
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ molten phase-assisted self-healing for maintaining fiber morphology during conversion from melamine diborate to boron nitride†
Chunzhi Wu a,
Bing Wang*a,
Nan Wub,
Cheng Hana,
Xiaoshan Zhanga and
Yingde Wang*a
a,
Bing Wang*a,
Nan Wub,
Cheng Hana,
Xiaoshan Zhanga and
Yingde Wang*a
aScience and Technology on Advanced Ceramic Fibers and Composites Laboratory, College of Aerospace Science and Engineering, National University of Defense Technology, Changsha, 410073, P. R. China. E-mail: bingwang@nudt.edu.cn; wangyingde@nudt.edu.cn
bDepartment of Materials Science and Engineering, College of Aerospace Science and Engineering, National University of Defense Technology, Changsha, 410073, P. R. China
First published on 17th March 2020
Abstract
C3N6H6·2H3BO3 (M·2B) is a highly promising precursor of boron nitride (BN) fibers due to its eco-friendly and low-cost fabrication. However, it is still unclear why the fibers can maintain their morphology in spite of drastic weight loss (nearly 80 wt%) during M·2B-to-BN pyrolysis. Herein, an interesting cracking and self-healing behavior of the heated M·2B fibers was observed at initial pyrolysis. In situ formed molten boron oxide (B2O3) was figured out to be the healing agent for the cracks and subsequently merged into the continuous matrix enclosing melamine/melem molecules, which subsequently acted as a nitrogen source. The B2O3 matrix helped to keep the fiber morphology undamaged under the second weight-loss stage in the pyrolysis process. This strategy of taking advantage of the in situ formed molten phase for healing cracks offers detailed guidance to prepare defect-free M·2B-derived BN fibers and would be significant in defect repair for other ceramics.
Introduction
Due to the excellent comprehensive performances of thermal stability, chemical corrosion tolerance, oxidation resistance, neutron absorption, high thermal conductivity, electrical insulation and low dielectric constant,1–4 hexagonal boron nitride (BN) holds great promise in various fields such as aerospace, high-temperature filtration, nuclear industry, and microelectronics. As typical BN materials, BN fibers similar to anisotropic BNNSs enjoy wide attention owing to their unique properties provided by their high-aspect-ratio morphology.5,6Up to now, numerous fabrication techniques have been developed for the high-efficiency production of BN fibers, which are mostly based on the spinning process that requires strict manipulations.7–11 In addition, the pyrolysis of a C3N6H6·2H3BO3 (M·2B) supra-molecule adduct is an alternative way and promising for large-scale production owing to the facile and eco-friendly fabrication of M·2B.12–14
Unfortunately, the ceramic yield of M·2B is much lower (∼20 wt%) than that of other reported polymeric precursors of BN,15–18 for example, 57 wt% yield of poly[2,4,6-tris(iso-propylamino)borazine].17 This implies that substantial release occurs during thermal conversion from M·2B to the final BN fibers. Moreover, M·2B is a supra-molecule constructed by low-strength hydrogen bonding19,20 that might fail to self-support at a high temperature, resulting in structural collapse. These two disadvantages may cause serious defects in the final BN fibers. The fact, astonishingly, is that the M·2B-derived BN fibers display intact morphology basically inherited from initial M·2B.13,21 It still remains unclear why the seemingly contradictory phenomenon occurs.
In this work, we observed unique self-healing behavior after morphological cracks were caused by huge weight loss. This self-healing behavior kept the fiber morphology intact, while its micro-structure was drastically broken and reconstructed during pyrolysis. The molten boron oxide (B2O3) generated in situ from the H3BO3 portion was proven to work for healing the cracks and repairing the fiber. Moreover, the boron oxide network could enclose melamine molecules and its condensate melem to prevent them from evaporation, which was crucial to keep the fibers intact as pyrolysis and nitridation proceeded in the subsequent process.
Experimental section
Synthesis of C3N6H6·2H3BO3
A mixture of C3N6H6 and H3BO3 (analytical reagents, Sinopharm Chemical Reagent Co., Ltd.) with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was added into de-ionized water and dissolved at 85 °C in a water bath with vigorous stirring; the concentration of C3N6H6·(H3BO3) was set as 0.08 mol L−1 (0.16 mol L−1) to ensure complete dissolution. Then, the hot aqueous solution was naturally cooled down to precipitate the C3N6H6·2H3BO3 (M·2B) flocculent. Subsequently, the white flocculent was frozen and dried using a vacuum freeze-dryer (LGJ-FD, Beijing Songyuan Huaxing Technology Develop Co., Ltd, China) to get well-dispersed M·2B fibers.
:2 was added into de-ionized water and dissolved at 85 °C in a water bath with vigorous stirring; the concentration of C3N6H6·(H3BO3) was set as 0.08 mol L−1 (0.16 mol L−1) to ensure complete dissolution. Then, the hot aqueous solution was naturally cooled down to precipitate the C3N6H6·2H3BO3 (M·2B) flocculent. Subsequently, the white flocculent was frozen and dried using a vacuum freeze-dryer (LGJ-FD, Beijing Songyuan Huaxing Technology Develop Co., Ltd, China) to get well-dispersed M·2B fibers.
Thermal treatment of C3N6H6·2H3BO3
The thermal behavior of M·2B was analyzed using thermogravimetry and differential scanning calorimetry (TG-DSC, NETZSCH STA449F5, Germany); the heating rate was set as 10 °C min−1. Based on the results, the M·2B fibers were separately heated to 125 °C, 150 °C, 200 °C, 400 °C, 550 °C, 650 °C, and 1000 °C at the same temperature ramping rate and immediately cooled down. The thermal treatments were conducted in a quartz tubular furnace under a protective N2 flow. The obtained samples were designated as M·2B125, M·2B150, M·2B200, M·2B400, M·2B550, M·2B650, and M·2B1000. For comparison, melamine and boric acid underwent the same TG-DSC testing and were also heated at 150 °C, 200 °C, and 400 °C. To explore the effect of the self-healing process on the morphology of the final BN fibers, the M·2B fibers were also heated under a stage-variable N2 or NH3 atmosphere. For example, the M·2B fiber was heated to 700 °C for 1 hour under NH3 and subsequently heated to 1000 °C under switched N2.Characterization and corrosion testing
The morphologies of the samples were observed by field-emission scanning electron microscopy (SEM, Hitachi S-4800, Japan), and the constituents were characterized through Fourier transform infrared spectroscopy (FT-IR, Frontier, PerkinElmer, USA), X-ray diffraction (XRD, Bruker AXS D8 Advance diffractometer, Germany), and 13C nuclear magnetic resonance (NMR) spectroscopy in a DMSO-d6 solvent (400 MHz Agilent 400/54/ASP, USA). The molten transition point of monophasic H3BO3 was measured using the melting point apparatus (WRS-2A, Shanghai INESA Physico-Optical Instrument Co., Ltd.).The corrosion testing of the M·2BT fiber (T = 150, 200, 400, and 550 °C) was conducted using ethanol (EA) and ethylene glycol (EG) as corrosive agents. Initially, two samples were separately immersed in EA and EG for 1 minute and then taken out for washing 3 times by de-ionized water via a vacuum filtration apparatus. After that, the treated samples were dried over-night in an oven at 80 °C for observing morphology. Furthermore, the M·2BT fibers (0.02 g) were dipped in 4 ml solvent (EA and EG) for 6 hours to estimate their solubility, which reflected the corrosion resistance of the fibers.
Results and discussion
Morphology transformation
Fig. 1a indicates the thermal behavior of M·2B using TG-DSC; the whole pyrolysis procedure can be divided into two obvious weight-loss stages. In the first stage, in the range of 125–200 °C, there is a sharp endothermic peak at 159.9 °C due to intense weight loss. During the second stage, the gravimetric curve slowly decreases from 500 to 1000 °C, and a broad weak endothermic peak is located at 535 °C. The insets show the morphology of the M·2B fiber and its thermal products after being heated separately at 150 °C, 200 °C, 400 °C, 650 °C, and 1000 °C; they are designated as M·2B150, M·2B200, M·2B400, M·2B650, and M·2B1000, respectively. It was obvious that a drastic variation in the fiber's morphology occurred at the first stage; the fiber cracked when heated at 150 °C and self-healed at 200 °C. After this, the fiber got smoother at 400 °C (inset (d) of Fig. 1a) and was intact in the whole second stage (from inset (d) to inset (f) of Fig. 1a). In the final stage, the M·2B1000 fiber displayed compact morphology. Thus, self-healing was proven to be the key to maintain the fiber's morphology in spite of the ultralow yield (∼20 wt%) of M·2B.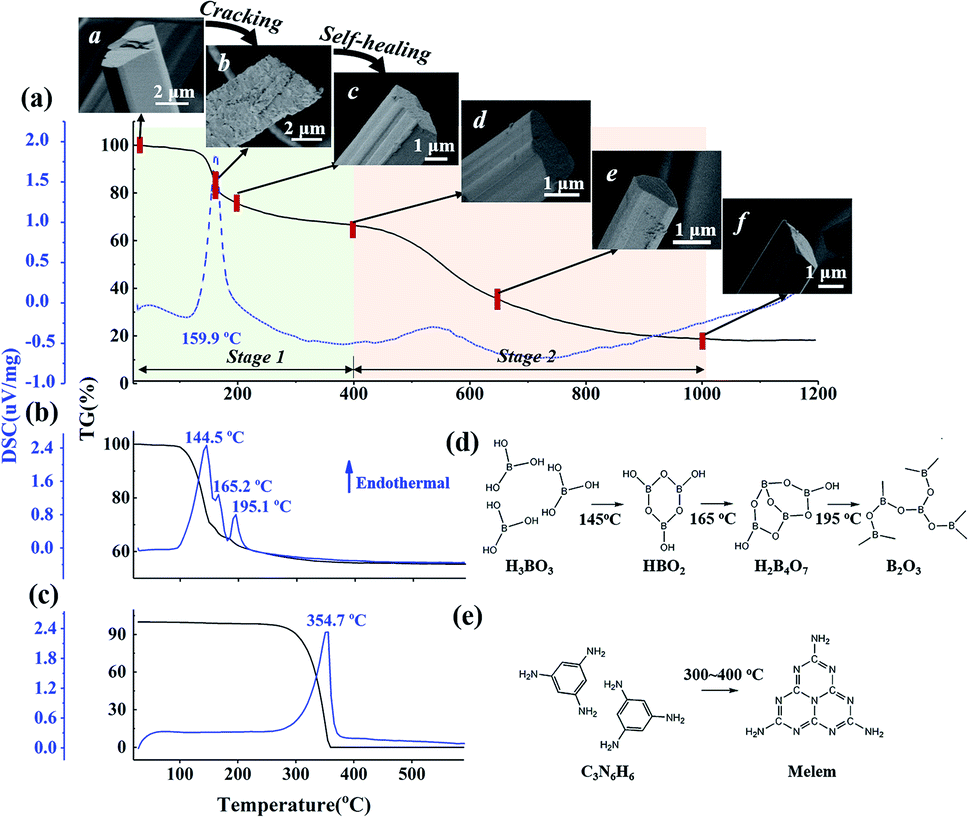 | ||
| Fig. 1 TG-DSC curves of (a) M·2B supramolecular fibers, (b) H3BO3 and (c) C3N6H6. In (a), the first weight-loss stage is in the range of 20–400 °C; the second weight-loss stage is in the range of 400–1000 °C, the inset SEM images demonstrate the morphological evolution of the C3N6H6·2H3BO3 fibers during thermal treatment: a M·2B, b M·2B150, c M·2B200, d M·2B400, e M·2B650, f M·2B1000. (d) Molecular transformation of H3BO3 and (e) C3N6H6 in the heating process. In (d), H3BO3 is converted to metaboric acid (HBO2) and further to tetraboric acid H2B4O7 and finally to a boron oxide structure. In (e), C3N6H6 condenses to melem at 300–400 °C.22 | ||
The fiber's cracking could undoubtedly be mainly attributed to the intense weight loss, revealing substantial decomposition at 150 °C, but it was not clear why the self-healing behavior of M·2B200 occurred. To explore the reason, the structural transformation during cracking-healing was analyzed.
Considering that M·2B is an adduct of H3BO3 and C3N6H6 connected via hydrogen bonding,20 we studied the thermal behavior of the two components separately. Fig. 1b and c display the TG-DSC curves of H3BO3 and C3N6H6, respectively. It can be found that the same-region weight loss of boric acid occurs (Fig. 1b) as the first-stage weight loss of M·2B. Meanwhile, C3N6H6 remained stable at 150–200 °C, as implied by the flat TG curve (Fig. 1c) and the XRD patterns of the heated products (Fig. S1b†).
The weight loss of boric acid could be divided into three stages according to three sharp endothermic peaks, separately corresponding to the different stages of the dehydration of H3BO3, as shown in Fig. 1d. These reaction equations were proved by the XRD patterns of the heating products (Fig. S1a†). Thus, the first-stage weight loss of M·2B and the corresponding morphology evolution were basically attributed to the dehydration of H3BO3 in M·2B. At the subsequent stage from 200 to 400 °C, there was no obvious weight loss against the sublimation and condensation of melamine (Fig. 1c and e), which would be discussed later. This stable stage ensured that the morphology of the M·2B400 fiber became more dense and smooth rather than broken.
Based on the analysis of the thermal transformation of the pure phases of C3N6H6 and H3BO3, the structural evolution of M·2B involving morphological cracking–healing was further studied using FT-IR spectroscopy and XRD. As shown in Fig. 2a, plenty of sharp peaks disappear after the conversion from M·2B to M·2B150, especially in the fingerprint region (marked by the light green rectangle areas) of the hydrogen-bonded structure. The FTIR peaks at 1186 cm−1 and 786 cm−1 that respectively represents bending and twisting vibrations of B–OH, became feeble as to M·2B150. This indicated the drastic removal of hydroxyl groups, which was in accordance with the dehydration of H3BO3. Meanwhile, the highly crystallized M·2B was converted into an unordered phase of M·2B150 characterized as double broad peaks (Fig. 2b).
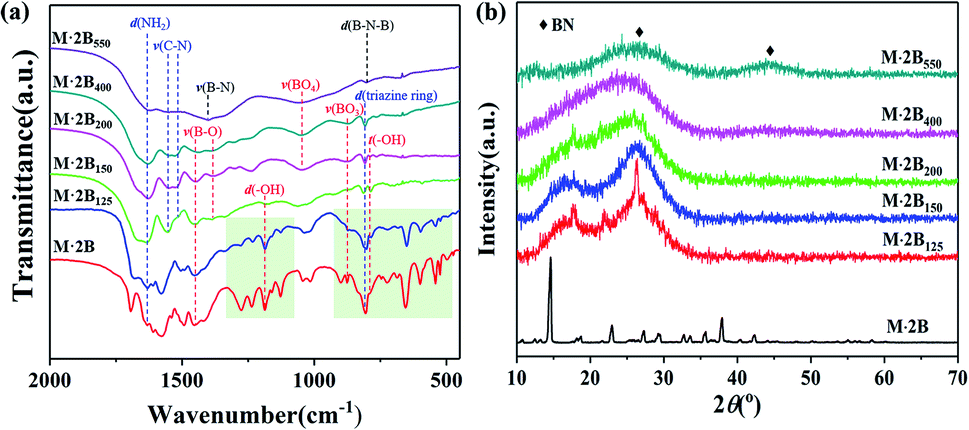 | ||
| Fig. 2 (a) FT-IR spectra and (b) XRD patterns of the M·2B supramolecule and its heated products M·2B125, M·2B150, M·2B200, M·2B400, M·2B550. The light green rectangle areas in (a) are the fingerprint region of the hydrogen-bonded adduct structure, and the absorption bands are labeled as follows: d(NH2): –NH2 bending, 1620 cm−1; ν(C–N): C–N stretching, 1552 cm−1 and 1525 cm−1; ν(B–O): 1450 cm−1 to B–O asymmetric stretching in BO3 and 1380 cm−1 to B–O stretching in B3O3 ring; ν(B–N): B–N stretching at 1400 cm−1; d(–OH): –OH bending, 1186 cm−1; ν(BO4): B–O stretching in the BO4 unit, 1050 cm−1; ν(BO3): B–O symmetric stretching in the BO3 unit, 873 cm−1; d(triazine ring): triazine ring bending at 808 cm−1; d(B–N–B): out-of-plane bending of B–N–B at 802 cm−1; t(–OH): out-of-plane –O–H twisting at 786 cm−1.23–25 | ||
M·2B125 displays an intermediate structure, revealing the transformation process from M·2B to unordered M·2B150, which was characterized as weaker peaks located mainly in the green fingerprint region in the IR spectrum and the co-existence of sharp and broad peaks in the XRD pattern. Therefore, the drastic removal of hydroxyl groups due to the dehydration of H3BO3 resulted in the breakage of the M·2B crystal and thus structural disordering. Morphology cracking could be attributed to the drastic structure transformation and also the huge release of the removed H2O. As for the micro-structure of the cracked fiber, it should be noted that the sharp XRD peaks of M·2B125 correspond well with the (−110), (012), (111) and (102) crystal planes of melamine (ICSD PDF#39-1950) but not M·2B (Fig. 3a). This means that there occurred the assembly of melamine molecules to form crystals, while disordering occurred in the initial M·2B crystal, allowing for the fact that C3N6H6 was molecularly isolated by H3BO3 in M·2B. Thus, the double broad peaks of M·2B150 were proved to result from the widening of the (−110) and (102) diffraction peaks and indicated the existence of melamine micro-crystals in the M·2B150 fiber.
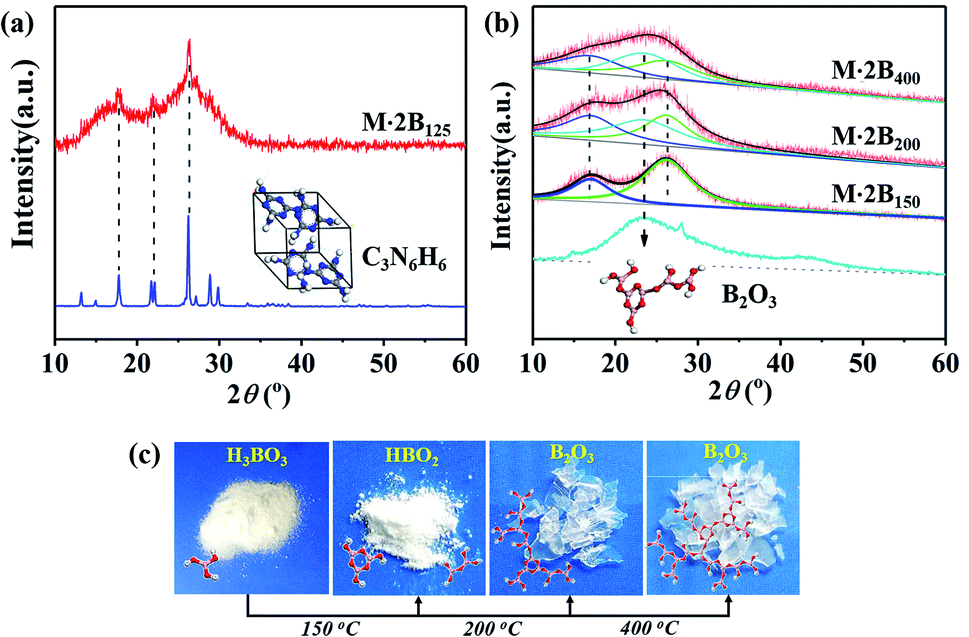 | ||
| Fig. 3 (a) Analysis of the XRD pattern of M·2B125 in comparison with that of melamine. (b) Peak fitting of the XRD patterns of M·2B150, M·2B200, and M·2B400 using B2O3 as a reference. (c) Photographs of the original H3BO3 and heat treatment products: HBO2 after 150 °C heating, B2O3 after 200–400 °C heating, the insets are the corresponding molecular structures. | ||
From the cracked M·2B150 to the healed M·2B200, there also occurred intense structural transformation. In addition to the IR peaks belonging to –NH2, C–N, BO3, triazine ring, and B3O3, the evolved peak of BO4 at 1050 cm−1 indicates the formation of a B2O3 network.23,26 Two undefined peaks at 1240 cm−1 and 1302 cm−1 existing for M·2B200–400 may be related to the C–O–B structure formed between melamine and boron oxide,27 which can keep melamine non-volatile in the range of 300–400 °C (Fig. 1a and c). The 13C NMR spectra of monophasic melamine, M·2B, M·2B150 and M·2B200 (Fig. S2†) also proved the consistent structural evolution, which especially revealed a slight difference between M·2B150 and M·2B200. The change may be caused by the newly formed carbonaceous structure.
Besides, the double broad XRD peaks of M·2B150 merged into an envelope peak of M·2B200–400. Interestingly, a broad peak belonging to g-B2O3 was located at 23° (Fig. S1a†) between the double broad peaks located at 16.3° and 26.2°. To study this in detail, we fitted the XRD peaks of M·2B150, M·2B200, and M·2B400 using that of g-B2O3 as a reference. As shown in Fig. 3b, the envelope peak for the healed M·2B200 fiber can be well fitted as the double peaks of M·2B150 and a broad peak of g-B2O3, and the envelope peak of smoothed M·2B400 becomes more integrated owing to the more intense peak of g-B2O3, while the double peaks belonging to melamine decrease. Therefore, it was confirmed that a g-B2O3 phase evolved in M·2B200–400, which may be related to the self-healing of the fiber. Fig. 3c displays the photos of H3BO3 and its heated products. Metaboric acid formed at 150 °C was a white powder just like boric acid, but g-B2O3 obtained at 200–400 °C turned into a glass-like solid, which revealed that it was melted. The result of the melting point measurement of H3BO3 also proved that a viscous phase evolved at 206.2–207.8 °C. Allowing for the same boron–oxygen network in M·2B200–400 as in pure B2O3, we have reason to believe that the viscous B2O3 formed in situ played a key role in healing the fiber's cracks. Besides, melamine and condensed melem displayed a powder state (Fig. S3†), indicating that the solid state remained during heating. This melamine/melem solid could prevent the excessive flow of viscous B2O3 and helped avoid the fusion of fibers. In the final stage, the self-healed M·2B200–400 fiber was composed of B2O3 glass and melamine/melem.
Moreover, to determine whether the B–O oligomer was uniformly distributed in M·2B150 and B2O3 acted as a matrix in M·2B200–400, the fibers underwent corrosion testing by EG (ethylene glycol) and EA (ethanol) considering that B2O3 could be rapidly dissolved in EG while remaining stable in EA for a long period. The testing went on merely for 1 minute for avoiding complete dissolution, and the corroded fibers were separately designated as M·2BT-EG and M·2BT-EA (T = 150, 200, 400 °C).
As shown in Fig. S5e and f,† all the M·2BT-EG fibers collapse into pieces due to the severe corrosion in EG. These results disclosed that the B–O oligomer or B2O3 was uniform throughout the whole fiber. After 6 hour corrosion, these fibers were completely dissolved, while the M·2B550 fiber remained undissolved due to the predominant BN phase (Fig. S4a†). In contrast, the samples in EA were partially dissolved or undissolved (Fig. S4b†). After being corroded for 1 minute in EA, M·2BT-EA maintained the fiber's profile (Fig. S5b and S5c†) despite slight etching on the surface of M·2B150-EA, which was composed of parallel short rods (Fig. S5a†) resulting from the decomposition of the intact M·2B fiber. The partial dissolution of the B–O oligomer at the interface between the rods disclosed this unique morphology.
The cracks in M·2B150 might be the gaps between the mismatched rod bundles. As for M·2B200-EA, the ultrafine fibers generated on the basis of the original M·2B200 fiber (Fig. S5b†) might be attributed to the assembly of C3N6H6 and H3BO3 (formed via the moisture absorption of superficial B2O3) in an ethanol solution. M·2B400-EA maintained the morphology for the initially smooth surface and the inactive melem. In summary, the different corrosion behaviors of the M·2BT fibers in EA compared with that in EG confirmed that the B–O oligomer or B2O3 was the dominant phase in affecting the fiber's morphology. In other words, B2O3 acted as the matrix in the M·2B200–400 fiber in which melamine/melem was enclosed.
On the basis of the above-mentioned discussion, the structural conversion corresponding to “cracking and self-healing” morphology transformation was discussed in detail. The schematic illustration in Fig. 4 shows structure evolution corresponding to the staged morphology. Initially, M·2B with intact fibrous morphology possessed a hydrogen-bonded lattice, in which C3N6H6 and H3BO3 were arranged uniformly and orderly. After heating at 150 °C, H3BO3 was dehydrated and formed a B–O oligomer, while the C3N6H6 molecules aggregated and assembled into tiny crystals. This resulted in the breakage of the ordered hydrogen-bonded structure and the intact fiber decomposed into short-rod bundles. However, the profile of the fiber was maintained integrated owing to the existence of the B–O oligomer at the interface of the rods. As a result, the cracked structure was derived from the mismatched gaps of the bundles. For M·2B200, the extended B2O3 network was formed as a result of further dehydration, while the C3N6H6 domain shrank and was isolated by the continuous B2O3 matrix. Thus, the cracked structure of M·2B150 was healed by the in situ viscous matrix. Our results provide a self-healing strategy for inorganic materials, which is different from that for organic polymers involving reversible bonding.28,29
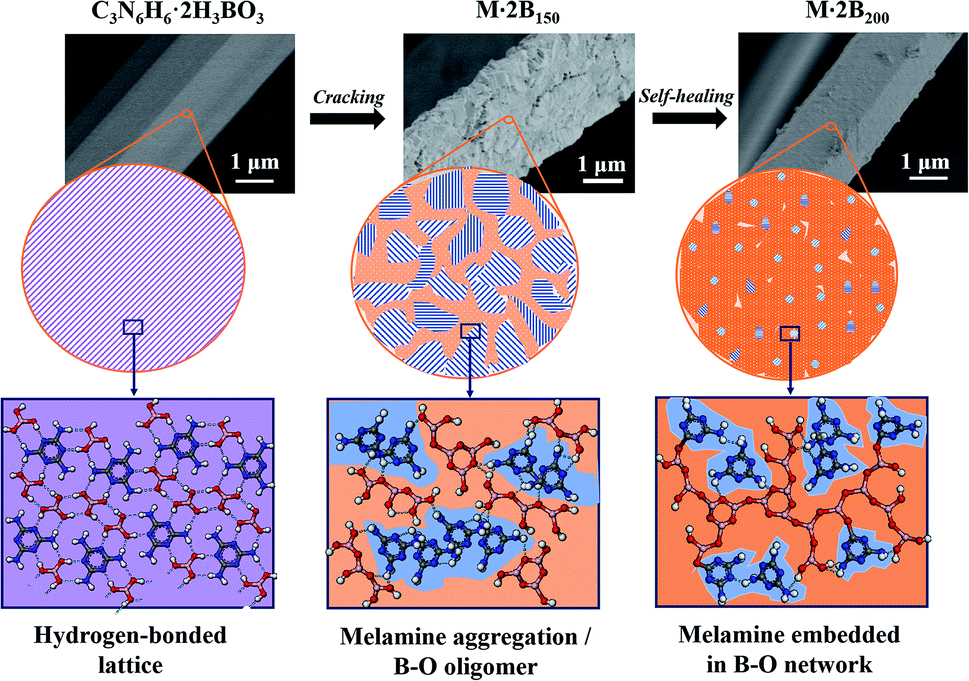 | ||
| Fig. 4 Schematic illustration of microstructural evolution corresponding to the morphological transformation: cracking and self-healing. | ||
To explore the effect of the “self-healing” process on the morphology of the final BN fiber, we treated the M·2B fibers under different pyrolysis conditions with a stage-variable atmosphere. As shown in Fig. 5, there are three sets of contrasts. In Fig. 5a and b, the BN fiber obtained after treatment under an NH3 atmosphere is defective, while that obtained under N2 is smooth in spite of undergoing the same heating process at 1000 °C for 1 hour. The difference in the fiber morphologies could be attributed to “self-healing” between 200 and 400 °C. As is well-known, the complex reaction between B2O3 and NH3 occurs at a temperature close to 200 °C, forming a non-melting (B2O3)n·NH3 compound,30 which prevents the cracked fiber from self-healing, while the molten B2O3 phase formed under a nitrogen atmosphere assists in healing the cracks.
 | ||
| Fig. 5 (a) BN fibers obtained after heating at 1000 °C for 1 hour under NH3 atmosphere; (b) heating at 1000 °C for 1 hour under N2 atmosphere; (c) heating at 700 °C for 1 hour under NH3 and subsequent heating at 1000 °C under N2; (d) heating at 700 °C for 1 hour in vacuum and subsequent heating at 1000 °C under N2; (e) heating at 400 °C for 1 hour under NH3 and heating to 1000 °C under N2; (f) heating at 400 °C for 1 hour under N2 and subsequently at 700 °C for 1 hour under NH3, and finally to 1000 °C under N2. | ||
The same comparisons were performed at temperatures lower than 1000 °C. As shown in Fig. 5c and d, the M·2B green fiber is separately heated under an NH3 atmosphere and vacuum at 700 °C. As shown in Fig. 5e and f, the M·2B green fiber is separately treated at 400 °C under the different combination of NH3/N2 atmosphere. Both these comparisons indicated the same results as those obtained from Fig. 5a and b; the fibers pyrolyzed under NH3 were defective, while the fibers pyrolyzed under N2 or vacuum were smooth. In summary, the significance of self-healing assisted by the molten B2O3 formed at 200 °C is crucial to obtain defect-free BN fibers. The result is of great significance to provide guidance to fabricate dense BN fibers when applying an active atmosphere such as NH3 for better nitridation and complete removal of carbon.
Conclusions
In summary, the dehydration of H3BO3 and evolution of B2O3 were figured out to be responsible for the unique “cracking and self-healing” morphological transformation. The resulting fiber was composed of a B2O3 matrix, in which melamine/melem was enclosed. This compound structure ensured stable morphology at the subsequent pyrolysis and nitridation stages and laid the foundation for the BN fiber. The self-healing mechanism assisted by the in situ molten phase can be adopted to guide the preparation of high-performance M·2B-derived BN fibers and can also be significant for other precursor-derived ceramic materials, especially those with low yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51773226, 61701514), the Natural Science Foundation of Hunan Province (2018JJ3603), Research Project of National University of Defense Technology (ZK17-XX-02) and Defense Industrial Technology Development Program (JCKY2016).Notes and references
- X. Duan, Z. Yang, L. Chen, Z. Tian, D. Cai, Y. Wang, D. Jia and Y. Zhou, J. Eur. Ceram. Soc., 2016, 36, 3725–3737 CrossRef CAS.
- J. Eichler and C. Lesniak, J. Eur. Ceram. Soc., 2008, 28, 1105–1109 CrossRef CAS.
- T. C. Doan, S. Majety, S. Grenadier, J. Li, J. Y. Lin and H. X. Jiang, Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip., 2014, 748, 84–90 CrossRef CAS.
- X. Yang, Y. Guo, Y. Han, Y. Li, T. Ma, M. Chen, J. Kong, J. Zhu and J. Gu, Compos. B Eng., 2019, 175, 1–8 CrossRef.
- Y. Han, X. Shi, X. Yang, Y. Guo, J. Zhang, J. Kong and J. Gu, Compos. Sci. Technol., 2020, 187, 107944 CrossRef.
- T. Ma, Y. Zhao, K. Ruan, X. Liu, J. Zhang, Y. Guo, X. Yang, J. Kong and J. Gu, ACS Appl. Mater. Interfaces, 2020, 12, 1677–1686 CrossRef CAS PubMed.
- Z. Liu, K. Zhao, J. Luo and Y. Tang, Scr. Mater., 2019, 170, 116–119 CrossRef CAS.
- V. Salles, S. Bernard, A. Brioude, D. Cornu and P. Miele, Nanoscale, 2010, 2, 215–217 RSC.
- Y. Qiu, J. Yu, J. Rafique, J. Yin, X. Bai and E. Wang, J. Phys. Chem. C, 2009, 113, 11228–11234 CrossRef CAS.
- Y. P. Lei, Y. De Wang and Y. C. Song, Ceram. Int., 2012, 38, 271–276 CrossRef CAS.
- C. Wang, C. Hong, L. Yan, X. Lin, M. Zhang, J. Wang and Q. Qu, Int. J. Appl. Ceram. Technol., 2018, 15, 660–667 CrossRef CAS.
- J. Lin, L. Xu, Y. Huang, J. Li, W. Wang, C. Feng, Z. Liu, X. Xu, J. Zou and C. Tang, RSC Adv., 2016, 6, 1253–1259 RSC.
- C. Wu, B. Wang and Y. Wang, Ceram. Int., 2018, 44, 5385–5391 CrossRef CAS.
- G. Li, M. Zhu, W. Gong, R. Du, A. Eychmüller, T. Li, W. Lv and X. Zhang, Adv. Funct. Mater., 2019, 29, 1–7 Search PubMed.
- P. Miele, S. Bernard, D. Cornu and B. Toury, Soft Mater., 2007, 4, 249–286 CrossRef.
- D. Cornu, S. Bernard, S. Duperrier, B. Toury and P. Miele, J. Eur. Ceram. Soc., 2005, 25, 111–121 CrossRef CAS.
- Y. P. Lei, Y. De Wang, Y. C. Song and C. Deng, Ceram. Int., 2011, 37, 3005–3009 CrossRef CAS.
- C. Deng, Y. Song, Y. Wang, Y. Li and Y. Lei, Chem. J. Chin. Univ., 2010, 31, 623–628 CAS.
- T. Kawasaki, Y. Kuroda and H. Nishikawa, J. Ceram. Soc. Jpn., 1996, 104, 935–938 CrossRef CAS.
- A. Roy, A. Choudhury and C. N. R. Rao, J. Mol. Struct., 2002, 613, 61–66 CrossRef CAS.
- J. Lin, X. Yuan, G. Li, Y. Huang, W. Wang, X. He, C. Yu, Y. Fang, Z. Liu and C. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 44732–44739 CrossRef CAS PubMed.
- H. B. Zheng, W. Chen, H. Gao, Y. Y. Wang, H. Y. Guo, S. Q. Guo, Z. L. Tang and J. Y. Zhang, J. Mater. Chem. C, 2017, 5, 10746–10753 RSC.
- X. Gouin, P. Grange, L. Bois, P. L'Haridon and Y. Laurent, J. Alloys Compd., 1995, 224, 22–28 CrossRef CAS.
- W. J. Jones and W. J. Orville-Thomas, Trans. Faraday Soc., 1959, 55, 203–210 RSC.
- C. Y. Panicker, H. T. Varghese, A. John, D. Philip and H. I. S. Nogueira, Spectrochim. Acta, Part A, 2002, 58, 1545–1551 CrossRef.
- X. Zhang, Z. Lu, H. Liu, J. Lin, X. Xu, F. Meng, J. Zhao and C. Tang, J. Mater. Chem. C, 2015, 3, 3311–3317 RSC.
- S. Wang, C. Bian, B. Jia, Y. Wang and X. Jing, Polym. Degrad. Stab., 2016, 130, 328–337 CrossRef CAS.
- X. Yang, Y. Guo, X. Luo, N. Zheng, T. Ma, J. Tan, C. Li, Q. Zhang and J. Gu, Compos. Sci. Technol., 2018, 164, 59–64 CrossRef CAS.
- C. Li, J. Tan, J. Gu, L. Qiao, B. Zhang and Q. Zhang, Compos. Sci. Technol., 2016, 123, 250–258 CrossRef CAS.
- J. Economy and R. Lin, in Boron and Refractory Borides, ed. V. I. Matkovich, Springer Berlin Heidelberg, Berlin, Heidelberg, 1977, pp. 552–564 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10292b |
| This journal is © The Royal Society of Chemistry 2020 |