
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of Cu+ impregnation of MOF-74 to improve CO/N2 and CO/CO2 separations†
Arwyn Evansa,
Matthew Cummingsb,
Donato Decarolis‡
a,
Diego Gianolioc,
Salman Shahid§
 a,
Gareth Law¶
b,
Martin Attfieldb,
David Lawd and
Camille Petit*a
a,
Gareth Law¶
b,
Martin Attfieldb,
David Lawd and
Camille Petit*a
aBarrer Centre, Department of Chemical Engineering, Imperial College London, UK. E-mail: camille.petit@imperial.ac.uk
bSchool of Chemistry, The University of Manchester, UK
cDiamond Light Source, Harwell Campus, Didcot, UK
dBP Chemicals Ltd Petrochemicals Technology, Saltend, Hull, UK
First published on 31st January 2020
Abstract
Carbon monoxide (CO) purification from syngas impurities is a highly energy and cost intensive process. Adsorption separation using metal–organic frameworks (MOFs) is being explored as an alternative technology for CO/nitrogen (N2) and CO/carbon dioxide (CO2) separation. Currently, MOFs' uptake and selectivity levels do not justify displacement of the current commercially available technologies. Herein, we have impregnated a leading MOF candidate for CO purification, i.e. M-MOF-74 (M = Co or Ni), with Cu+ sites. Cu+ allows strong π-complexation from the 3d electrons with CO, potentially enhancing the separation performance. We have optimised the Cu loading procedure and confirmed the presence of the Cu+ sites using X-ray absorption fine structure analysis (XAFS). In situ XAFS and diffuse reflectance infrared Fourier Transform spectroscopy analyses have demonstrated Cu+–CO binding. The dynamic breakthrough measurements showed an improvement in CO/N2 and CO/CO2 separations upon Cu impregnation. This is because Cu sites do not block the MOF metal sites but rather increase the number of sites available for interactions with CO, and decrease the surface area/porosity available for adsorption of the lighter component.
1. Introduction
The production of carbon monoxide (CO) as an industrial reagent requires downstream separation processes to achieve the high purity level required for CO to be used as a chemical feedstock (i.e. >99 mol%).1,2 During the CO purification process, by-product impurities such as CH4, CO2 and N2 must be removed. While current CO capture and gas separation technologies exist for purification,2,3 numerous energy-intensive steps are required which contribute to large capital and operating costs.4 The design of a single adsorption process to purify CO from all impurities would bring energy and cost savings.Activated carbons (AC), zeolites and metal–organic frameworks (MOFs) have been identified for CO purification.5 Yet, their adsorption performance, especially ACs' and zeolites', fall below the CO capacity and selectivity levels required to justify the displacement of the current benchmark technology, i.e. cryogenic distillation.2 MOFs have shown the most promising performance to date,6 recording the highest CO adsorption uptakes and theoretical CO/X selectivities (where X = H2, N2 and CH4).5,7
MOFs are formed by organic linkers coordinating to metal nodes (or polynuclear clusters).8 Their metal sites can provide coordinatively unsaturated metal sites, also known as open-metal sites, which have an affinity for CO.5 Hence, the best performing MOFs are highly dependent on the MOFs' metal sites as they determine the metal–CO bond strength through both σ-bond formation and π-backdonation.9 A comprehensive study by Bloch et al. reported high CO capacities and CO/H2 and CO/N2 selectivities for MOF-74 (also known as CPO-27 or M2(DOBDC), DOBDC = 2,5-dioxido-1,4-benzenedicarboxylate).7 The study highlighted trends between the Irving–Williams series and the M2+ centres' affinity to CO, demonstrating the importance of the metal ion's electronic configuration upon CO separation.
To further improve MOFs' CO uptake, one can use metal impregnation, a method tested on ACs and zeolites.9–18 This approach increases selectivity by decreasing the uptake of the undesired light product (e.g. N2) whose affinity with the impregnated metal is low. The introduction of Cu+ metal sites is the most common example used for ACs and zeolites. The electronic configuration of Cu+ allows strong π-complexation from the 3d electrons.9 Several groups have produced Cu+-impregnated zeolites and activated carbons either through the use of a Cu+-containing salt or that of a Cu2+-containing salt with subsequent reduction.9–18 A similar route has also been conducted on Na–zeolite Y, initially through metal cation exchange with Cu2+ ions.19,20 These studies demonstrate that it is favourable to initially perform Cu2+ impregnation or ion-exchange, before reducing to Cu+. The Cu+ ions are prone to oxidation and reduction and therefore cannot be employed for direct ion-exchange or impregnation. While Cu impregnated materials typically perform better for CO separation than the unimpregnated materials, their CO capacity and theoretical selectivities still fall short of the best performing MOFs.5,7 Impregnation has also been performed on MOFs to improve their CO separation performance. Cu+ metal sites have been incorporated on Fe-MIL-100 for CO/N2 via impregnation with Cu2+-based salts followed by reduction to Cu+.21 The same method has also been employed on Fe-MIL-100 for CO/CO2 separation,22–24 and on MIL-101 for CO/N2 separation.25 The CuAlCl4 complex used in the COSORB process3 has been loaded onto Cr-MIL-101 to improve CO/N2 separation and Cu+ stability. HKUST-1 has been used as a support for CuCl to improve CO/N2 and CO/H2 selectivity.26 A table comparing the performance of all these Cu impregnated MOFs is provided in the ESI (Table S1†).
Despite improving the CO purification ability, the CO capacity of Cu impregnated MOFs remains lower than those of most M-MOF-74 structures.7 Interestingly, Cu impregnation of MOF-74 has never been reported. Provided the new Cu sites do not block the MOF metal sites, this approach would increase the number of sites available for interactions with CO, while decreasing the surface area/pore size available for adsorption of the lighter component, i.e. N2. The combination of such effects should enhance CO uptake while increasing selectivity. Cu impregnation, particularly Cu+ impregnation, is notably challenging due to the unstable nature of the metal species. Monitoring the state of the Cu sites becomes important to understand the adsorption mechanism. To date, there has been no in situ analysis of the Cu2+ to Cu+ reduction procedure, and there has been no confirmation of Cu+–CO binding. X-ray photoelectron spectroscopy has been the sole characterisation technique used to confirm the presence of Cu+ by the absence of Cu2+ satellite peaks.16,21 Yet, this approach can only evaluate the surface, and not the bulk, chemistry of the Cu-impregnated MOFs. Furthermore, the absence of Cu2+ satellite peaks may be caused by Cu2+ to Cu0 over reduction rather than the formation of Cu+.
The present study aims to improve on the performance of MOF-74 for CO purification while bringing fundamental insight into Cu+ impregnation and Cu+–CO interactions in MOFs. To achieve this, we have performed a systematic Cu impregnation study on Ni-MOF-74 and Co-MOF-74.5,7 We have: (i) synthesized Cu+-impregnated MOF-74 structures to simultaneously use the metal–CO binding strength of the framework M2+ and introduce additional Cu+ ions, (ii) optimised the Cu impregnation and monitored in situ the Cu2+ to Cu+ reduction process using X-ray absorption near edge structure (XANES) analysis, (iii) verified Cu+–CO and M2+–CO binding using in situ DRIFTS analysis and (iv) tested Cu@Ni-MOF-74 for dynamic CO/N2 and CO/CO2 separation.
2. Experimental section
2.1 Materials synthesis
For 200 mg of M-MOF-74, a total of 1.6 mL of Cu solution was used for this step and was kept constant for each synthesis. Aqueous solutions of the Cu salts were prepared by dissolving the salts in MeOH (for CuCl2) or MeCN (for CuCl and Cu(HCO2)2) through sonication. The Cu concentration depended on the Cu loading desired using 1.6 mL Cu solution. For Cu impregnation procedures with two salts (e.g. CuCl2 and Cu(HCO2)2), an equimolar solution was prepared.
The Cu solution and M-MOF-74 were sonicated in a 20 mL scintillation vial for 1 h, with the vial shaken by hand every 15 min to mix the suspension further. After sonication and allowing the suspension to settle, the excess solution was decanted and the remaining slurry was dried with flowing N2 overnight. The solvent removal for MOF activation and reduction of the Cu2+ to Cu+ sites are referred to as separate procedures throughout (activation and reduction), with the conditions varying depending on the procedure desired. The Cu impregnated M-MOF-74 structures are labelled X-Cu@M-MOF-74, where M = Ni, Co, and X represents the ratio of Cu over Ni in the samples, rounded to the nearest integer.
2.2 Materials characterisation
2.3 In situ characterisation of Cu impregnated M-MOF-74
2.4 Dynamic adsorption measurements
The Cu impregnation and reduction procedure was optimised using flux response technology (FRT). This allowed us to calculate the dynamic CO adsorption capacity after in situ Cu reduction and evaluate the Cu@M-MOF-74 CO purification performance. We conducted binary CO/N2 and CO/CO2 adsorption measurements using a breakthrough apparatus to test the best performing Cu@Ni-MOF-74 sample.Samples (∼80 mg of dry sample) were loaded in a 4 mm ID quartz tube and packed using quartz wool. The column was pressurised and sample activation and Cu reduction was performed depending on the requirements, outlined below. After activation and reduction, the He and CO line was switched to N2 to purge and remove He and CO in preparation for analysis. For analysis, the CO flow-rate was set to 5 mL min−1 (N2 flow-rate at 10 mL min−1) and the CO perturbation gas was switched using the 3-port valve. An adsorption peak was recorded along the new baseline due to the pressure response upon adsorption. After equilibration, the system side flow-rate was measured using a bubble flow-meter, and the CO perturbation gas was then switched off to induce desorption.
An example of an FRT profile is shown in Fig. S2.† The integrated area under adsorption peak (with aid of the curve fitting tool on Origin) was subtracted from an area of the graph with a known quantity of flowing adsorbate molecules after perturbation. This provided the number of adsorbed molecules from which the dynamic CO adsorption capacity could be determined. The standard deviation for measurements on the instrument is ±5%, and error also arises from experimental variance of the MOF synthesis and Cu impregnation and reduction procedure.
2.4.1.1 Cu impregnation loading and Cu salt precursors analyses. For M-MOF-74 and Cu@M-MOF-74 (M = Ni or Co) activation and Cu2+ to Cu+ reduction, the samples were heated for 6 h at 250 °C with 10% CO (in He) flowing at 1 bar on the FRT instrument.
2.4.1.2 Cu reduction analysis. For Ni-MOF-74 activation and Cu2+ to Cu+ reduction, Ni-MOF-74 and Cu@Ni-MOF-74 samples were subjected to three different activation and Cu reduction procedures: (i) heating at 250 °C with He flowing for 16 h, (ii) heating at 250 °C with He flowing for 16 h, followed by 10% CO (in He) at 250 °C for 6 h and (iii) heating at 250 °C with 10% CO (in He) for x h (where x = 2, 6 and 16).
Samples (∼200 mg) were loaded in a 4 mm ID quartz tube and packed using quartz wool. The packed tube was loaded and the system pressurized to 1 bar gauge pressure. Sample activation and Cu reduction was performed by flowing 10% CO in He with a total flow-rate of 20 mL min−1 for 6 h at 250 °C using heating tape (Omega). The temperature was controlled using a temperature probe (RS components) and a Proportional–Integral–Derivative (PID) controller.
For analysis, the volumetric flow-rates (total flow-rate of 20 mL min−1) were set for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v CO:X (X = N2, CO2) mixture composition and a back-pressure regulator was used to control the inlet feed pressure. The flow-rates were controlled by Bronkhorst mass-flow controllers and outlet flow-rate controlled by a Bronkhorst mass-flow meter. The four-port switch valve was switched to send the binary mixture to the sample column before reaching the MS detector at the outlet.
:50 v/v CO:X (X = N2, CO2) mixture composition and a back-pressure regulator was used to control the inlet feed pressure. The flow-rates were controlled by Bronkhorst mass-flow controllers and outlet flow-rate controlled by a Bronkhorst mass-flow meter. The four-port switch valve was switched to send the binary mixture to the sample column before reaching the MS detector at the outlet.
Analysis of the outlet feed concentration was performed by an ESS GeneSys Quadrupole Mass Spectrometer (MS) 200, with data acquisition using a Quadstar 32 bit software. Digital pressure gauges at the inlet and outlet were used to monitor the pressure drop in the column, which did not exceed 0.1 bar. Hexagonal boron nitride (Saint Gobain, Tres BN) was used as a non-adsorbing material to measure the dead volume of the instrument using He. The breakthrough curves have been normalised to eliminate the inlet dead volume time, so the plotted curves are a direct reflection of dynamic adsorption of the given sample. The relative concentration, C/C0, on the y-axis is standardised to the mole fractions of the inlet feed. Here, the breakthrough capacity (adsorption uptake when C/C0 = 0.01) is presented as opposed to the saturation capacity (adsorption uptake when C = C0). An industrial adsorption cycle would not enable the adsorbent bed to reach equilibrium with the inlet feed, therefore breakthrough capacity is a more useful metric. The standard deviation for measurements on the instrument is ±5%, and error also arises from experimental variance of the MOF synthesis and Cu impregnation and reduction procedure.
3. Results and discussion
We impregnated Ni-MOF-74 and Co-MOF-74 with Cu and carried out a systematic Cu impregnation study, with the aim to produce an improved CO purification adsorbent. We present our findings below.3.1 Characterisation of Cu impregnated samples
Initially, we synthesized Cu@Ni-MOF-74 samples using CuCl2 and Cu(HCO2)2 as Cu precursors and used the results to optimise the Cu loading. We selected CuCl2, a commonly used precursor,17,18,21–23 because of its ability to be reduced to CuCl leading to strong Cu+–CO π-complexation. Cu(HCO2)2 was chosen due to its ability to simultaneously provide a reducing agent and a Cu+ source.21,22 Indeed, upon heating, the decomposition of the formate ion produces H2 for Cu2+ to Cu+ reduction.32We initially impregnated Ni-MOF-74 with equimolar CuCl2:Cu(HCO2)2 ratios while varying the Cu loading. We used ICP-MS and XPS analyses to quantify the loading (Table S2 in the ESI, Section 3.1†). ICP-MS probed the bulk Cu loading, while XPS measured the surface loading and as a result, the values from XPS analyses surpass those from ICP-MS. From the ICP-MS analysis, the estimated Cu loadings are 1.77, 3.50 and 7.07% wt and the samples are subsequently referred to as 2-, 4- and 7-Cu@Ni-MOF-74 throughout. These percentages represent the ratios of Cu over Ni in the samples.
Using XRD, we confirmed that the impregnation did not significantly alter the MOF structure and that there was no major Cu salt particle aggregation (Fig. 1(a)). Porosity measurements allowed us to study the effect of impregnation on pore blocking with increasing Cu amount (Fig. 1(b) and Table S2†). Increasing Cu loading reduced the BET surface area and pore volume as anticipated, due to the Cu sites inducing pore blocking and reducing accessibility to pores. As the Cu loading approximately doubled from 4- to 7-Cu@Ni-MOF-74, there was no additional reduction in porosity. This observation suggests that Cu accumulated on the external surface beyond 3.50% wt Cu loading.
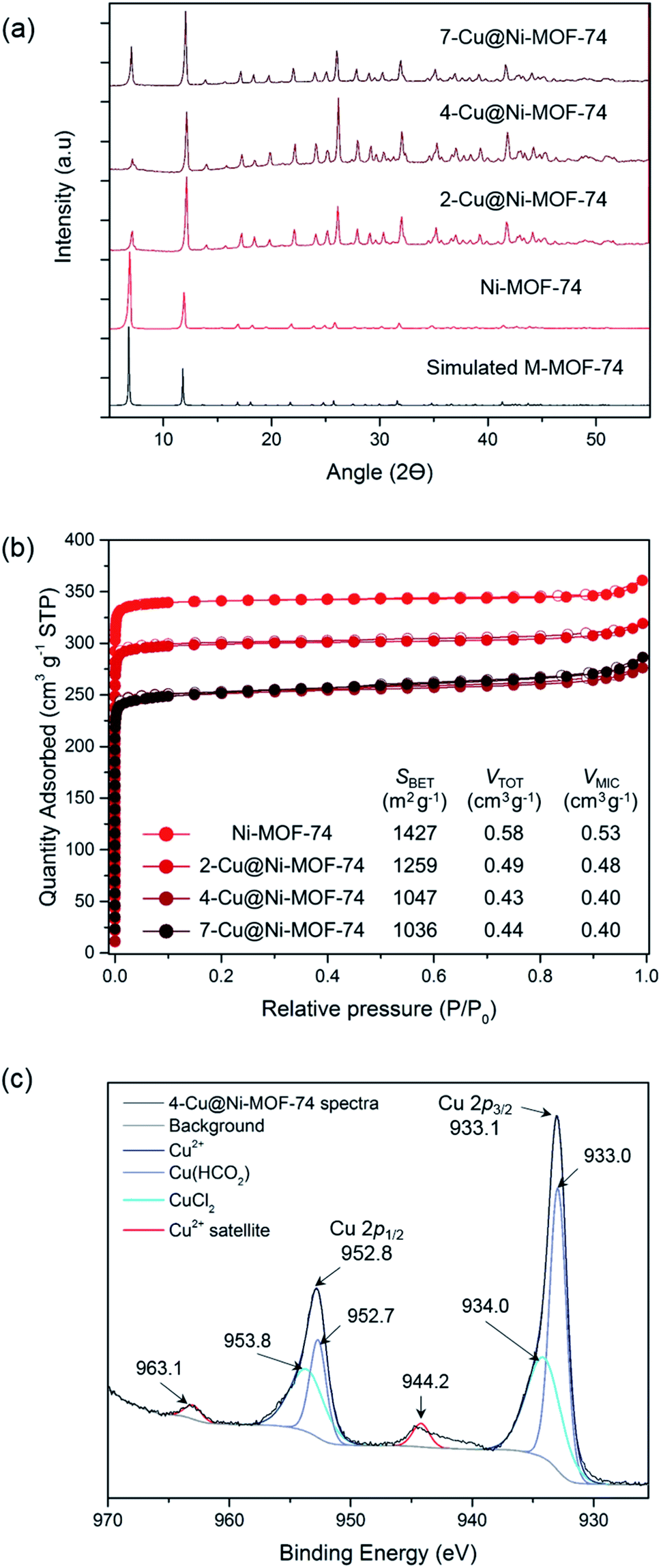 | ||
| Fig. 1 Characterisation of the Cu-impregnated MOFs: (a) X-ray diffraction patterns of simulated MOF-74, Ni-MOF-74, 2-Cu@Ni-MOF-74, 4-Cu@Ni-MOF-74 and 7-Cu@Ni-MOF-74. (b) N2 sorption isotherms at −196 °C of Ni-MOF-74, 2-Cu@Ni-MOF-74, 4-Cu@Ni-MOF-74 and 7-Cu@Ni-MOF-74. The textural parameters derived from the isotherms are shown on the table. (c) Cu 2p spectra of 4-Cu@Ni-MOF-74. The deconvoluted Cu peaks and energy levels are shown alongside the characteristic Cu2+ satellites. | ||
VT-PXRD and TG analyses enabled us to establish an activation and reduction temperature that would not damage the Ni-MOF-74 and Co-MOF-74 structures (see ESI, Fig. S4–S6†). The thermogravimetric curves indicated solvent release <350 °C, thermal decomposition at 350–400 °C and total decomposition at ∼550 °C. The VT-PXRD spectra (Fig. S5 and S6†) pointed to MOF degradation at 400–450 °C. Therefore, we chose 250 °C for this study to maintain structure stability during the thermally-activated Cu reduction.
We confirmed the oxidation state of surface Cu post activation and reduction using XPS. The full XPS survey of 4-Cu@Ni-MOF-74 is shown in Fig. S7.† Fig. 1(c) shows the XPS 2p spectra of 4-Cu@Ni-MOF-74. The reference 2p spectra of the Cu salts are presented in Fig. S8.† The Cu 2p subshell is split into doublets: 2p3/2 and 2p1/2. The presence of Cu(CHO2)2 and CuCl2 is confirmed by comparison against the spectra of the pure Cu salts. These peaks are assigned on the basis that the maximum Cu 2p3/2 and Cu 2p1/2 peaks of the CuCl2 Cu 2p XPS spectra (Fig. S8†) are more energetic than the Cu(CHO2)2 Cu 2p peaks. The negative charge of the oxygen formate ions is dispersed across two atoms and therefore the CuCl2 chloride ions result in a slightly more energetic Cu 2p peaks. The small peaks at 944.2 and 963.1 eV in Fig. 1(c) are due to the characteristic Cu2+ satellite peaks.16 Their weak intensities result from Cu2+ reduction to Cu+ (or possibly Cu0) facilitated by the heat treatment under vacuum during sample preparation. Overall, the XPS results indicate the possible reduction of Cu2+ to Cu+ but they are only representative of the surface chemistry of the materials. The bulk chemistry of the materials is investigated later using XANES.
3.2 Effect of Cu impregnation on CO adsorption
Having produced Cu@Ni-MOF-74 and Cu@Co-MOF-74 samples of varying Cu loadings, we then evaluated their dynamic CO capacity using FRT. This technique allowed us to activate and reduce the samples in situ under flowing He or CO atmosphere prior to testing.:50 v/v CO:N2) are presented in Fig. 2(a), 25 °C and 1 bar. The uptakes are presented per gram of Ni-MOF-74 and Cu@Ni-MOF-74. Overall, Cu impregnation brings limited gains and appears to be detrimental over 3.50% wt Cu loading, as the difference in values from unimpregnated Ni-MOF-74 falls within the ±5% standard deviation. The increased quantity of Cu induces pore blockage and reduces its capacity. Furthermore, some Cu2+ may remain and limit access to the more strongly binding Ni2+ open-metal sites.
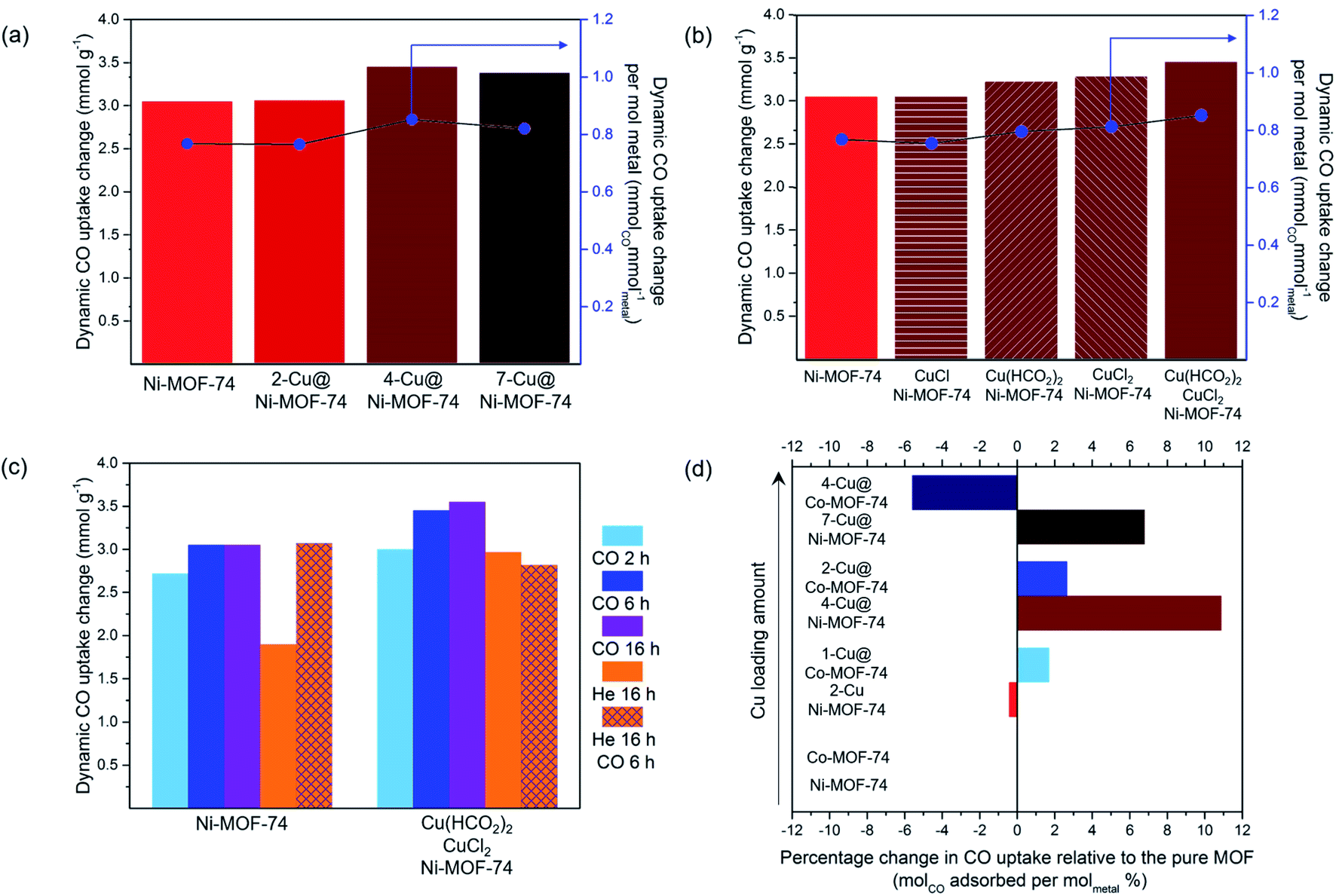 | ||
| Fig. 2 Dynamic CO capacities (50 : 50 v/v CO : N2) measured at 25 °C and 1 bar on the FRT set-up: (a) effect of Cu loading; (b) effect of the type of Cu salts; (c) effect of activation and reduction procedures; (d) case of Cu@Co-MOF-74. | ||
To accurately study the influence of the Cu loading, one must consider the increased metal density of the structure. Therefore, we normalised the CO uptake per mol of metal in the adsorbent, taking into account both the Cu loading and the Ni content (Table S2†). The results normalised to Cu only are also presented in Table S2.†
Thermal activation and Cu reduction were performed for 6 h at 250 °C with flowing 10% CO (in He) unless stated otherwise. The dynamic CO capacity per mol of metal was calculated using the measured values on the FRT and the Cu loading obtained from the ICP-MS analysis. (a) Dynamic CO capacities at varied Cu loadings using equimolar CuCl2:Cu(HCO2)2 salt precursors. (b) Dynamic CO capacities using different Cu salts for impregnation. (c) Dynamic CO capacities using CuCl2:Cu(HCO2)2 salts with thermal activation and Cu reduction performed using He or CO atmosphere. (d) Dynamic CO capacities of Cu@Ni-MOF-74 and Cu@Co-MOF-74 at varied Cu loadings using equimolar CuCl2:Cu(HCO2)2 salt precursors.
These results demonstrate that increasing the Cu loading above 3.50% wt is detrimental (Fig. 2(a) and Table S2†). The normalised values show 4-Cu@Ni-MOF-74 (3.50% wt) achieves the highest CO uptake per gram of metal and per mol of Cu. Increasing Cu loading above 3.50% wt reduces the normalised CO uptake. For this reason, we identified 4-Cu@Ni-MOF-74 as the optimised Cu loading for Ni-MOF-74.
Activating Ni-MOF-74 under CO atmosphere produced a higher dynamic CO capacity than activating under He. We hypothesize that at elevated temperatures, CO reacts with residual MeOH bound to the MOF, thereby increasing the number of open-metal sites.33 Activation via a combination of heating and CO flow led to the highest CO uptake for the equimolar CuCl2:Cu(HCO2)2 impregnated sample. Overall, the results indicate that both He and CO behave as reducing agents to different degrees, and dictate the duration of heat treatment required. Following this study, we chose to conduct Cu2+ to Cu+ reduction via heating at 250 °C with 10% CO (in He) for 6 h as the activation and Cu reduction procedure for the subsequent experiments. These conditions compromise between minimising the time of CO used at elevated temperature while maximising the amount of Cu+ generated.
:Cu(HCO2)2 mixture for impregnation. The ICP-MS analyses indicated Cu loading of 1.28, 2.42 and 4.22% wt The corresponding samples are referred to as 1-, 2- and 4-Cu@Co-MOF-74. The CO uptakes were normalised to account for the variance in Cu loading. 1-Cu@Co-MOF-74 and 2-Cu@Co-MOF-74 adsorbed similar amounts of CO, with marginal improvement compared to Co-MOF-74 (Fig. 2(d)). Cu loading above 2.42% wt was detrimental due to pore blocking and reduced accessibility to framework open-metal sites for strong Co2+–CO binding. We attribute the differences in uptake limit between Co- and Ni-MOF-74 to differences in particle size and defect sites, as shown by the unexpected measured mesopore volume of Ni-MOF-74 (Table S2†). 4-Cu@Ni-MOF-74 and 2-Cu@Co-MOF-74 exhibit similar degrees of reduction in porosity (Table S2†) compared to the unimpregnated samples, excluding any porosity effect upon these discrepancies.3.3 In situ Cu@M-MOF-74 XAFS characterisation
XAFS experiments were conducted on the best performing Cu impregnated M-MOF-74 samples, i.e. 4-Cu@Ni-MOF-74 and 2-Cu@Co-MOF-74. The objectives were: (i) to investigate further the Cu reduction step since XPS only provided information on surface Cu sites, (ii) to assess the impact of the Cu impregnation on the states of the Ni and Co sites and (iii) to monitor changes in the Cu, Ni and Co sites upon CO adsorption.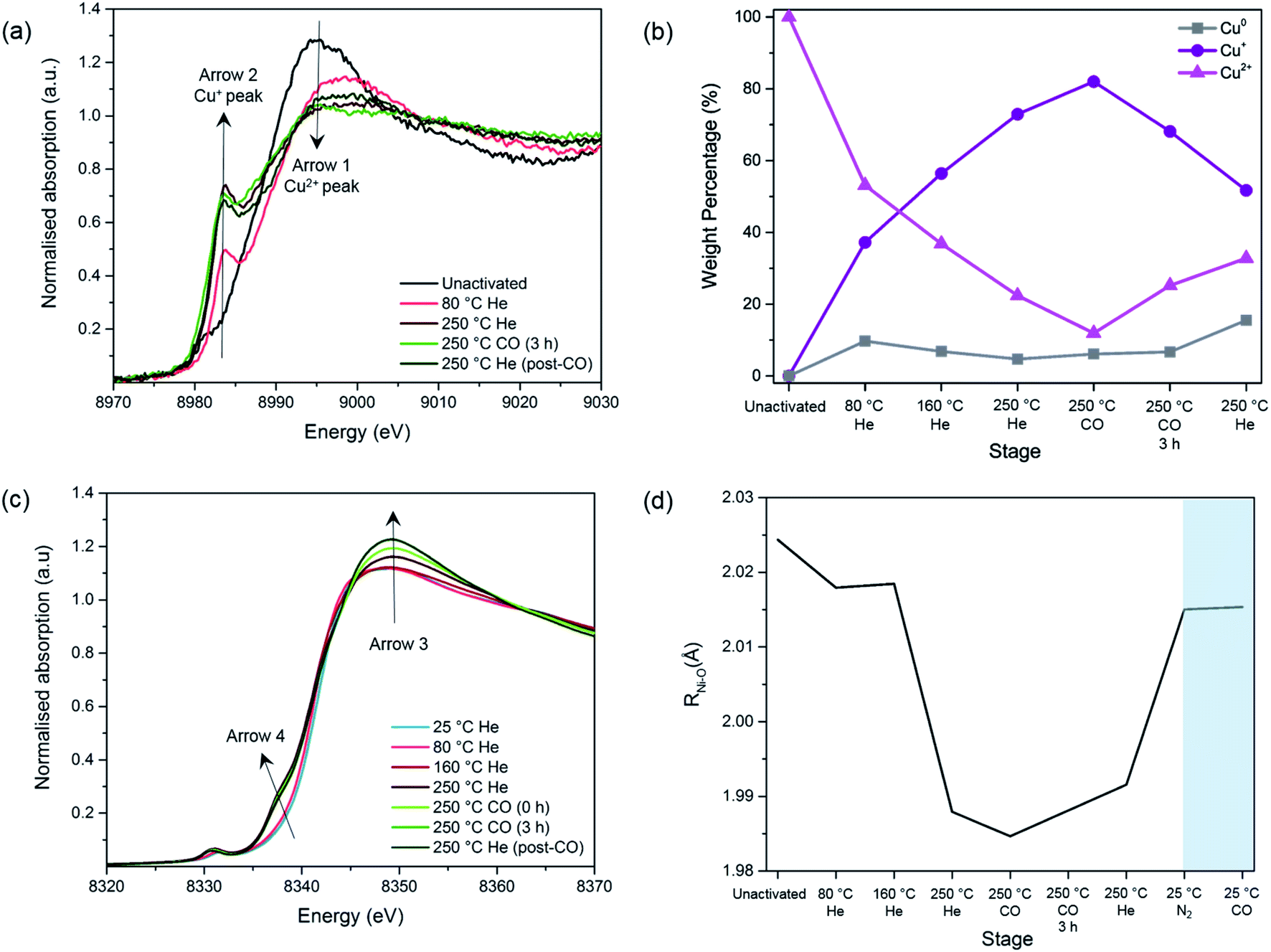 | ||
| Fig. 3 Study of the MOF activation and Cu reduction procedure of 4-Cu@Ni-MOF-74: (a) Cu-edge XANES spectra. Arrow 1 = Cu2+ peak; Arrow 2 = Cu+ peak. (b) Linear Combination Fit analysis of Cu species. (c) Ni-edge XANES spectra. (d) Ni–O bond lengths obtained from the EXAFS fit during the activation and Cu reduction procedure and the room temperature adsorption of N2 and CO (shaded). The average error for the bond length is 0.01 Å. | ||
Before the activation/reduction process, 4-Cu@Ni-MOF-74 exhibits the characteristic intense white line peak of Cu2+ (i.e. intense absorption in the near-edge) at 8995 eV (Fig. 3(a)). As the temperature increases, the intensity of the white line decreases and is accompanied by the appearance of a feature at 8983.7 eV. This feature is attributed to the first transition peak of Cu+, and is confirmed when its first derivative is overlaid with the CuCl standard (Fig. S13†). This observation indicates reduction from Cu2+ to Cu+ through oxidation of the formate (eqn (1) and (2)) proceeds at temperatures as low as 80 °C.
| 2COOH− ↔ H2 + CO2 | (1) |
| 2Cu2+ + H2 ↔ 2Cu+ + 2H+ | (2) |
By applying a linear combination fit in Athena, similar to that used by Lamberti et al.,34 we determined the percentage of each Cu species present (Cu2+, Cu+ and Cu0) at various stages of activation and reduction using the spectra from the pure standards as references (Fig. 3(b)). Overall, as the temperature increases, the amount of Cu+ increases while that of Cu2+ decreases. At the end of the activation/reduction procedure, some Cu2+ species are present (32.8%) but the majority of the copper is Cu+ (51.7%), with a small but significant amount of Cu0 (15.5%) due to over reduction. We hypothesize that disproportionation of Cu+ also occurs, as the amount of Cu2+ increases when flowing with pure CO and He at 250 °C.
A switch from a pure flow of He to a pure flow of CO at 250 °C decreases the intensity of the peak at 8983 eV (see Fig. S11†). This observation is attributed to the formation of [Cu(I)(CO)n]+ adducts in the sample, as previously shown by Prestipino et al. and Yamashita et al. in the case of Cu+ in ZSM-5.35,36 The formation of these adducts reduces the amount of quantifiable Cu+ species, explaining the decrease of the Cu+ peak upon the introduction of CO at 250 °C (Fig. 3(b)). However, as reduction proceeds the CO adduct decomposes and the Cu+ peak at 8983 eV increases again (Fig. S11†).
A similar analysis of the Cu+ evolution through the activation/reduction process was performed for the Co-MOF-74 based samples and similar conclusions are derived. The amount of Cu0 species present for this sample remains negligible (Fig. S14†).
The following changes are observed as the temperature and the gas composition change: (i) an increase in the intensity of the white line position at ∼8348 eV with a shift towards higher energies (Arrow 3), (ii) the appearance of a pre-edge feature assigned to 1s–4pz transition at 8338 eV (Arrow 4) and (iii) an increase in intensity of the pre-edge feature with a shift towards lower energies assigned to 1s–3d transition at 8333 eV. These observations are mostly coherent with the previously reported results regarding the pre-edge feature of the Ni- and Co-edge of the MOFs during heating.37,38 During MOF activation, the symmetry changes from octahedral to square-pyramidal as the solvent and water molecules attached to the Ni2+ and Co2+ open-metal sites desorb and provide accessibility for CO adsorption. This causes the removal of the degeneration of p levels and the loss of the inversion centre37,38 and allows for the transition 1s–4pz to appear,39,40 as well as for the increase in intensity in the 1s–3d electronic transition which is forbidden in a perfect octahedral symmetry.39,41
Upon addition of N2, the M–O bond length increases from 1.991 to 2.015 Å for Ni–O and from 2.014 to 2.044 Å in the case of Co–O (Fig. S16†). This suggests that the system has moved back to a 6-fold coordination state.38,43
Upon addition of CO, no further changes can be seen, which is in disagreement with the previously reported data.43 This could be due to the short k-range of the data that does not allow a fine distinction between the O atoms of the ligands and the CO species. To summarise, we observed no unexpected trends of the framework geometry, demonstrating negligible interference from the Cu species in the adsorption of CO on the Ni and Co sites.
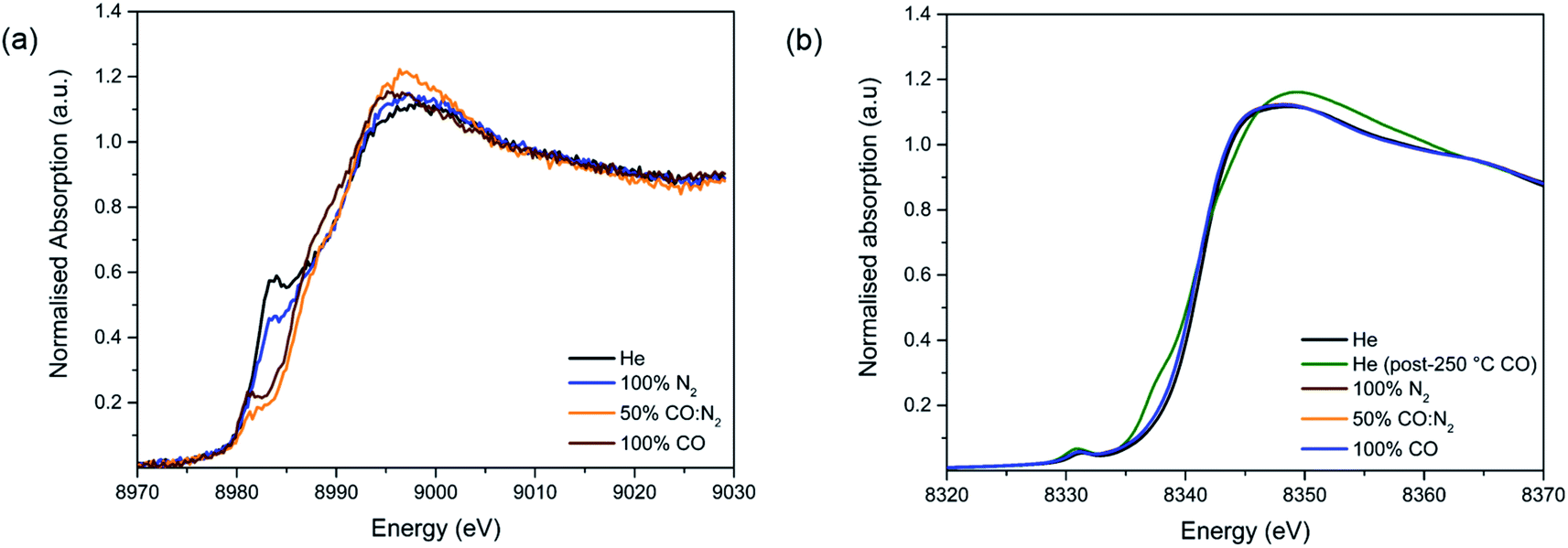 | ||
| Fig. 4 Study of CO adsorption on 4-Cu@Ni-MOF-74: (a) Cu-edge XANES spectra. (b) Ni-edge XANES spectra. | ||
Upon the addition of CO, the white line intensity decreases and shifts towards lower energies (Fig. S18†). This pattern has been previously seen for CO and NO interacting with Cu+ cations in zeolites. The results suggest much stronger interactions between Cu+ and CO than the one with N2 as in a 50:50 N2:CO mixture the only ligand present appears to be CO.
3.4 In situ Cu@M-MOF-74 DRIFTS characterization
We used diffuse reflectance infrared Fourier Transform spectroscopy (DRIFTS) to further study the metal–CO interactions of both Ni and Cu of 4-Cu@Ni-MOF-74 and pure Ni-MOF-74 (Fig. 5). We consider first the 4-Cu@Ni-MOF-74 sample. The Ni2+–CO stretch appears at 2171 cm−1, and corresponds to a non-classical interaction.7 The spectrum shows that Cu+ impregnation left the framework Ni2+ sites unaffected and accessible to CO binding. This aligns with the findings from the XAFS study. We observe Cu–CO interaction at 2119 cm−1. A free CO stretch would be expected 2143 cm−1, therefore the band at 2119 cm−1 indicates a classical Cux+–CO interaction.45 This stretch is assigned to [Cu(I)(CO)]+ species, previously observed in Cu-ZSM-5 and at defect sites in Cu-MOF-74.46,47 Ni-MOF-74 spectrum is similar to that of 4-Cu@Ni-MOF-74, minus the Cu–CO interactions. We also note that the small band observed at 2115–2130 cm−1 is part of the background noise.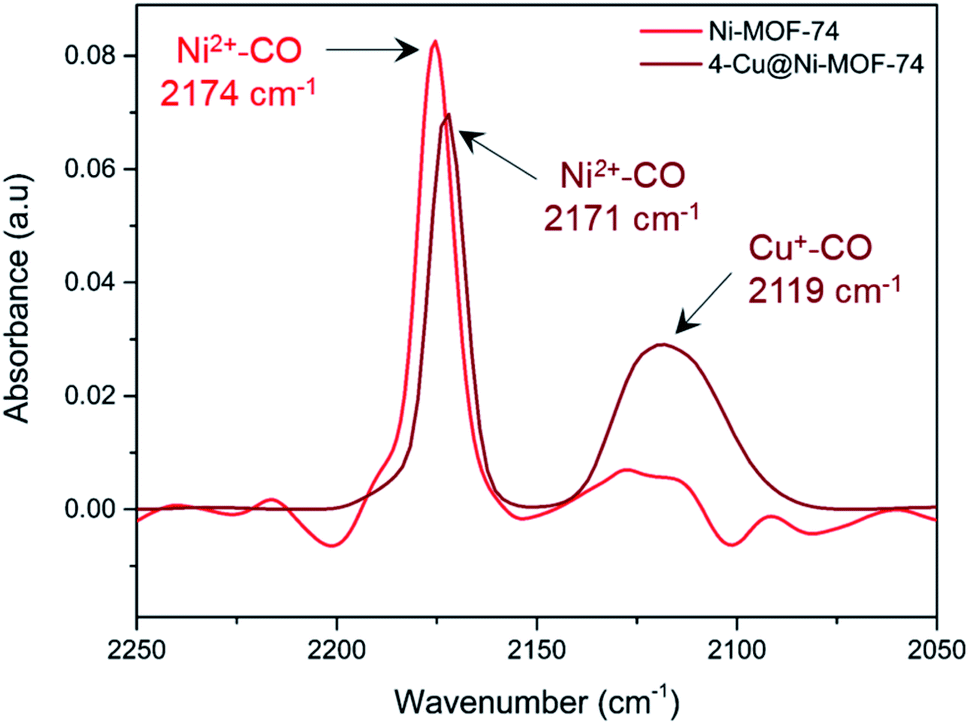 | ||
| Fig. 5 In situ DRIFTS measurement of chemisorbed CO on Ni-MOF-74 and 4-Cu@Ni-MOF-74 at 25 °C and 1 bar. Each spectrum has been background corrected with respect to the instrument and sample holder. | ||
3.5 Dynamic CO/N2 and CO/CO2 separation testing
We tested the dynamic separation performance of 4-Cu@Ni-MOF-74 for CO/N2 and CO/CO2 using a dynamic breakthrough column apparatus. We compared the results to those of Ni-MOF-74 to evaluate the effect of Cu impregnation. The breakthrough curves for 50:50 v/v binary mixtures are shown on Fig. 6.
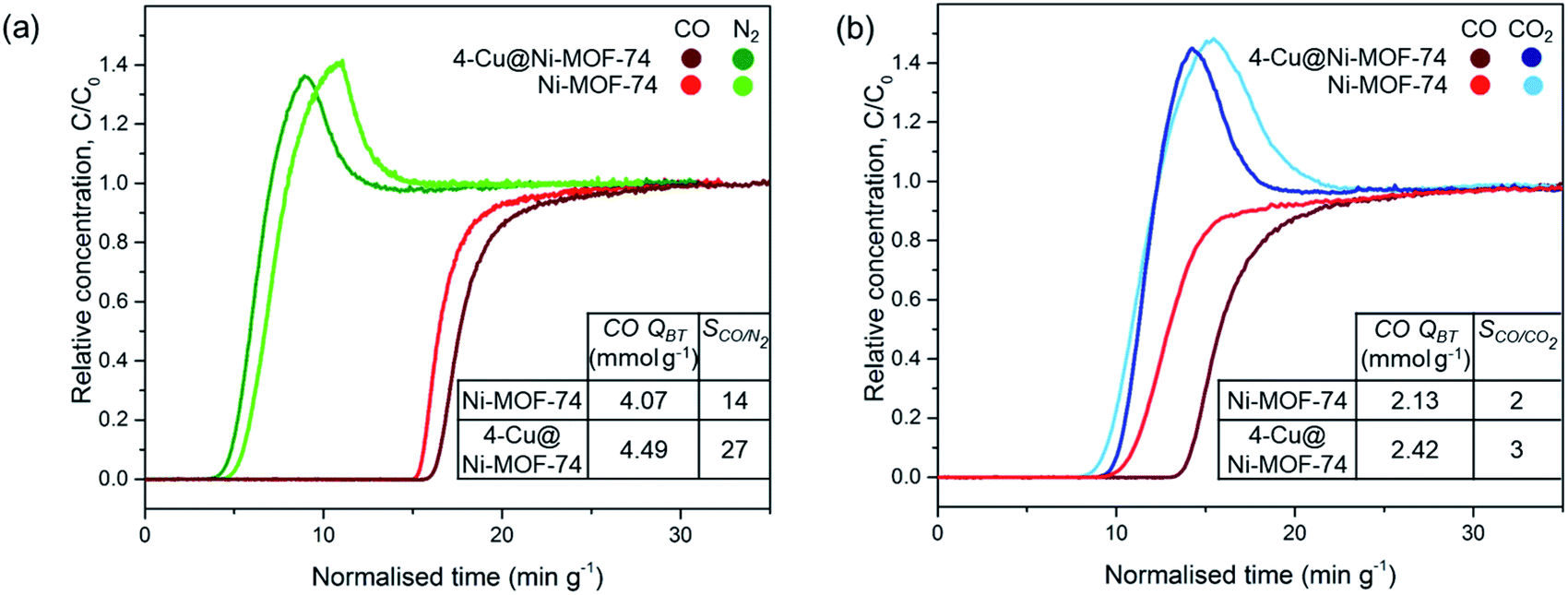 | ||
| Fig. 6 Breakthrough curves of Ni-MOF-74 and 4-Cu@Ni-MOF-74 for 50:50 v/v binary gas mixtures on a breakthrough adsorption column at 25 °C and 1 bar: (a) CO:N2 mixture and (b) CO:CO2 mixture. (CO QBT = CO breakthrough capacity, SCO/X = CO/X ideal selectivity at equilibrium where X = N2 or CO2). | ||
For the 50:50 v/v CO:N2 mixture, N2 breaks through first at similar times for both samples because of the weaker physisorption forces responsible for adsorbing to non-polar N2. Upon CO breakthrough, the N2 concentration exceeds the inlet feed concentration and produces a roll-up effect. The competitive binding with CO induces a spike in concentration as CO replaces the weakly adsorbed N2 molecules. Cu impregnation resulted in an increased breakthrough capacity (from 4.07 to 4.49 mmol g−1) and CO/N2 selectivity at equilibrium after saturation (from 14 to 27). The increase in CO/N2 separation performance is attributed to: (i) the increased number of open-metal sites binding to CO and (ii) the lower porosity of the Cu impregnated samples reducing N2 sorption.21 Introducing Cu+ sites with no affinity to N2 results in greater CO/N2 selectivity. We did not observe the same degree of improvement earlier with the FRT. Indeed, in the FRT tests, the sample is pre-saturated with N2 before CO adsorption, and the instrument is unable to distinguish the CO selectivity and separation performance.
Fig. 6(b) exhibits similar breakthrough times for CO and CO2 for Ni-MOF-74 because of similar binding strength between the open-metal sites and polar CO and CO2 adsorbates. The CO breakthrough capacity is smaller than the amount in a CO/N2 stream because of the MOFs greater affinity to CO2 over N2.21,22 Cu impregnation improved the CO/CO2 separation performance by increasing the number of Cu+ open-metal sites which undergo strong π-complexation to CO, but do not interact with CO2 through this mechanism.22 Therefore, both the CO breakthrough capacity and CO/CO2 selectivity increased upon impregnation. Cu impregnation replaced the accessibility of Ni2+ open-metal sites for CO and CO2 binding with Cu+ open-metal sites solely for CO binding. CO/N2 and CO/CO2 separation performance was also improved after Cu impregnation of Fe-MIL-100 and Cr-MIL-101,21–26 but was improved to a smaller degree with M-MOF-74 because of the highly active Ni2+ sites compared to Fe3+ and Cr3+.
In an industrial setting, an adsorbent must maintain high adsorption capacity and selectivity over multiple cycles and require minimal energy for regeneration. Previous studies have tested the cyclability of Ni-MOF-74 (ref. 6) and Cu impregnated MOFs for CO purification,22,23,25 with He purging22 and vacuum swing25 regeneration techniques favoured to minimise any potential overreduction which would be associated with thermal regeneration. Yet, vacuum swing and He purging would not regenerate the Ni2+ sites of the MOF. A high regeneration temperature is required due to the high CO isosteric heat of adsorption value of Ni-MOF-74 (ref. 6) (−54.4 kJ mol−1 at low loading) potentially leading to undesired chemisorbing Cu0 sites. Hence, further monitoring and optimisation of the Cu oxidation state is needed for cyclic testing with varying regeneration conditions, while simultaneously measuring its CO separation performance.
4. Conclusion
We have optimised the Cu impregnation procedure for M-MOF-74 (M = Ni or Co) to improve the CO purification ability of the adsorbent. The optimisation has focused on identifying the type of Cu salt, the Cu loading as well as the MOF activation and Cu reduction process required to maximise CO uptake. A mixture of CuCl2 and Cu(HCO2)2 salts with a 4% wt Cu loading led to the largest CO uptake. In terms of activation and Cu reduction, we have established that heating at 250 °C with 10% CO (in He) for 6 h allowed the best compromise between minimising the time of CO used at elevated temperature while maximising the amount of Cu+ generated.We have performed in situ XAFS analysis of the Cu impregnated Ni- and Co-MOF-74 samples during the activation/reduction procedure as well as during CO adsorption. This type of in situ analysis has not been performed on Cu impregnated MOFs before. Using this technique, we have shown that Cu+ is generated with He flowing at temperatures as low as 80 °C and becomes the dominant Cu species at the end of the activation/reduction procedure (between 57% and 70% depending on the MOF). Some Cu2+ and Cu0 still remain in the activated samples. Using this same technique, we have confirmed Cu+–CO binding for the first time on Cu impregnated MOFs. EXAFS analysis has confirmed no interference from the Cu species on CO adsorption on the Ni and Co sites. DRIFTS analysis has further supported this finding.
Finally, we have performed dynamic CO/N2 and CO/CO2 separation measurements 50:50 v/v gas mixtures (i.e. CO/N2 and CO/CO2) to assess any enhancement in CO purification upon Cu impregnation. CO uptake and CO/N2 selectivity increased by 10% and 93%, respectively, for CO/N2 separation. This is due to a combined increase in the number of open-metal sites for metal–CO binding and decrease in porosity reducing N2 sorption. CO uptake and CO/CO2 selectivity increased by 14% and 50%, respectively. This is due to the Cu sites preferentially interacting with CO rather than CO2. Overall, this study provides the first confirmation of Cu+–CO binding in Cu impregnated MOFs after optimising the synthesis and reduction. This impregnation was demonstrated to improve CO separation ability in CO/N2 and CO/CO2 mixtures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr L. W. Bolton for his technical input. The authors would also like to thank Dr R. Luebke for his technical input and support in the operation of the FRT. The authors would also like to acknowledge the funding and technical support from BP through the BP International Centre for Advanced Materials (BP-ICAM) as well as EPSRC funding through an iCASE award (EP/N509206/1).References
- A. Briglia, A. Darde, N. Haik-Beraud and M.-K. Khy, US Pat., 9410738, 2009.
- N. N. Dutta and G. S. Patil, Gas Sep. Purif., 1995, 9, 277–283 CrossRef CAS.
- D. J. Haase and D. G. Walker, Chem. Eng. Prog., 1974, 5, 74–77 Search PubMed.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–438 CrossRef PubMed.
- A. Evans, R. Luebke and C. Petit, J. Mater. Chem. A, 2018, 6, 10570–10594 RSC.
- A. D. Evans, M. S. Cummings, R. Luebke, M. S. Brown, S. Favero, M. P. Attfield, F. Siperstein, D. Fairen-Jimenez, K. Hellgardt, R. Purves, D. Law and C. Petit, Ind. Eng. Chem. Res., 2019, 58, 18336–18344 CrossRef CAS.
- E. D. Bloch, M. R. Hudson, J. A. Mason, S. Chavan, V. Crocellà, J. D. Howe, K. Lee, A. L. Dzubak, W. L. Queen, J. M. Zadrozny, S. J. Geier, L.-C. Lin, L. Gagliardi, B. Smit, J. B. Neaton, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 10752–10761 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- N. A. Khan and S. H. Jhung, J. Hazard. Mater., 2017, 325, 198–213 CrossRef CAS PubMed.
- Y. Xie, J. Zhang, J. Qiu, X. Tong, J. Fu, G. Yang, H. Yan and Y. Tang, Adsorption, 1997, 3, 27–32 CrossRef CAS.
- F. Gao, Y. Wang and S. Wang, Chem. Eng. J., 2016, 290, 418–427 CrossRef CAS.
- H. Hirai, K. Wada and M. Komiyama, Chem. Lett., 1983, 12, 361–364 CrossRef.
- H. Hirai, M. Komiyama and K. Wada, Chem. Lett., 1982, 11, 1025–1028 CrossRef.
- H. Hirai, K. Wada and M. Komiyama, Bull. Chem. Soc. Jpn., 1986, 59, 2217–2223 CrossRef CAS.
- L.-q. Zhu, J.-l. Tu and Y.-j. Shi, Gas Sep. Purif., 1991, 5, 173–176 CrossRef CAS.
- Y. Huang, Y. Tao, L. He, Y. Duan, J. Xiao and Z. Li, Adsorption, 2015, 21, 373–381 CrossRef CAS.
- J. Ma, L. Li, J. Ren and R. Li, Sep. Purif. Technol., 2010, 76, 89–93 CrossRef CAS.
- F. Gao, S. Wang, W. Wang, J. Duan, J. Dong and G. Chen, RSC Adv., 2018, 8, 39362–39370 RSC.
- Y.-Y. Huang, J. Catal., 1973, 30, 187–194 CrossRef CAS.
- T. M. Salama, A. H. Ahmed and Z. M. El-Bahy, Microporous Mesoporous Mater., 2006, 89, 251–259 CrossRef CAS.
- J. Peng, S. Xian, J. Xiao, Y. Huang, Q. Xia, H. Wang and Z. Li, Chem. Eng. J., 2015, 270, 282–289 CrossRef CAS.
- A.-R. Kim, T.-U. Yoon, S.-I. Kim, K. Cho, S.-S. Han and Y.-S. Bae, Chem. Eng. J., 2018, 348, 135–142 CrossRef CAS.
- T. K. Vo, Y.-S. Bae, B.-J. Chang, S.-Y. Moon, J.-H. Kim and J. Kim, Microporous Mesoporous Mater., 2019, 274, 17–24 CrossRef CAS.
- Y.-X. Li, Y.-N. Ji, M.-M. Jin, S.-C. Qi, S.-S. Li, D.-M. Xue, M. B. Yue, X.-Q. Liu and L.-B. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 40044–40053 CrossRef CAS PubMed.
- Y. Wang, C. Li, F. Meng, S. Lv, J. Guo, X. Liu, C. Wang and Z. Ma, Front. Chem. Sci. Eng., 2014, 8, 340–345 CrossRef CAS.
- Y. Yin, Z.-H. Wen, X.-Q. Liu, L. Shi and A.-H. Yuan, J. Porous Mater., 2018, 1–7 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- D. A. Gómez-Gualdrón, P. Z. Moghadam, J. T. Hupp, O. K. Farha and R. Q. Snurr, J. Am. Chem. Soc., 2016, 138, 215–224 CrossRef PubMed.
- S. G. Chen and R. T. Yang, Langmuir, 1994, 10, 4244–4249 CrossRef CAS.
- C. Palmer, A. Sasegbon and K. Hellgardt, Adsorption, 2011, 17, 783–794 CrossRef CAS.
- S. Luidold and H. Antrekowitsch, JOM, 2007, 59, 20–26 CrossRef CAS.
- Y. Matsumura, K. Tanaka, N. Tode, T. Yazawa and M. Haruta, J. Mol. Catal. A: Chem., 2000, 152, 157–165 CrossRef CAS.
- C. Lamberti, S. Bordiga, F. Bonino, C. Prestipino, G. Berlier, L. Capello, F. D'Acapito, F. X. Llabrés i Xamena and A. Zecchina, Phys. Chem. Chem. Phys., 2003, 5, 4502–4509 RSC.
- C. Prestipino, L. Capello, F. D'Acapito and C. Lamberti, Phys. Chem. Chem. Phys., 2005, 7, 1743–1746 RSC.
- H. Yamashita, M. Matsuoka, K. Tsuji, Y. Shioya, M. Anpo and M. Che, J. Phys. Chem., 1996, 100, 397–402 CrossRef CAS.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- S. Chavan, J. G. Vitillo, E. Groppo, F. Bonino, C. Lamberti, P. D. C. Dietzel and S. Bordiga, J. Phys. Chem. C, 2009, 113, 3292–3299 CrossRef CAS.
- C. Prestipino, G. Berlier, F. X. Llabrés i Xamena, G. Spoto, S. Bordiga, A. Zecchina, G. Turnes Palomino, T. Yamamoto and C. Lamberti, Chem. Phys. Lett., 2002, 363, 389–396 CrossRef CAS.
- J. Estephane, E. Groppo, A. Damin, J. G. Vitillo, D. Gianolio, C. Lamberti, S. Bordiga, C. Prestipino, S. Nikitenko, E. A. Quadrelli, M. Taoufik, J. M. Basset and A. Zecchina, J. Phys. Chem. C, 2009, 113, 7305–7315 CrossRef CAS.
- C. Prestipino, P. L. Solari and C. Lamberti, J. Phys. Chem. B, 2005, 109, 13132–13137 CrossRef CAS PubMed.
- E. Groppo, C. Prestipino, F. Cesano, F. Bonino, S. Bordiga, C. Lamberti, P. C. Thüne, J. W. Niemantsverdriet and A. Zecchina, J. Catal., 2005, 230, 98–108 CrossRef CAS.
- S. Chavan, F. Bonino, J. G. Vitillo, E. Groppo, C. Lamberti, P. D. C. Dietzel, A. Zecchina and S. Bordiga, Phys. Chem. Chem. Phys., 2009, 11, 9811–9822 RSC.
- I. J. Drake, Y. Zhang, D. Briggs, B. Lim, T. Chau and A. T. Bell, J. Phys. Chem. B, 2006, 110, 11654–11664 CrossRef CAS PubMed.
- S. H. Strauss, J. Chem. Soc., Dalton Trans., 2000, 1–6, 10.1039/a908459b.
- K. I. Hadjiivanov, M. M. Kantcheva and D. G. Klissurski, J. Chem. Soc., Faraday Trans., 1996, 92, 4595–4600 RSC.
- N. Drenchev, M. H. Rosnes, P. D. C. Dietzel, A. Albinati, K. Hadjiivanov and P. A. Georgiev, J. Phys. Chem. C, 2018, 122, 17238–17249 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials synthesis procedures, testing equipment schematics, full textural parameters, X-ray absorption fine structure (XAFS) analysis procedure and supplementary spectra, thermal analysis and full XPS survey. See DOI: 10.1039/c9ra10115b |
| ‡ Current address: School of Chemistry, Cardiff University, UK. |
| § Current address: Centre for Advanced Separations Engineering, Department of Chemical Engineering, University of Bath, UK. |
| ¶ Current address: Radiochemistry Unit, Dept. of Chemistry, The University of Helsinki, Helsinki, Finland, 00014. |
| This journal is © The Royal Society of Chemistry 2020 |