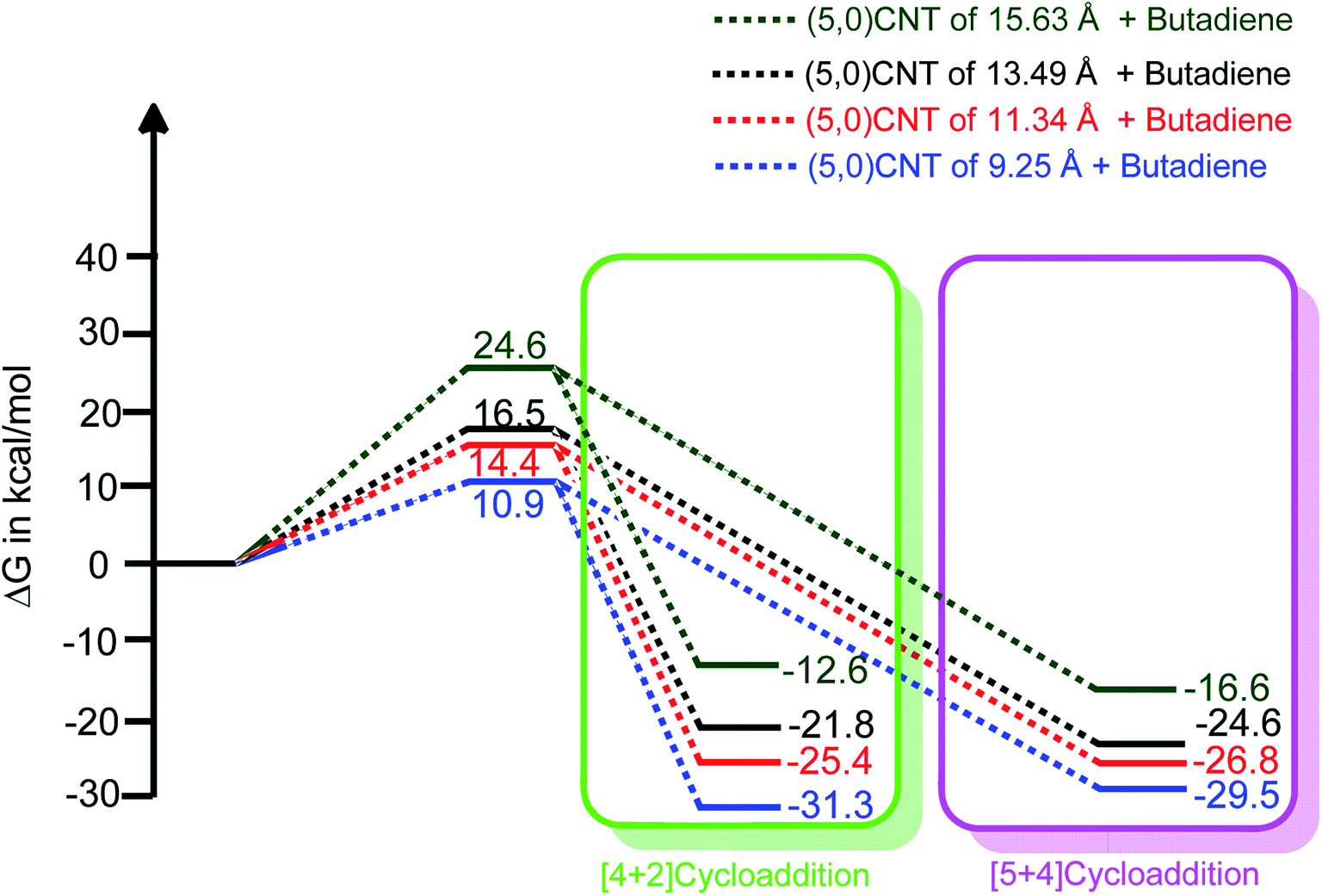 | ||
| Fig. 4 The length dependent reaction free energy profiles of (5,0)CNT with butadiene for both the [4 + 2] and [5 + 4]-cycloaddition reactions (using B3LYP-D3/6-311++G**:6-31G*). | ||
It should be noted from Fig. 4 that an increment in the tube length enhances the energy barrier significantly. Typically, the calculated energy barrier of the different lengths of (5,0)CNT range from 10.9 to 24.6 kcal mol−1. The enhancement in the energy barrier with the tube length may be attributed to the extended π-electron cloud in conjugation (extended conjugation) for the larger nanotubes.
It is interesting to observe that the adducts obtained from the cycloaddition reaction of the 9.25 Å length CNT show an exceptional stability trend compared with the same nanotube with a longer length. The exceptional behaviour of (5,0)CNT with a length of 9.25 Å may be attributed to the small length and small radius of the tube, which leads to reduced conjugation of the π-electron cloud.
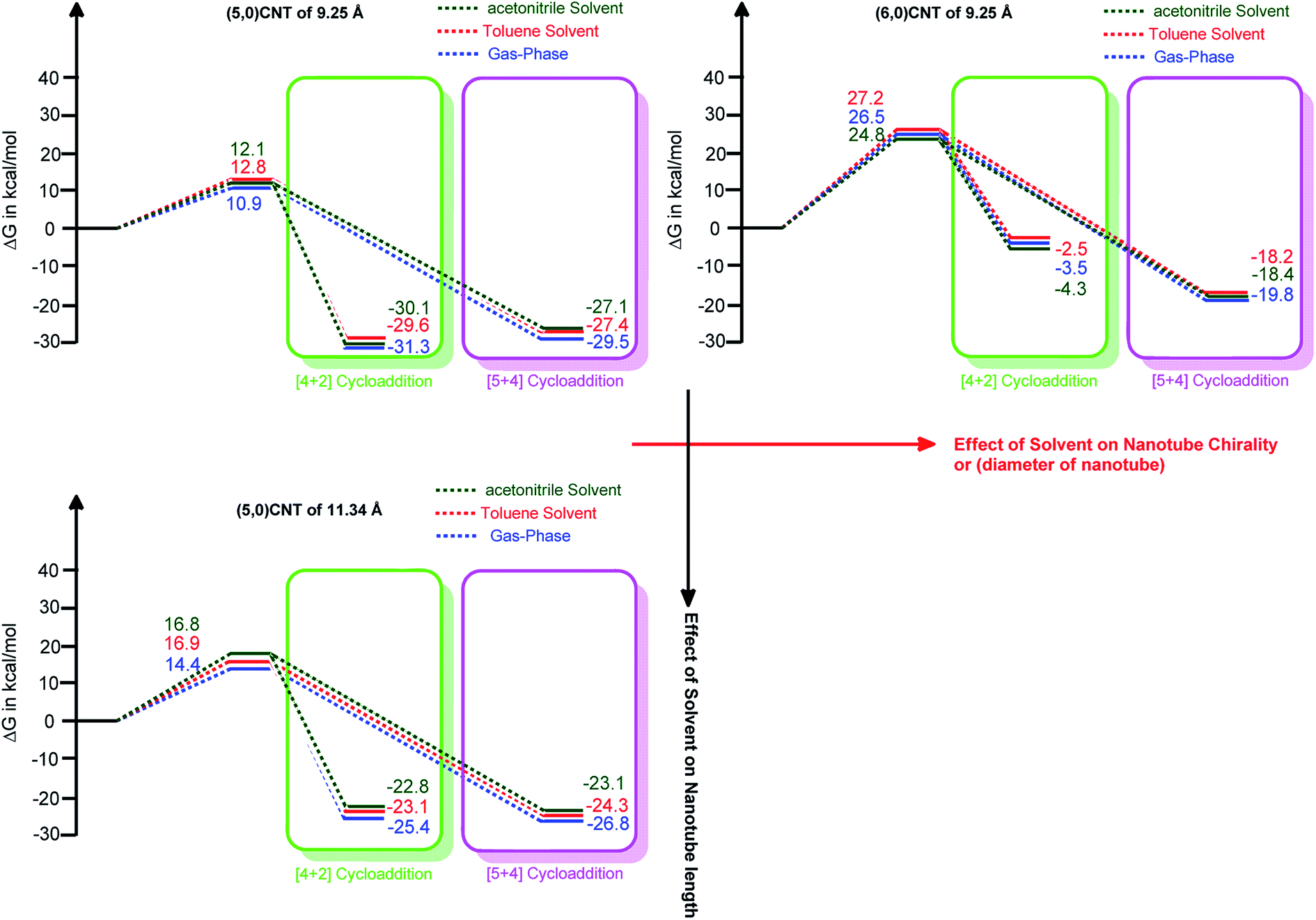 | ||
| Fig. 5 The relative free energy profiles of the [5 + 4] and [4 + 2]-cycloaddition reactions of various carbon nanotubes with 1,3-butadiene in the presence of different solvents along with the gas-phase reaction (using B3LYP-D3/6-311++G**:6-31G*). | ||
It should be noted that the change in the polarity of the solvent only marginally alters the energy barrier height. The change in the thermodynamic stability attained owing to the change of solvent is of the order of ∼1 kcal mol−1. Therefore, it is worthwhile mentioning that the solvent effect on the formation of the ambimodal transition structure, as well as the thermodynamic stability of the adduct formed, is only marginal.
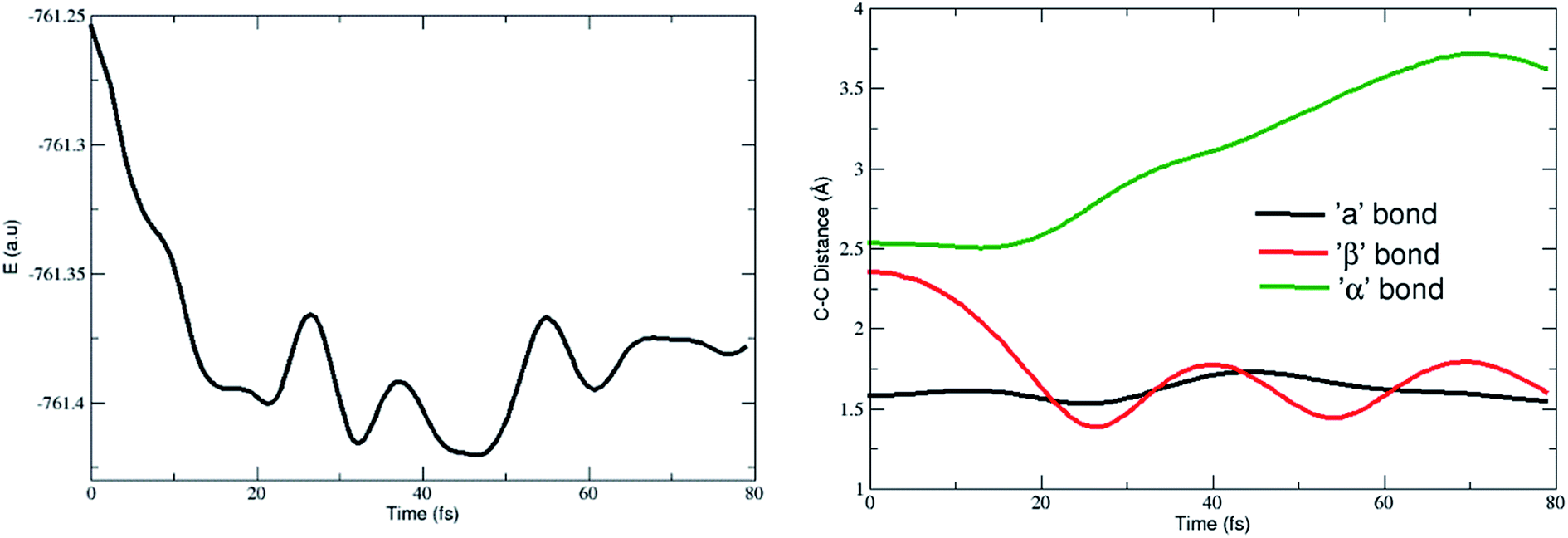 | ||
| Fig. 6 Potential energy and geometric variations with time. | ||
The calculated geometric parameters clearly emphasize that the large trajectories were found to be similar to the conventional [4 + 2]-cycloadduct. The feasibility of formation of the [5 + 4]-cycloadduct in a singlet ground state is significantly low when compared with the [4 + 2]-counterpart. Therefore, the singlet ground state dynamics indicate that more of the [4 + 2]-cycloadduct is formed from the ambimodal TS. The results are in agreement with the thermodynamic stability obtained for both of the adducts.
Conclusions
DFT based calculations clearly indicate that the cycloaddition reactions of various CNTs with 1,3-butadiene involve the formation of two thermodynamically stable products via an ambimodal transition state. Bifurcation of the PES leads to a thermodynamically more stable [5 + 4]-cyclic adduct compared to the conventional [4 + 2]-cyclic adduct. As the nanotube length increases, the energy barrier increases and the stability of the adducts decreases. The effect of solvents with different polarities on the reaction is only marginal.Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
This work is supported by SERB (ECR/2018/002346 and EEQ/2019/000656). The authors are grateful to Dr V. Subramanian, CSIR-CLRI, Prof. P. Fleurat-Lessard and CCUB at the Universite de Bourgogne for computational calculations. AAS thanks SERB for JRF.References
- D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Çelebi-Ölçüm and K. N. Houk, Angew. Chem., Int. Ed., 2008, 47, 7592–7601 CrossRef CAS PubMed.
- J. Rehbein and B. K. Carpenter, Phys. Chem. Chem. Phys., 2011, 13, 20906–20922 RSC.
- S. R. Hare and D. J. Tantillo, Pure Appl. Chem., 2017, 89, 679–698 CAS.
- S. Chen, P. Yu and K. N. Houk, J. Am. Chem. Soc., 2018, 140, 18124–18131 CrossRef CAS PubMed.
- L. Toma, S. Romano, P. Quadrelli and P. Caramella, Tetrahedron Lett., 2001, 42, 5077–5080 CrossRef CAS.
- P. Caramella, P. Quadrelli and L. Toma, J. Am. Chem. Soc., 2002, 124, 1130–1131 CrossRef CAS PubMed.
- S. Pratihar, X. Ma, Z. Homayoon, G. L. Barnes and W. L. Hase, J. Am. Chem. Soc., 2017, 139, 3570–3590 CrossRef CAS PubMed.
- B. K. Carpenter, Annu. Rev. Phys. Chem., 2005, 56, 57 CrossRef CAS PubMed.
- J. B. Thomas, J. R. Waas, M. Harmata and D. A. Singleton, J. Am. Chem. Soc., 2008, 130, 14544–14555 CrossRef CAS PubMed.
- N. Çelebi-Ölçüm, D. H. Ess, V. Aviyente and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 4528–4529 CrossRef PubMed.
- X. S. Bogle and D. A. Singleton, Org. Lett., 2012, 14, 2528–2531 CrossRef CAS PubMed.
- B. K. Carpenter, J. Am. Chem. Soc., 1995, 117, 6336–6344 CrossRef CAS.
- B. R. Ussing, C. Hang and D. A. Singleton, J. Am. Chem. Soc., 2006, 128, 7594–7607 CrossRef CAS PubMed.
- Z. Wang, J. S. Hirschi and D. A. Singleton, Angew. Chem., Int. Ed., 2009, 48, 9156 CrossRef CAS PubMed.
- P. Yu, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 13518–13523 CrossRef CAS PubMed.
- A. Patel, Z. Chen, Z. Yang, O. Gutiérrez, H. W. Liu, K. N. Houk and D. A. Singleton, J. Am. Chem. Soc., 2016, 138, 3631–3634 CrossRef CAS PubMed.
- P. Yu, T. Q. Chen, Z. Yang, C. Q. He, A. Patel, Y. H. Lam, C. Y. Liu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 8251–8258 CrossRef CAS PubMed.
- J. M. Burns, Org. Biomol. Chem., 2018, 16, 1828–1836 RSC.
- J. M. Burns and E. D. Boittier, J. Org. Chem., 2019, 84, 5997–6005 CrossRef CAS PubMed.
- N. Mandal and A. Datta, J. Phys. Chem. B, 2018, 122, 1239–1244 CrossRef CAS PubMed.
- N. Mandal and A. Datta, J. Org. Chem., 2018, 83, 11167–11177 CrossRef CAS PubMed.
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial College Press, London, 1998 Search PubMed.
- P. J. F. Harris, Carbon Nanotubes and Related Structures, Cambridge University Press, Cambridge, U. K., 1999 Search PubMed.
- P. Politzer, J. S. Murray, P. Lane and M. C. Concha, in Handbook of Semiconductor Nanostructures and Devices, ed. A. A. Balandin and K. L. King, American Scientific Publishers, Los Angeles, 2006, vol. 2, p. 215 Search PubMed.
- P. M. Ajayan, Chem. Rev., 1999, 99, 1787–1800 CrossRef CAS PubMed.
- J. Hu, T. W. Odom and C. M. Lieber, Acc. Chem. Res., 1999, 32, 435–445 CrossRef CAS.
- M. F. Yu, B. S. Files, S. Arepalli and R. S. Ruoff, Phys. Rev. Lett., 2000, 84, 5552–5555 CrossRef CAS PubMed.
- P. G. Collins, M. S. Arnold and P. Avouris, Science, 2001, 292, 706–709 CrossRef CAS PubMed.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS PubMed.
- Y. P. Sun, K. Fu, Y. Lin and W. Huang, Acc. Chem. Res., 2002, 35, 1096–1104 CrossRef CAS PubMed.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- P. Ravinder, R. Kumar Mahesh and V. Subramanian, J. Phys. Chem. A, 2012, 116, 5519–5528 CrossRef CAS PubMed.
- P. Ravinder and V. Subramanian, J. Phys. Chem. C, 2013, 117, 5095–5100 CrossRef CAS.
- E. T. Michelson, C. B. Huffman, A. G. Rinzler, R. E. Smalley, R. H. Hauge and J. L. Margrave, Chem. Phys. Lett., 1998, 296, 188–194 CrossRef.
- S. Niyogi, H. Hu, M. A. Hamon, P. Bhowmik, B. Zhao, S. M. Rozenzhak, J. Chen, M. E. Itkis, M. S. Meier and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 733–734 CrossRef CAS PubMed.
- B. Zhao, H. Hu, S. Niyogi, M. E. Itkis, M. A. Hamon, P. Bhowmik, M. S. Meier and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 11673–11677 CrossRef CAS PubMed.
- Y.-P. Sun, W. Huang, Y. Lin, K. Fu, A. Kitaygorodskiy, L. A. Riddle, Y. J. Yu and D. L. Carroll, Chem. Mater., 2001, 13, 2864–2869 CrossRef CAS.
- K. Fu, W. Huang, Y. Lin, L. A. Riddle, D. L. Carroll and Y.-P. Sun, Nano Lett., 2001, 1, 439–441 CrossRef CAS.
- Y.-. Ping, S. Kefu, F. Yi and L. J. Huang, Acc. Chem. Res., 2002, 35, 1096–1104 CrossRef PubMed.
- A. Christopher, J. Dyke and M. Tour, J. Phys. Chem. A, 2004, 108, 11151–11159 CrossRef.
- M. Q. L. Cuong, X. T. Cao, T. K. T. Rang, W. K. Lee and K. T. Lim, Appl. Surf. Sci., 2018, 450, 122–129 CrossRef.
- Y. Li, S. Osuna, M. G.-B. Xiaotian, Q. S. Liu, K. N. Houk and Y. Lan, Chem.–Eur. J., 2016, 22, 12819–12824 CrossRef CAS PubMed.
- D. Mata, M. Amaral, A. J. S. Fernandes, B. Colaço, A. Gama, M. C. Paiva, P. S. Gomes, R. F. Silva and M. H. Fernandes, Nanoscale, 2015, 7, 9238–9251 RSC.
- P. Ravinder and V. Subramanian, J. Phys. Chem. A, 2012, 116, 6870–6878 CrossRef CAS PubMed.
- P. Ravinder and V. Subramanian, Theor. Chem. Acc., 2012, 131, 1128 Search PubMed.
- J. L. Delgado, P. Cruz, F. Langa, A. Urbina, J. Casadoc and J. T. López Navarretec, Chem. Commun., 2004, 1734–1735 RSC.
- L. Zhang, J. Yang, C. L. Edwards, L. B. Alemany, V. N. Khabashesku and A. R. Barron, Chem. Commun., 2005, 3265–3267 RSC.
- C. Menard-Moyon, F. Dumas, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2006, 128, 14764–14765 CrossRef CAS PubMed.
- X. Lu, F. Tian, N. Wang and Q. Zhang, Org. Lett., 2002, 4, 4313–4315 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S. W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149–5160 CrossRef CAS.
- R. Jasinski, React. Kinet., Mech. Catal., 2016, 119, 49–57 CrossRef CAS.
- R. Jasinski, Monatsh. Chem., 2016, 147, 1207–1213 CrossRef CAS PubMed.
- R. Jasinski, J. Mol. Graphics Modell., 2017, 75, 55–61 CrossRef CAS PubMed.
- R. Jasinski, J. Fluorine Chem., 2018, 206, 1–7 CrossRef CAS.
- R. Jasinski, Tetrahedron Lett., 2015, 56, 532–535 CrossRef CAS.
- R. Jasinski, K. Wasik, M. Mikulska and A. Baranski, J. Phys. Org. Chem., 2009, 22, 717–725 CrossRef CAS.
- A. A. Fokin, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, J. Am. Chem. Soc., 2012, 134, 13641–13650 CrossRef CAS PubMed.
- P. Ravinder and V. Subramanian, J. Phys. Chem. C, 2012, 116, 16815–16822 CrossRef CAS.
- L. R. Domingo and J. A. Saez, Org. Biomol. Chem., 2009, 7, 3576–3583 RSC.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Periodic calculation details and results, the role of the basis set, biradical formation, IRC calculations, the PES bifurcation model, the optimized geometries of various TSs along with the important geometrical parameters, and all of the optimized coordinates of various pristine, TS, and functionalized nanotubes are given. See DOI: 10.1039/c9ra10252c |
| This journal is © The Royal Society of Chemistry 2020 |