
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes
Lifen Peng *a,
Zhifang Hua,
Hong Wanga,
Li Wua,
Yinchun Jiaoa,
Zilong Tanga and
Xinhua Xu*b
*a,
Zhifang Hua,
Hong Wanga,
Li Wua,
Yinchun Jiaoa,
Zilong Tanga and
Xinhua Xu*b
aKey Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, Hunan Provincial Key Laboratory of Controllable Preparation and Functional Application of Fine Polymers, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, P. R. China. E-mail: 1060137@hnust.edu.cn
bState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China
First published on 10th March 2020
Abstract
In this review, direct cyanation, hydrocyanation, dicyanation, cyanofunctionalization and other cyanation reactions of alkynes were highlighted. Firstly, the use of nitriles and development of cyanation was simply introduced. After presenting the natural properties of alkynes, cyanation reactions of alkynes were classified and introduced in detail. Transition metal catalysed direct cyanation and hydrocyanation of alkynes gave alkynyl cyanides and alkenyl nitriles in good yields. Dicyanation of alkynes produced 1,2-dicyano adducts. Cyanofunctionalization of alkynes afforded functional cyanated compounds. Thiocyanation and selenocyanation yielded the expected functional vinylthiocyanates and vinylselenocyanates. A plausible reaction mechanism is presented if available.
Introduction
The nitrile group, an equivalent to carbonyl, carboxyl, amino and hydroxymethyl groups, has been considered as not only a key building block in many pharmaceuticals and bioactive compounds but also an important and versatile group that can be easily transformed into various derivatives such as nitrogen-containing heterocycles, amides, amines and so on.1 Meanwhile, nitriles, occurring in lots of natural products and important precursors for amides, amines, esters, carboxylic acids, ketones, aldehydes and alcohols, are versatile intermediates for the preparation of pharmaceuticals, pesticides and organic materials.2 For example, acrylonitrile3 is an important monomer for manufacture of plastics, rubbers, fiber, agrochemicals and pharmaceutical, while cyanamides4 have been widely used as ambidentate ligands in coordination chemistry. Nitriles could be readily obtained by traditional cyanation reactions including Sandmeyer reaction, Rosenmund-von Braun reaction, and other transition-metal5 catalyzed cyanation reactions applying metal cyanides or metalloid cyanides as cyanide source.6 But these above protocols usually used toxic metal cyanation reagents and also required prefunctionalization of these cyanation reagents.7 According to the 12 principles of green chemistry,8 a green chemical process should avoid the use of toxic reagents. Thus, many less toxic or green cyanation reagents were discovered, including safe reagent 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ),9 benzyl cyanide,10 acetonitrile,11 N,N-dimethylformamide (DMF),12 azobisisobutyronitrile (AIBN),13 trimethylsilylcyanide (TMSCN),14 ary(cyano)iodonium triflates,15 N-cyano-N-phenyl-p-toluenesulfonamide (NCTS),16 bis(dialkylamino)cyanoboranes,17 malononitrile,18 and so on.Alkynes are fundamentally useful unsaturated compounds which are omnipresent in many natural products, and pharmaceuticals, agrochemicals and organic materials because of their unique physical, chemical and biological properties.19 And inherently nucleophilic alkynes are also important intermediates in organic synthesis for preparation of biological active compounds and organic functional compounds used in organic field-effect transistors, organic light-emitting diodes, liquid crystals and dye-sensitised solar cells.20 Direct cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes, useful protocols for synthesis of interesting nitriles such as acrylonitriles, alkynyl cyanides, dicyano-substituted and cyanofunctionalized derivatives, have attracted more and more attention.21 In this review, direct cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes are presented. Numerous less toxic or green cyanation reagents are efficient for direct cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes.
Direct cyanation and hydrocyanation of alkynes
In 2013, Okamoto and Ohe reported copper catalysed cyanation of terminal alkynes with cyanogen iodide (ICN). When CuOTf-toluene was applied as a catalyst and tetramethylpiperidine (TEMP) was used as a base, direct cyanation of terminal acetylene 1 with ICN proceeded smoothly and gave the desired alkynyl cyanides 2 in moderate to good yields (Scheme 1).22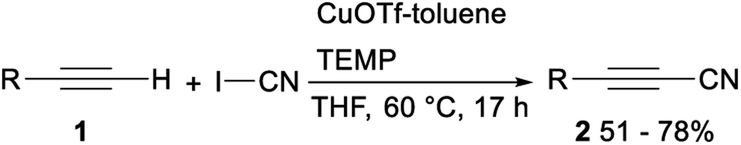 | ||
| Scheme 1 Copper catalysed cyanation of terminal alkynes with ICN. | ||
Cu-catalyzed direct cyanation of terminal alkynes with AMBN (azobisisoamylonitrile) or AIBN (azobisisobutyronitrile) was demonstrated by Mao and Xu in 2015. As shown in Scheme 2, the desired alkynyl cyanides 4 were obtained in 45–92% yield when terminal acetylene 3 was treated with AMBN and Cu(OAc)2 in acetonitrile at 80 °C under air. While under argon atmosphere, the direct cyanation of terminal alkynes 5 with AIBN occurred and gave the desired addition products isobutyronitriles 6 in moderate yields. The above direct cyanation reactions of terminal alkynes with AMBN or AIBN had remarkable advantages including a less toxic cyanation reagent and wide substrate scope.23
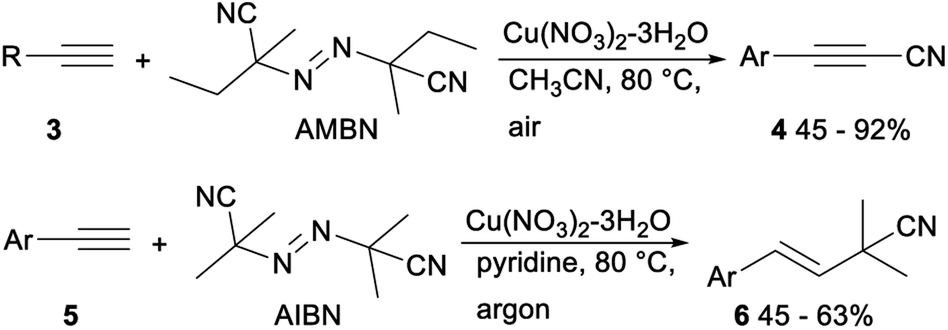 | ||
| Scheme 2 Cu catalysed direct cyanation of terminal alkynes with AMBN or AIBN. | ||
Silver-mediated direct cyanation of terminal alkynes for the synthesis of propionitrile derivatives had been achieved by Wang and Bi. Direct cyanation reaction of terminal alkynes 7 with N-isocyanoiminotriphenylphosphorane (NIITP) in the presence of a quantitative amount of AgOTf in DMF produced propionitriles 8 in moderate to good yields (Scheme 3).24
 | ||
| Scheme 3 Silver-mediated direct cyanation of terminal alkynes. | ||
A plausible mechanism for this cyanation reaction was shown in Scheme 4, firstly, ethynylbenzene reacted with AgOTf to generate the silver acetylide A. Then, an acetylenic imido complex B was produced by insertion of NIITP into the silver–carbon bond of A. Finally, the complex B converted to target 3-phenylpropiolonitrile with the release of AgOH and triphenylphosphine azaylide. This procedure, applying NIITP as a nontoxic, facile “CN” source, was characterized by its operational simplicity, high efficiency with excellent yields, broad substrate scope, and greater functional group tolerance.24
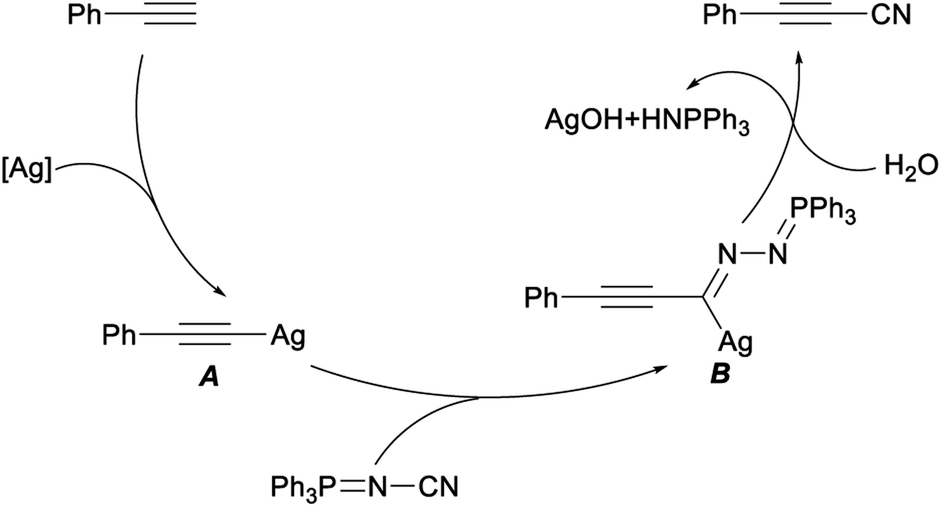 | ||
| Scheme 4 Plausible mechanism for silver-mediated direct cyanation of terminal alkynes. | ||
In addition, copper-catalyzed direct cyanation of terminal alkynes with benzoyl cyanide was reported by Li in 2018. This protocol using less toxic, stable and easy to handle benzoyl cyanide as a cyanide source and air as an oxidant provided a good alternative to the preparation of 3-arylpropiolonitriles under mild condition.25
The first highly stereo- and regioselective hydrocyanation of terminal alkynes with acetone cyanohydrin was reported by Ritter. Treatment of aromatic terminal alkynes 9 with acetone cyanohydrin, dppf, and TpRh(COD) (Tp = (tris(1-pyrazolyl)borohydride)) in CH3CN and heating at 110 °C afforded the desired anti-Markovnikov alkenyl nitriles 10 in 51–88% yield without the formation of Markovnikov product. Instead of dppf, DPE-phos enabled facile hydrocyanation of aliphatic terminal alkynes with acetone cyanohydrin. Mixing of aliphatic terminal alkynes 11, acetone cyanohydrin, DPE-phos, and TpRh(COD) (Tp = (tris(1-pyrazolyl)borohydride)) in CH3CN and heating at 110 °C afforded the desired alkenyl nitriles 12 in 62–85% yield with the ratio of anti-Markovnikov products (a-M) to Markovnikov products (M) from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to >20:1 (Scheme 5).26
:1 to >20:1 (Scheme 5).26
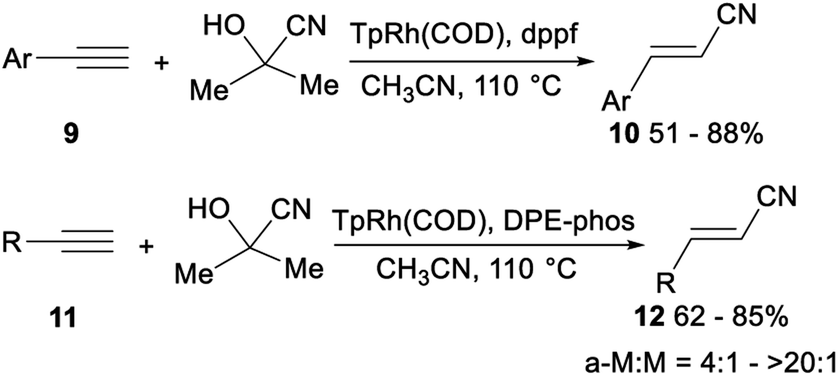 | ||
| Scheme 5 Stereo- and regioselective hydrocyanation of terminal alkynes with acetone cyanohydrin. | ||
The first efficient and general nickel catalysed hydrocyanation of terminal alkynes with Zn(CN)2 was developed by Liu in 2018. When water was used as a hydrogen source, Ni(acac)2 was applied as a catalyst and Mn was employed as an additive, hydrocyanation of terminal aryl alkynes 13 with Zn(CN)2 proceeded smoothly to afforded the desired Markovnikov addition products 14 in moderate to good yields. Under the above standard reaction conditions of aryl alkynes, the hydrocyanation reactions of terminal aliphatic alkynes with Zn(CN)2 did not proceed completely. While addition of neocuproine as ligand finished the desired functionalized vinyl nitriles 16 in 60–83% yields (Scheme 6). This protocol, avoiding use of the volatile and hazardous reagent like HCN, offered a regioselective method for preparation of functionalized vinyl nitriles with a range of structural diversity under mild reaction conditions.27
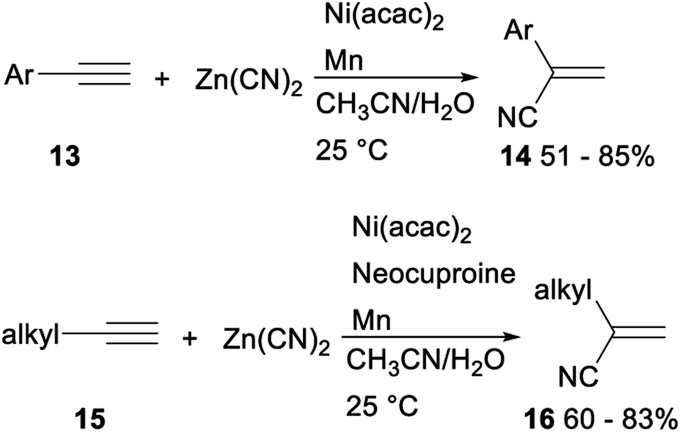 | ||
| Scheme 6 Nickel catalysed hydrocyanation of terminal alkynes with Zn(CN)2. | ||
A reaction mechanism for the above hydrocyanation of terminal alkynes with Zn(CN)2 was tentatively propose. As depicted in Scheme 7, firstly, reduction of Ni(acac)2 with Mn formed Ni(0) species. Then Ni(II) intermediate C was generated by oxidative addition of water to Ni(0). The alkenyl nickel complex D was produced by insertion of alkyne to C via cis-addition of Ni–H bond (hydronickelation). Possibly, the stability of the alkenyl nickel species influenced the regioselectivity of this step. The desired nitriles was formed via transmetalation of D with Zn(CN)2 followed by reductive elimination. Alternatively, the same intermediate D could be also afforded by reaction of nickel π-alkyne complex F with H2O. The detailed process for this transformation was not clear yet, it may proceed through oxidative addition of water with F or cleavage of nickelacyclopropene intermediate G by water.27
 | ||
| Scheme 7 Plausible mechanism for nickel catalysed hydrocyanation of terminal alkynes with Zn(CN)2. | ||
In 2019, Yang and Chang reported the first example of Ni-mediated dehydration of formamide to form “CN” and its subsequent catalytic applications in the hydrocyanation of alkynes (Scheme 8). Treatment of acetylenes 17 and formamide with Ni(acac)2/Co(acac)2/Xantphos catalytic system gave the corresponding acrylonitriles 19 in 70–96%. The above reaction generated “CN” unit from readily available nontoxic organic precursors via nickel catalysis, was an efficient protocol for synthesis of vinyl nitriles.28
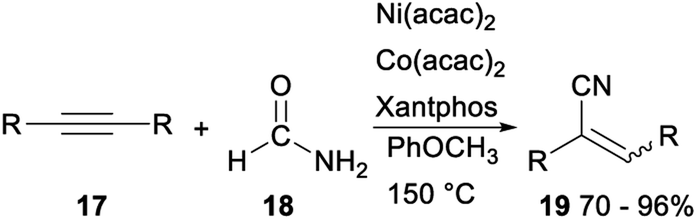 | ||
| Scheme 8 Ni mediated hydrocyanation of alkynes. | ||
In the same year, Lan and Xiao demonstrated the first asymmetric propargylic radical cyanation via a synergetic photoredox/copper catalysis strategy. After irradiating acetylenes 20 and trimethylsilyl cyanide (TMSCN) with visible light (2 × 3 W, purple LEDs, 390 nm) for 24 h in the presence of organic photocatalyst Ph-PTZ, catalyst Cu(MeCN)4BF4 and chiral bisoxazoline (box) ligand L′, the desired propargyl cyanide products 21 was detected in moderate to excellent yields with 83–98% ee. The above strategy, providing an unprecedented access to optically enriched propargyl cyanides with generally high reaction efficiency and enantioselectivity under mild conditions, not only offered a new way for catalytic asymmetric propargylic functionalization, but also provided a novel dual catalysis system for visible-light-induced asymmetric chemical bond formation (Scheme 9).29
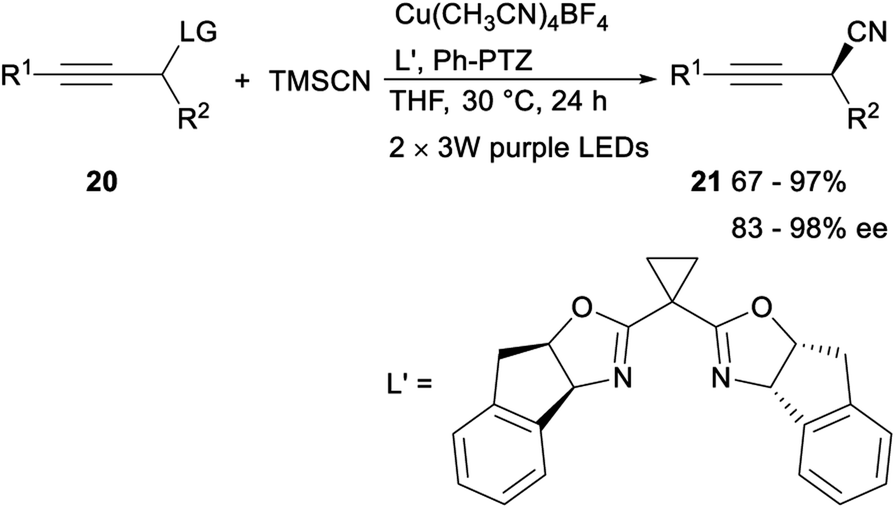 | ||
| Scheme 9 Asymmetric propargylic radical cyanation. | ||
Dicyanation of alkynes
In 2009, Arai reported a palladium(II) catalysed stereoselective 1,2-dicyanation of various alkynes with TMSCN under aerobic conditions. Treatment of alkynes 22 with PdCl2, TMSCN and O2 in toluene gave the corresponding 1,2-dicyano adducts 23 in 50–82% yields (Scheme 10).30 | ||
| Scheme 10 Palladium(II) catalysed stereoselective 1,2-dicyanation of alkynes. | ||
The same research group investigated palladium catalysed dicyanative [4 + 2] cycloaddition of various ene–enynes. As shown in Scheme 11, Pd(CN)2 catalysed 1,2-dicyanation of alkynes 24 with TMSCN in toluene under O2 proceeded smoothly and gave a mixture of cis- and trans-adducts or trans only adducts 25 in 56–78% yields.31
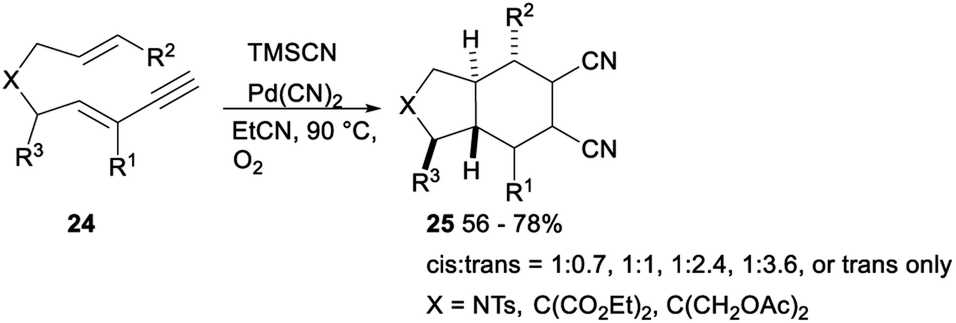 | ||
| Scheme 11 Palladium catalysed dicyanative [4 + 2] cycloaddition of ene–enynes. | ||
Palladium catalysed dicyanation of carbon–carbon triple bonds under aerobic conditions was also investigated. As shown in Scheme 12, subjection acetylenes 26 to PdCl2 or Pd(CN)2 catalysed dicyanation with TMSCN gave the corresponding products 27 in moderate to good yields.21c This above simple and basic method provided a new approach to 1,2-dicyano olefin.
 | ||
| Scheme 12 Palladium catalysed dicyanation of carbon–carbon triple bonds. | ||
Cyanofunctionalization of alkynes
Regio- and stereoselective chlorocyanation of alkynes was reported by Alcarazo in 2017. The reaction of alkynes 28 with thioimidazolium salt 29 in the presence of boron trichloride (BCl3) was able to provide the desired product 30 in moderate to excellent yields. The stereo- and regioselectivity observed in the above transformation was remarkable. The cyanide group was exclusively incorporated at the unsubstituted carbon atom of terminal alkynes, or at the alkyl-substituted carbon atom of 1-aryl-2-alkyl alkynes. This regioselectivity is typical for an electrophilic mechanism. In both cases, only the Z isomer was formed, thus suggesting a syn-addition pathway for the reaction (Scheme 13).32 | ||
| Scheme 13 Regio- and stereoselective chlorocyanation of alkynes. | ||
The following mechanism for the chlorocyanation reaction was proposed. Initially, Lewis adduct H which was already partially fluorinated at boron was formed by activation of the cyanating reagent 29. Vinyl cation I was generated by regioselective attack of H to the corresponding alkyne 28. The syn transfer of one chloride from boron to the carbocationic center gave iminoborane J, which then produced the desired chlorocyanated products 30 by elimination of the imidazolium–thioborane fragment (Scheme 14).32
 | ||
| Scheme 14 Plausible mechanism of chlorocyanation of alkynes. | ||
Okamoto and Ohe investigated copper catalysed regio- and stereoselective iodocyanation of alkynes with cyanogen iodide. Subjection of alkyne 31 to Cu(OAc)2 catalysed iodocyanation with ICN afforded the desired iodocyanoalkene 32 in 41–94% (Scheme 15). This reaction, including diiodide formation, selective monocyanation, and second cyanation processes, shew high regio- and stereoselectivity.33
 | ||
| Scheme 15 Regio- and stereoselective iodocyanation of alkynes. | ||
In 2016, Chien demonstrated CuI catalysed intramolecular aminocyanation of terminal alkynes. A series of 1-sulfonyl-3-cyanoindoles 34 was obtained in 53–83% yields when aminocyanation ring closure of N-(2-ethynylphenyl)-N-sulfonylcyanamides 33 was carried out in 1,4-dioxane with CuI as the catalyst, and Na2CO3 as the base under argon at 80 °C, and the reaction could be completed within 3 hours (Scheme 16).34
 | ||
| Scheme 16 CuI catalysed intramolecular aminocyanation of terminal alkynes. | ||
The plausible mechanism was proposed (Scheme 17). First, p-coordination of alkyne and nitrile moieties to the copper centre was occurred to form a substrate–copper complex K. Subsequently, the N-tosyl-2-alkynylanilide cyanocopper complex L was obtained through the heterolytic cleavage of the N–CN bond by the migration of the electrophilic cyano group to the copper centre. Transaminometalation of L across the alkyne and the following cyanation produced the desired 1-tosyl-3-cyanoindoles 34 via the intermediary of the 1-tosylindol-3-yl cyano–copper complex M. Another plausible mechanism was also proposed to start with the formation of Cu–acetylide N with the increase of π-electron density at the β-carbon. The β-cyano Cu–vinylidene complex O was formed via intramolecular nucleophilic attack of the β-carbon to the electrophilic cyanamide carbon accompanied by the N–CN bond cleavage. M was afforded by intramolecular nucleophilic attack of the remaining tosyl amide at the α-carbon of the Cu–vinylidene complex O. Protonation of M furnished the desired product 34 and regained the Cu(I) catalyst to continue the catalytic cycle.34
 | ||
| Scheme 17 Plausible mechanism of intramolecular aminocyanation of terminal alkynes. | ||
A visible-light-mediated direct cyanomethylation of aryl alkynoates for synthesis of 3-cyanomethylated coumarins was demonstrated by Li in 2018. In the presence of fac-Ir(ppy)3 as a photocatalyst and NaHCO3 as a base under blue-light irradiation in acetone at room temperature, direct cyanomethylation of aryl alkynoates 35 with 2-bromoacetonitrile proceeded smoothly to afford the desired 3-cyanomethylated coumarins 36 in moderate to good yields. This reaction, using cheap and easily available bromoacetonitrile as the cyanomethylation reagent, provided a new method for synthesis of 3-cyanomethylated coumarins (Scheme 18).35
 | ||
| Scheme 18 Cyanomethylation of aryl alkynoates. | ||
A possible reaction mechanism for the above cyanomethylation was propose. Firstly, fac-Ir(III)(ppy)3 was irradiated to the excited state  , which was then oxidatively quenched by BrCH2CN with the formation of a [fac-Ir(IV)(ppy)3]+ complex and a highly reactive radical P. The radical intermediate Q, generated by addition of radical P to the triple bond of aryl alkynoates 35, underwent intramolecular spirocyclization and gave the spiroradical intermediate R. The coumarin radical T was generated by selective ester migration via a carboxyl radical S. Then, oxidation of T by [fac-Ir(IV)(ppy)3]+ formed the cyclohexadienyl cation U and regenerated fac-Ir(III)(ppy)3. Finally, the desired product 36 was produced by deprotonation of U assisted by the base (Scheme 19).35
, which was then oxidatively quenched by BrCH2CN with the formation of a [fac-Ir(IV)(ppy)3]+ complex and a highly reactive radical P. The radical intermediate Q, generated by addition of radical P to the triple bond of aryl alkynoates 35, underwent intramolecular spirocyclization and gave the spiroradical intermediate R. The coumarin radical T was generated by selective ester migration via a carboxyl radical S. Then, oxidation of T by [fac-Ir(IV)(ppy)3]+ formed the cyclohexadienyl cation U and regenerated fac-Ir(III)(ppy)3. Finally, the desired product 36 was produced by deprotonation of U assisted by the base (Scheme 19).35
 | ||
| Scheme 19 Plausible mechanism for cyanomethylation of aryl alkynoates. | ||
Nickel catalysed carbocyanative cyclization of 1,6-enynes was described by Arai (Scheme 20). When acetone cyanohydrin was used as an inexpensive and easy-to-handle HCN source and Ni[P(OPh3)]4 was applied as a catalyst, carbocyanative cyclization of 1,6-enynes 37 proceeded smoothly in toluene and gave the corresponding products 38 in moderate yields.36
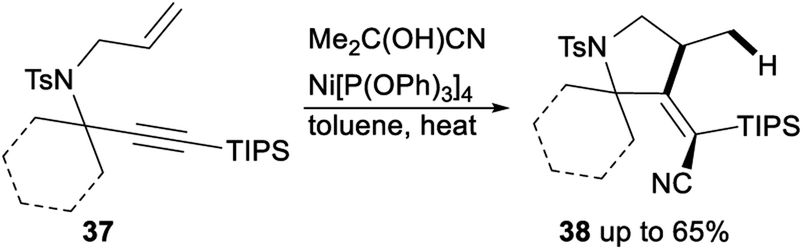 | ||
| Scheme 20 Carbocyanative cyclization of 1,6-enynes. | ||
Sc(OTf)3 catalysed formal acylcyanation of electron-rich alkynes for synthesis of fully-substituted acrylonitriles was investigated by Zhao and Sun in 2018. The formal acylcyanation of electron-rich alkynes 39 with cyanide 40 proceeded smoothly in the presence of Sc(OTf)3 in CHCl3 under N2 and gave the corresponding ynamides, thioalkynes or ynol ethers 41 in moderate to excellent yields with remarkable Z selectivity. This reaction, featuring mild conditions, high regio- and stereoselectivity, and a broad scope, offered an efficient protocol to fully-substituted acrylonitriles (Scheme 21).37
 | ||
| Scheme 21 Sc(OTf)3 catalysed formal acylcyanation of electron-rich alkynes. | ||
A plausible mechanism for the above formal acylcyanation was proposed in Scheme 22. Sc(OTf)3 could serve to activate the acylnitrile by lowering its LUMO level, thereby increasing its electrophilicity and giving intermediate V. The β-carbon of the ynamide attacked the carbonyl of V followed by cyclization (i.e., [2 + 2] cycloaddition) to form the oxetane intermediate W with high regioselectivity because of the high polarization of the alkyne. The desired product 42 was produced by the subsequent electrocyclic opening of the oxetane W. Conrotatory torquoselection in the ring-opening process governed the syn selectivity. During the conrotatory ring opening, the CN group rotates inward, so that its π* orbital can favourably interact with the breaking σ(C–O) orbital, thus preferentially delivering the (Z)-olefin product.37
 | ||
| Scheme 22 Plausible mechanism for Sc(OTf)3 catalysed formal acylcyanation of alkynes. | ||
Regio- and stereoselective cyanotriflation of alkynes using aryl(cyano)iodonium triflates was reported by Studer in 2016. When 3,5-di(trifluoromethyl)-phenyl(cyano)iodonium triflate 43 was used as the triflate and cyanide source, Fe(OAc)2 in combination with phenanthroline as the catalyst, direct vicinal alkyne cyanotriflation of alkynes 44 occurred with regioselective syn-addition of both the CN and OTf groups of 43 to alkynes 44 to give the desired tetrasubstituted alkenes 45 in moderate to excellent yields. This cyanotriflation of alkynes possessed some advantages such as mild conditions, complete regioselectivity, excellent stereoselectivity, and a wide range of functional groups tolerances. And products tetrasubstituted alkenes were useful building blocks in a series of stereospecific palladium catalysed cross-coupling reactions such as Suzuki coupling, Sonogashira reaction and a Buchwald–Hartwig amidation (Scheme 23).38
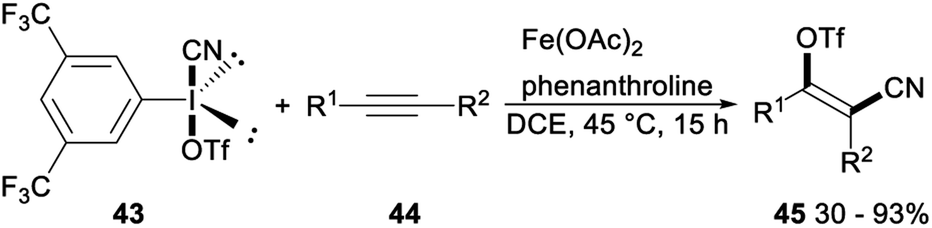 | ||
| Scheme 23 Regio- and stereoselective cyanotriflation of alkynes. | ||
The CuCN-mediated cyclization–cyanation reactions of β-hydroxyalkynes and o-alkynylphenol and -aniline derivatives was developed by Pyne. Treatment of β-hydroxyalkynes or o-alkynylphenol or -aniline derivatives 46 or 48 with CuCN in DMF afforded the desired 3-cyanobenzofurans 47 or 3-cyanoindoles 49 in moderate to good yields, respectively (Scheme 24).39
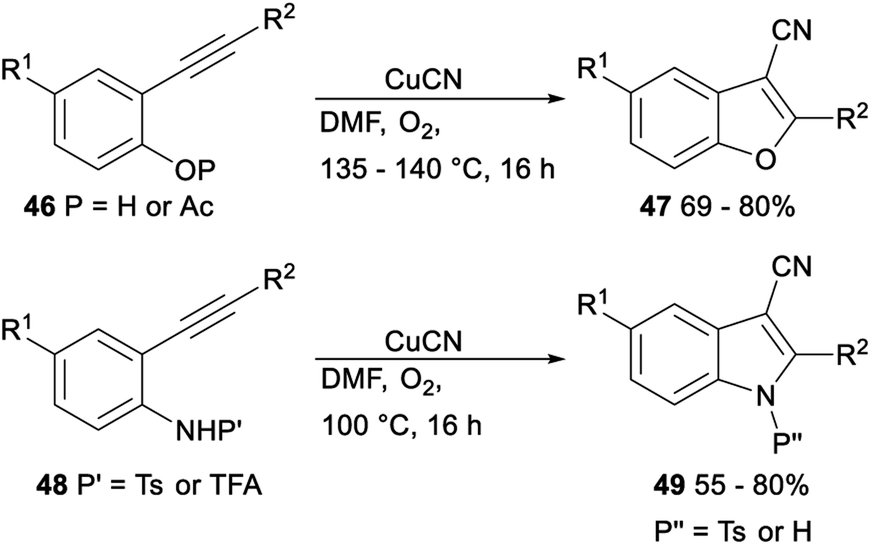 | ||
| Scheme 24 Cyclization–cyanation reactions of β-hydroxyalkynes and o-alkynylphenol and -aniline derivatives. | ||
In 2012, Hiyama demonstrated nickel/Lewis acid catalysed polyfluoroarylcyanation of alkynes. When Ni(cod)2 was used as a catalyst and DPEphos/BPh3 was employed as a ligand, polyfluoroarylcyanation of polyfluorobenzonitriles 50 with 4-octyne proceeded smoothly and produced the desired adducts 51 in excellent yields (Scheme 25).40
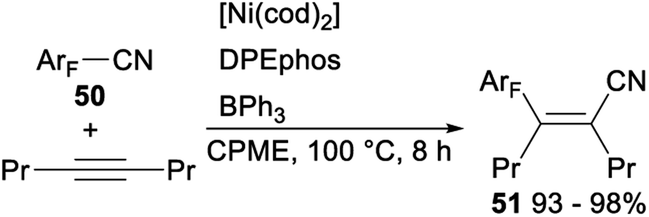 | ||
| Scheme 25 Nickel/Lewis acid catalysed polyfluoroarylcyanation of alkynes. | ||
A plausible mechanism for the above nickel/Lewis acid catalysed polyfluoroarylcyanation of alkynes was shown in Scheme 26. The catalytic cycle should be initiated by the formation of the η2-complex Y or Z. The following oxidative addition of the C–CN bond to nickel(0) gave A′ after the cyano group nitrogen atom is bound to BPh3. Insertion of 4-octyne into the ArF–Ni bond in A′ afforded C′ via the alkyne-coordinated complex B′. Steric repulsion between the bulkier group Pr and the polyfluorophenyl group on the Ni center in B′ was assumed to be minimal.40 Finally, C–C bond-forming reductive elimination of 51 from C′ generated a nickel(0) complex to complete the catalytic cycle.40
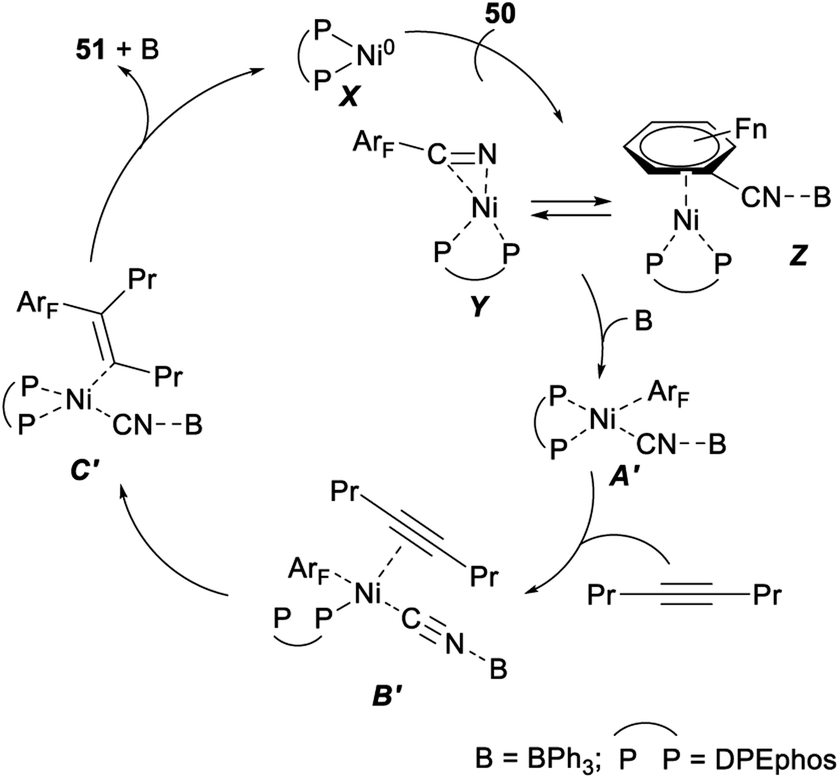 | ||
| Scheme 26 Plausible mechanism for nickel/Lewis acid catalyzed polyfluoroarylcyanation of alkynes. | ||
Nickel catalysed hydrocyanative cyclization and three-component cross-coupling reaction between allenes and alkynes was reported by Arai in 2013 (Scheme 27). Subjection of 52 to Me2C(OH)CN/[Ni{P(OPh3)}4]/P(OPh)3 catalytic system afforded the corresponding products 53 in 43–70% yields. This protocol offered a new method for the addition of a CN functionality to C–C triple bonds and provided tetra substituted alkenes with highly regio- and stereoselective.21g
 | ||
| Scheme 27 Nickel catalyzed hydrocyanative cyclization and three-component cross-coupling reaction between allenes and alkynes. | ||
The possible mechanism for the above nickel catalysed hydrocyanative cyclization and three-component cross-coupling reaction between allenes and alkynes was described in Scheme 28. Firstly, a regioselective hydronickelation of the allene occurred and generated a Ni–H species. Then, hydride attacked at the central carbon atom of the allene 52 and would give a π-allylnickel intermediate E′. The following 5-exo cyclization of E′ gave F′. A subsequent elimination would give the tetrasubstituted alkene 53 stereoselectively together with Ni0. When the initial C–H bond formation occurred at the terminal allenyl carbon atom, the resulting organonickel species G′ was not suitable for cyclization and produced by-products 54 through reductive elimination. Since a cyano functionality was added exclusively to a C–C triple bond in a regio- and stereoselective fashion, it was unlikely that the two vinyl–nickel bonds in D′, which could be generated by the oxidative cyclization of 52, reacted specifically with AC through protonation to give F′.21g
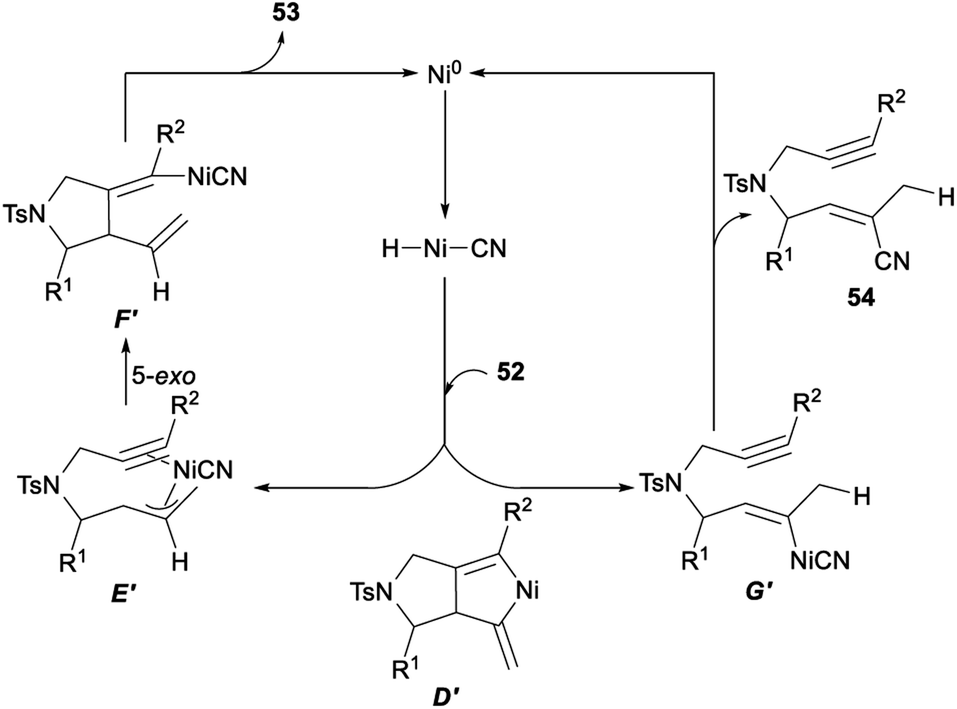 | ||
| Scheme 28 Plausible mechanism for hydrocyanative cyclization and three-component cross-coupling reaction between allenes and alkynes. | ||
Other cyanation of alkynes
In 2018, He reported ultrasound-promoted Brønsted acid ionic liquid catalysed hydrothiocyanation of activated alkynes (Scheme 29). When alkynes 55 was treated with KSCN, BAIL-1 and water at room temperature in minimal solvent under ultrasonic radiation (40 kHz/40 W), the corresponding vinyl thiocyanates 56 was afforded in 78–94% yields. And the catalytic system could be repeated five times without significant influence on the yield. This reaction provided an eco-friendly and practical protocol for the synthesis of Z-vinyl thiocyanates, having some features including the abundance and accessibility of raw materials, the usage of recyclable and reusable catalysts, a wide substrate scope with good to high yields, ease of scale-up and high energy efficiency.41 | ||
| Scheme 29 Ultrasound-promoted Brønsted acid ionic liquid catalysed hydrothiocyanation of activated alkynes. | ||
A plausible mechanism for the above hydrothiocyanation was proposed in Scheme 30. Firstly, a cationic intermediate G′ was generated by protonation of the activated alkyne with BAIL-1. Then H′ was further tautomerized into the zwitterionic complex I′. Intermediate J′ was produced by the nucleophilic thiocyanate ion attacked the β-carbon of intermediate I′. Finally, the capture of a proton dissociated from water by ultrasonic radiation proceeded from the less hindered face of intermediate J′, trans to SCN, thus leading to product Z-vinyl thiocyanates.41
 | ||
| Scheme 30 Plausible mechanism for ultrasound-promoted Brønsted acid ionic liquid catalyzed hydrothiocyanation of activated alkynes. | ||
Ultrasonic multicomponent synthesis of (Z)-β-iodo vinylthiocyanates via hydrothiocyanation of alkynes was also studied. When alkynes 57 was treated with KSCN and I2 in the presence of K2S2O8 as the oxidant in MeCN at room temperature, the corresponding (Z)-β-iodo vinylthiocyanates 58 was yielded in 82–94% yield with Z/E from 11:1 to 28:1 (Scheme 31).42
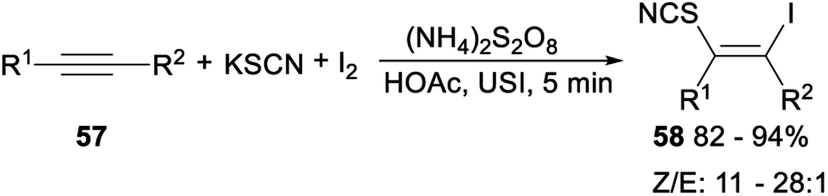 | ||
| Scheme 31 Ultrasonic multicomponent synthesis of (Z)-β-iodo vinylthiocyanates via hydrothiocyanation of alkynes. | ||
In the plausible mechanism of the above ultrasonic multicomponent synthesis of (Z)-β-iodo vinylthiocyanates, an activated iodoirenium ion K′ was formed through the molecular iodine electrophilic addition to alkyne with the release an iodine anion which was oxidized by (NH4)2S2O8 to regenerate I2 for continuing participation in the reaction. Then, an energetically favourable six-membered ring intermediate L′ was yielded by the coordination of HOAc to intermediate K′. Finally, the thiocyanate anion attacked the less steric hindered carbon atom to give the corresponding product (Z)-β-iodo vinylthiocyanates with concomitant release of HOAc to complete the catalytic cycle (Scheme 32).42
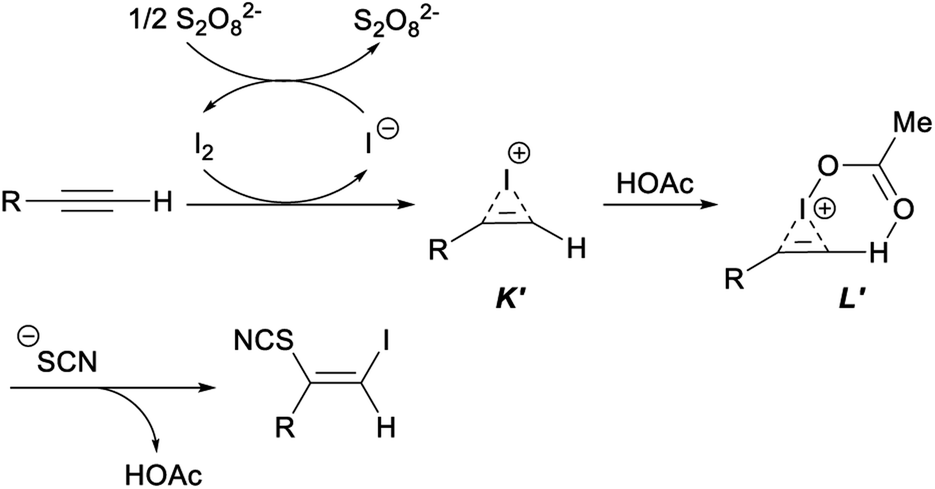 | ||
| Scheme 32 Plausible mechanism for ultrasonic multicomponent synthesis of (Z)-β-iodo vinylthiocyanates. | ||
Synthesis of a variety of Z-β-thiocyanate alkenyl esters via lactic acid catalysed multicomponent hydrothiocyanation of alkynes was established (Scheme 33). After treating alkynes 59 with KSCN in the presence of natural lactic acid under ultrasound conditions for 30 min, the expected Z-β-thiocyanate alkenyl esters 60 was formed in excellent yields.43
 | ||
| Scheme 33 Synthesis of Z-β-thiocyanate alkenyl esters via lactic acid catalyzed hydrothiocyanation of alkynes. | ||
Iodine-mediated regio- and stereoselective iodothiocyanation of alkynes was reported by Zeng and Chen in 2018 (Scheme 34). Iodothiocyanation of alkyne 61 with NH4SCN in the presence of I2 as the iodine source and EtOH/H2O as the solvent at 80 °C under air afforded series of functional β-iodo vinylthiocyanates 62 in good to excellent yields. This protocol proceeding in aqueous ethanol media under mild conditions with good functional group tolerance was a new environmentally friendly, economical and straightforward approach for versatile synthesis of vinyl thiocyanates from readily available and inexpensive NH4SCN and I2.44
 | ||
| Scheme 34 Iodine-mediated iodothiocyanation of alkynes. | ||
The plausible reaction pathway for the above iodothiocyanation of alkynes was shown in Scheme 35. First, an electrophilic addition of I2 to the C–C triple bond of substrate 61 led a cyclic iodonium ion M′. And then, nucleophilic attack of SCN− on a more substituted site of M′ resulted succedent ring opening, which produced the difunctionalized product 62.44
 | ||
| Scheme 35 Plausible mechanism for iodothiocyanation of alkynes. | ||
In 2019, He investigated natural deep eutectic solvent catalysed selenocyanation of activated alkynes (Scheme 36). Under ultrasonic radiation, treatment of alkynes 63 with H2O and KSeCN in natural deep eutectic solvent (ChCl/glycolic acid) afforded the corresponding Z-vinylselenolates 64 in 78–94% yields with Z/E ratio from 14:1 to 39:1.45
 | ||
| Scheme 36 Deep eutectic solvent catalysed selenocyanation of activated alkynes. | ||
The plausible mechanism for deep eutectic solvent catalysed selenocyanation of activated alkynes was depicted in Scheme 37. First, two hydrogen bonds formed between the oxygen atom of the carboxyl group in alkynes and the H atom of the hydroxyl groups (glycolic acid) in DES, resulting in enhanced polarization of carbonyl group of alkynes (N′), which was in resonance with a zwitterionic intermediate O′. Then, an intermediate P′, in which a hydrogen bond formed between the nitrogen-atom of “SeCN” and the H atom of the hydroxyl group (ChCl) in ChCl/glycolic acid, was generated by the attachment of selenocyanate anion to intermediate P′. Finally, the intermediate P′ captured a proton (in situ generated by ultrasound-assisted self-ionization of H2O) on the reverse side of the sterically hindered H-bond activated atom to produce the desired Z-vinyl selenocyanates with concomitant release of the DES to fulfil the catalytic cycle.45
 | ||
| Scheme 37 Plausible mechanism for deep eutectic solvent catalysed selenocyanation of activated alkynes. | ||
A green selenocyanation of activated alkynes catalysed by lactic acid was achieved by He in 2019. Treatment of a mixture of alkynes 65, H2O, KSeCN and lactic acid at ambient temperature under ultrasonic conditions (30 W/44 KHz/25–32 °C) in open air led to the expected Z-3-selenocyanatoacrylates and analogues 66 in excellent yields. This protocol, using a minimal amount of lactic acid as reaction media, entirely avoided the usage of organic volatile compounds. And the reaction also had other advantages such as the conversion of substrate was almost quantitative, the pure products could be conveniently collected through water precipitation and the cheap biomass lactic acid could be re-used for five consecutive runs without obvious decrease in catalytic activity (Scheme 38).46
 | ||
| Scheme 38 Green selenocyanation of activated alkynes catalysed by lactic acid. | ||
Stereoselective difunctionalization of alkynes with NaSCN or KSeCN was also reported (Scheme 39). When NaSCN was used as a [SCN] source and oxone was employed as an oxidant, dithiocyanation of alkynes 67 proceeded smoothly in DCM and produced the desired alkenyl dithiocyanates 68 in 72–87% yield. The expected alkenyl diselenocyanates 69 was obtained in good yield when alkynes 67 was treated with KSeCN [SeCN] source and PhI(OAc)2 oxidant in H2O.47
 | ||
| Scheme 39 Stereoselective difunctionalization of alkynes with NaSCN or KSeCN. | ||
A possible mechanism for the difunctionalization of alkynes was proposed in Scheme 40. Firstly, in the presence of an oxidant oxone or PhI(OAc)2, NaSCN or KSeCN was converted into the SCN radical Q′ or the SeCN radical S′. Then, the anti- Markovnikov addition of Q′ or S′ to alkyne 67 produced the alkenyl radical R′ or T′. Finally, the radical Q′ or S′ attacked more favourably the sterically less hindered side of the alkenyl radical R′ or T′ which could result the formation of desired products 68 and 69, respectively.47
 | ||
| Scheme 40 Plausible mechanism for difunctionalization of alkynes with NaSCN or KSeCN. | ||
Conclusions
In summary, nitriles were versatile intermediates for preparation of pharmaceuticals, pesticides and organic materials. Inherently nucleophilic alkynes were important intermediates in organic synthesis for preparation of biological active compounds and organic functional compounds. Cyanation reactions of alkynes had attracted more and more attention. Transition metal catalysed direct cyanation and hydrocyanation of alkynes gave the corresponding alkynyl cyanides and alkenyl nitriles in high yields. And dicyanation of alkynes could produce 1,2-dicyano adducts. Cyanofunctionalization of alkynes afforded functional cyanated compounds. Other cyanation of alkynes such as thiocyanation and selenocyanation yielded the expected functional vinylthiocyanates and vinylselenocyanates.Limitations and perspectives
Although these above cyanation reactions were useful and convenient for synthesis of cyanates, expensive transition metal catalysts like rhodium and palladium complexes were often used to realize reasonable yields in direct cyanation of alkynes. Thus, establishment of cheap catalytic system should be focused in future research. And developing other kind cyanation of alkynes was still in high need.Conflicts of interest
No conflict of interest exists in this paper, and the paper is approved by all authors for publication. I and my co-authors would like to declare that the work described is original and has not been published previously, and not under consideration for publication elsewhere, in whole or in part. All the authors listed have approved the contents of the paper.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21802040, 21877034), the Natural Science Fund Youth Project of Hunan Province (No. 2018JJ3145), the General project of Hunan Education Department (17C0629), and the Open Foundation of Key Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, Hunan University of Science and Technology (No. E21843) for financial support.Notes and references
- (a) J. M. Sansano, Angew. Chem., 2009, 121, 2488 CrossRef; (b) B.-B. Liu, W.-B. Cao, F. Wang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2018, 83, 11118 CrossRef CAS PubMed; (c) C. Nájera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 48, 2452 CrossRef PubMed; (d) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappoport, Wiley-VCH, New York, 1983 Search PubMed; (e) X. Wang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2014, 79, 8577 CrossRef CAS PubMed; (f) E. N. Bess and M. S. Sigman, Linear free energy relationships (LFERs) in asymmetric catalysis, in Asymmetric Synthesis II: More Methods and Applications, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, Germany, 2012, p. 363 Search PubMed; (g) Z. Rappoport, in Chemistry of the Cyano Group, John Wiley & Sons, London, 1970, p. 121 Search PubMed; (h) P. Xu, Y.-M. Zhu, F. Wang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2019, 21, 683 CrossRef CAS PubMed; (i) J. W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892 CrossRef CAS; (j) X.-D. Xiong, C.-L. Deng, X.-S. Peng, Q. Miao and H. N. C. Wong, Org. Lett., 2014, 16, 3252 CrossRef CAS PubMed; (k) C. Cheng, Z. Cai, X.-S. Peng and H. N. C. Wong, J. Org. Chem., 2013, 78, 8562 CrossRef CAS PubMed; (l) X. Wang, X.-P. Xu, S.-Y. Wang, W.-Q. Zhou and S.-J. Ji, Org. Lett., 2013, 15, 4246 CrossRef CAS PubMed.
- (a) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Appl. Organomet. Chem., 2001, 15, 726 CrossRef; (b) S. Krautwald and E. M. Carreira, in Comprehensive Organic Transformations: a Guide to Functional Group Preparations, ed. C. R. Larock, Wiley-VCH, Weinheim, 1989, p. 819 Search PubMed; (c) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed; (d) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed.
- (a) N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628 CrossRef CAS; (b) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597 RSC; (c) Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers, London, 1970 Search PubMed; (d) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 1989 Search PubMed; (e) UllmannQs Encyclopedia of Industrial Chemistry: Acrylonitrile, ed. P. W. Langvardt, Wiley-VCH, Weinheim, 2011, vol. 1, p. 365 Search PubMed; (f) Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, Wiley, Hoboken, 3rd edn, 2007 Search PubMed.
- (a) M. H. Larraufie, G. Maestri, M. Malacria, C. Ollivier, L. Fensterbank and E. Lacote, Synthesis, 2012, 1279 CAS; (b) D. D. Nekrasov, Russ. J. Org. Chem., 2004, 40, 1387 CrossRef CAS; (c) R. J. Crutchley, Coord. Chem. Rev., 2001, 219, 125 CrossRef.
- (a) L.-F. Peng, Z.-F. Hu, Z.-L. Tang, Y.-C. Jiao and X.-H. Xu, Chin. Chem. Lett., 2019, 30, 1481 CrossRef CAS; (b) K.-J. Liu, X.-L. Zeng, Y. Zhang, Y. Wang, X.-S. Xiao, H. Yue, M. Wang, Z. Tang and W.-M. He, Synthesis, 2018, 50, 4637 CrossRef CAS; (c) C. Wu, J. Wang, X.-Y. Zhang, G.-K. Jia, Z. Cao, Z. Tang, X. Yu, X. Xu and W.-M. He, Org. Biomol. Chem., 2018, 16, 5050 RSC; (d) R.-N. Yi, Z.-J. Wang, Z.-W. Liang, M. Xiao, X.-H. Xu and N.-B. Li, Appl. Organomet. Chem., 2019, 33, 4917 CrossRef; (e) L.-Y. Xie, S. Peng, L.-L. Jiang, X. Peng, W. Xia, X. Yu, X.-X. Wang, Z. Cao and W.-M. He, Org. Chem. Front., 2019, 2, 167 RSC; (f) L.-Y. Xie, Y.-J. Li, J. Qu, Y. Duan, J. Hu, K.-J. Liu, Z. Cao and W.-M. He, Green Chem., 2017, 19, 5642 RSC; (g) L.-F. Peng, R.-Z. Li, Z.-L. Tang, J.-Y. Chen, R.-N. Yi and X.-H. Xu, Tetrahedron, 2017, 73, 3099 CrossRef CAS; (h) L.-F. Peng, Z.-H. Hu, Q.-C. Lu, Z.-L. Tang, Y.-C. Jiao and X.-H. Xu, Chin. Chem. Lett., 2019, 30, 2151 CrossRef CAS; (i) L.-Y. Xie, S. Peng, F. Liu, Y.-F. Liu, M. Sun, Z. Tang, S. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 7193 CrossRef CAS.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef; (b) C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS; (c) K. W. Rosenmund and E. Struck, Ber. Dtsch. Chem. Ges., 1919, 52, 1749 CrossRef; (d) J. Lindley, Tetrahedron, 1984, 40, 1433 CrossRef CAS; (e) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (f) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC; (g) Y.-Y. Ping, Q.-P. Ding and Y.-Y. Peng, ACS Catal., 2016, 6, 5989 CrossRef CAS; (h) B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709 CrossRef CAS PubMed; (i) G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068 CrossRef CAS.
- (a) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed; (b) J. Ramnauth, N. Bhardwaj, P. Renton, S. Rakhit and S. P. Maddaford, Synlett, 2003, 14, 2237 Search PubMed; (c) M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS; (d) R. Chidambaram, Tetrahedron Lett., 2004, 45, 1441 CrossRef CAS; (e) J. M. Veauthier, C. N. Carlson, G. E. Collis, J. L. Kiplinger and K. D. John, Synthesis, 2005, 16, 2683 Search PubMed; (f) H. J. Cristau, A. Ouali, J. F. Spindler and M. Taillefer, Chem.–Eur. J., 2005, 11, 2483 CrossRef CAS PubMed; (g) R. S. Jensen, A. S. Gajare, K. Toyota, M. Yoshifuj and F. Ozawa, Tetrahedron Lett., 2005, 46, 8645 CrossRef CAS; (h) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; (i) F. G. Buono, R. Chidambaram, R. H. Mueller and R. E. Waltermire, Org. Lett., 2008, 10, 5325 CrossRef CAS PubMed; (j) X. Jia, D. Yang, S. Zhang and J. Cheng, Org. Lett., 2009, 11, 4716 CrossRef CAS; (k) H. Q. Do and O. Daugulis, Org. Lett., 2010, 12, 2517 CrossRef CAS; (l) A. V. Ushkov and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 10999 CrossRef CAS PubMed; (m) G. Zhang, L. Zhang, M. Hu and J. Cheng, Adv. Synth. Catal., 2011, 353, 291 CrossRef CAS; (n) J. Chen, Y. Sun, B. Liu, D. Liu and J. Cheng, Chem. Commun., 2012, 48, 449 RSC; (o) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 52, 1749 CrossRef; (p) X. Jia, D. Yang, W. Wang, F. Luo and J. Chen, J. Org. Chem., 2009, 74, 9470 CrossRef CAS PubMed; (q) D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202 CrossRef CAS; (r) G.-Y. Zhang, J.-T. Yu, M.-L. Hu and J. Chen, J. Org. Chem., 2013, 78, 2710 CrossRef CAS.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; (b) A. S. Matlack, Introduction to Green Chemistry, Marcel Dekker, New York, 2001 Search PubMed; (c) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS; (d) L.-Y. Xie, J. Qu, S. Peng, K.-J. Liu, Z. Wang, M.-H. Ding, Y. Wang, Z. Cao and W.-M. He, Green Chem., 2018, 20, 760 RSC; (e) K.-J. Liu, S. Jiang, L.-H. Lu, L.-L. Tang, S.-S. Tang, H.-S. Tang, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3038 RSC; (f) M. Lancaster, Green Chemistry: An Introductory Text, RSC Publishing, Cambridge, 2002 Search PubMed; (g) K.-J. Liu, S. Jiang, L.-H. Lu, L.-L. Tang, S.-S. Tang, H.-S. Tang, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3683 RSC; (h) L.-H. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 1237–1240 CrossRef CAS; (i) W.-H. Bao, M. He, J.-T. Wang, X. Peng, M. Sung, Z. Tang, S. Jiang, Z. Cao and W.-M. He, J. Org. Chem., 2019, 84, 6065–6071 CrossRef CAS PubMed; (j) W.-H. Bao, C. Wu, J.-T. Wang, W. Xia, P. Chen, Z. Tang, X. Xu and W.-M. He, Org. Biomol. Chem., 2018, 16, 8403 RSC; (k) L.-H. Lu, S.-J. Zhou, W.-B. He, W. Xia, P. Chen, X. Yu, X. Xu and W.-M. He, Org. Biomol. Chem., 2018, 16, 9064 RSC; (l) L.-Y. Xie, Y. Duan, L.-H. Lu, Y.-J. Li, S. Peng, C. Wu, K.-J. Liu, Z. Wang and W.-M. He, ACS Sustainable Chem. Eng., 2017, 5, 10407 CrossRef CAS; (m) L.-Y. Xie, S. Peng, J.-X. Tan, R.-X. Sun, X. Yu, N.-N. Dai, Z.-L. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 16976 CrossRef CAS; (n) C. Wu, P.-P. Yang, Z.-M. Fu, Y. Peng, X. Wang, Z.-Z. Zhang, F. Liu, W.-Y. Li, Z.-Z. Li and W.-M. He, J. Org. Chem., 2016, 81, 10664 CrossRef CAS PubMed; (o) L.-Y. Xie, S. Peng, L.-H. Lu, J. Hu, W.-H. Bao, F. Zeng, Z. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 7989 CrossRef CAS; (p) C. Wu, H.-J. Xiao, S.-W. Wang, M.-S. Tang, Z.-L. Tang, W. Xia, W.-F. Li, C. Zhong and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 2169 CrossRef CAS; (q) H. Jiang, X.-Y. Tang, Z.-H. Xu, H.-X. Wang, K. Han, X.-L. Yang, Y.-Y. Zhou, Y.-L. Feng, X.-Y. Yu and Q.-W. Gui, Org. Biomol. Chem., 2019, 17, 2715 RSC.
- G. Zhang, S. Chen, H. Fei, J. Cheng and F. Chen, Synlett, 2012, 23, 2247 CrossRef CAS.
- Q. Wen, J. Jin, B. Hu, P. Lu and Y. Wang, RSC Adv., 2012, 2, 6167 RSC.
- (a) Y.-M. Zhu and Z.-M. Shen, Adv. Synth. Catal., 2017, 359, 3515 CrossRef CAS; (b) C.-D. Pan, H.-M. Jin, P. Xu, X. Liu, Y.-X. Cheng and C.-J. Zhu, J. Org. Chem., 2013, 78, 9494 CrossRef CAS PubMed; (c) F.-H. Luo, C.-I. Chu and C.-H. Cheng, Organometallics, 1998, 17, 1025 CrossRef CAS.
- J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed.
- H. Xu, P.-T. Liu, Y.-H. Li and F.-S. Han, Org. Lett., 2013, 15, 3354 CrossRef CAS PubMed.
- (a) N. Chatani and T. Hanafusa, J. Chem. Soc., Chem. Commun., 1985, 838 RSC; (b) N. Chatani, T. Takeyasu, N. Horiuchi and T. Hanafusa, J. Org. Chem., 1988, 53, 3539 CrossRef CAS.
- (a) Z. Shu, W. Ji, X. Wang, Y. Zhou, Y. Zhang and J.-B. Wang, Angew. Chem., Int. Ed., 2014, 53, 2186 CrossRef CAS PubMed; (b) M. V. Vita, P. Caramenti and J. Waser, Org. Lett., 2015, 17, 5832 CrossRef CAS PubMed; (c) J. T. Reeves, C. A. Malapit, F. G. Buono, K. P. Sidhu, M. A. Marsini, C. A. Sader, K. R. Fandrick, C. A. Busacca and C. H. Senanayake, J. Am. Chem. Soc., 2015, 137, 9481 CrossRef CAS PubMed; (d) G. Talavera, J. Peña and M. Alcarazo, J. Am. Chem. Soc., 2015, 137, 8704 CrossRef CAS PubMed; (e) R. Frei, T. Courant, M. D. Wodrich and J. Waser, Chem.–Eur. J., 2015, 21, 2662 CrossRef CAS PubMed; (f) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS PubMed; (g) Y.-F. Wang, J. Qiu, D. Kong, Y. Gao, F. Lu, P. G. Karmaker and F.-X. Chen, Org. Biomol. Chem., 2015, 13, 365 RSC; (h) Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 8677 CrossRef CAS PubMed; (i) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (j) C. Zhu, J.-B. Xia and C. Chen, Org. Lett., 2014, 16, 247 CrossRef CAS PubMed; (k) T.-J. Gong, B. Xiao, W.-M. Cheng, W. Su, J. Xu, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS PubMed; (l) T. Hoshikawa, S. Yoshioka, S. Kamijo and M. Inoue, Synthesis, 2013, 45, 874 CrossRef CAS; (m) J. P. Brand, D. F. González, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC; (n) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928 CrossRef CAS PubMed; (o) P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS PubMed; (p) Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 5608 CrossRef CAS PubMed; (q) T. Dohi, K. Morimoto, N. Takenaga, A. Goto, A. Maruyama, Y. Kiyono, H. Tohma and Y. Kita, J. Org. Chem., 2007, 72, 109 CrossRef CAS PubMed; (r) P. Anbarasan, H. Neumann and M. Beller, Chem.–Eur. J., 2010, 16, 4725 CrossRef CAS PubMed; (s) Y.-Q. Wu, D. C. Limburg, D. E. Wilkinson and G. S. Hamilton, Org. Lett., 2000, 2, 795 CrossRef CAS PubMed.
- (a) J. Cui, J. Song, Q. Liu, H. Liu and Y.-H. Dong, Chem.–Asian J., 2018, 13, 482 CrossRef CAS PubMed; (b) R. Lopez and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170 CrossRef CAS PubMed.
- (a) M. Suginome, A. Yamamoto and Y. Ito, Chem. Commun., 2002, 1392 RSC; (b) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef CAS PubMed; (c) B. Jiang, Y. Kan and A. Zhang, Tetrahedron, 2001, 57, 1581 CrossRef CAS; (d) W. R. Jackson and C. G. Lovel, J. Chem. Soc., Chem. Commun., 1982, 1231 RSC; (e) N. Chatani and T. Hanafusa, J. Chem. Soc., Chem. Commun., 1985, 838 RSC; (f) N. Chatani, T. Takeyasu, N. Horiuchi and T. Hanafusa, J. Org. Chem., 1988, 53, 3539 CrossRef CAS; (g) M. Suginome, H. Kinugasa and Y. Ito, Tetrahedron Lett, 1994, 35, 8635 CrossRef CAS; (h) Y. Obora, A. S. Baleta, M. Tokunaga and Y. Tsuji, J. Organomet. Chem., 2002, 660, 173 CrossRef CAS; (i) K. Nozaki, N. Sato and H. Takaya, J. Org. Chem., 1994, 59, 2679 CrossRef CAS.
- X. Wang, S. Y. Wang and S. J. Ji, Org. Lett., 2013, 15, 1954 CrossRef CAS PubMed.
- (a) P. J. Stang and F. Diederich, Modern Acetylene Chemistry, VCH, Weinheim, 1995 CrossRef; (b) F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry, Willey-VCH Verlag GmbH & CO. KgaA, Weinheim, 2005 Search PubMed; (c) L.-F. Peng, S.-W. Zhang, B.-H. Wang, M.-S. Xun, Z.-L. Tang, Y.-C. Jiao and X.-H. Xu, Chin. J. Org. Chem., 2018, 38, 519 CrossRef CAS; (d) L.-F. Peng, B.-H. Wang, M. Wang, Z.-L. Tang, Y.-Z. Jiang, Y.-C. Jiao and X.-H. Xu, J. Chem. Res., 2018, 42, 235 CrossRef CAS; (e) Z. Wang, L. Yang, H.-L. Liu, W.-H. Bao, Y.-Z. Tan, M. Wang, Z. Tang and W.-M. He, Chin. J. Org. Chem., 2018, 38, 2639 CrossRef CAS; (f) L.-F. Peng, J.-Y. Lei, L. Wu, Z.-L. Tang, Z.-P. Luo, Y.-C. Jiao and X.-H. Xu, J. Chem. Res., 2018, 42, 271 CrossRef CAS; (g) W.-Y. Li, G.-X. Yin, L. Huang, Y. Xiao, Z.-M. Fu, X. Xin, F. Liu, Z.-Z. Li and W.-M. He, Green Chem., 2016, 18, 4879 RSC; (h) C. Wu, Z. Wang, Z. Hu, F. Zeng, X.-Y. Zhang, Z. Cao, Z. Tang, W.-M. He and X.-H. Xu, Org. Biomol. Chem., 2018, 16, 3177 RSC; (i) L.-F. Peng, C. Peng, M. Wang, Z.-L. Tang, Y.-C. Jiao and X.-H. Xu, Chin. J. Org. Chem., 2018, 38, 3048 CrossRef CAS; (j) L. Wu, L.-F. Peng, Z.-F. Hu, H. Wang, Z.-L. Tang, Y.-C. Jiao and X.-H. Xu, J. Chem. Res., 2019, 43, 503 CrossRef CAS.
- (a) G. Mao, A. Orita, L. Fenenko, M. Yahiro, C. Adachi and J. Otera, Mater. Chem. Phys., 2009, 115, 378 CrossRef CAS; (b) L. Fenenko, G. Shao, A. Orita, M. Yahiro, J. Otera, S. Svechnikov and C. Adachi, Chem. Commun., 2007, 2278 RSC; (c) D. Matsuo, X. Yang, A. Hamada, K. Morimoto, T. Kato, M. Yahiro, C. Adachi, A. Orita and J. Otera, Chem. Lett., 2010, 39, 1300 CrossRef CAS; (d) T. Oyamada, G. Shao, H. Uchiuzou, H. Nakanotani, A. Orita, J. Otera, M. Yahiro and C. Adachi, Jpn. J. Appl. Phys., Part 2, 2006, 45, 46 CrossRef; (e) X. Yang, S. Kajiyama, J.-K. Fang, F. Xu, Y. Uemura, N. Koumura, K. Hara, A. Orita and J. Otera, Bull. Chem. Soc. Jpn., 2012, 85, 687 CrossRef CAS; (f) X. Yang, J.-K. Fang, Y. Suzuma, F. Xu, A. Orita, J. Otera, S. Kajiyama, N. Koumura and K. Hara, Chem. Lett., 2011, 40, 620 CrossRef CAS.
- (a) Y.-N. Cheng, Z. Duan, L.-J. Yu, Z.-X. Li, Y. Zhu and Y.-J. Wu, Org. Lett., 2008, 10, 901 CrossRef CAS PubMed; (b) Y. Nakao, T. Yukawa, Y. Hirata, S. Oda, J. Satoh and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 7116 CrossRef CAS PubMed; (c) S. Arai, T. Sato, Y. Koike, M. Hayashi and A. Nishida, Angew. Chem., Int. Ed., 2009, 48, 4528 CrossRef CAS PubMed; (d) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070 CrossRef CAS PubMed; (e) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428 CrossRef CAS PubMed; (f) N. R. Rondla, S. M. Levi, J. M. Ryss, R. A. V. Berg and C. J. Douglas, Org. Lett., 2011, 13, 1940 CrossRef CAS PubMed; (g) S. Arai, Y. Amako, X. Yang and A. Nishida, Angew. Chem., Int. Ed., 2013, 52, 8147 CrossRef CAS PubMed; (h) Y. Obora, A. S. Baleta, M. Tokunaga and Y. Tsuji, J. Organomet. Chem., 2002, 660, 173 CrossRef CAS; (i) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef CAS PubMed; (j) M. Suginome, A. Yamamoto and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 2380 CrossRef CAS PubMed; (k) J. Kamiya, S. Kawakami, A. Yano, A. Nomoto and A. Ogawa, Organometallics, 2006, 25, 3562 CrossRef; (l) M. Murai, R. Hatano, S. Kitabata and K. Ohe, Chem. Commun., 2011, 47, 2375 RSC; (m) D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS PubMed; (n) N. Chatani, T. Morimoto, M. Toyoshige and S. Murai, J. Organomet. Chem., 1994, 473, 335 CrossRef CAS; (o) I. Kamiya, J. Kawakami, S. Yano, A. Nomoto and A. Ogawa, Organometallics, 2006, 25, 3562 CrossRef CAS; (p) C. Najera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 45, 2452 CrossRef PubMed; (q) Y. Nakao, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2004, 126, 13904 CrossRef CAS PubMed; (r) Y. Nakao, K. Kanyiva, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 8146 CrossRef CAS PubMed; (s) Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874 CrossRef CAS PubMed; (t) Y. Cheng, Z. Duan, L. Yu, Z. Li, Y. Zhu and Y. Wu, Org. Lett., 2008, 10, 901 CrossRef CAS PubMed; (u) K. Nozaki, N. Sato and H. Takaya, J. Org. Chem., 1994, 59, 2679 CrossRef CAS; (v) Y. Kobayashi, H. Kamisaki, R. Yanada and Y. Takemoto, Org. Lett., 2006, 8, 2711 CrossRef CAS PubMed; (w) Y. Nishihara, Y. Inoue, S. Izawa, M. Miyasaka, K. Tanemura, K. Nakajima and K. Takagi, Tetrahedron, 2006, 62, 9872 CrossRef CAS; (x) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef CAS PubMed; (y) M. Suginome, A. Yamamoto and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 2380 CrossRef CAS PubMed; (z) M. Sun, J. Jiang, J.-L. Chen, Q. Yang and X.-Y. Yu, Tetrahedron, 2019, 75, 130456 CrossRef.
- K. Okamoto, M. Watanabe, N. Sakata, M. Murai and K. Ohe, Org. Lett., 2013, 15, 5810 CrossRef CAS PubMed.
- G.-W. Rong, J.-C. Mao, Y. Zheng, R.-W. Yao and X.-F. Xu, Chem. Commun., 2015, 51, 13822 RSC.
- H.-N. Wang, P.-B. Mi, W.-J. Zhao, R. Kumar and X.-H. Bi, Org. Lett., 2017, 19, 5613 CrossRef CAS PubMed.
- Y. Du and Z. Li, Tetrahedron Lett., 2018, 59, 4622 CrossRef CAS.
- F. Ye, J.-T. Chen and T. Ritter, J. Am. Chem. Soc., 2017, 139, 7184 CrossRef CAS PubMed.
- X.-J. Zhang, X. Xie and Y.-H. Liu, J. Am. Chem. Soc., 2018, 140, 7385 CrossRef CAS PubMed.
- L. Yang, Y.-T. Liu, Y. Park, S. W. Park and S. Chang, ACS Catal., 2019, 9, 3360 CrossRef CAS.
- F.-D. Lu, D. Liu, L. Zhu, L.-Q. Lu, Q. Yang, Q.-Q. Zhou, Y. Wei, Y. Lan and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167 CrossRef CAS PubMed.
- S. Arai, T. Sato and A. Nishida, Adv. Synth. Catal., 2009, 351, 1897 CrossRef CAS.
- S. Arai, Y. K. Koike, H. Hada and A. Nishida, J. Org. Chem., 2010, 75, 7573 CrossRef CAS PubMed.
- A. G. Barrado, A. Z. Ski, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 13401 CrossRef CAS PubMed.
- N. Sakata, K. Sasakura, G. Matsushita, K. Okamoto and K. Ohe, Org. Lett., 2017, 19, 3422 CrossRef CAS PubMed.
- Z.-Y. Liao, P.-Y. Liao and T.-C. Chien, Chem. Commun., 2016, 52, 14404 RSC.
- W. Zhang, C. Yang, Y.-L. Pan, X. Li and J.-P. Cheng, Org. Biomol. Chem., 2018, 16, 5788 RSC.
- T. Igarashi, S. Arai and A. Nishida, J. Org. Chem., 2013, 78, 4366 CrossRef CAS PubMed.
- B. Liu, Y. Wang, Y. Chen, Q. Wu, J. Zhao and J.-W. Sun, Org. Lett., 2018, 20, 3465 CrossRef CAS PubMed.
- X. Wang and A. Studer, J. Am. Chem. Soc., 2016, 138, 2977 CrossRef CAS PubMed.
- N. K. Swamy, A. Yazici and S. G. Pyne, J. Org. Chem., 2010, 75, 3412 CrossRef CAS PubMed.
- Y. Minami, H. Yoshiyasu, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2013, 52, 883 CrossRef CAS PubMed.
- C. Wu, L.-H. Lu, A.-Z. Peng, G.-K. Jia, C. Peng, Z. Cao, Z.-L. Tang, W.-M. He and X.-H. Xu, Green Chem., 2018, 20, 3683 RSC.
- L.-H. Lu, S. J. Zhou, M. Sun, J.-L. Chen, W. Xia, X.-Y. Yu, X.-H. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 1574 CrossRef CAS.
- C. Wu, L. Hong, H. Shu, Q.-H. Zhou, Y. Wang, N. Su, S.-i. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 8798 CrossRef CAS.
- X.-H. Zeng and L. Chen, Org. Biomol. Chem., 2018, 16, 7557 RSC.
- C. Wu, H.-J. Xiao, S.-W. Wang, M.-S. Tang, Z.-L. Tang, W. Xia, W.-F. Li, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 2169 CrossRef CAS.
- L.-H. Lua, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 1237 CrossRef.
- L.-H. Lu, S.-J. Zhou, W.-B. He, W. Xia, P. Chen, X.-Y. Yu, X.-H. Xu and W.-M. He, Org. Biomol. Chem., 2018, 16, 9064 RSC.
| This journal is © The Royal Society of Chemistry 2020 |