DOI:
10.1039/C9RA09959J
(Paper)
RSC Adv., 2020,
10, 10245-10253
The protective effects of GLP-1 receptor agonist lixisenatide on oxygen-glucose deprivation/reperfusion (OGD/R)-induced deregulation of endothelial tube formation
Received
28th November 2019
, Accepted 25th January 2020
First published on 10th March 2020
Abstract
Acute myocardial infarction (AMI) is a complication of atherosclerosis that takes place in coronary arteries. Cardiac endothelial cells play a significant role in the pathogenesis of AMI. Oxygen-glucose deprivation/reperfusion (OGD/R) is widely used as a model to simulate AMI in vitro. Recently, antidiabetic GLP-1 receptor agonists have been shown to exert pleiotropic effects that modulate cardiovascular complications. In this study, we investigated the vascular effect of lixisenatide. We show that pre-treatment of endothelial cells with lixisenatide protected them from OGD/R-induced cytotoxicity and improved their viability. Pre-treatment with lixisenatide ameliorated OGD/R-induced ROS accumulation and disturbed endothelial tube formation. At the molecular level, lixisenatide mitigated OGD/R-induced reduced eNOS expression and NO production but further promoted the expression of the anti-oxidant regulators Nrf2 and HO-1. Mechanistically, we confirmed that the PI3K/Akt pathway is essential for mediating the effects of lixisenatide, and blockage of PI3K/Akt using the inhibitor LY294002 abolished the ameliorative effect of lixisenatide on ROS production and impaired tube formation. These data indicate that lixisenatide possesses a beneficial effect on the vasculature in a model of ischemia-induced endothelial injury. We conclude that the GLP-1 receptor agonist lixisenatide has pleiotropic properties that can modulate vascular function independent of its anti-glycemic effect.
1. Introduction
Acute myocardial infarction (AMI) is commonly referred to as a heart attack. AMI occurs when the blood flow from a coronary artery is suddenly blocked, and cardiac muscles are starved to death due to prolonged exposure to ischemic conditions. According to data from the World Health Organization (WHO), AMI accounts for about 12% of mortality worldwide and is the leading cause of hospital admissions and mortality in many countries.1 From the view of its pathogenesis, AMI is a complication resulting from atherosclerosis in the coronary arteries. The majority of AMI results from the rupture or erosion of atherosclerotic lesions. The rupture of coronary plaque refers to the separation of the fibrous cap from the lipid core, while lesion erosion features the disruption of surface smooth muscle cells and proteoglycans without a lipid core.2 From the initial stage of development to the late stage of cardiovascular complications, endothelial dysfunction has been recognized as an important mechanism of atherosclerosis.3 Cardiac endothelial cells play an important role in the remodeling of injured cardiac myocytes after cardiac tissue injury.4 It is estimated that the number of cardiac endothelial cells is almost triple that of cardiomyocytes.5 This large population of cardiac endothelial cells plays a critical role in the formation of microvessels (angiogenesis) in the early stages of remodeling after AMI. Targeted intervention maybe able to prevent the transition to heart failure.6 cardiomyocytes exposed to oxygen-glucose deprivation/reperfusion (OGD/R) injury is extensively used as a model to simulate myocardial ischemia-reperfusion injury.7 OGD/R-induced cellular oxidative stress has been well described,8 and the accumulation of ROS in cardiac tissues constitutes a great risk to cardiac endothelial cells.9 In OGD/R conditions, the PI3K (phosphatidylinositol 3-kinase)/Akt pathway and the Nrf2 (nuclear factor erythroid 2-related factor 2)/HO-1 (heme oxygenase-1)-dependent antioxidant system have been shown to be activated and to act as pro-survival pathways.10
Glucagon-like peptide-1 (GLP-1) is a key factor that can enhance the secretion of insulin and reduce blood glucose levels. GLP-1 analogs and GLP-1 receptor agonists have been intensively investigated as potential treatments for type 2 diabetes. Based on their pharmacodynamic profile, GLP receptor agonists are divided into two classes: short-acting agonists and long-acting agonists. Long-acting agonists are effective in plasma for 24 hours or longer, while short-acting agonists are active for only a few hours.11 Recent studies have shown that GLP-1 agonists have pleiotropic effects that can modulate cardiovascular complications, such as increasing heart rate, reducing blood pressure, and improving myocardial infarction and vascular endothelial function.12–14 Lixisenatide is an improved short-acting GLP-1 receptor agonist widely used in clinical practice. Lixisenatide was developed based on the structure of the earlier GLP-1 analog exendin-4. Lixisenatide contains 44 amino acids and shares 97% homology with native GLP-1, with a plasma half-life of 9 to 14 hours.15 Lixisenatide exhibited consistent cardioprotective effects in animal models.16 We hypothesized that this agent would have vascular protective effects in endothelial cells. In this study, we tested its biological effect in OGD/R-challenged human endothelial cells.
2. Materials and methods
2.1 Reagents
Human umbilical vascular endothelial cells (HUVECs), culture medium (EGM-2), growth factor BulletKits, and 0.25% trypsin–EDTA were purchased from Lonza (USA). Lixisenatide was purchased from Sinopharm Chemical Reagents (Shanghai, China). The fluorescent probes dihydroethidium (DHE) and DAF-FM DA were purchased from Themo Fisher Scientific (USA). LY294002, MTT, DMSO and other chemicals were purchased from Sigma-Aldrich (USA). The primary and secondary antibodies used in this study were purchased from Cell Signaling Technology (USA).
2.2 Endothelial cell culture, OGD/R, and treatment
HUVECs were cultured in complete EGM-2 media with 2% FBS. The cells were maintained at 37 °C in a 5% CO2 humidified incubator and used within passage 3–7 at healthy status. For pretreatment, two doses of lixisenatide (10, 20 nM)17,18 were added to the culture media for 6 hours. For OGD/R conditioning, 95% confluent HUVECs were washed, and the culture medium was replaced with EGM media without glucose. Then, the cells were transferred to an anaerobic chamber with 95% N2/5% CO2 (MIC-101) and incubated at 37 °C for 6 hours. Afterwards, the cells were switched to glucose-containing media and incubated for another 24 hours under normal culture conditions. To block the activity of PI3K/Akt, the cells were pretreated with 20 μM LY294002 for 6 hours.
2.3 MTT assay
Cell viability was measured using an MTT assay. Briefly, 1 × 104 HUVECs were placed in a 96-well plate. After necessary treatment, cells were reacted with 20 μL of MTT solution (Sigma-Aldrich, USA), and incubated at 37 °C for 4 hours in darkness. The purple formazan product was then solubilized by incubating the cells with 150 μL of DMSO. The OD values were read by a microplate reader at a wavelength of 490 nm. Cell viability was expressed as a percentage of the control group.
2.4 Lactate dehydrogenase (LDH) assay
Cell cytotoxicity was determined by the measurement of released LDH. Briefly, 1 × 104 HUVECs were placed in a 96-well plate. After necessary treatment, cell culture media was collected and centrifuged to obtain supernatant. The released LDH was detected using a commercial LDH assay kit (Thermo Fisher Scientific, USA) in accordance with the manufacturer's manual. The reacted plate was read at 490 nm, and the rate of LDH release was normalized by subtracting the blank wells and comparing to non-treated cells.
2.5 Endothelial tube formation assay
Endothelial tube formation was assessed by culturing HUVECs on a commercial Matrigel matrix (BD Biosciences). In brief, 5000 HUVECs were plated in a 24-well plate precoated with 6 mg mL−1 cold Matrigel membrane. After 72 hours of incubation at 37 °C, 10 random areas were imaged and analyzed to quantify tube formation using the software Image J (NIH, USA).
2.6 ROS measurement
Oxidative stress in HUVECs was measured using the ROS probe DHE (Life Science, Inc). After OGD/R or lixisenatide treatment, HUVECs were washed with PBS and incubated with 10 μM DHE at 37 °C for 30 minutes in darkness. Cells were then immediately imaged using a laser-scanning confocal microscope (Leica TCS SP2). The fluorescence intensity was quantitated using Image J software (NIH, USA).
2.7 Determination of intracellular nitric oxide (NO)
The intracellular NO level was measured based on the results of staining with the fluorescent probe DAF-FM DA. In brief, HUVECs were washed and reacted with DAF-FM DA solution for 30 minutes. The released DAF-FM captured intracellular NO and formed visible green-fluorescence. The fluorescence intensity of DAF-FM was digitally quantified using a Leica fluorescence microscope.
2.8 Western blot analysis
To obtain cell lysates, HUVECs were lysed in ice-cold RIPA buffer (20 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate) plus proteinase and phosphatase inhibitor tablets for half an hour. The lysates were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C to remove debris, and the protein concentration was quantified using a BCA Protein Assay Kit (Sigma-Aldrich, USA). About 20 μg of protein was mixed with loading buffer and boiled at 95 °C for 5 minutes. Equal amounts of protein were separated on a 10% SDS-PAGE and transferred to PVDF membranes. After blocking with 5% non-fat milk, the membranes were incubated at 4 °C overnight with primary antibodies and further incubated with horseradish peroxidase-conjugated secondary antibodies. The blots were developed with enhanced chemiluminescence reagents (ECL) and scanned with a Canon scanner. The density of each band was normalized to GAPDH using Image J software (NIH). The following antibodies were used in this study: NRF2 (1:1000, #ab137550, Abcam, USA); HO-1 (1:1000, #82206, Cell Signaling Technology, USA); p-PI3K (1:1000, #17366, Cell Signaling Technology, USA); PI3K (1:5000, #4292, Cell Signaling Technology, USA); p-Akt (1:1000, #4060, Cell Signaling Technology, USA); Akt (1:3000, #4691, Cell Signaling Technology, USA); eNOS (1:1000, #9586, Cell Signaling Technology, USA); p-eNOS (1:1000, #9570, Cell Signaling Technology, USA); β-actin (1:10000, #3700, Cell Signaling Technology, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked antibody (1:2000, #7076, Cell Signaling Technology, USA).
000 rpm at 4 °C to remove debris, and the protein concentration was quantified using a BCA Protein Assay Kit (Sigma-Aldrich, USA). About 20 μg of protein was mixed with loading buffer and boiled at 95 °C for 5 minutes. Equal amounts of protein were separated on a 10% SDS-PAGE and transferred to PVDF membranes. After blocking with 5% non-fat milk, the membranes were incubated at 4 °C overnight with primary antibodies and further incubated with horseradish peroxidase-conjugated secondary antibodies. The blots were developed with enhanced chemiluminescence reagents (ECL) and scanned with a Canon scanner. The density of each band was normalized to GAPDH using Image J software (NIH). The following antibodies were used in this study: NRF2 (1:1000, #ab137550, Abcam, USA); HO-1 (1:1000, #82206, Cell Signaling Technology, USA); p-PI3K (1:1000, #17366, Cell Signaling Technology, USA); PI3K (1:5000, #4292, Cell Signaling Technology, USA); p-Akt (1:1000, #4060, Cell Signaling Technology, USA); Akt (1:3000, #4691, Cell Signaling Technology, USA); eNOS (1:1000, #9586, Cell Signaling Technology, USA); p-eNOS (1:1000, #9570, Cell Signaling Technology, USA); β-actin (1:10000, #3700, Cell Signaling Technology, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked antibody (1:2000, #7076, Cell Signaling Technology, USA).
2.9 Statistical analysis
Data were analyzed using the commercial SPSS software package. The test significance of the mean difference was determined by one-way analysis of variance (ANOVA) followed by Tukey's test or by a two-tailed type 2 Student's t-test. P values less than 0.05 were determined to be statistically significant.
3. Results
3.1 Lixisenatide protects against OGD/R-induced endothelial cell death
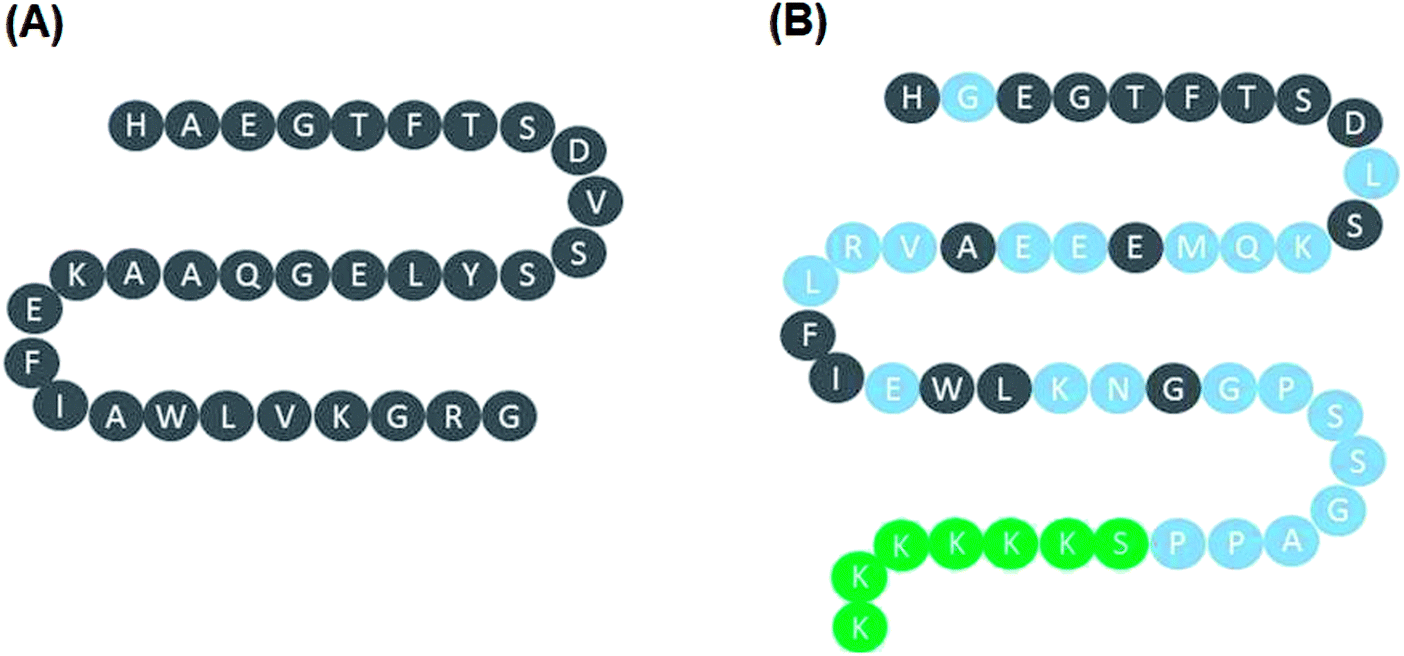
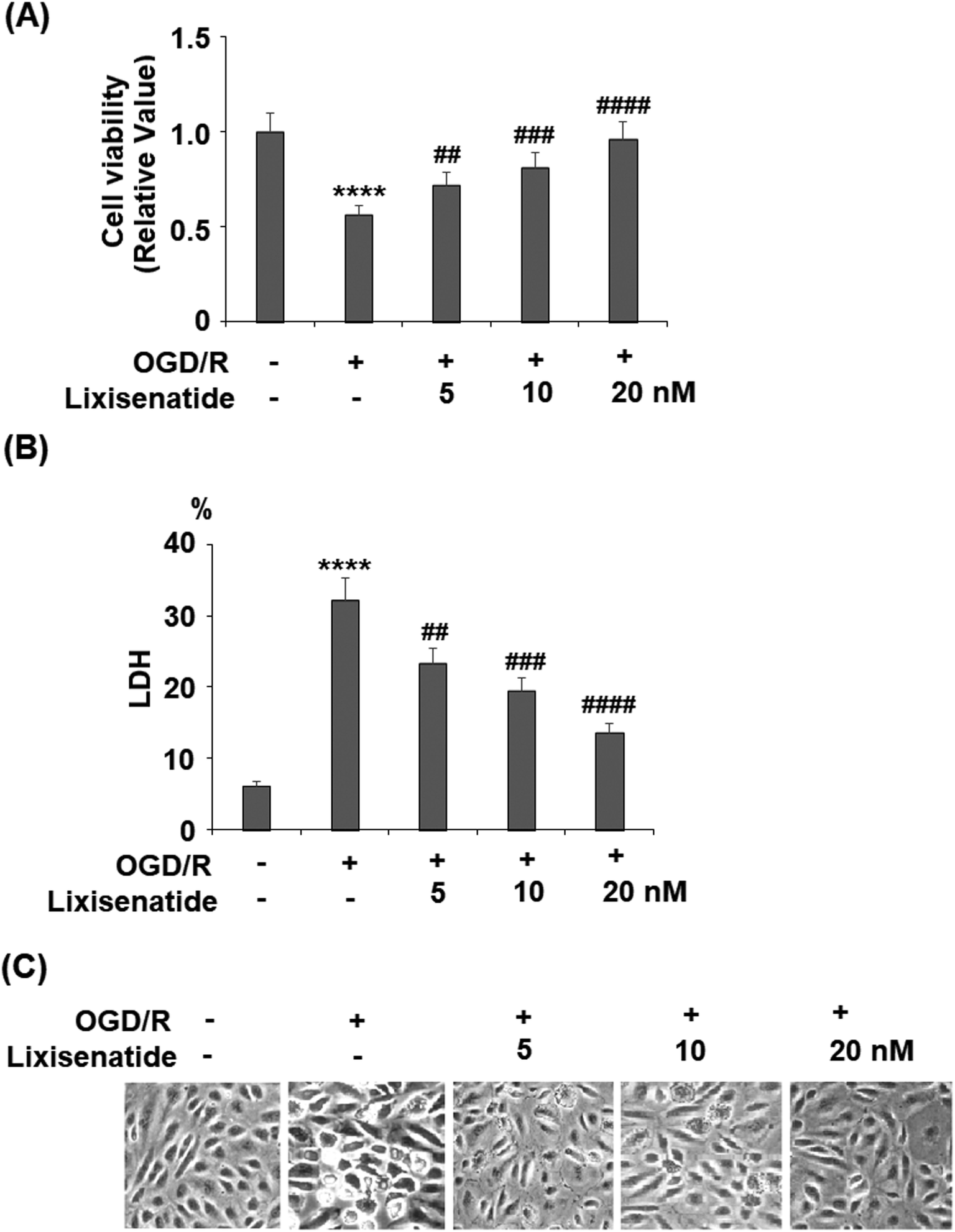
Lixisenatide is an analog of human GLP-1. The peptide sequences of native human GLP-1 and lixisenatide are displayed in Fig. 1A and B, respectively. To investigate the effect of lixisenatide in vascular endothelial cells, we pretreated HUVECs with lixisenatide prior to OGD/R conditioning. The aim of our experiment was to assess whether lixisenatide pretreatment had any influence on OGD/R-induced endothelial injury. Using a MTT assay, we found that pretreatment with the three doses of lixisenatide (5, 10, 20 nM) showed a protective effect against OGD/R-induced reduced cell viability and this protection was dose-responsive as 20 nM lixisenatide protected nearly the entire population of HUVECs from OGD/R-induced death (Fig. 2A). Meanwhile, we found that pretreatment with the three doses of lixisenatide (5, 10, 20 nM) exerted a suppressive effect on OGD/R-induced release of LDH (Fig. 2B). The protective effect of lixisenatide on cell morphology is shown in the representative images (Fig. 2C).
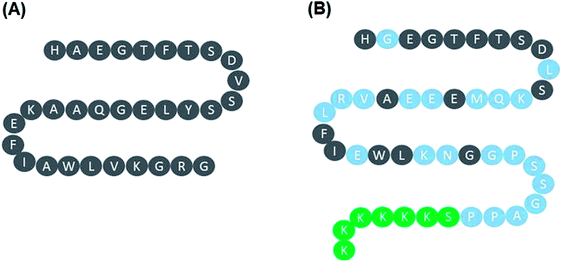 |
| | Fig. 1 Peptide sequence of native human GLP-1 and lixisenatide. (A) Native human GLP-1; (B) lixisenatide. | |
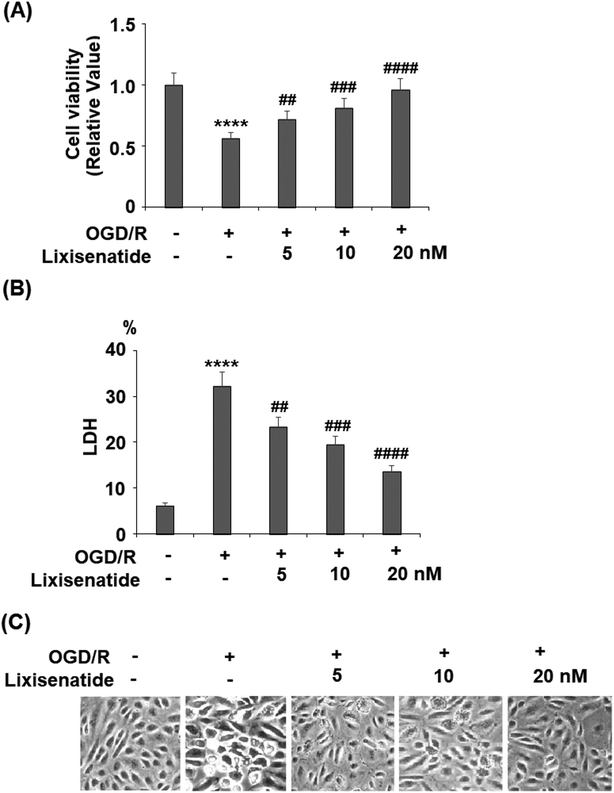 |
| | Fig. 2 Lixisenatide protects oxygen-glucose deprivation/reperfusion (OGD/R)-induced reduction of cell viability and release of LDH in HUVECs. Cells were treated with lixisenatide (5, 10, 20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). (A) Cell viability of HUVECs; (B) release of lactate dehydrogenase (LDH); (C) cell morphology of HUVECs (****, P < 0.0001 vs. control group; ##, ###, ####, P < 0.01, 0.001, 0.0001 vs. OGD/R treatment group, n = 5). | |
3.2 Lixisenatide protects against OGD/R-induced ROS production
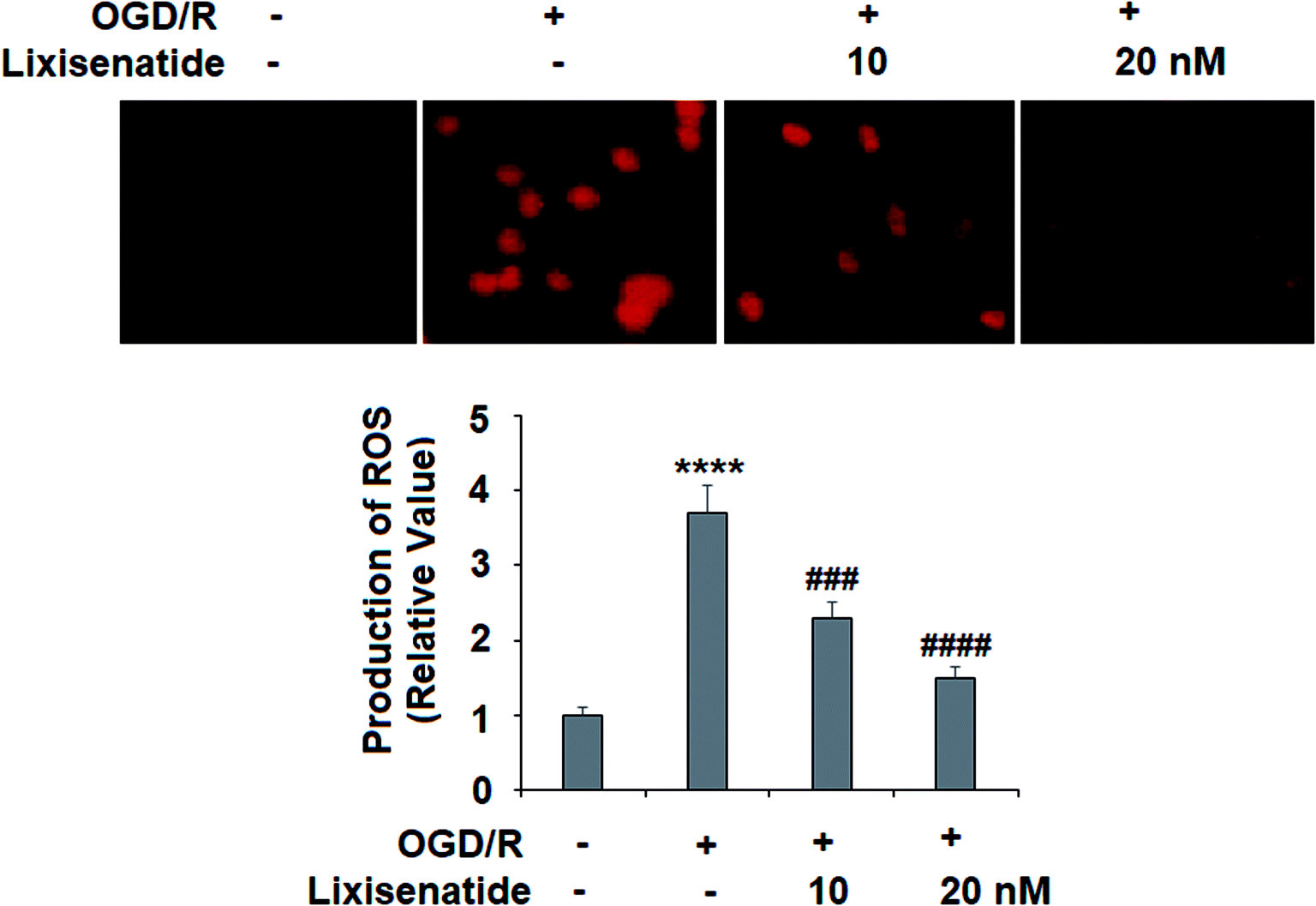
OGD/R-induced production of ROS is critical to endothelial injury, and so we measured the effect of lixisenatide on cellular ROS levels. By staining treated cells with dihydroethidium (DHE), we found that pretreatment with lixisenatide dose-responsively suppressed OGD/R-mediated ROS generation (Fig. 3).
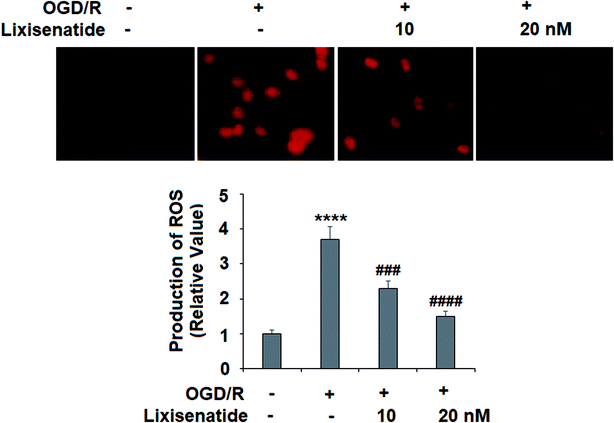 |
| | Fig. 3 Lixisenatide reduced OGD/R-induced generation of ROS in HUVECs. Cells were treated with lixisenatide (10, 20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). Generation of ROS was measured by dihydroethidium (DHE) staining (****, P < 0.0001 vs. control group; ###, ####, P < 0.001, 0.0001 vs. OGD/R treatment group, n = 5). | |
3.3 Lixisenatide promotes OGD/R-induced cellular Nrf2 and HO-1 expression
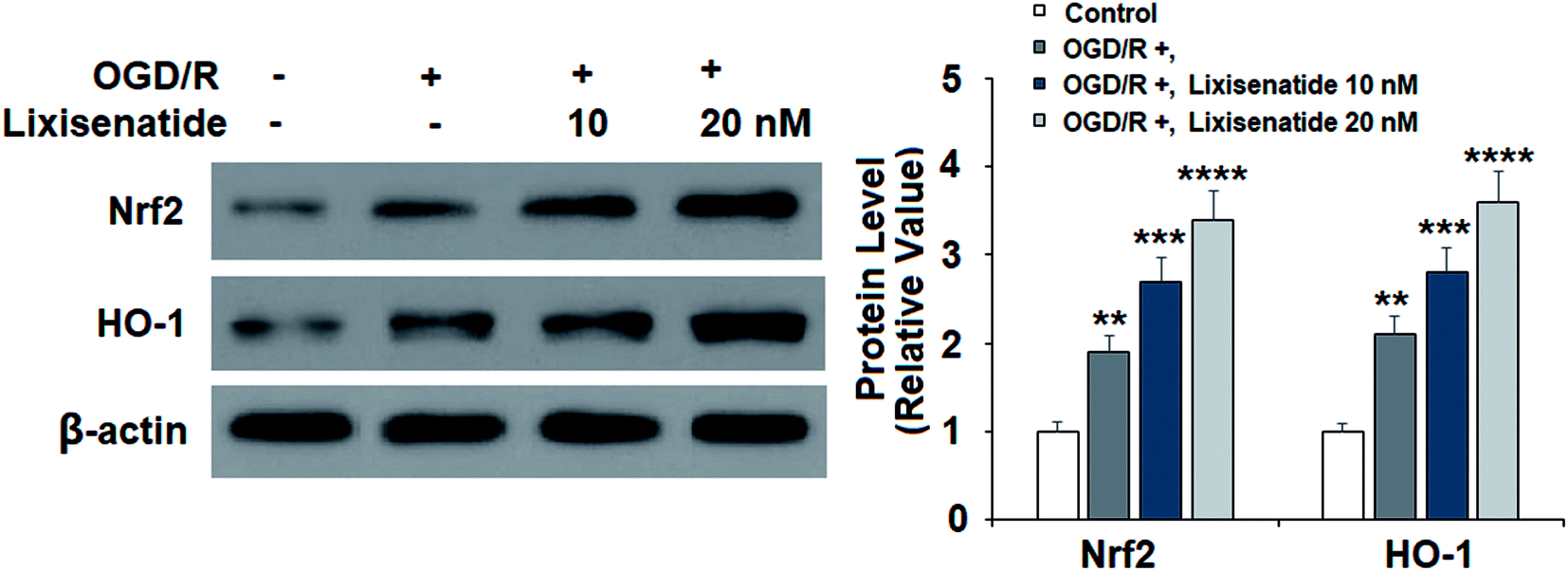
Next, we evaluated the expression of two key anti-oxidant proteins in our treatment experiment. Compared with non-treated cells, OGD/R induced about two-fold expression of both Nrf2 and HO-1 proteins, while pretreatment with lixisenatide significantly promoted this increase, and high dose of lixisenatide pretreatment increased the expression of both Nrf-2 and HO-1 more than three-fold (Fig. 4).
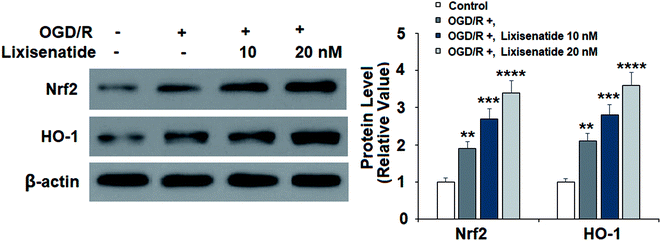 |
| | Fig. 4 Lixisenatide activated the expression of Nrf2 and HO-1 under OGD/R in HUVECs. Cells were treated with lixisenatide (10, 20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). Expression of Nrf2 and HO-1 was measured by western blot analysis (**, ***, ****, P < 0.01, 0.001, 0.0001 vs. control group, n = 4). | |
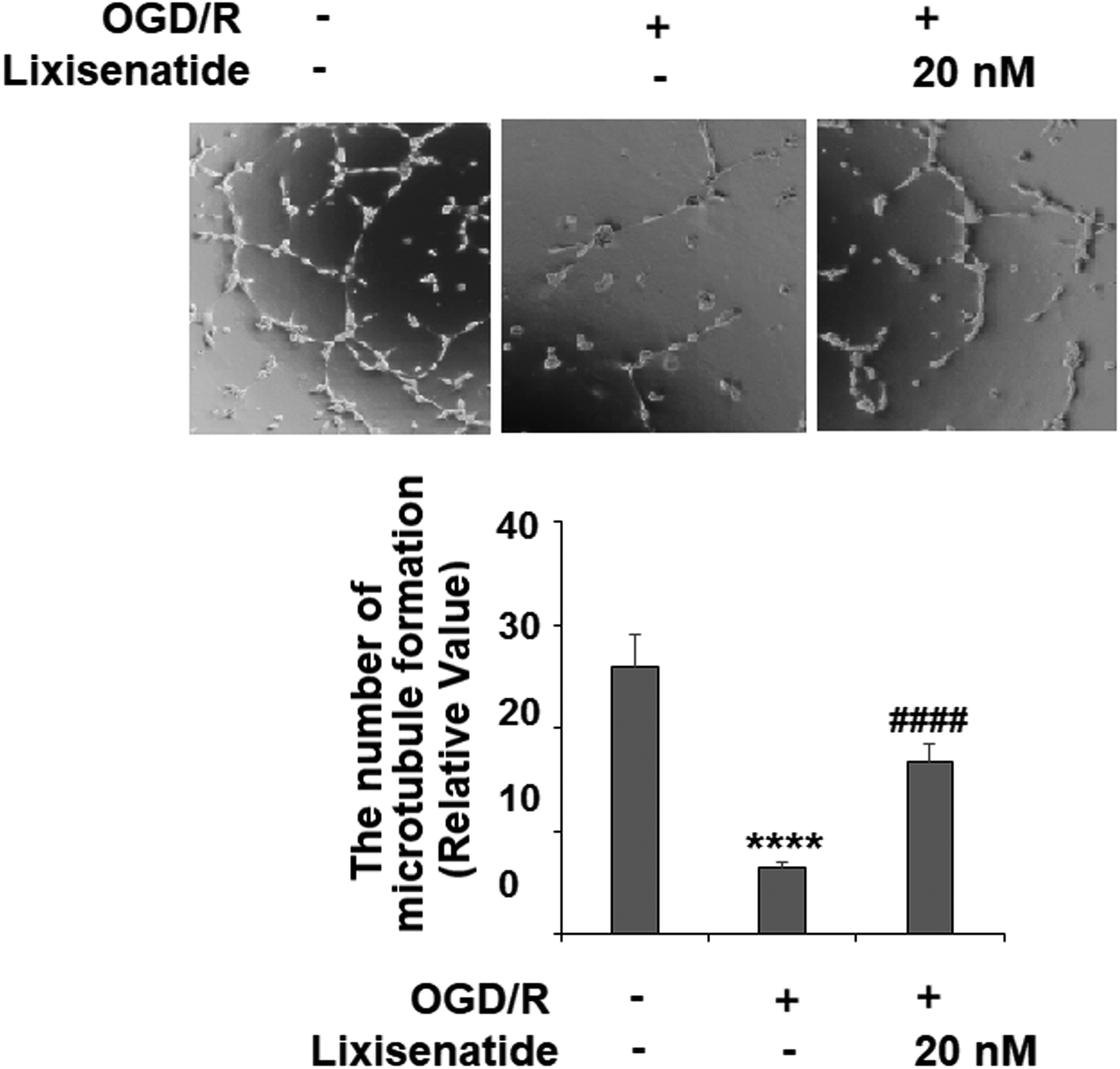
3.4 Lixisenatide restored OGD/R-induced reduced endothelial tube formation
We also measured the influence of lixisenatide on endothelial angiogenesis in vitro via tube formation assay. Our experiments revealed that OGD/R treatment suppressed the formation of capillary-like tubes by about 80% in vitro, but pretreatment with 20 nM lixisenatide relieved the impairment of capillary-like tube formation by almost half (Fig. 5).
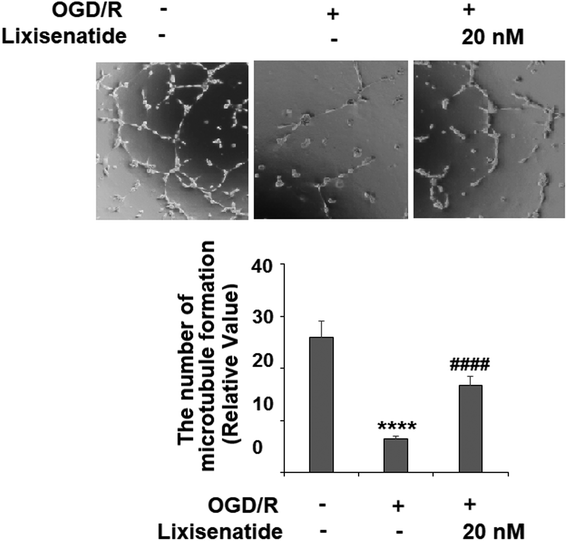 |
| | Fig. 5 Lixisenatide prevented OGD/R-induced reduced angiogenesis in HUVECs. Cells were treated with lixisenatide (20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). Microtubule formation was quantified by Matrigel assay (****, P < 0.0001 vs. control group; ####, P < 0.0001 vs. OGD/R treatment group, n = 5). | |
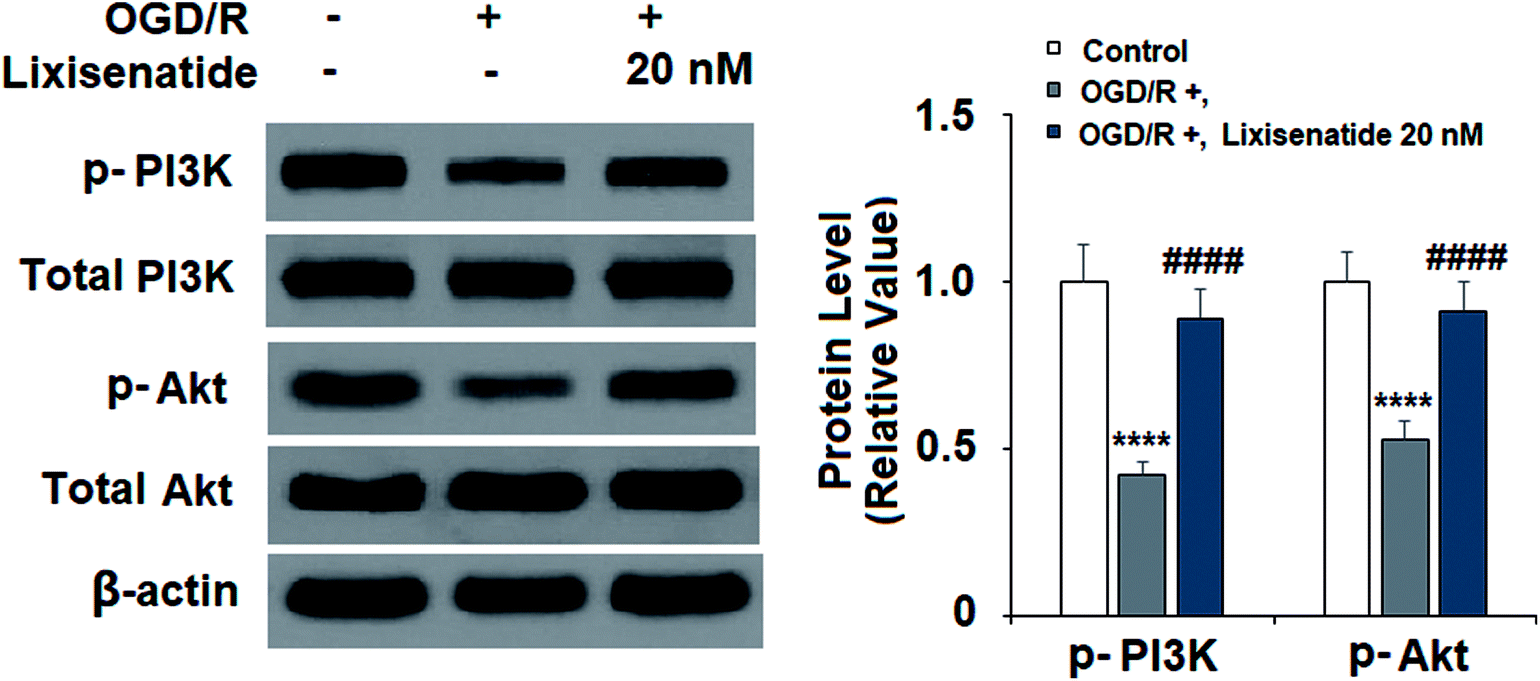
3.5 Lixisenatide alleviates OGD/R-mediated inactivation of the PI3K/Akt pathway
The PI3K/Akt pathway has been reported to play an important role in regulating angiogenesis and cell survival. We explored the effect of lixisenatide on the PI3K/Akt pathway. Compared to non-treated cells, OGD/R treatment reduced both active PI3K (phospho-PI3K) and Akt (phospho-Akt) levels, but pretreatment with 20 nM lixisenatide robustly alleviated OGD/R-mediated reduced active PI3K and Akt (Fig. 6).
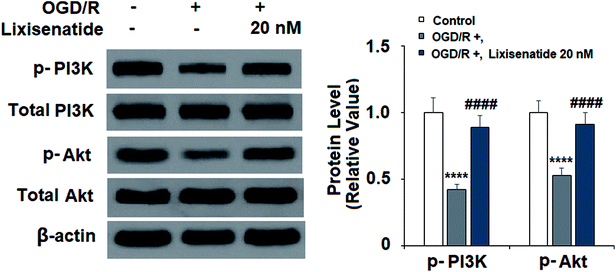 |
| | Fig. 6 Lixisenatide prevented OGD/R-induced dephosphorylation of PI3K and Akt in HUVECs. Cells were treated with lixisenatide (20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). Phosphorylated and total levels of PI3K and Akt were measured by western blot analysis (****, P < 0.0001 vs. control group; ####, P < 0.0001 vs. OGD/R treatment group, n = 4). | |
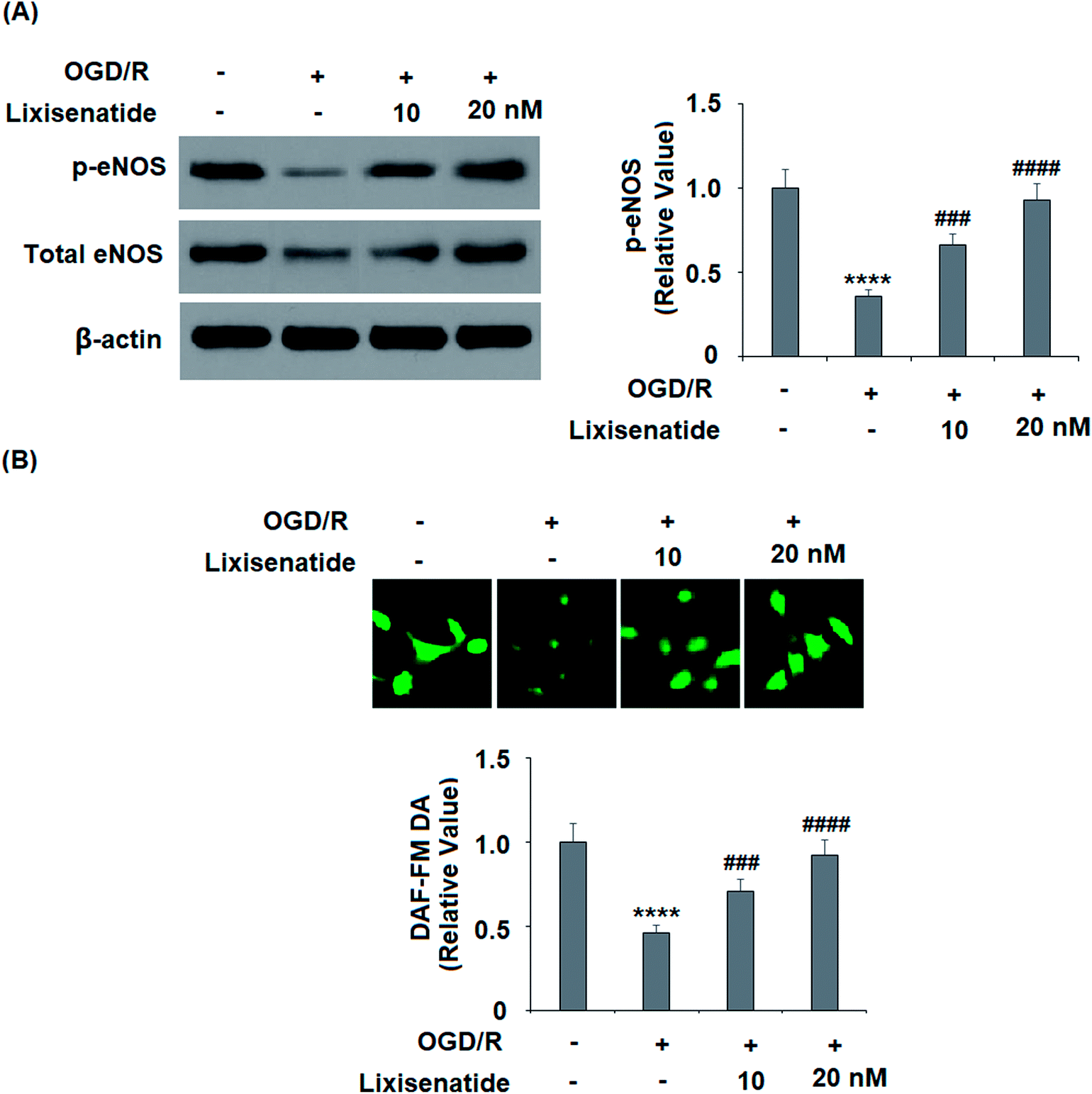
3.6 Lixisenatide alleviates OGD/R-induced reduced eNOS phosphorylation and NO production
Endothelial nitric oxide (NO) produced by endothelial NO synthase (eNOS) is a critical regulator of vascular protection. We examined the active level of eNOS (phosphorylated eNOS) and the cellular NO level. As expected, pretreatment with lixisenatide dose-responsively alleviated OGD/R-induced reduced phosphorylated eNOS (Fig. 7A) and intracellular NO production (Fig. 7B).
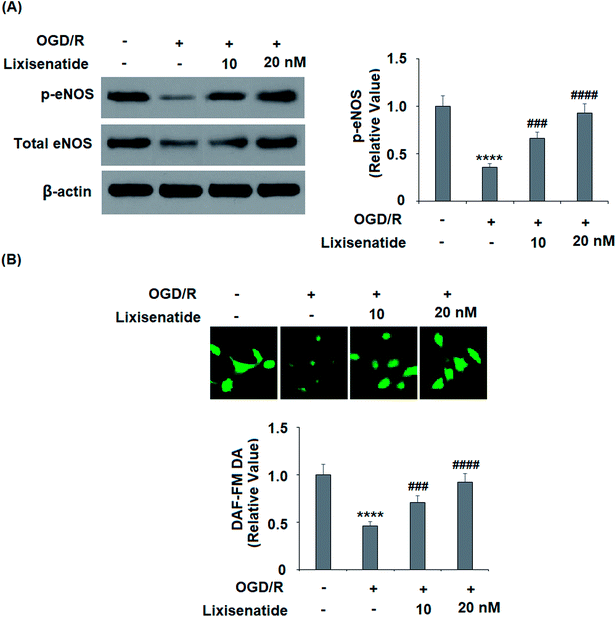 |
| | Fig. 7 Lixisenatide prevented OGD/R-induced dephosphorylation of eNOS and production of nitric oxide (NO) in HUVECs. Cells were treated with lixisenatide (20 nM) for 6 h, followed by exposure to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). (A) Phosphorylated and total levels of eNOS were measured by western blot analysis. (B) Intracellular nitric oxide (NO) was measured by diaminofluorescein-FM diacetate (DAF-FM DA) (****, P < 0.0001 vs. control group; ###, ####, P < 0.01, 0.001, 0.0001 vs. OGD/R treatment group, n = 5). | |
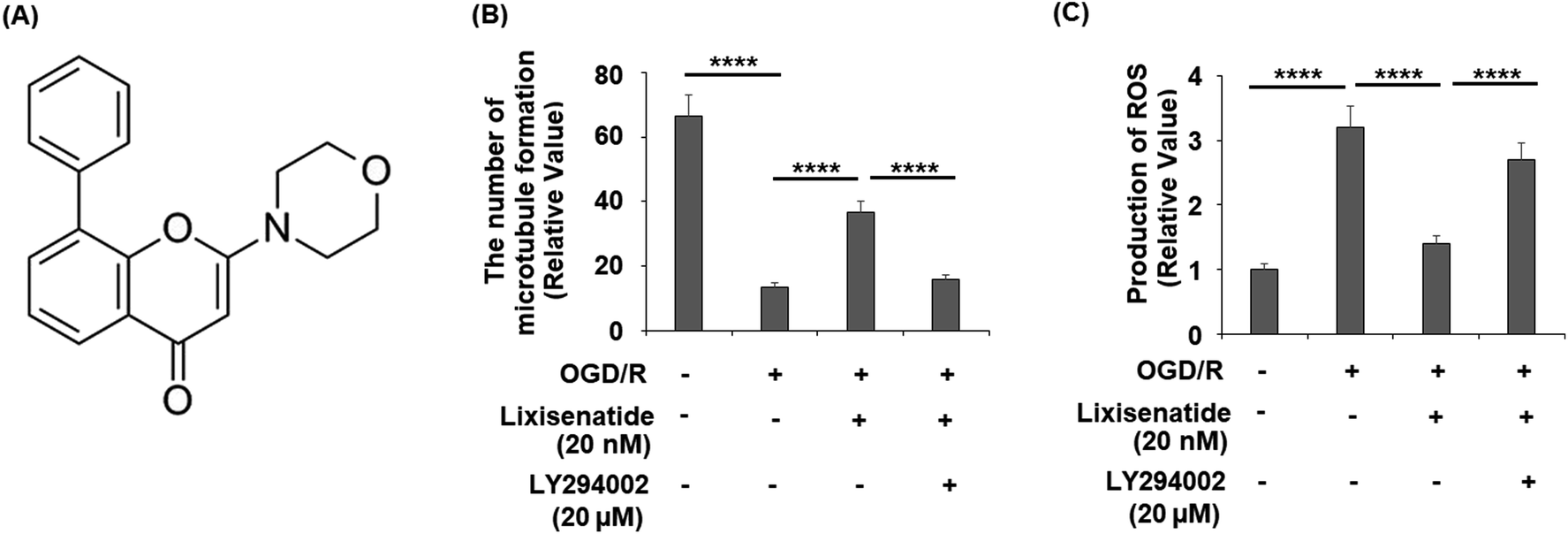
3.7 The effect of lixisenatide is dependent on the PI3K/Akt pathway
Finally, we assessed whether the activation of PI3K/Akt is necessary for the protective action of lixisenatide. We employed the specific inhibitor LY294002 to block the PI3K/Akt pathway in our OGD/R and lixisenatide treatment experiment. The molecular structure of LY294002 is shown in Fig. 8A. The results show that the presence of LY294002 completely abolished the ability of lixisenatide to relieve OGD/R-induced reduced tube formation (Fig. 8B) and increased ROS (Fig. 8C).
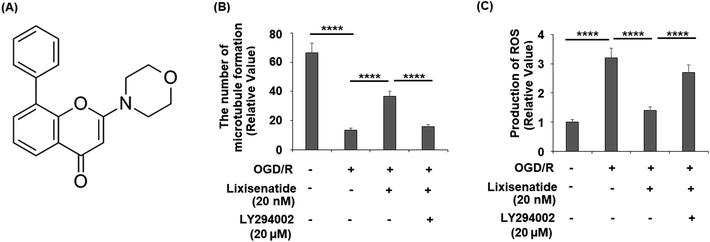 |
| | Fig. 8 Blockage of the PI3K/Akt signaling pathway using LY294002 abolished the protective effects of lixisenatide on promoting angiogenesis in HUVECs. Cells were treated with lixisenatide (20 nM) in the presence or absence of LY294002 (20 μM) for 6 h, followed by exposed to oxygen-glucose deprivation (6 h)/reperfusion (24 h) (OGD/R). (A) Molecular structure of LY294002; (B) microtubule formation was quantified by Matrigel assay; (C) production of ROS (****, P < 0.0001, n = 5). | |
4. Discussion
Endothelial cells play a significant role in cardiac ischemia/reperfusion (I/R) injury.6 OGD/R-induced hypoxia and metabolic stress in cultured endothelial cells is used to mimic I/R injury in vivo. OGD/R-induced endothelial cytotoxicity changes the cellular pH balance, increases oxidative stress, and reduces the production of endothelial nitric oxide.19 Native GLP-1 and several GLP-1 receptor agonists have been shown to have beneficial impacts on vascular function.20,21 Our study aimed to investigate the effect of the commonly used GLP-1 agonist lixisenatide in OGD/R-challenged endothelial cells. We found that lixisenatide exhibited protective effects against OGD/R-induced cytotoxicity and improved endothelial survival. Lixisenatide also exhibited an alleviative effect on OGD/R-induced ROS production, indicating that it may relieve OGD/R-induced oxidative stress by suppressing cellular ROS production. Furthermore, we found that lixisenatide exerted a protective effect on OGD/R-induced reduced endothelial tube formation, indicating that this agonist possesses a pro-angiogenic role. Upon damage from ischemia, the heart undergoes extensive revascularization.22 The pro-angiogenic effect of lixisenatide suggests that this agent may promote revascularization in the ischemia-injured heart. At the molecular level, we found that lixisenatide promoted the production of the anti-oxidative regulators Nrf2 and HO-1. Nrf2 is a critical regulator of cellular ROS production as well as the endogenous antioxidant defense system, thereby playing an active role in the resistance to intracellular ROS. HO-1 is the direct target of Nrf2, and its induction is critical to preventing vascular inflammation.23
GLP-1, identified as an endogenous peptide hormone released by intestinal L-cells with a short half-life of 3–5 minutes, is easily degraded by dipeptidyl peptidase-4 (DPP-4).24 Lixisenatide is the fourth GLP-1R agonist. It contains 44 amino acids with a single proline substitution and a modified C-terminus of six lysine molecules, which make it relatively resistant to degradation by DPP-4 and provides an extended half-life, allowing for once daily dosing.25 Administration of lixisenatide has been shown to improve glycemic control and lower HbA1c levels in patients with type 2 diabetes. Lixisenatide has displayed appropriate pharmacodynamic actions by reducing blood glucose, improving glycemic control, and lowering body weight with 20 μg once-daily dosing.26 Interestingly, lixisenatide possesses pleiotropic effects in different types of cells and tissues. For example, treatment with lixisenatide inhibited the inflammatory response through the downregulation of proinflammatory cytokines and matrix metalloproteinases in human fibroblast-like synoviocytes.17 Importantly, lixisenatide has shown a protective effect against free fatty acids (FFA)-induced endothelial dysfunction by suppressing the activation of the NF-κB pathway in endothelial cells,18 which is consistent with the findings in our study.
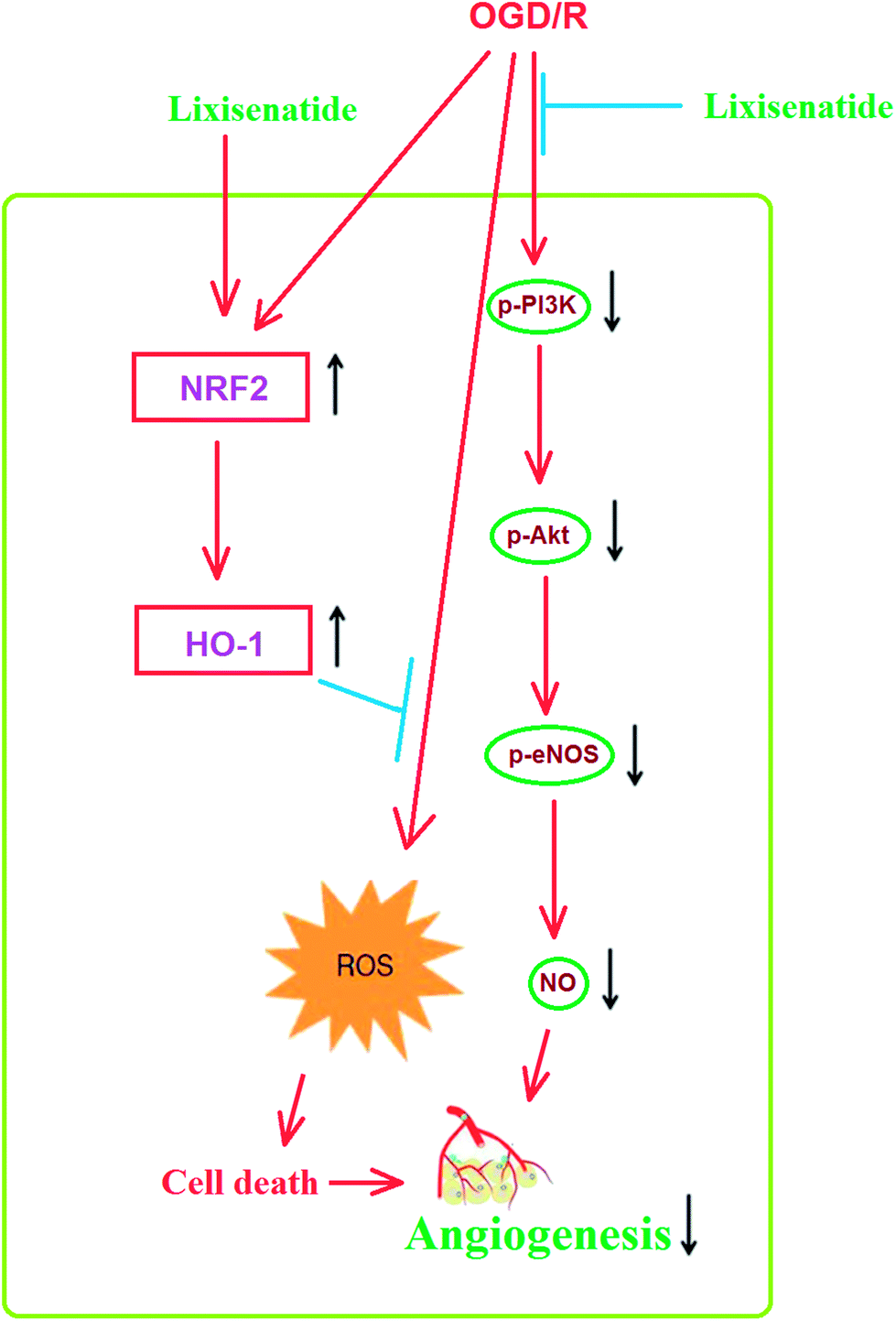
Mechanistically, we showed that the PI3K/Akt pathway is required for the effect of lixisenatide in endothelial cells. Multiple studies have demonstrated the central role of PI3K/Akt in endothelial cell protection. Firstly, the PI3K/Akt pathway has been shown to be involved in the anti-inflammatory response in OGD/R-induced endothelial injury models.27 It has been shown that blockage of endothelial PI3K/Akt activation increases Nrf2/HO-1 expression.28 Secondly, a previous study has shown that endothelial production of NO is also dependent on PI3K/Akt signaling. Thirdly, the PI3K/Akt pathway is also essential for pro-angiogenic factor-induced endothelial tube formation.29,30 We hypothesize that the induction of the anti-oxidative regulator Nrf2/HO-1 by lixisenatide and its amelioration of OGD/R-induced NO production and tube formation is dependent on the PI3K/Akt pathway. To verify this dependence, we blocked the kinase activity of PI3K/Akt by its specific inhibitor LY294002 and found that the loss of PI3K/Akt kinase activity completely abolished the dual effect of lixisenatide on tube formation and ROS production. Thus, we conclude that the PI3K/Akt pathway is essential for the beneficial actions of lixisenatide in endothelial cells. In accordance with our statement, a recent study shows that lixisenatide administration relieves endothelial dysfunction by increasing eNOS expression and reduces ROS production in an ischemic stroke rat model.31 Therefore, we conclude that lixisenatide plays an anti-oxidative, anti-inflammatory, and pro-angiogenic role in ischemic endothelial injury. A graphical representation of the underlying mechanism is shown in Fig. 9.
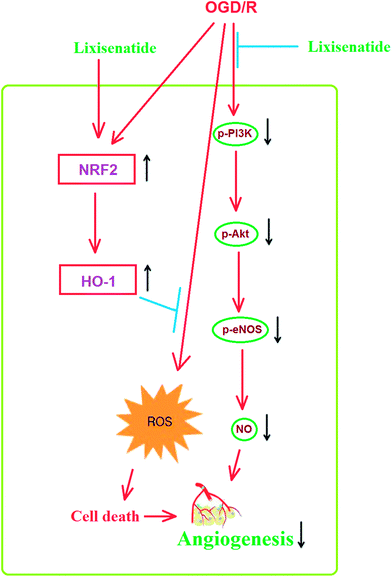 |
| | Fig. 9 A graphical representation of the underlying mechanism. | |
In summary, our study provides evidence that the molecular mechanism of lixisenatide provides beneficial vascular effects in an acute ischemic injury model and implies that lixisenatide could have a pleiotropic role in modulating cardiovascular function independent of its glucose lowing capacity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study is funded by (1) “Heilongjiang Provincial Scientific Research Program (201813). The mechanism of adipocytokines on atrial fibrosis through focal adhesion kinase signal pathway. (2) “Heilongjiang Province Youth Science Foundation Project” (QC2016122). The early diagnosis of contrast-induced nephropathy through blood cystatin C and urinary trace protein series (MA, TRU, IGU, AIM)”.
References
- World Health Organization, The Global Burden of Disease: 2004 Update, Geneva, World Health Organization, 2008 Search PubMed.
- R. Virmani, A. P. Burke and A. Farb, Thromb. Haemostasis, 1999, 82(suppl. 1), 1–3 Search PubMed.
- M. A. Gimbrone Jr and G. García-Cardeña, Circ. Res., 2016, 118(4), 620–636 CrossRef PubMed.
- V. F. M. Segers, D. L. Brutsaert and G. W. De Keulenaer, Front. Physiol., 2018, 9, 382 CrossRef PubMed.
- A. R. Pinto, A. Ilinykh, M. J. Ivey, J. T. Kuwabara, M. L. D'Antoni, R. Debuque, A. Chandran, L. Wang, K. Arora, N. A. Rosenthal and M. D. Tallquist, Circ. Res., 2016, 118, 400–409 CrossRef PubMed.
- C. Cochain, K. M. Channon and J. S. Silvestre, Antioxid. Redox Signaling, 2013, 18, 1100–1113 CrossRef PubMed.
- G. Z. Yang, F. S. Xue, Y. Y. Liu, H. X. Li, Q. Liu and X. Liao, China Med. J., 2018, 131(19), 2277–2286 CrossRef PubMed.
- Y. Liu, X. D. Song, W. Liu, T. Y. Zhang and J. Zuo, J. Cell. Mol. Med., 2003, 7(1), 49–56 CrossRef PubMed.
- P. Song and M. H. Zou, Cell. Mol. Life Sci., 2014, 71(17), 3219–3239 CrossRef PubMed.
- H. Qi, Y. Han and J. Rong, Neuropharmacology, 2012, 62(4), 1659–1670 CrossRef PubMed.
- A. Uccellatore, S. Genovese, I. Dicembrini, E. Mannucci and A. Ceriello, Diabetes Ther., 2015, 6(3), 239–256 CrossRef PubMed.
- M. H. Davidson, Am. J. Cardiol., 2011, 108, 33B–41B CrossRef PubMed.
- V. Gupta, Indian J. Endocrinol. Metab., 2013, 17(3), 413–421 CrossRef PubMed.
- M. I. Del Olmo-Garcia and J. F. Merino-Torres, J. Diabetes Res., 2018, 2018, 4020492 Search PubMed.
- T. Forst and A. Pfützner, Expert Opin. Pharmacother., 2013, 14(16), 2281–2296 CrossRef PubMed.
- P. Wohlfart, W. Linz, T. Hübschle, D. Linz, J. Huber, S. Hess, D. Crowther, U. Werner and H. Ruetten, J. Transl. Med., 2013, 11, 84 CrossRef PubMed.
- X. Du, H. Zhang, W. Zhang, Q. Wang, W. Wang and G. Ge, Int. Immunopharmacol., 2019, 75, 105732 CrossRef PubMed.
- Q. Zhao, H. Xu, L. Zhang, L. Liu and L. Wang, Artif. Cells, Nanomed., Biotechnol., 2019, 47(1), 2325–2332 CrossRef PubMed.
- Q. Yang, G. W. He, M. J. Underwood and C. M. Yu, Am. J. Transl. Res., 2016, 8(2), 765–777 Search PubMed.
- A. Sjöholm, Diabetes, Obes. Metab., 2009, 11(suppl. 3), 19–25 CrossRef PubMed.
- M. Almutairi, R. Al Batran and J. R. Ussher, Peptides, 2019, 111, 26–32 CrossRef.
- K. Iwasaki, World J. Cardiol., 2014, 6(4), 130–139 CrossRef.
- S. R. McSweeney, E. Warabi and R. C. Siow, Hypertension, 2016, 67(1), 20–29 CrossRef.
- T. D. Müller, B. Finan, S. R. Bloom, D. D'Alessio, D. J. Drucker and P. R. Flatt, et al., Mol. Metab., 2019, 30, 72–130 CrossRef PubMed.
- S. C. Bain, Diabetes Ther., 2014, 5(2), 367–383 CrossRef PubMed.
- A. N. Okere, J. Montesdeoca, A. Glasper and V. Diaby, Curr. Diabetes Rev., 2018, 14(4), 363–375 CrossRef.
- S. Zhang, X. Hu, S. Guo, L. Shi, Q. He, P. Zhang, S. Yu and R. Zhao, J. Pharmacol. Sci., 2019, 140(1), 62–72 CrossRef.
- T. Zhang, X. Liang, L. Shi, L. Wang, J. Chen, C. Kang, J. Zhu and M. Mi, PLoS One, 2013, 8(11), e79075 CrossRef.
- J. Karar and A. Maity, Front. Mol. Neurosci., 2011, 4, 51 CrossRef.
- N. Gu, J. Wang, Z. Di, Z. Liu, X. Jia, Y. Yan, X. Chen, Q. Zhang and Y. Qian, Front. Neurol., 2019, 10, 383 CrossRef.
- R. G. Abdel-Latif, G. H. Heeba, A. Taye and M. M. A. Khalifa, Naunyn-Schmiedeberg's Arch. Pharmacol., 2018, 391(7), 705–717 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d and
Dong Chang*e
*d and
Dong Chang*e