
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective doping effect, phase separation, and structure evolution in 1:1:1 triple-cation B-site ordered perovskites Ca4−xSrxGaNbO8†
He Huanga,
Pengfei Jiang *ab,
Wenliang Gaoa,
Rihong Conga and
Tao Yang*a
*ab,
Wenliang Gaoa,
Rihong Conga and
Tao Yang*a
aCollege of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 401331, P. R. China. E-mail: pengfeijiang@cqu.edu.cn; taoyang@cqu.edu.cn
bCollege of Physics, Chongqing University, Chongqing, 401331, P. R. China
First published on 8th January 2020
Abstract
Oxygen-deficient perovskites are a family of important materials that may exhibit oxide ionic conductivities. We attempted to introduce oxygen-vacancy disordering in perovskite Ca4GaNbO8 (Ca4-type) by substituting Ca2+ with larger Sr2+. Sr2+-to-Ca2+ substitution did not lead to oxygen-vacancy ordering–disordering transition but an interesting Ca4-to-Sr4 type structure transition. Rietveld refinements revealed that the two-type structures exhibit similar oxygen-vacancy ordering and identical 1:1:1 triple-cation B-site ordering. Close inspection of the two-type structures revealed the subtle structure difference lies in the orientations of GaO4 tetrahedra, which is the origin of the formation of the narrow two-phase region (0.3 ≤ x < 0.65) in Ca4−xSrxGaNbO8. More importantly, the A- and B-site cavities with large differences in size for both structures resulted in a site-selective doping behaviour for Sr2+ in Ca4−xSrxGaNbO8. These structural changes found in Ca4−xSrxGaNbO8 will provide a broad route approaching new oxygen-deficient phases with oxide ionic conductivities.
Introduction
Perovskites with the general formula ABO3 can accommodate cations with a wide range of ionic radii and charge, thus giving rise to rich chemical compositions accompanied with diverse physical properties such as magnetism,1,2 superconductivity,3,4 dielectricity,5,6 ferroelectric.7–9 Such diversity in chemical compositions as well as physical properties for perovskites have motivated an intense and enduring interesting for chemists to design new materials and modify the physical properties of a specific phase by chemical substitution.Among the various perovskites, 1:1 double-cation B-site ordering perovskites usually exhibit physical properties different from the disordered ones distinctively. For example, B-site ordered perovskites Sr2FeMoO6 and Sr2CrOsO6 exhibit colossal magnetoresistance and high temperature ferrimagnetism, respectively.10,11 In perovskites, B-site rock-salt ordering is commonly observed, because such an arrangement manner of B-site cations is benefit for keeping local charge neutrality and release of structure strain. In contrast, the layered ordered and columnar ordered perovskites are relatively rare, and only a few specific phases have been reported up to now.12,13
Owing to the structural flexibility, perovskite can accommodate various defects including A-site and oxygen vacancies, which can result in ionic diffusion at high temperature. A-site ionic diffusion was observed indeed in Li3xLa2/3−xTiO3, in which the pre-existing A-site vacancies allow for easy Li+ diffusion.14–16 In contrast to A-site ionic conductivity, oxygen ionic conductivity or mixed ionic and electronic conductivities for oxygen deficient perovskites are much more widely investigated due to their potential applications in solid oxide fuel cells (SOFCs) as electrolyte or cathode. For example, Sr2+ and Mg2+ co-doped LaGaO3 (LSGM) is the one of the best oxygen ionic conducting electrolytes.17–19 Much efforts have been devoted to improve the oxygen ionic conductivities of various perovskites by increasing the number of oxygen vacancies. However, increasing the number of oxygen vacancies may result in completely ordering of oxygen vacancies, which in turn lead to a significant decrease of oxide ionic conductivity. For example, oxygen-vacancy ordered perovskite Ba2In2O5 exhibits poor ionic conductivity in low temperature range (<900 °C), though there exists a large number of oxygen vacancies in the structure.20–22 Oxygen-vacancy ordering/disordering in perovskite is closely related to the A-site cationic size (or tolerance factor, t). For example, with an increase of the Sr2+-content (increase of tolerance factor) in Ca2−xSrxFeCoO6−δ resulted in an oxygen-vacancy ordering to disordering transition.23 Therefore, the strategy of increasing the A-site cationic size of anion-ordered perovskites might be utilized to improve the oxide ionic conductivity.
Herein this contribution, our attention is turned to the newly discovered oxygen-deficient perovskite Ca4GaNbO8, where the complex 1:1:1 triple-cation B-site ordering is coupled with the oxygen-deficient ordering.24 We attempted to incorporate larger Sr2+ cations into Ca4GaNbO8 to bring in oxygen-vacancy disordering, so as to obtain new oxide ionic conductors. Substitution of Ca2+ with Sr2+ led to a Ca4-type to the Sr4-type structure transition as we expected. Unfortunately, Rietveld refinements manifested that the oxygen vacancies are also ordered in the Sr4-type structure, which was further confirmed by AC impedance spectroscopy measurements on selected compositions due to the absence of oxide ionic conductivity. The subtle structural differences between the Ca4-and Sr4-type structures lie in the orientations of GaO4 tetrahedra, which is the origin of the coexistence of two phases in a narrow region 0.3 ≤ x < 0.65. Moreover, a site-selective doping behaviour was observed for Sr2+ in Ca4−xSrxGaNbO8, however the A-site cationic ordering is only in short-range for all compositions.
Experimental section
Synthesis
The polycrystalline samples Ca4−xSrxGaNbO8 (x = 0–4) were prepared by high temperature solid-state reactions using calcium carbonate (CaCO3, 99.99%), strontium carbonate (SrCO3, 99.99%), gallium oxide (Ga2O3, 99.99%), niobium oxide (Nb2O5, 99.99%) as raw materials. The starting materials were preheated at 500 °C for 10 h to remove the absorbed moisture. Stoichiometric starting materials with a total weight of 1.2 g were weighted and mixed in an agate mortar by hands and then preheated at 950 °C to decompose the carbonates. After this calcination, the resultant white powders were further grinded and pressed into pellet with 13 mm diameter and then calcinated between 1200 °C and 1300 °C for 60 hours with intermediate grinding and pressing.X-ray diffraction
The phase purity of the samples was investigated by powder X-ray diffraction (PXRD). PXRD were performed on the PANalytical Empyrean diffractometer equipped with a PXIcel 1D detector (Cu Kα radiation). The operation voltage and current were 40 kV and 40 mA, respectively. The PXRD data for phase identification were collected with the setting of 30 s/0.0262°. High quality PXRD data, which were used for Rietveld refinements with the TOPAS-Academic V6 software,25 were collected with a setting of 200 s/0.0131°.AC impedance spectroscopy
AC impedance spectroscopy measurements for Ca4−xSrxGaNbO8 (x = 0, 2, and 4) were carried out over temperature range from room temperature to 800 °C using a Solartron 1260A impedance phase analyzer with the frequency from 10−1 to 107 Hz. Before the measurements, the pellets were coated with platinum pasts and then heated at 800 °C for 1 h to form electrodes.Raman spectroscopy
The Raman spectroscopy measurements were performed on the LabRAM HR Evolution spectrometer. The room temperature Raman spectra for Ca4GaNbO8 and Sr4GaNbO8 were measured over the range of 100–4000 cm−1 using the 325 nm line and with the light focus on the sample trough an optical lens. The spectral resolution was about 1–2 cm−1.Results and discussion
Phase identification
PXRD data for Ca4−xSrxGaNbO8 (x = 0–4) are elucidated in Fig. 1, where the diffraction peaks continuous evolves to lower angles upon increasing Sr2+-content. This observation indicates the cell volume expansion of Ca4−xSrxGaNbO8 upon substituting Ca2+ with Sr2+, which is consistent with the larger cationic radii of Sr2+ (1.44 Å in 12-fold coordination) in comparison with Ca2+ (1.34 Å in 12-fold coordination).26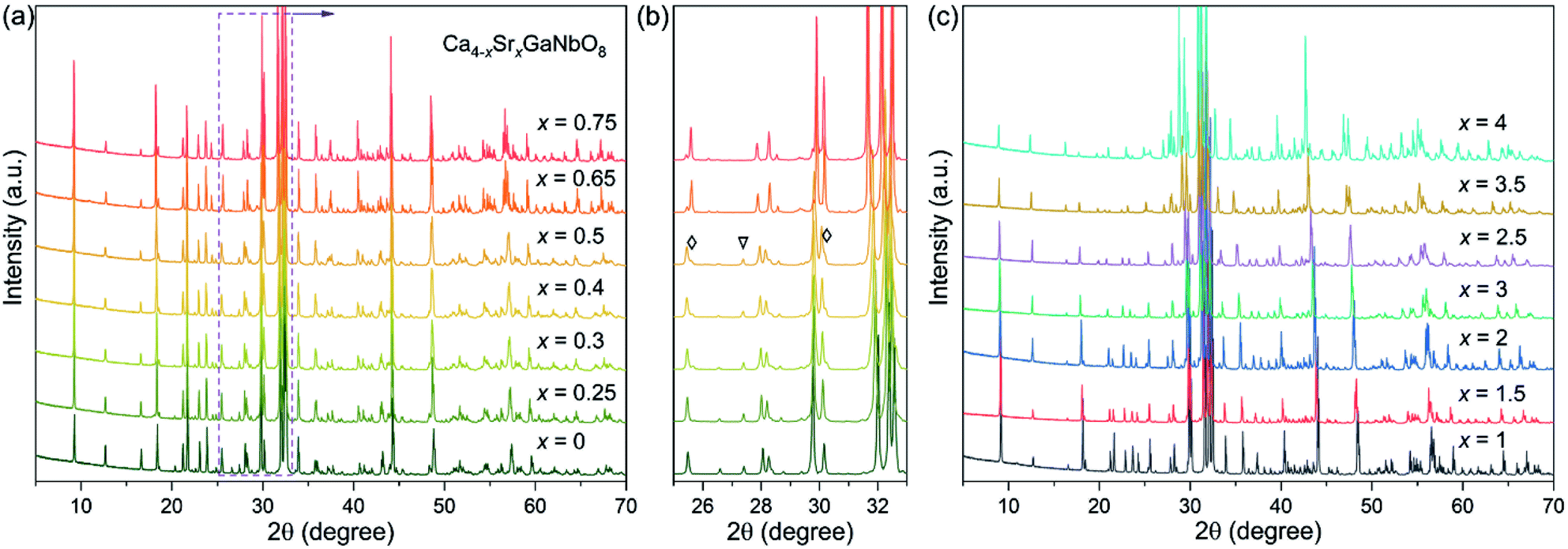 | ||
| Fig. 1 (a and c) Powder XRD patterns for Ca4−xSrxGaNbO8 (x = 0–4). (b) The enlargement of 25–33° range for compositions x ≤ 0.65. The triangle represents the specific reflection ascribe to Ca4-type phase, the diamonds represent the reflections ascribe to Sr4-type phase. | ||
Previous study on Ca4GaNbO8 revealed that this compound adopts a triple-cation B-site ordered perovskite-type structure and crystallizes in P21/c with lattice parameters a ≈ 11.18 Å, b ≈ 5.59 Å, c ≈ 14.07 Å, and β ≈ 121.55°.24 Our preliminary Le-bail fitting performed on Ca4−xSrxGaNbO8 manifested that the XRD data for compositions with x < 0.65 could be indexed by this monoclinic cell. However, a group of weak reflections for compositions with x ≥ 0.65, which is not ascribe to impurity phases, could not be fitted with this monoclinic cell any more, indicating Sr2+-doping induced a change of lattice parameters. Indexing the XRD data of Ca3SrGaNbO8 yield a monoclinic cell with lattice parameters a ≈ c1/2, b ≈ b1, c ≈ √3a1, β ≈ 97°, where a1, b1, and c1 represent the cell dimension of Ca4GaNbO8 (denoted as Ca4-type phase). Similar unit cell was also observed for Sr4AlNbO8 (P21/c), which also adopt a triple-cation B-site ordered perovskite-type structure with anionic vacancy ordering, suggesting Ca4−xSrxGaNbO8 (x ≥ 0.65) (denoted as Sr4-type phase) is isostructural to Sr4AlNbO8.27
Preliminary Rietveld refinements performed on Ca4−xSrxGaNbO8 (x = 0.5) using Ca4GaNbO8 as the initial structure model resulted in unreasonable structural parameters, i.e. too short interatomic distances. Close inspection of the XRD data revealed that some reflections from the Sr4-type phase were observed for compositions x = 0.3, 0.4 and 0.5. To clarify this clearly, the diffraction components from Cu Kα2 were striped for comparison. As shown Fig. 1b, small shoulders, corresponding to contribution of the Sr4-type phases, of some characteristic reflections were visually observed. Moreover, a representative Le-bail fitting pattern for x = 0.5 presented in Fig. S1† demonstrated that all the reflections could only be well reproduced by a two-phase model fitting. These results indicate that the compositions for x = 0.3, 0.4 and 0.5 comprise two phases. More importantly, single-phase for compositions x = 0.3, 0.4 and 0.5 was not attainable by neither calcination at elevate temperature nor prolongation of calcination time (Fig. S2†), suggesting the two phases are thermodynamically favorable. Therefore, the compositions for Ca4−xSrxGaNbO8 (x = 0–4) can be divided into three regions: (i) a single Ca4-type phase region (x < 0.3), (ii) a narrow two-phase region contains both Ca4-type and Sr4-type phases (0.3 ≤ x < 0.65), and (iii) a single Sr4-type phase region (x ≥ 0.65). We should note that the change of the relative content for the two phases within the two-phase region was slight (Fig. 1b), which is distinctly differ from commonly observed two-phase regions for perovskites e.g. the two-phase region observed in Ba3−xSrxZnSb2O9 (0.3 ≤ x ≤ 1.0).28 Such a difference is attribute to the subtle distinction in crystal structures between Ca4-type and Sr4-type structures, which will be discussed later.
The evolution of normalized lattice parameters against Sr2+-content in Ca4−xSrxGaNbO8 is presented in Fig. 2, where the linear expansion of the cell volume for single-phase compositions is in good agreement with the PXRD patterns. Interestingly, the lattice parameters for both Ca4-type and Sr4-type phases also showed a linear increase within the two-phase region, which opposite to commonly observed constant lattice parameters for the two phases in the two-phase region. This uncommon phenomenon further corroborated that the co-existence of two phases in Ca4−xSrxGaNbO8 is thermodynamically favorable.
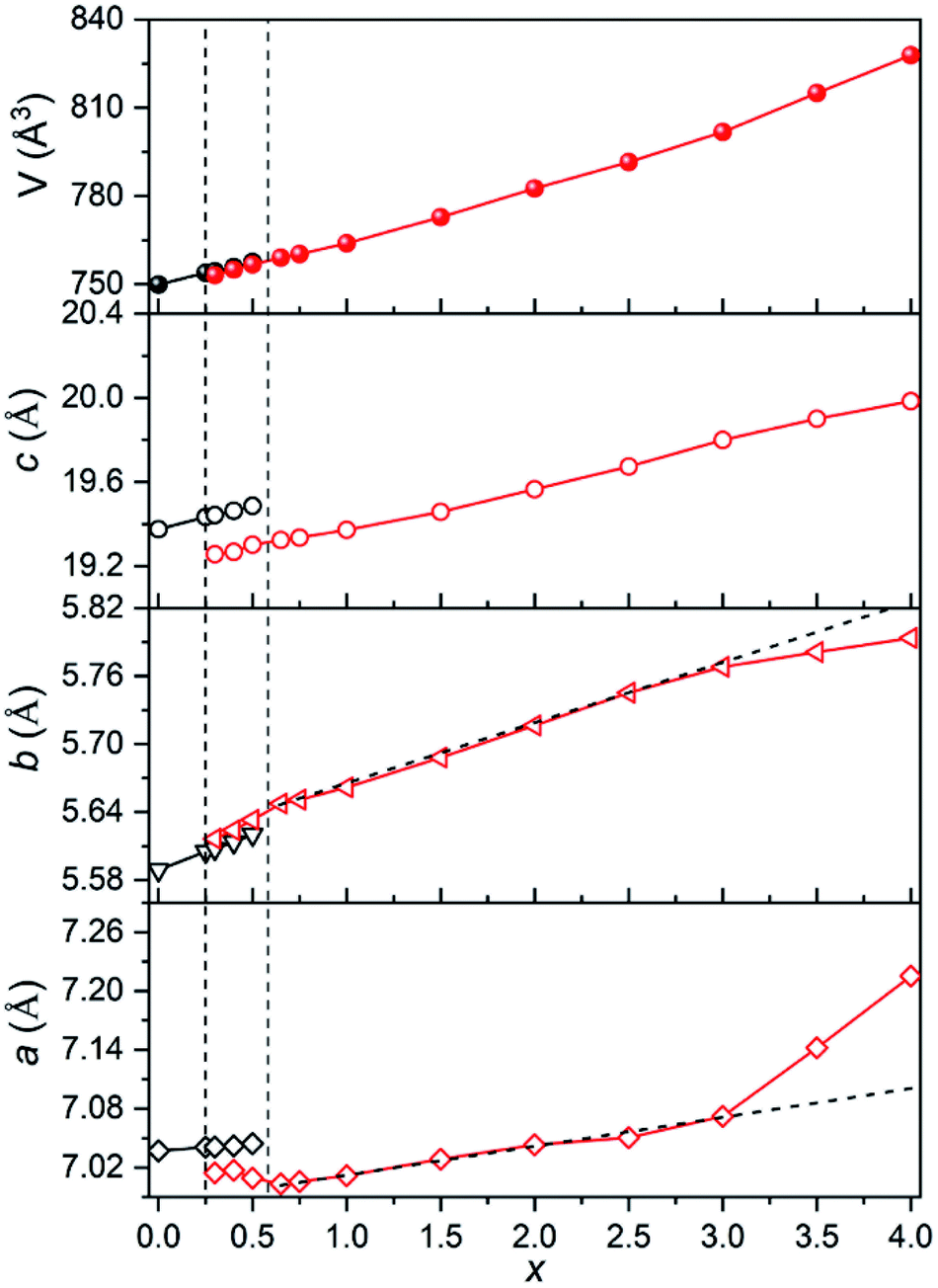 | ||
| Fig. 2 Plots of lattice parameters against Sr2+-content in Ca4−xSrxGaNbO8. | ||
Rietveld refinements
As described above, both Ca4-type and Sr4-type phases adopt oxygen-deficient perovskite-type structure with 1:1:1 triple-cation B-site ordering. Owing to their low structure symmetry (P21/c), both structures exhibit three distinctly different A-sites (denoted as A1, A2, and A3, respectively), which could be readily discerned through their surrounding environments. As shown in Fig. 3, A1 cation is surrounded by three B1O6 octahedra (B1 represents the B-site occupied by alkali earth cation), four NbO6 octahedra, and one GaO4 tetrahedron; A2 is surrounded by three B1O6 octahedra, one NbO6 octahedron, and four GaO4 tetrahedra; A3 is surrounded by two B1O6 octahedra, three NbO6 octahedra, and three GaO4 tetrahedra. Such difference in surrounding environments for A-site cations results in the A1-site is much larger than A2-and A3-site, suggesting Sr2+ is prone to occupy the A1-site at first.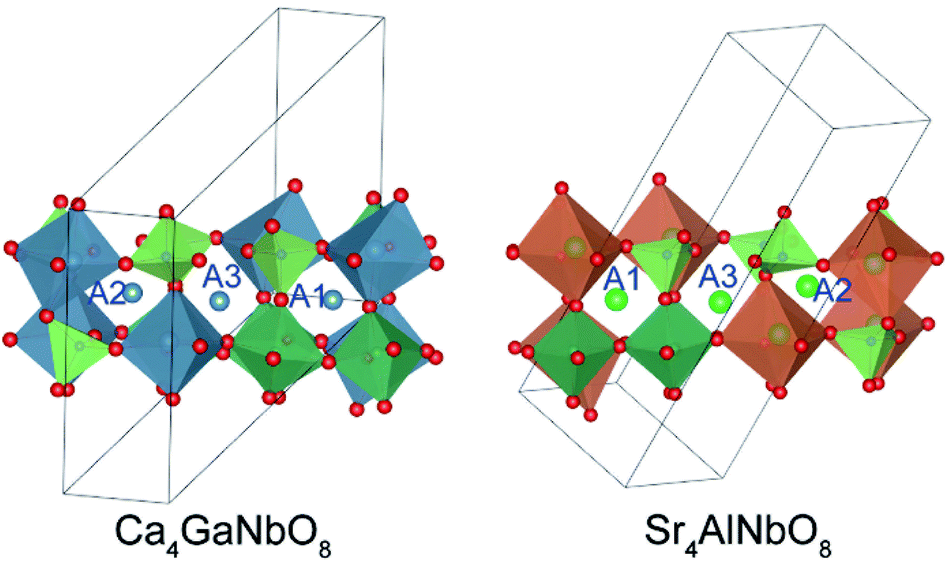 | ||
| Fig. 3 The surrounding environments for A-site cations in Ca4-type and Sr4-type structures. | ||
Careful Rietveld refinements against laboratory XRD data were further performed on selected compositions Ca4−xSrxGaNbO8 (x = 0, 1, 1.5, 2, 2.5, 3, 3.5 and 4) to get an insight picture of the site occupancy preference for Sr2+. The refined structure parameters for Ca4GaNbO8 is in good agreement with that reported by Yang et al. using combined refinements against neutron and synchrotron data (Table S1†). For compositions with x ≥ 1.0, Sr4AlNbO8 was used as the starting structure model for Rietveld refinements. At first, an A-site ordered structure model was constructed for preliminary refinements, for instance, Sr2+ occupies A1-site exclusively and the remaining two A-sites were occupied by Ca2+ in Ca3SrGaNbO8. The refinement proceeded smoothly with this completely ordered model, resulting in reliable agreement factors (Rwp = 7.625%, Rp = 5.592%) and structural parameters. However, some peaks with large discrepancies between the observed and calculated were observed, suggesting the A-site cations are not completely ordering in Ca3SrGaNbO8. Consequently, the occupancy factor for Ca2+ and Sr2+ cations at B1-site and three A-sites were refined freely during the subsequent refinements. It turned out that A1-site was dominantly by Sr2+, and A2, A3, and B1 sites were mainly occupied by Ca2+. Despite the occupancies for Ca2+ and Sr2+ at four independent sites were refined freely, the refined composition Ca2.92Sr1.08GaNbO8 agrees well with the nominal formula Ca3SrGaNbO8. Moreover, the reliable factors were improved significantly to Rwp = 5.543% and Rp = 4.074%. These results manifest that the Ca2+ and Sr2+ are partially ordered in Ca3SrGaNbO8. Rietveld refinements performed on other compositions further demonstrated that all the compositions with mixed A-site cations were partially ordered. We should note that the inversion between A- and B-sites, which is commonly observed in spines,29 was not considered in Ca4−xSrxGaNbO8 because of the large differences in cationic size and coordination environment preference between (Ca, Sr) and (Ga, Nb). The final crystallographic parameters and selected bond lengths for Ca4−xSrxGaNbO8 are summarized in Table S1 and S2.† The Rietveld refinement patterns are presented in Fig. 4 and S3.†
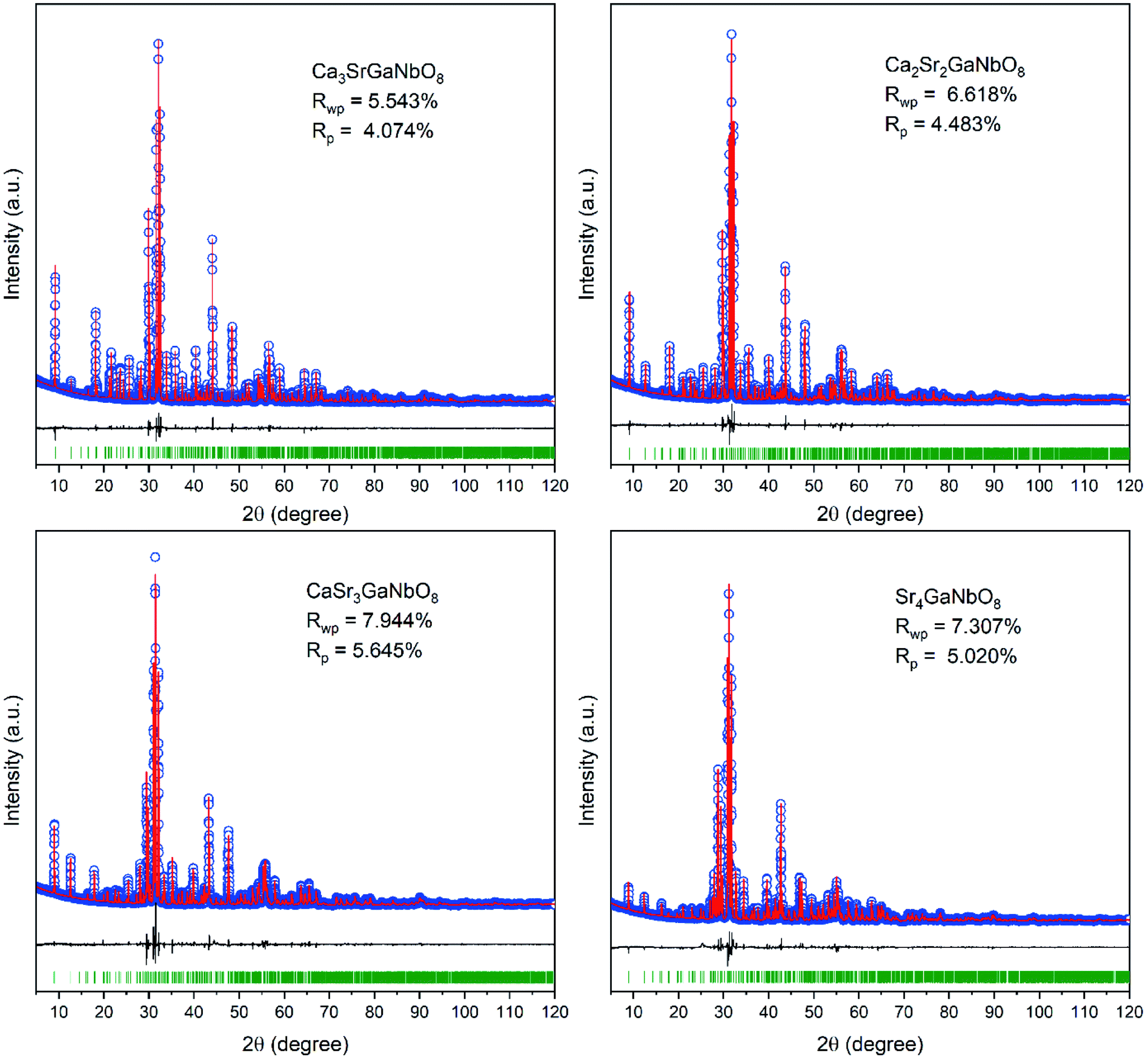 | ||
| Fig. 4 Rietveld refinement plots of XRD data for Ca4−xSrxGaNbO8 (x = 0, 1, 2, 3, 4). | ||
Structure evolution of Ca4−xSrxGaNbO8
The refined crystal structures for Ca4GaNbO8 and Sr4GaNbO8 are presented in Fig. 5, where both structures view along the same cubic-perovskite direction [110]p. Ca4GaNbO8 and Sr4GaNbO8 share the similar structure features with 1:2 layer-ordered hexagonal perovskites i.e. Ca4Nb2O9, however the structure for Ca4GaNbO8 and Sr4GaNbO8 are more complex because there is an another B-site cation (Ga3+) and ordered anionic deficiency.30 As shown in Fig. 5, the removal of oxygen within the AO2 layers creates tetrahedral cavities that could only accommodate smaller Ga3+ cations, which resulted in layered stacking sequence of (Ca/Sr)–(Ga1/2Nb1/2)–(Ga1/2Nb1/2)–(Ca/Sr) for B-site cations along the closed-packing direction [111]p. Owing to the ordered removal of every other oxygen-only O2 columns within the AO2 layers, Ga3+ and Nb5+ within (Ga1/2Nb1/2)–(Ga1/2Nb1/2) layers are column ordered along [110]p.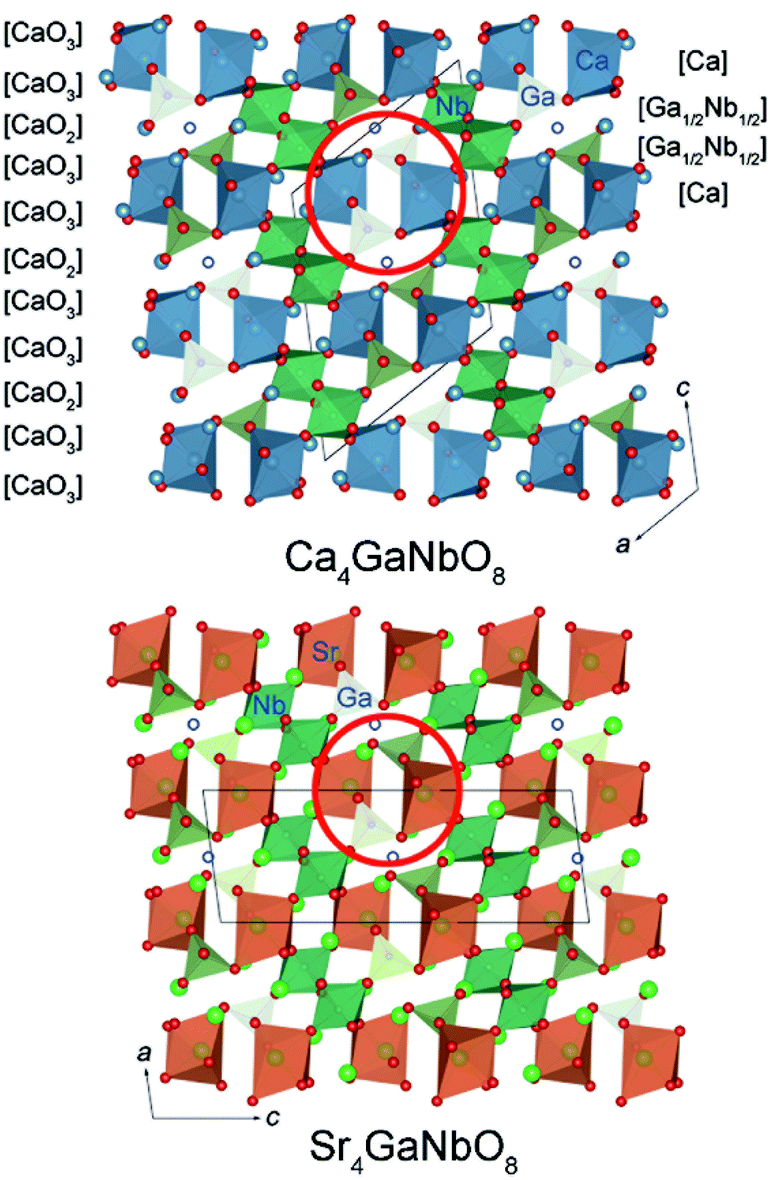 | ||
| Fig. 5 Comparison of crystal structures for Ca4GaNbO8 and Sr4GaNbO8. The open blue circles represent the oxygen-column defects. | ||
Given the same cationic ordering and oxygen deficient manner, the crystal structures for Ca4GaNbO8 and Sr4GaNbO8 seem identical at first glance. Close inspecting the structures revealed that the structural difference between Ca4GaNbO8 and Sr4GaNbO8 stems from the distinct orientations of GaO4 tetrahedra along-side of (Ca/Sr)O6 octahedra. As highlighted in Fig. 5, the GaO4 tetrahedra in Ca4GaNbO8 point to the same direction, however, the GaO4 tetrahedra in Sr4GaNbO8 point to opposite directions. Such a difference resulted in a doubled cell dimension along [111]p for Ca4GaNbO8 in comparison with Sr4GaNbO8 (see Fig. 5). The structural transformation between two-type structures requires the rearrangement of the orientations of GaO4 tetrahedra along [110]p, which is much more difficult than collective octahedra-tilting observed universally in cubic-type perovskites. Consequently, a narrow two-phase region is observed in Ca4−xSrxGaNbO8, and continuous structural transition is usually observed for simple cubic-type perovskites. We thus can speculate that this subtle structural difference in GaO4 orientations is the origin of the formation of the two-phase region in Ca4−xSrxGaNbO8.
The evolution of occupancy factors for Sr2+ cations at both A and B sites in Ca4−xSrxGaNbO8 is elucidated in Fig. 6a, where a site-selective doping behaviour is observed clearly. In detail, Sr2+ prefers to occupy the A1-site, which can be deduced from the sharp increase of occupancy factor to 0.926(6) for Sr2+ at A1-site when x ≤ 1.5, whereas the increase of occupancy factors at A2 and A3 sites are relatively slow. The occupancy factors for Sr2+ at A2 and A3 sites show a synchronized increase behaviour in the range of 1.5 ≤ x ≤ 3.0, where the Sr2+-occupancy at A1 site manifests a slight increase. Further incorporation of Sr2+ into Ca4−xSrxGaNbO8 lead to a sharp increase of occupancy factor for Sr2+ at B1 site when x ≥ 3.0, whereas a slight increase is observed for x ≤ 3.0. Such a sharp increase of occupancy of Sr2+ at B1-site lead to a significant deviation of the lattice parameters, especially for a, from the Vegard's law (see Fig. 2). Given the layered structure nature of Ca4−xSrxGaNbO8, when viewed along [110]p, the sharp increase of Sr2+-content in B1-site would unambiguously result in a sharp expansion along [111]p, namely a-axis of Sr4-type structure (see Fig. 5).
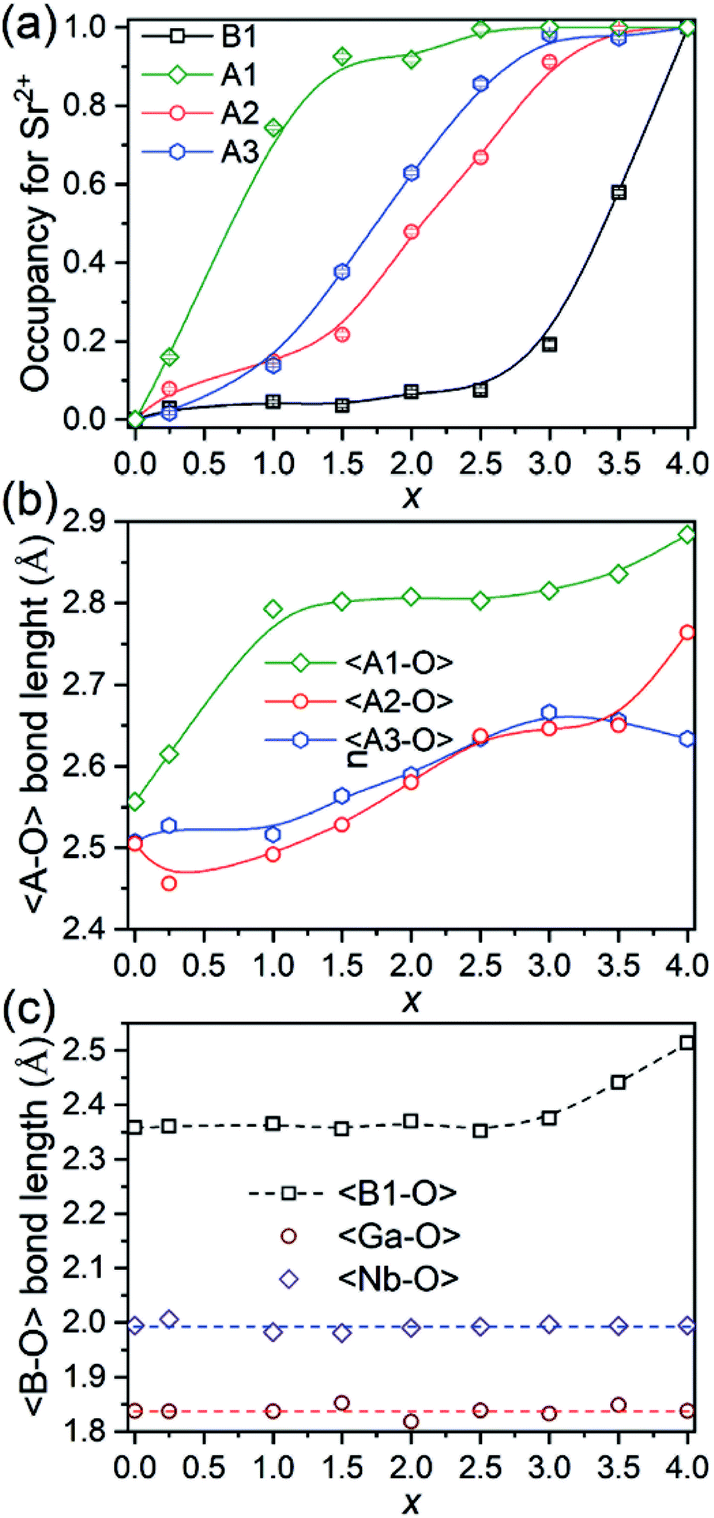 | ||
| Fig. 6 (a) Plots of occupancy factors for Sr2+, (b) average 〈A–O〉 bond lengths, and (c) average 〈B–O〉 bond length along with Sr2+-content in Ca4−xSrxGaNbO8. | ||
As described above, Sr2+ showed a site-selective doping behaviour due to the distinctly large differences in size for A1, A2/A3, and B1 sites. The evolutions of the average 〈A–O〉 and 〈B–O〉 bond lengths are elucidated in Fig. 6b and c, where the change trends for both 〈A–O〉 and 〈B1–O〉 bonds are in good agreement with that of occupancy factors. The 〈Ga–O〉 and 〈Nb–O〉 bond lengths are almost kept in constant at ∼1.85 Å and ∼2.0 Å, respectively, for all compositions (Fig. 6c). The Ga–O bond lengths in all compounds are in the range of 1.76–1.90 Å, which are comparable with four-coordinated Ga3+ in LaAGa3O7 (A = Ca2+, Sr2+, Ba2+).31–33 Detailed inspection of the Nb–O bonds revealed that the Nb5+ exhibits a distorted coordination environment with three Nb–O atomic distances shorter than 2.0 Å and the remaining three bond lengths longer than 2.0 Å (Table S2†), indicating Nb5+ displaced from the centre of the octahedral cavity due to the second-order Jahn–Teller (SOJT) effect.34,35 Though both Ga3+ and Nb5+ cations can adopt four- and six-fold coordinations, incorporation of Sr2+ into A- and B-sites did not lead to anti-site disordering between Ga- and Nb-sites for all compositions, which should unambiguous attribute to their large differences in charge and cationic size.
It is well known that Raman scattering is sensitive to local structural changes, including cationic ordering, structure symmetry change, and John–Teller distortion.36 To gain an insight of structural change induced by Sr2+-doping, Raman spectra for Ca4GaNbO8 and Sr4GaNbO8 were measured. Raman spectra of Ca4GaNbO8 and Sr4GaNbO8 show similar features (Fig. S4†), which is consistent with their similar crystal structures. The intensive peaks with wavelength numbers higher than 700 cm−1, especially for the strongest peaks with frequency near 800 cm−1, are characteristic features for the complex perovskites with B-site ordering.30 Moreover, the broad band in the middle frequency range of 530–610 cm−1 is assigned to be the stretching vibration of Nb–O bonds due to the displacement of Nb5+ from the centre of NbO6 octahedra.37 All these observations from the Raman spectra are coherent with the crystal structures for Ca4GaNbO8 and Sr4GaNbO8 obtained by Rietveld refinements.
Transport properties
Typical AC impedance spectra at different temperatures for Ca4−xSrxGaNbO8 (x = 0, 2, and 4) are presented in Fig. S5† and 7a, where the well-resolved semicircle in the high frequency rang can be modelled with parallel resistance (R) and capacity (C) element. This large semicircle is associated with a capacity close to 1 × 10−12 F cm−1, implying the contribution of bulk response. The absence of inclined-like spike in low frequency range indicates the absence of oxide ionic conductivity. This observation is consistent with the structural features that there are no terminal oxygens, which is capable of immigration at high temperature, boned to only one B-site cation in both Ca4-and Sr4-type structures. Moreover, the oxygen-vacancy ordering is coupled with the B-site cation ordering, which could not be disrupted upon warming. Therefore, the absence of oxide ionic conductivity for Ca4−xSrxGaNbO8 is understandable. The plots of conductivities for Ca4−xSrxGaNbO8 (x = 0, 2, and 4) against temperature is elucidated in Fig. 7b. All the compounds exhibit typical semi-conductive behaviour with comparable bulk conductivities (10−8–10−4 S cm−1) in the measured temperature range. Fitting the σ–T curves with Arrhenius equation gives active energy of ∼ 0.85 eV for all compounds, which in turn corroborated that the B-site cationic ordering is not disrupted by Sr2+-to-Ca2+ replacement.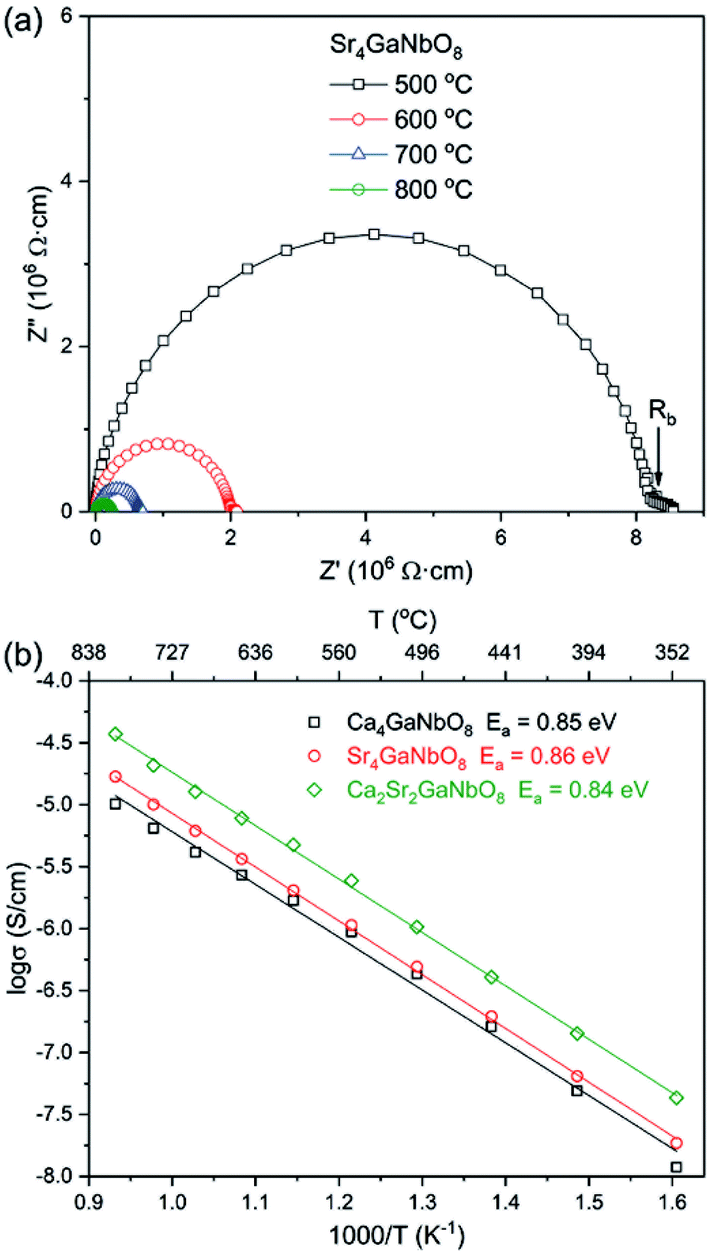 | ||
| Fig. 7 (a) Typical ac impedance spectra for Sr4GaNbO8 at different temperatures. (b) Arrhenius plots of bulk conductivities for Ca4−xSrxGaNbO8 (x = 0, 2, and 4). | ||
Conclusions
Substitution of Ca2+ in Ca4−xSrxGaNbO8 with larger Sr2+ did not lead to oxygen-vacancy and B-site cation ordering–disordering transition but a Ca4-type (x < 0.3) to Sr4-type (x ≥ 0.65) structure transition across a narrow two-phase region (0.3 ≤ x < 0.65). Rietveld refinements revealed that two-type structures possess similar anionic ordering and identical B-site ordering. The structural difference only lies in different orientations of GaO4 tetrahedra, which is responsible for the formation of the narrow two-phase region. In the process of changing the Ca4-phase to Sr4-phase, the Sr2+ cations were mainly incorporated into A1-site for x < 1.5, and then doped into A2/A3-site for x < 3, and finally doped into B1-site (x ≥ 3). Such site-selective doping behaviour observed in Ca4−xSrxGaNbO8 was driven by the distinctly large difference in size for A1, A2/A3, and B1 sites. Incorporation of Sr2+ cations into both A and B sites had no influence on the arrangement of Ga3+/Nb5+ cations and oxygen defects, thus no significant change in electronic properties of Ca4−xSrxGaNbO8 was detected.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Science Foundation of China (21805020, 21671028, 21771027), Natural Science Foundation of Chongqing (cstc2019jcyj-msxmX0330), Fundamental Research Funds for Central Universities (2019CDQYWL009), Chongqing Special Postdoctoral Science Foundation (XmT2018004), and Postdoctoral Research Foundation of China (2018M643402). We also acknowledge the support of sharing fund of large-scale equipment of Chongqing University (201903150092).Notes and references
- N. Hayashi, T. Yamamoto, H. Kageyama, M. Nishi, Y. Watanabe, T. Kawakami, Y. Matsushita, A. Fujimori and M. Takano, Angew. Chem., Int. Ed., 2011, 50, 12547–12550 CrossRef CAS PubMed.
- P. Kayser, M. J. Martinez-Lope, J. A. Alonso, M. Retuerto, M. Croft, A. Ignatov and M. T. Fernández-Díaz, Inorg. Chem., 2013, 52, 11013–11022 CrossRef CAS PubMed.
- R. J. Cava, B. Batlogg, J. J. Krajewski, R. Farrow, L. W. Rupp Jr, A. E. White, K. Short, W. F. Peck and T. Kometani, Nature, 1998, 332, 814–816 CrossRef.
- R. J. Cava, B. Batlogg, R. B. Van Dover, J. J. Krajewski, J. V. Waszczak, R. M. Fleming, W. F. Peck Jr, L. W. Rupp Jr, P. Marsh, A. C. W. P. James and L. F. Schneemeyer, Nature, 1990, 345, 602–604 CrossRef CAS.
- Y. Q. Tan, M. Yan and Y. M. Hao, J. Phys. D: Appl. Phys., 2013, 46, 015303 CrossRef.
- W. Z. Yang, M. M. Mao, X. Q. Liu and X. M. Chen, J. Appl. Phys., 2010, 107, 124102 CrossRef.
- B.-W. Li, M. Osada, T. C. Ozawa and T. Sasaki, Chem. Mater., 2012, 24, 3111–3113 CrossRef CAS.
- B. H. Park, B. S. Kang, S. D. Bu, T. W. Noh, J. Lee and W. Jo, Nature, 1999, 401, 682–684 CrossRef CAS.
- C. A. de Araujo, J. D. Cuchiaro, L. D. McMillan, M. C. Scott and J. F. Scott, Nature, 1995, 374, 627–629 CrossRef CAS.
- S. Ray, A. Kumar, S. Majumdar, E. V. Sampathkumaran and D. D. Sarma, J. Phys.: Condens. Matter, 2001, 13, 607–616 CrossRef CAS.
- Y. Krockenberger, K. Mogare, M. Reehuis, M. Tovar, M. Jansen, G. Vaitheeswaran, V. Kanchana, F. Bultmark, A. Delin, F. Wilhelm, A. Rogalev, A. Winkler and L. Alff, Phys. Rev. B, 2007, 75, 020404 CrossRef.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785–5796 RSC.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- H. X. Geng, J. L. Lan, A. Mei, Y. H. Lin and C. W. Nan, Electrochim. Acta, 2011, 56, 3406–3414 CrossRef CAS.
- K.-Y. Yang, J.-W. Wang and K.-Z. Fung, J. Alloys Compd., 2008, 458, 415–424 CrossRef CAS.
- A. Várez, J. Ibarra, A. Rivera, C. León, J. Santamarıía, M. A. Laguna, M. L. Sanjuán and J. Sanz, Chem. Mater., 2003, 15, 225–232 CrossRef.
- T. Ishihara, H. Matsuda and Y. Takita, J. Am. Chem. Soc., 1994, 116, 3801–3803 CrossRef CAS.
- T. Ishihara, J. A. Kilner, M. Honda and Y. Takita, J. Am. Chem. Soc., 1997, 119, 2747–2748 CrossRef CAS.
- F. Blanc, D. S. Middlemiss, Z. H. Gan and C. P. Grey, J. Am. Chem. Soc., 2011, 133, 17662–17672 CrossRef CAS PubMed.
- G. B. Zhang and D. M. Smyth, Solid State Ionics, 1995, 82, 161–172 CrossRef CAS.
- J. F. Shin, A. Orera, D. C. Apperley and P. R. Slater, J. Mater. Chem., 2011, 21, 874–879 RSC.
- J. F. Shin, L. Hussey, A. Orera and P. R. Slater, Chem. Commun., 2010, 46, 4613–4615 RSC.
- R. K. Hona, A. Huq and F. Ramezanipour, Inorg. Chem., 2017, 56, 14494–14505 CrossRef CAS PubMed.
- T. Yang, J. B. Claridge and M. J. Rosseinsky, Inorg. Chem., 2013, 52, 3795–3802 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- E. Lee and S.-T. Hong, J. Solid State Chem., 2008, 181, 2930–2934 CrossRef CAS.
- J. Li, P. F. Jiang, W. L. Gao, R. H. Cong and T. Yang, Inorg. Chem., 2017, 56, 14335–14344 CrossRef CAS PubMed.
- J.-M. Li, X.-L. Zeng and Z.-A. Xu, Appl. Phys. Lett., 2013, 103, 232410 CrossRef.
- I. Levin, J. Y. Chan, R. G. Geyer, J. E. Maslar and T. A. Vanderah, J. Solid State Chem., 2001, 156, 122–134 CrossRef CAS.
- J. M. S. Skakle and R. Herd, Powder Diffr., 1999, 14, 195–202 CrossRef CAS.
- J. G. Xu, J. H. Wang, X. Tang, X. J. Kuang and M. J. Rosseinsky, Inorg. Chem., 2017, 56, 6897–6905 CrossRef CAS PubMed.
- C. I. Thomas, X. J. Kuang, Z. Q. Deng, H. J. Niu, J. B. Claridge and M. J. Rosseinsky, Chem. Mater., 2010, 22, 2510–2516 CrossRef CAS.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 10–20 CrossRef PubMed.
- C. J. Howard and M. A. Carpenter, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 40–50 CrossRef CAS PubMed.
- J.-M. Li, C. H. A. Huan, Y.-W. Du, D. Feng and Z. X. Shen, Phys. Rev. B, 2000, 63, 024416 CrossRef.
- Y. I. Yuzyuk, E. Gagarina, P. Simon, L. A. Reznitchenko, L. Hennet and D. Thiaudière, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144105 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld refinement patterns, crystallographic parameters, selected interatomic distances for Ca4−xSrxGaNbO8 (x = 0, 1, 1.5, 2, 2.5, 3, 3.5, and 4). See DOI: 10.1039/c9ra09970k |
| This journal is © The Royal Society of Chemistry 2020 |