
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and amide 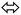 imidic prototropic tautomerization in thiophene-2-carbohydrazide: XRD, DFT/HSA-computation, DNA-docking, TG and isoconversional kinetics via FWO and KAS models†
imidic prototropic tautomerization in thiophene-2-carbohydrazide: XRD, DFT/HSA-computation, DNA-docking, TG and isoconversional kinetics via FWO and KAS models†

Nabil Al-Zaqri a,
Tamer Khatibb,
Ali Alsalmea,
Fahad A. Alharthia,
Abdelkader Zarroukc and
Ismail Warad*de
a,
Tamer Khatibb,
Ali Alsalmea,
Fahad A. Alharthia,
Abdelkader Zarroukc and
Ismail Warad*de
aDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
bDepartment of Energy Engineering and Environment, An-Najah National University, Nablus 97300, Palestine
cLaboratory of Materials, Nanotechnology and Environment, Faculty of Science, Mohammed V University, 4Av. Ibn Battuta, B. P. 1014, Rabat, Morocco
dDepartment of Chemistry and Earth Sciences, Qatar University, PO Box 2713, Doha, Qatar. E-mail: ismail.warad@qu.edu.qa
eDepartment of Chemistry, Science College, An-Najah National University, P. O. Box 7, Nablus, Palestine
First published on 10th January 2020
Abstract
Thiophene-2-carbohydrazide as a novel small-molecule amide tautomer has been synthesized with an acceptable yield under microwave radiation (MW) conditions. The amide  imidic thiophene-2-carbohydrazide prototropic tautomerization via single proton intramigration was computed using the DFT B3LYP/6-311G(d,p) level of theory. The endo-isomer amide structure of thiophene-2-carbohydrazide was proved by XRD and is considered to be the kinetically favored isomer. The DFT-structure parameters were compared to their corresponding XRD-experimental parameters. Several H-bond interactions were detected in the crystal lattice experimentally using the XRD-packing model then correlated to MEP and HSA calculations. The manual and calculated electronic parameters such as, frontier molecular orbital energies, excitation energy, absorption, dipole moment, DOS, GRD quantum parameters and TD-SCF/B3LYP were DFT computed. The thiophene-2-carbohydrazide isomers together with their prototropic (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were docked against 1BNA DNA. FWO and KAS isoconversional kinetic methods were applied, and the thermal behavior and estimated Ea–α relations were determined.
imidic thiophene-2-carbohydrazide prototropic tautomerization via single proton intramigration was computed using the DFT B3LYP/6-311G(d,p) level of theory. The endo-isomer amide structure of thiophene-2-carbohydrazide was proved by XRD and is considered to be the kinetically favored isomer. The DFT-structure parameters were compared to their corresponding XRD-experimental parameters. Several H-bond interactions were detected in the crystal lattice experimentally using the XRD-packing model then correlated to MEP and HSA calculations. The manual and calculated electronic parameters such as, frontier molecular orbital energies, excitation energy, absorption, dipole moment, DOS, GRD quantum parameters and TD-SCF/B3LYP were DFT computed. The thiophene-2-carbohydrazide isomers together with their prototropic (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were docked against 1BNA DNA. FWO and KAS isoconversional kinetic methods were applied, and the thermal behavior and estimated Ea–α relations were determined.
1. Introduction
Methods to prepare hydrazides/hydrazide-derivatives of –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–NH–N
O)–NH–N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) as general framework and active intermediate compounds have been recently reported for the synthesis and development of several types of N–N donor ligands.1–3 Thiophene-2-carbohydrazide as a N–S–O-chelate small molecule ligand and its metal ion complexes have some wide applications, mainly catalytic, biological and industrial.2–4 The presence of oxygen, nitrogen and sulfur as electron-donor atoms in the backbone of thiophene-2-carbohydrazide exhibit broad types of hydrogen bonds which enhance the structural therapeutic effects and variety of its pharmaceutical activities like: antitumor, antifungal and anti-HIV.5–8 Moreover, the carbohydrazide small molecule function as an essential pharmacophore in many therapeutically valuable materials, for example, the antiviral, antibacterial and antitubercular biological activities were dramatically increased by inserting the thiophene-carbohydrazide in the structure of big molecules.5–12 Therefore, a huge number of heterocyclic carbohydrazide derivatives have been made available.6–15
as general framework and active intermediate compounds have been recently reported for the synthesis and development of several types of N–N donor ligands.1–3 Thiophene-2-carbohydrazide as a N–S–O-chelate small molecule ligand and its metal ion complexes have some wide applications, mainly catalytic, biological and industrial.2–4 The presence of oxygen, nitrogen and sulfur as electron-donor atoms in the backbone of thiophene-2-carbohydrazide exhibit broad types of hydrogen bonds which enhance the structural therapeutic effects and variety of its pharmaceutical activities like: antitumor, antifungal and anti-HIV.5–8 Moreover, the carbohydrazide small molecule function as an essential pharmacophore in many therapeutically valuable materials, for example, the antiviral, antibacterial and antitubercular biological activities were dramatically increased by inserting the thiophene-carbohydrazide in the structure of big molecules.5–12 Therefore, a huge number of heterocyclic carbohydrazide derivatives have been made available.6–15
The prototropic tautomers are compounds that can be interconverted from molecule to another via single proton migration from group to the close neighbor atom.16 It's known that there is no pure separating line between such tautomers and isomers: tautomer is obviously isomer that transform with a comparatively depressed activation energy <20 kcal mol−1.17–24
The most well-known single-proton prototropic tautomerization reactions are illustrated in Scheme 1a–d, such reactions may play a vital role in biological suits.18 For instance, the hydrogen-bonds which connected bases pairs like G–C and Genol–T together (Scheme 1) to build the DNA is a remarkable example emphasizes the importance of such proposition.17 In mother nature, tautomerization in nitrogenous bases likely cases genetic mutation via H-migration, therefore, in organic molecules prototropic tautomerization toward more stable or (and) active forms play essential role in structural shape selectivity (SSS).15–19
 | ||
| Scheme 1 (a–d) Prototropic tautomerization reactions and G–C and Genol–T DNA-bases pairs (e and f). | ||
Thiophene-2-carbohydrazide can be pointed as an excellent model simple-small molecule, for example, Balachandran and coworkers have geometrically optimized thiophene-2-carbohydrazide without having its XRD-crystal structure as trustable structural reference.10 Moreover, theoretical amide  imidic tautomerization of thiophene-2-carbohydrazide to thiophene-2-carbohydrazonic acid via single proton intra-migration has been never studied before; such tautomerization process was DFT-computed in this study, the transition states energies and structures were estimated by QST2 method of calculation. Additionally, the XRD-structure of thiophene-2-carbohydrazide confirmed the endo-isomer existence of molecule as kinetic favored isomer has been matched to the DFT-calculated parameters. Appling of KAS and FWO isoconversional kinetic methods resulting the multistep reaction decomposition. To establish the intermolecular forces in crystal lattice, MPE and HSA computed result were compared to experimental XRD-packing result. The TD-SCF/DFT/B3LYP calculation, FMO, DOS and GRD quantum parameters were matched with its relative's experimental electronic one. The thiophene-2-carbohydrazide endo and exo isomers reflected no DNA-docking effect, meanwhile its (E)/(Z)-thiophene-2-carbohydrazonic acid isomers showed cisplatin like DNA-binding mode.
imidic tautomerization of thiophene-2-carbohydrazide to thiophene-2-carbohydrazonic acid via single proton intra-migration has been never studied before; such tautomerization process was DFT-computed in this study, the transition states energies and structures were estimated by QST2 method of calculation. Additionally, the XRD-structure of thiophene-2-carbohydrazide confirmed the endo-isomer existence of molecule as kinetic favored isomer has been matched to the DFT-calculated parameters. Appling of KAS and FWO isoconversional kinetic methods resulting the multistep reaction decomposition. To establish the intermolecular forces in crystal lattice, MPE and HSA computed result were compared to experimental XRD-packing result. The TD-SCF/DFT/B3LYP calculation, FMO, DOS and GRD quantum parameters were matched with its relative's experimental electronic one. The thiophene-2-carbohydrazide endo and exo isomers reflected no DNA-docking effect, meanwhile its (E)/(Z)-thiophene-2-carbohydrazonic acid isomers showed cisplatin like DNA-binding mode.
2. Materials and methods
2.1. XRD
A colorless block single-crystal of thiophene-2-carbohydrazide with the dimension of 0.71 × 0.12 × 0.08 mm was used. The structure was solved using SHELXL and SHELXS programs, respectively (Table 1).25| Empirical formula | C5H6N2OS |
| CCDC | 936463 |
| Temperature | 293(2) K |
| Formula weight | 284.36 |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, P21/c |
| Volume | 639.55(5) Å3 |
| Unit cell dimensions | a = 6.06900(4) Å |
| b = 8.50100(2) Å | |
| c = 12.51800(3) Å | |
| β = 98.048(4)° | |
| Crystal size | 0.71 × 0.12 × 0.08 mm3 |
| Z | 4 |
| Absorption coefficient | 0.42 mm−1 |
| F000 | 296 |
| Reflections collected/unique | 1131/[R(int) = 0.021] |
| Refinement method | Full-matrix least-squares on F2 |
| Final R indices [I > 2sigma(I)] | R1 = 0.038, wR2 = 0.094, |
| Largest diff. peak and hole | 0.21, −0.22 e Å−3 |
2.2. Computations
The DFT-computations were performed at the DFT/B3LYP level of theory in gaseous phase using Gaussian09 software.26 AUTO-DOCK version 4.5 was used for docking, CRYSTAL EXPLORER 3.1 program was used for HSA analysis.272.3. Kinetic FWO and KAS methods
In FWO and KAS methods, the correlation between the heating rate βi and temperature Tαi is given by eqn (1) (ref. 28) and (2), respectively.29| ln(βi) = −1.052(Eα/RTαi) + constant | (1) |
| ln(βi/Tαi2) = −Eα/RTαi + constant | (2) |
2.4. Synthesis of thiophene-carbohydrazide
Methyl thiophene-2-carboxylate (1.0 mmol) and hydrazine monohydrated (5.0 mmol) was dissolved in 40 ml of methanol in 250 ml round bottom flask containing 5 small pieces of boiling cheeps, to a void solvent evaporation during radiation an effective reflux system with two considers was setup, the reaction mixture in the bottom flask only was subjected to MW radiation for 5 min, the white precipitate was collected then filtered off after direct cooling of the reaction in ice bath, yield: 86.7%. The mp = 136–138 °C, EI-MS m/z: found 143.2 [M+ + 1], cal.: 142.3 [M+]. CHN-EA for C5H6N2OS, obs. C, 42.24; H, 4.25; N, 19.70%, cal.: C 42.12; H 4.11; N 19.58%.3. Result and discussion
3.1. Synthesis
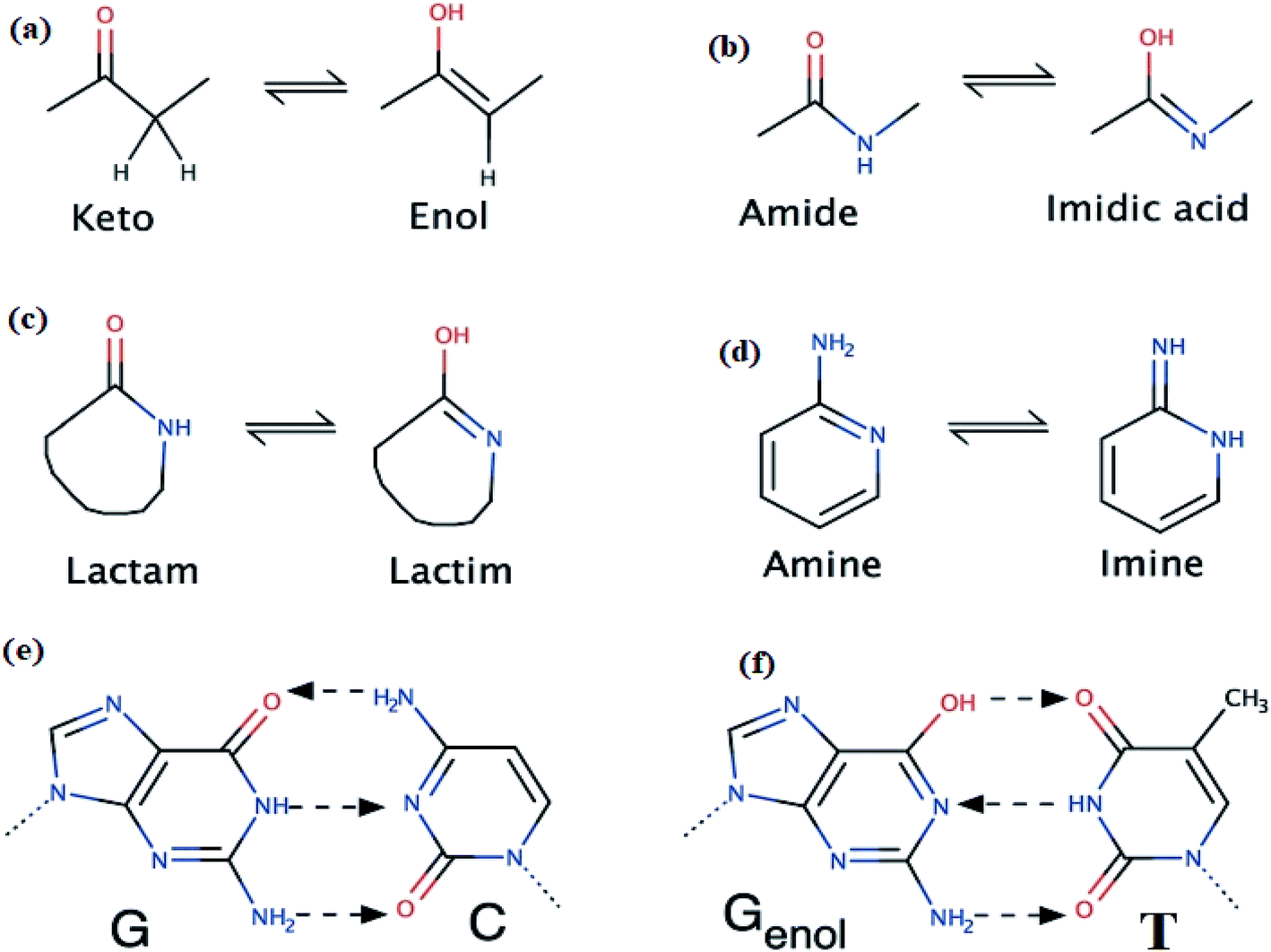
A one-pot eco-synthesis method has been developed for the preparation of thiophene-2-carbohydrazide simple molecule using water–methanol as solvents and cheap kitchen 1500 W normal microwave as source of radiation. In the eco-synthesis one-pot time process was performed by subjecting a combination of hydrazine monohydrated with methyl thiophene-2-carboxylate to 5 min microwave period radiation, as seen in Scheme 2.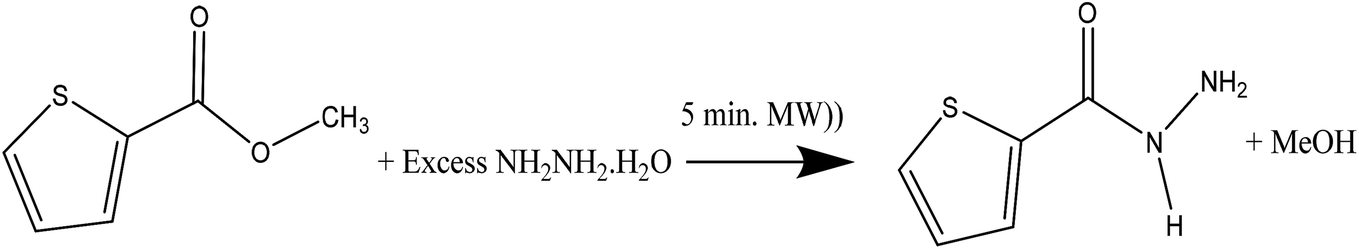 | ||
| Scheme 2 Synthesis of thiophene-2-carbohydrazide. | ||
The well-known thiophene-2-carbohydrazide compound was prepared in an excellent yield, short time reaction and without side products, moreover; the excess hydrazine monohydrated was allowed to be evaporated under the heat of MW radiation. The formation of the thiophene-2-carbohydrazide compound was proved by XRD.
3.2. XRD analysis
Novel conformal thiophene-2-carbohydrazide endo isomer (H of amide trans to O of carbonyl, exo) has been proven by XRD single crystal measurement, as seen in Scheme 3. The rotation around Csp2–Nsp3 single bond exactly at 180° led to the less stable conformational exo isomer (H of amide cis to O of carbonyl, endo) was not detected by XRD yet. | ||
| Scheme 3 Endo/exo-thiophene-2-carbohydrazide isomerization. | ||
The details crystal structure parameters of the endo thiophene-2-carbohydrazide isomer were illustrated in Table 2, moreover, Fig. 1 showed the numbering molecular structure scheme together with its packing unit cell. The thiophene-2-carbohydrazide, C5H7N2OS, crystallized as a monoclinic//P21/c space-group, four molecules were crystalline in the unit cell (Z = 4), as seen in Fig. 1b. The structure is look like planer around the carbonyl and the thiophene centers, the carbonyl oxygen atom is in cis to the S of the thiophene with dihedral angle = 4.7° and trans to the H of the amide with dihedral angle = 170.9°, such seen raised up the polarity of the compound to more than 3.00 D and enhance its metal ions chelate effect.
| Bond no. | Bonds | Exp. XRD | DFT | Angles no. | Angles (°) | Exp. XRD | DFT | |||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | S1 | C4 | 1.713 | 1.7431 | 1 | C4 | S1 | C1 | 91.4 | 91.25 |
| 2 | S1 | C1 | 1.698 | 1.7276 | 2 | O1 | C5 | N1 | 122.4 | 121.76 |
| 3 | C5 | O1 | 1.234 | 1.2287 | 3 | O1 | C5 | C4 | 122.3 | 122.57 |
| 4 | C5 | N1 | 1.327 | 1.378 | 4 | N1 | C5 | C4 | 115.3 | 115.68 |
| 5 | C5 | C4 | 1.474 | 1.4789 | 5 | C5 | N1 | N2 | 122 | 119.62 |
| 6 | N1 | N2 | 1.411 | 1.4141 | 6 | C5 | N1 | H1A | 122 | 118.72 |
| 7 | N1 | H1A | 0.84 | 1.0111 | 7 | N2 | N1 | H1A | 113 | 113.2 |
| 8 | C4 | C3 | 1.376 | 1.3773 | 8 | S1 | C4 | C5 | 118.1 | 117.79 |
| 10 | C3 | C2 | 1.408 | 1.4222 | 9 | S1 | C4 | C3 | 111.4 | 111.15 |
| 11 | N2 | H2A | 0.88 | 1.0207 | 10 | C5 | C4 | C3 | 130.5 | 131.04 |
| 12 | N2 | H2B | 0.89 | 1.023 | 11 | C4 | C3 | C2 | 111.9 | 112.99 |
| 14 | C2 | C1 | 1.347 | 1.3716 | 12 | N1 | N2 | H2A | 107 | 106.93 |
| 13 | N1 | N2 | H2B | 107 | 106.48 | |||||
| 14 | H2A | N2 | H2B | 104 | 105.03 | |||||
| 15 | C3 | C2 | C1 | 112.7 | 112.31 | |||||
| 16 | S1 | C1 | C2 | 112.6 | 112.29 | |||||
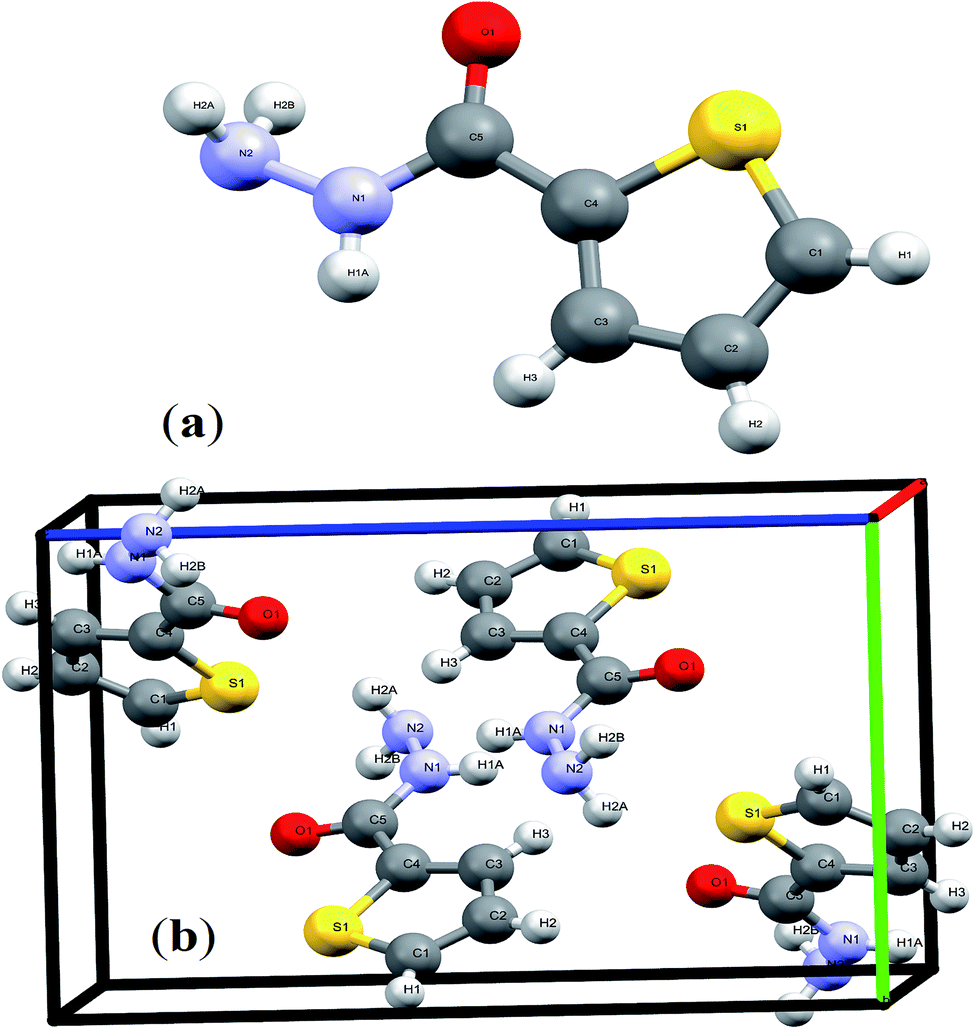 | ||
| Fig. 1 (a) Endo-isomer structure and (b) packing unit cell with Z = 4 in thiophene-2-carbohydrazide. | ||
3.3. Structure optimizations
The endo isomer structure of thiophene-2-carbohydrazide which was confirmed by XRD-single crystal analysis was optimized at B3LYP/6-311G(d,p) theory. All the structural parameters like bond lengths and angles were compared to the XRD/exp. collected data. An excellent matching between calculated and theoretical (excepting N–H bonds) was illustrated in Fig. 2, the correlation between the calculated/experimental bond lengths is 0.9783 (Fig. 2a and b). Similarly, the correlation between the calculated/experimental angles is 0.9857 (Fig. 2c and d).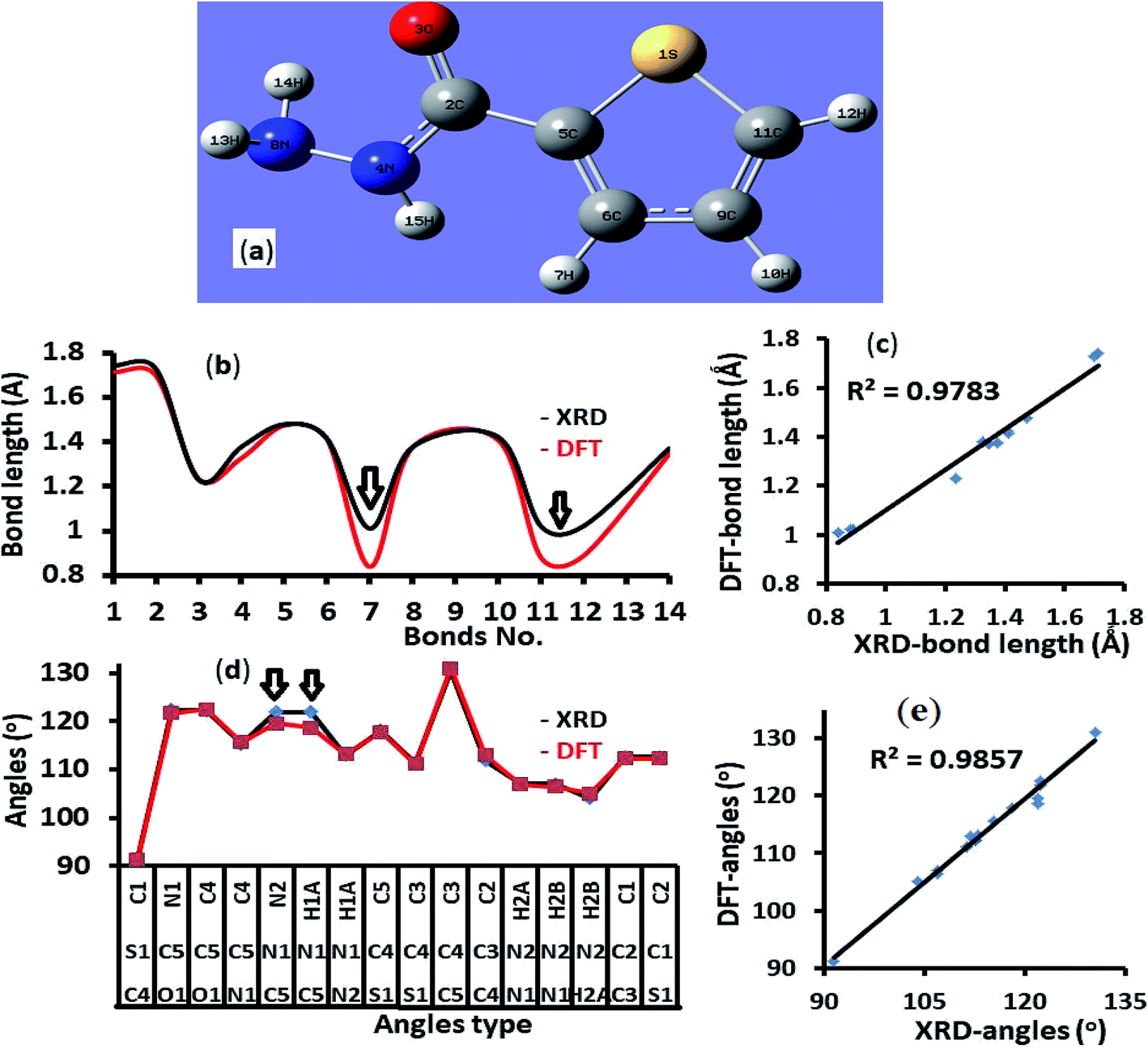 | ||
| Fig. 2 (a) B3LYP/6-311G(d) structure, (b) XRD/DFT bond lengths histogram, (c) XRD/DFT bond lengths graphical correlation, (d) XRD/DFT angles histogram, and (e) XRD/DFT angles graphical correlation. | ||
3.4. Crystal interactions, HSA, 2D fingerprint (FP) and MEP investigation
In the crystal lattice, several interactions shorter than 3 Å were detected experimentally, since four heteroatoms 2N, 1O and 1S in addition to three polar H atoms cited in the backbone of ligand, therefore, four shorter head-to-tail H-bonds as –HN–H⋯O = 2.138 Å and –N–H⋯NH3 = 2.163 Å strongly dimerized molecules with its neighbors as illustrated in Fig. 3a. Moreover, two longer H-bonds were detected as CO⋯H = 2.702 Å and –(CH2)2–S⋯HNH– = 2.993 Å (Fig. 3b).
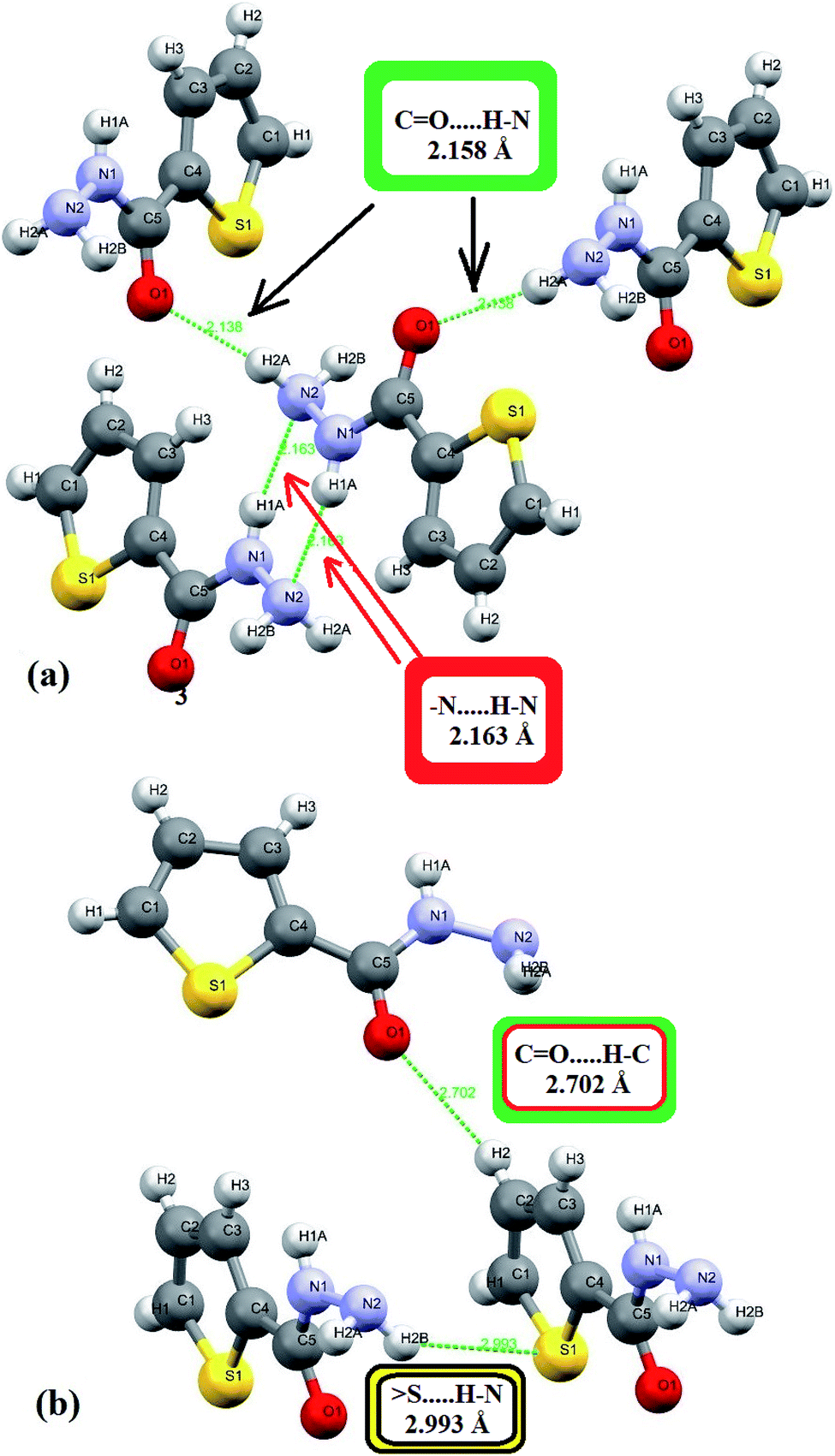 | ||
| Fig. 3 H-bonds interactions types and lengths: (a) short (N–H⋯O and –N–H⋯N) and (b) long (CO⋯H and S⋯H–N) interactions. | ||
In HSA, three big red spots were detected close to the O and N atoms on the dnorm surface supporting their roles in formation of several types of H-bonds (Fig. 4a). Moreover, each molecule is connected with surrounding molecules via three types of H-bonds (Fig. 4b), which built a 3D network around the center molecule.2,30–35 The shortest H-bonds are identified as CO⋯H–N type with 2.138 Å length, followed by two –N–H⋯N types with 2.336 and 2.546 Å which is consistent with the XRD result. No true C–H⋯Sthiophene H-bond was detected by the Hirshfeld surface; meanwhile, XRD analysis reflected such bond as H-bond with 2.993 Å bond lengths.
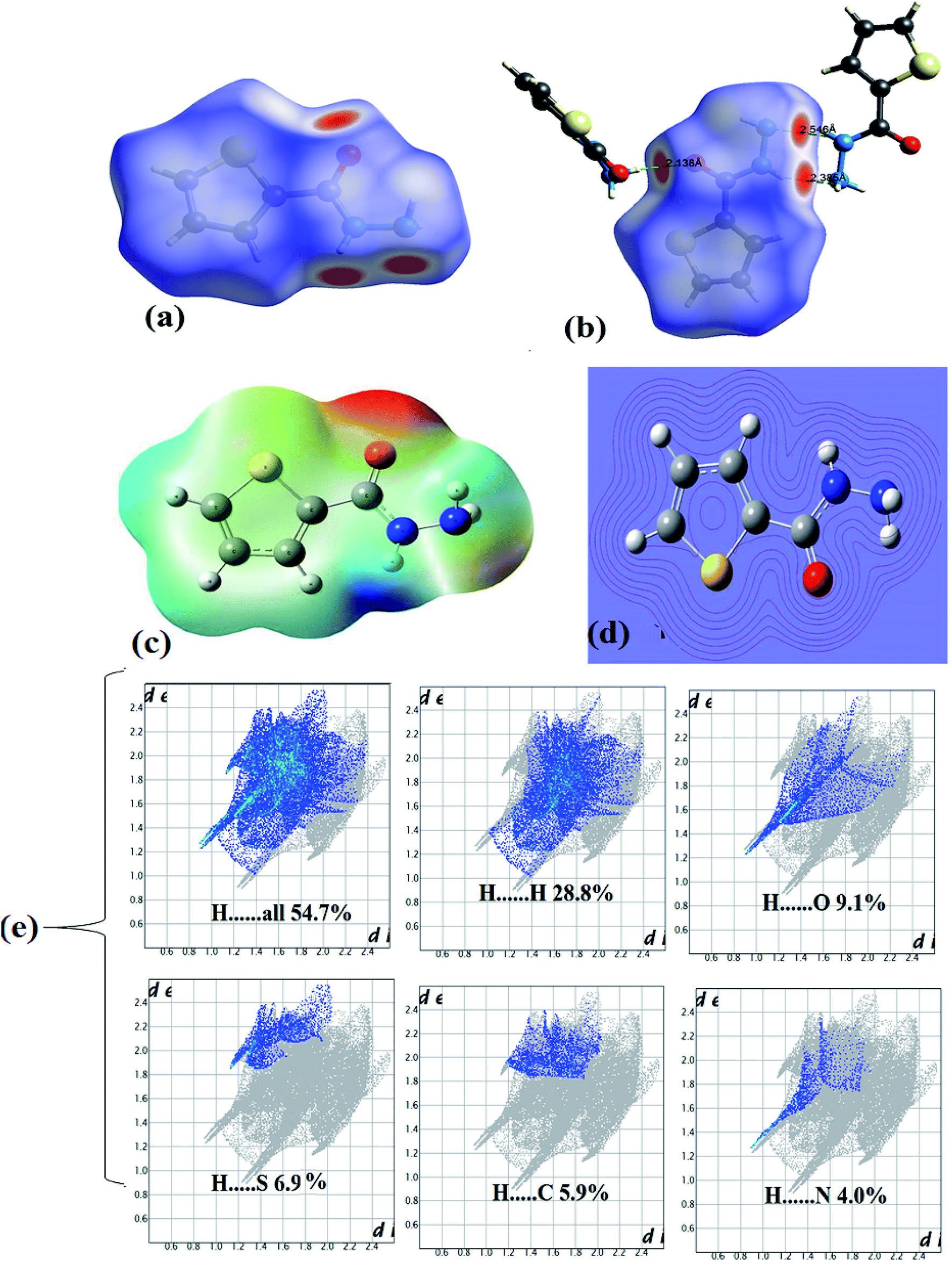 | ||
| Fig. 4 (a) dnorm, (b) H-bonds interactions, (c) MEP, (d) contour and (e) FP inside⋯outside maps. | ||
The MEP/B3LYP reflecting electrophilic/nucleophilic atoms in the molecule as indicated by colors [red (highest) > orange > yellow > green > blue (lowest)]; the electrostatic potential ratios are illustrated not only by colors also by different in lines computing intensity, as seen in Fig. 4c and d. The MEP proved the presence of both e-poor/e-rich positions in the ligand, for example, the line intensity and the red color reflected the O atom of the carbonyl as strong nucleophile site, on the other hand, the blue color of amide H atom showed a strong electrophilic.30,31 Since both red and blue colors detected by MEP in the same molecule this proved probability of H⋯O hydrogen bonds formation. The result is consistent with XRD experimental and computed HSA result of having such NH⋯OC H-bonds.
The 2D-fingerprint plots over the HSA computed surface molecule reflected the presence of inter-contacts as: H⋯H (28.8%) > H⋯O (9.1%) > H⋯S (6.9%) > H⋯C (5.9%) > H⋯N (4.0%) with H⋯overall connections (54.7%), as depicted in Fig. 4e.
3.5. Amide 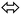 imidic tautomerization
imidic tautomerization
In prototropic tautomerization the proton atom migrated from functional group to the neighbor one casing H-bond shift via pseudo four, five or six (favor) membered ring transition state.2–8 This tautomeric behavior can place between enol → keto, lactim → lactam, imine → amine, and amide → imidic functionality resulted in single sigma bond migration followed mostly by double bond shift.15–19

All the XRD and DFT-B3LYP/6-311G(d)-calculations supported the endo-thiophene-2-carbohydrazide as favor amide isomer (Fig. 1). Therefore, endo-isomer considered to be the most stable tautomeric-isomer (kinetic favored isomer) refer to zero-point-reference-energy with global total DFT energy = −777.0233007 a.u.
The gaseous DFT calculation showed that two steps are needed to tautomerize for A → C (Scheme 4), when crossing amide → imidic CO oxygen to OH parallel to N in NH to CN bonds functional groups performed prototropic transformed.15,16 The first reaction-path involves the conversion of A → B, the H of amine should be in the same direction of carbonyl O atom, for that, A performed simple flip-rotation around Csp2–Nsp3 with O–C–N–H dihedral angle = 0.43° to produce the B isomer. The transition state (T.S1) of this step is very simple, a perpendicular Csp2–Nsp3 rotation around C–N single bond to make the O–C–N–H dihedral angle around ∼90° was detected, as seen in Scheme 4.
 | ||
Scheme 4 Amide  imidic prototropic tautomerization reaction. imidic prototropic tautomerization reaction. | ||
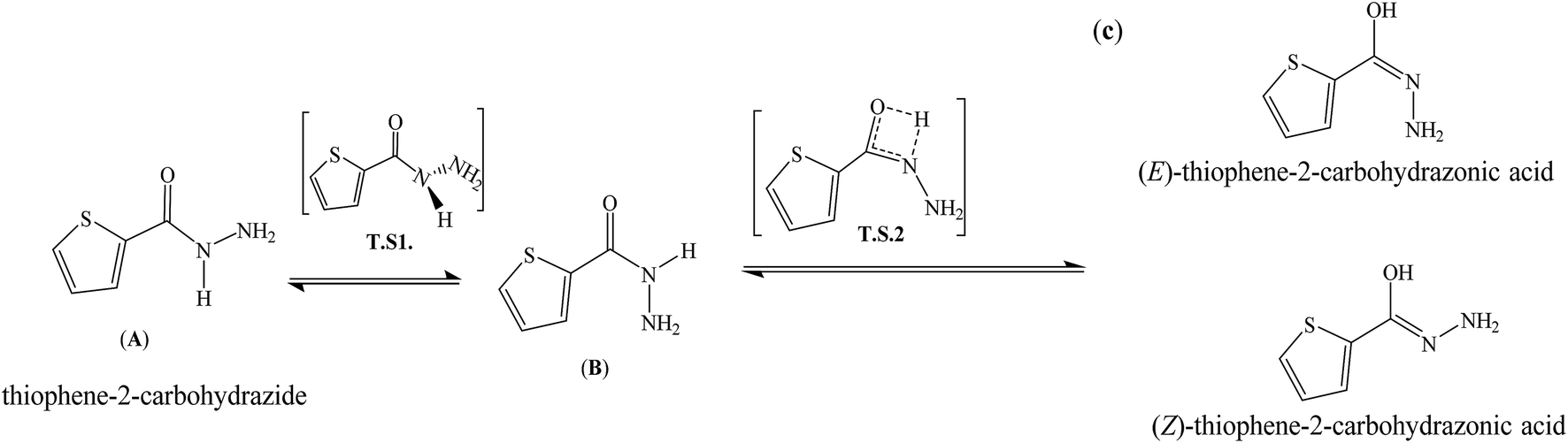
The second reaction-path involves the conversion of B to C (E or Z isomer as final product) via single proton-transfer, the proton is intra-migrated from N to O atoms establishing a pseudo-four-membered-ring O⋯C⋯N⋯H as transition state T.S2 (Fig. 5). The QST2-computed method of the T.S2 demonstrated the H-transfer inside a group containing a CO moiety in a direct neighborhood of the N–H group, at this level Hammond's postulate was applied since the proton lost its N-attractive correlation and be trapped by O forming O–H new bond, therefore the T.S2 structure should be closed to the product shape. This seen is consistent with T.S2 computed structure which generated with O1⋯H1 and N1⋯H1 distances 1.31 and 1.37 Å, respectively.
 | ||
Fig. 5 Structures and energies profiles of amide  imidic prototropic tautomerization. imidic prototropic tautomerization. | ||
The energy profiles in gaseous-phase calculation of amide  imidic prototropic tautomerism via the single-proton transfer mode is illustrated in Fig. 5. The XRD and DFT-computation revealed the amide as favored isomer over imidic tautomer one libeled with zero-point-energy. The ΔE tautomerization energy was estimated for each step as the energy differences between the tautomer and its transition state. The ET.S1 calculated to 64.14 kJ mol−1, the energy difference between A and B tautomers were found to be 20.90 kJ mol−1. The ET.S2 calculated to 188.72 kJ mol−1, the energy difference between B and C(Z) tautomer was found to be 41.45 kJ mol−1, the energy difference between B and C(E) tautomer was found to be 61.44 kJ mol−1, the energy difference between C(E) and C(Z) was found to be 19.29 kJ mol−1. In conclusion, the energy calculations in gaseous-phase supported the amide
imidic prototropic tautomerism via the single-proton transfer mode is illustrated in Fig. 5. The XRD and DFT-computation revealed the amide as favored isomer over imidic tautomer one libeled with zero-point-energy. The ΔE tautomerization energy was estimated for each step as the energy differences between the tautomer and its transition state. The ET.S1 calculated to 64.14 kJ mol−1, the energy difference between A and B tautomers were found to be 20.90 kJ mol−1. The ET.S2 calculated to 188.72 kJ mol−1, the energy difference between B and C(Z) tautomer was found to be 41.45 kJ mol−1, the energy difference between B and C(E) tautomer was found to be 61.44 kJ mol−1, the energy difference between C(E) and C(Z) was found to be 19.29 kJ mol−1. In conclusion, the energy calculations in gaseous-phase supported the amide  imidic prototropic tautomerism possibility under mild condition.
imidic prototropic tautomerism possibility under mild condition.
In this process, the amide tautomer of thiophene-2-carbohydrazide was detected by XRD as endo-isomer where it classified as kinetic favored isomer, the DFT-calculation supported the probability of amide  imidic prototropic tautomerization, since the energy required is not so high it can be easily provided from surrounding temperature or solvents. Therefore, imidic acid isomer form in this can be isolated under suitable conditions, this fact was seen in similar work, the imidic acid tautomer in methanol solvent reaction was isolated by XRD as predominant product.28
imidic prototropic tautomerization, since the energy required is not so high it can be easily provided from surrounding temperature or solvents. Therefore, imidic acid isomer form in this can be isolated under suitable conditions, this fact was seen in similar work, the imidic acid tautomer in methanol solvent reaction was isolated by XRD as predominant product.28
3.6. HOMO–LUMO, DOS, exp. electronic transfer, TD-SCF-B3LYP, Mulliken/NPA and GRD
HOMO → LUMO and HOMO−1 → LUMO+1 shapes and energy diagram reflected the electron donation capacity in the ultraviolet region with ΔEHOMO/LUMO = 5.301 eV (233.88 nm) and ΔEHOMO−1/LUMO+1 = 7.646 eV (162.15 nm) as illustrated in Fig. 6a.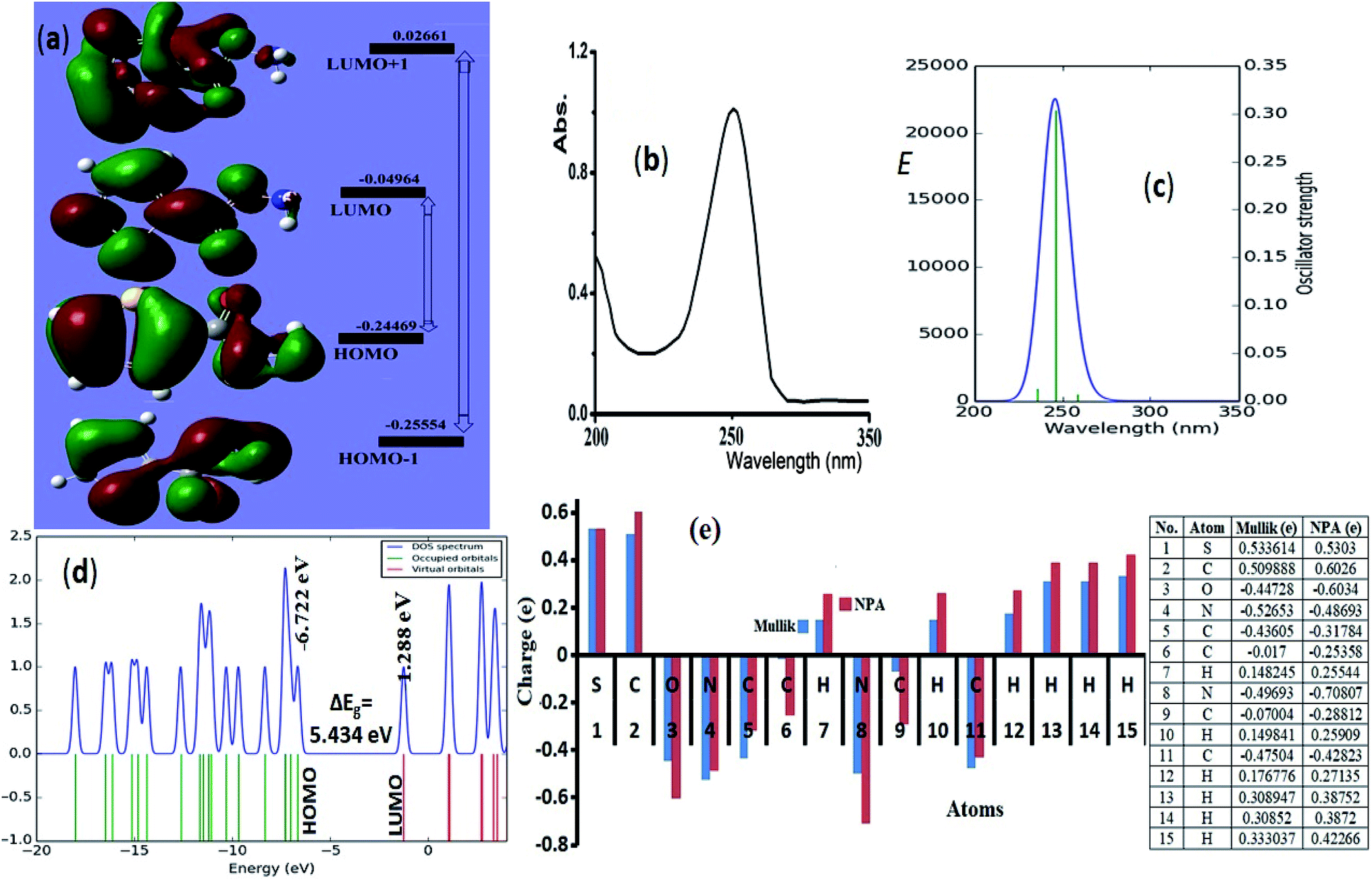 | ||
| Fig. 6 (a) HOMO/LUMO FMO, exp. (b) and calculated electronic spectra in methanol (c), (d) DOS and (e) NPA (---) and Mulliken (---) atomic charge-distribution. | ||
Experimental-spectral analysis of the thiophene-2-carbohydrazide was carried out in methanol. One broad peak λmax = 250 nm was recorded, which was attributed to π → π* e-transition. No other peaks in the visible region were detected (Fig. 6b). The computed TD-SCF/DFT-B3LYP/6-311G(d,p) in the same solvent exhibited same electronic behavior, a mean abroad band at λmax = 245.8 nm corresponding mainly to HOMO to LUMO (87%) e-transition (Fig. 6c). The other minor e-transfer together with the molecular contribution ratios were illustrated in Table 3.
| No. | Wavelength (nm) | Osc. strength | Major contribs | Minor contribs |
|---|---|---|---|---|
| 1 | 258.65 | 0.0072 | H−3 → LUMO (91%) | H−1 → LUMO (3%), HOMO → LUMO (4%) |
| 2 | 245.85 | 0.3041 | HOMO → LUMO (87%) | H−3 → LUMO (4%), H−2 → LUMO (5%) |
| 3 | 235.76 | 0.0127 | H−1 → LUMO (92%) | H−3 → LUMO (2%) |
In general, the molecular orbital energy levels together with their energy gap (5.301 eV) are strongly agreed with energy gap generated from DOS (5.434 eV) as well as the UV-experimental result as seen in Fig. 6d, the small bathochromic shift (∼5 nm) detected between exp./DFT can be resonated to solute–solvent interactions behavior.17,30–35
NPA and Mulliken charges play a critical role in theoretical quantum-charge distribution; it revealed information about electrophilic and nucleophilic sited on molecule.17,30 B3LYP/6-31G(d) Mulliken population and NPA charge analysis of the desired compound were illustrated in Fig. 6e. In general, the Mulliken showed lower atomic-charges compared to NPA, the NPA and Mulliken proved the 2N, O and all carbons atoms except C2 as nucleophilic atoms. The electrophilic sites are S, C2 carbonyl carbon and all the hydrogen atoms, the highest electrophilic hydrogen were sited to the amide proton (H15). The NPA and Mulliken charge result is strongly consistent with XRD-packing, MEP and HSA results.
The global reactivity descriptors (GRD) like: hardness (η), the chemical potential (μ), electrophilicity (ω), softness (σ) and electronegativity (χ) of the molecule were elaborated by using the following equations:
| I: ionization potential = −EHOMO | (3) |
| A: electron affinity = −ELUMO | (4) |
| ΔEgap: the energy gap (eV) = ELUMO − EHOMO | (5) |
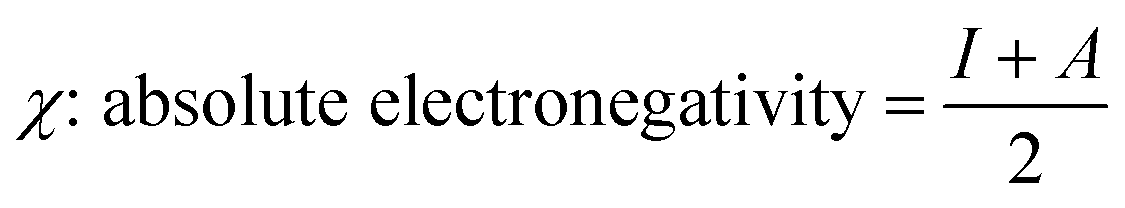 | (6) |
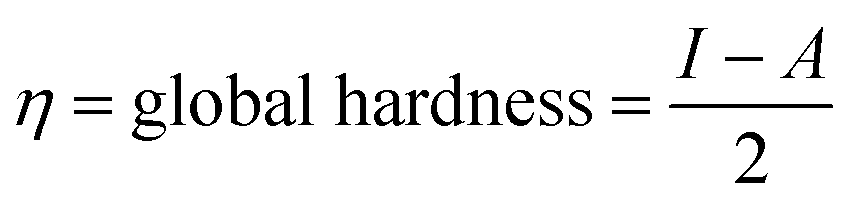 | (7) |
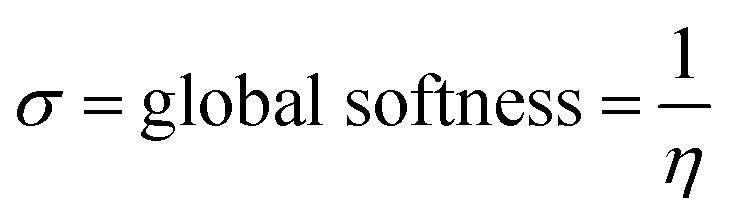 | (8) |
| μ: chemical potential = −χ | (9) |
| ω: electrophilicity = μ2/2η | (10) |
The GRD data values were collected in Table 4.
| GRD | Value | |
|---|---|---|
| Global total energy | ET | 777.0233007 a.u. |
| Low unoccupied molecular orbital | LUMO | −1.3516 eV |
| High occupied molecular orbital | HOMO | −6.6524 eV |
| Energy gap | ΔEgap | 5.3008 eV |
| Electron affinity | A | 1.3516 eV |
| Ionization potential | I | 6.6524 eV |
| Global hardness | η | 2.6504 eV |
| Global softness | σ | 0.3773 eV |
| Chemical potential | μ | −4.0020 eV |
| Absolute electronegativity | χ | 4.0020 eV |
| Electrophilicity | ω | 1.5099 eV |
| Dipole moment | 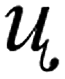 |
2.9936 D |
3.7. Molecular docking
Model small-molecule, endo–exo thiophene-2-carbohydrazide and its (E)/(Z)-thiophene-2-carbohydrazonic acid tautomers were theoretically molecular docked under the same level of theory using DNA (PDB ID: 1BNA) available data.Interestingly, endo/exo thiophene-2-carbohydrazide isomers (see Scheme 3) reflected no docking effect with 1BNA, meanwhile the (E)/(Z)-thiophene-2-carbohydrazonic acid resemble an excellent docking by crosslinking both chains of DNA double helix like the cisplatin binding mode. Several hydrogen bonds between DNA:(E/Z)drug were detected meanwhile, no non-covalent π–π stacking interactions were observed (Fig. 7a). The greater binding affinity of E-isomer reflected close contact with the surface of molecule through minor groove intercalation mode (Fig. 7a). Three short hydrogen bonds: DNA DG16:H22⋯O with 1.98 Å, DNA DG10:H21⋯N with 1.86 Å and DNA DA17:O4⋯H with 2.084 Å were detected (Fig. 7b and c). Usually docking data are good when the Root Mean Square Deviation (RMSD) value is close or less than 2 Å.36
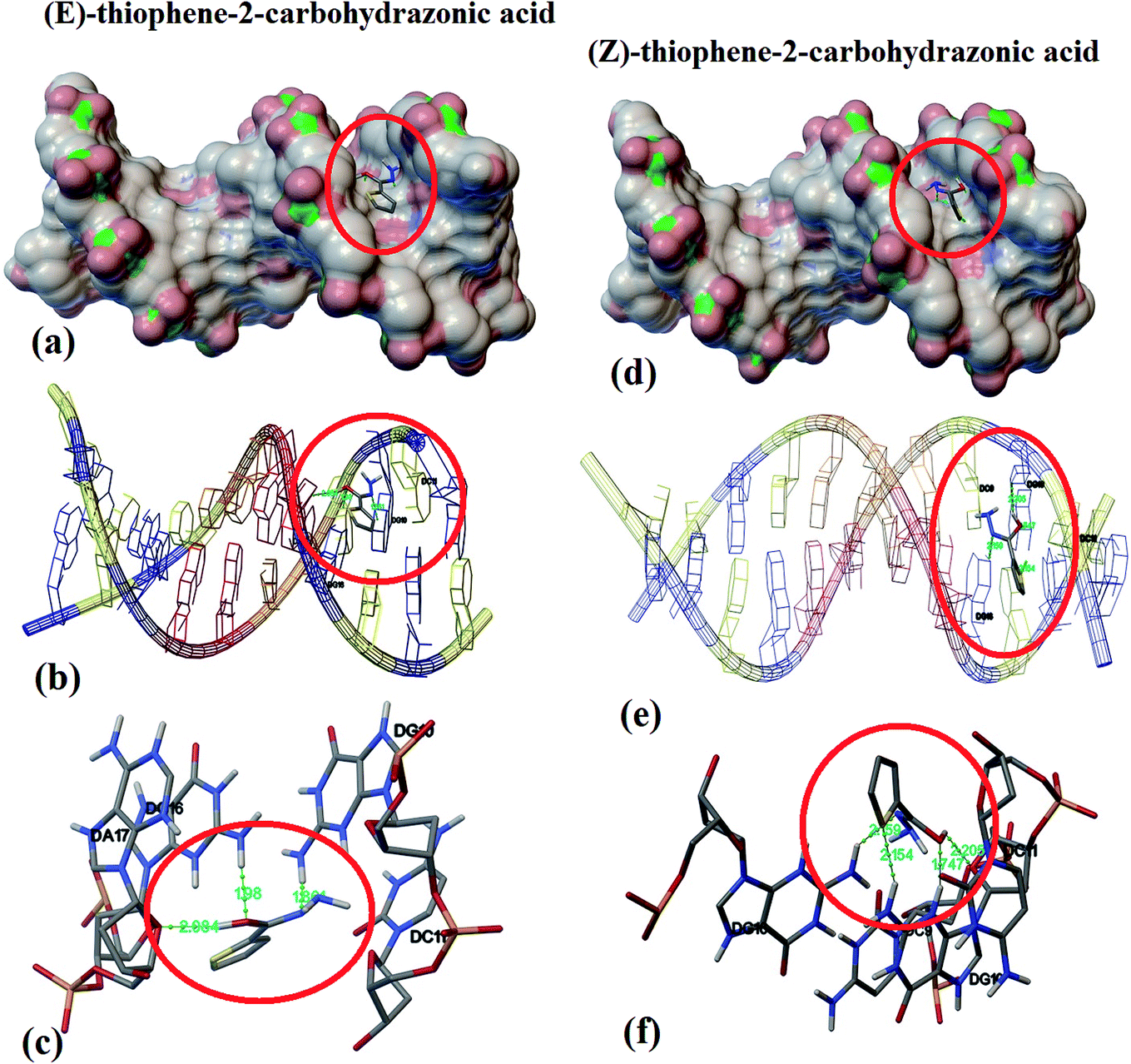 | ||
| Fig. 7 DNA binding position for E (a) and for Z-isomer (d), nuclides-binding types for E (b) and for Z-isomer (e), and DNA:drug H-bonds interactions for E (c) and for Z-isomer (f). | ||
In the Z-thiophene-2-carbohydrazonic acid tautomer, small structural conformation with ∼20 kJ mol−1 isomerization energy difference compared to E-isomer, a dramatically changes in the DNA binding behavior was detected. Due to the structural appropriate of Z-isomer it moved deeper between DNA double helix, therefore deep groove intercalation mode with new binding position was observed as in Fig. 7d and e. Moreover, four short hydrogen bonds like: DNA-DG10:H21⋯S with 2.154 Å (new bond), DNA-DG16:H22⋯N with 2.159 Å, DNA-DG10:H3⋯O with 1.747 Å and DNA-DG10:O4⋯H with 2.205 Å (Fig. 7f). Since Z-isomer energetically and structurally more favored over E one, therefore, it can be claimed that (Z)-thiophene-2-carbohydrazonic acid is better DNA-binder; this result is consistent with structural shape selectivity method.36
3.8. Thermal and isoconversional kinetics analysis via FWO and KSA
The TG/DTG-curves in the temperature range 0 to 400 °C under various heating rates in an open atmosphere revealed the desired compound with good thermal stability, it reflected an acceptable stability up to To = 110 °C under heat rate = 5 °C min−1, increased to 155 °C by increasing the heat rate to reach up to 20 °C min−1. In all the trials 1–4, the compound decayed from 100% weight to ∼zero residue weight via one broad step with Toff 250–270 °C and full thermal decomposition (Fig. 8a and b).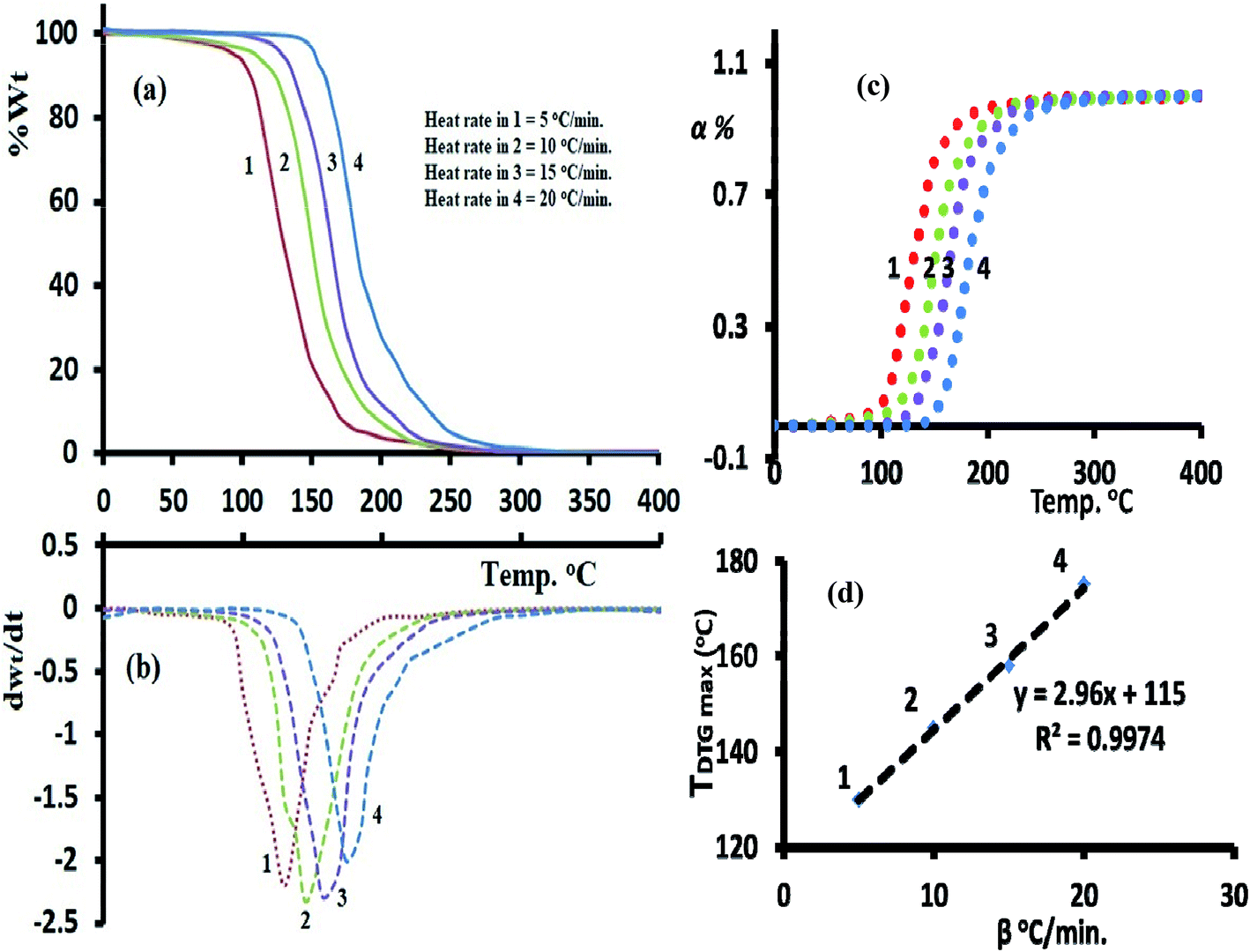 | ||
| Fig. 8 (a) TG, (b) DTG, (c) conversions and (d) β–T curves at four different heating rates. | ||
The TG-DTG-thermograms not served only for thermal analysis of the desired ligand, but also to study its isoconversional kinetic-thermal decomposition. Accordingly, the FWO/KAS isoconversional kinetics models performed TG-analysis at four various heating rates: 20, 15, 10 and 5 °C min−1 to record enough data for Ea's activations. The TDTG max was shifted to high temperature by rising up β's as seen Fig. 8b. A perfect linear relation with R2 = 0.997 between β's and the TDTG max values were recorded as seen Fig. 8d.
Both FWO and KAS as model-free isoconversional methods assumed that the conversion% (α) dependence directly on the temperature values, therefore the activation energy (Ea) of the thermal decomposition reaction can be estimated via plotting ln(β) or ln(β/T2) versus 1000/T using FWO Fig. 9a and KAS model Fig. 9b respectively.
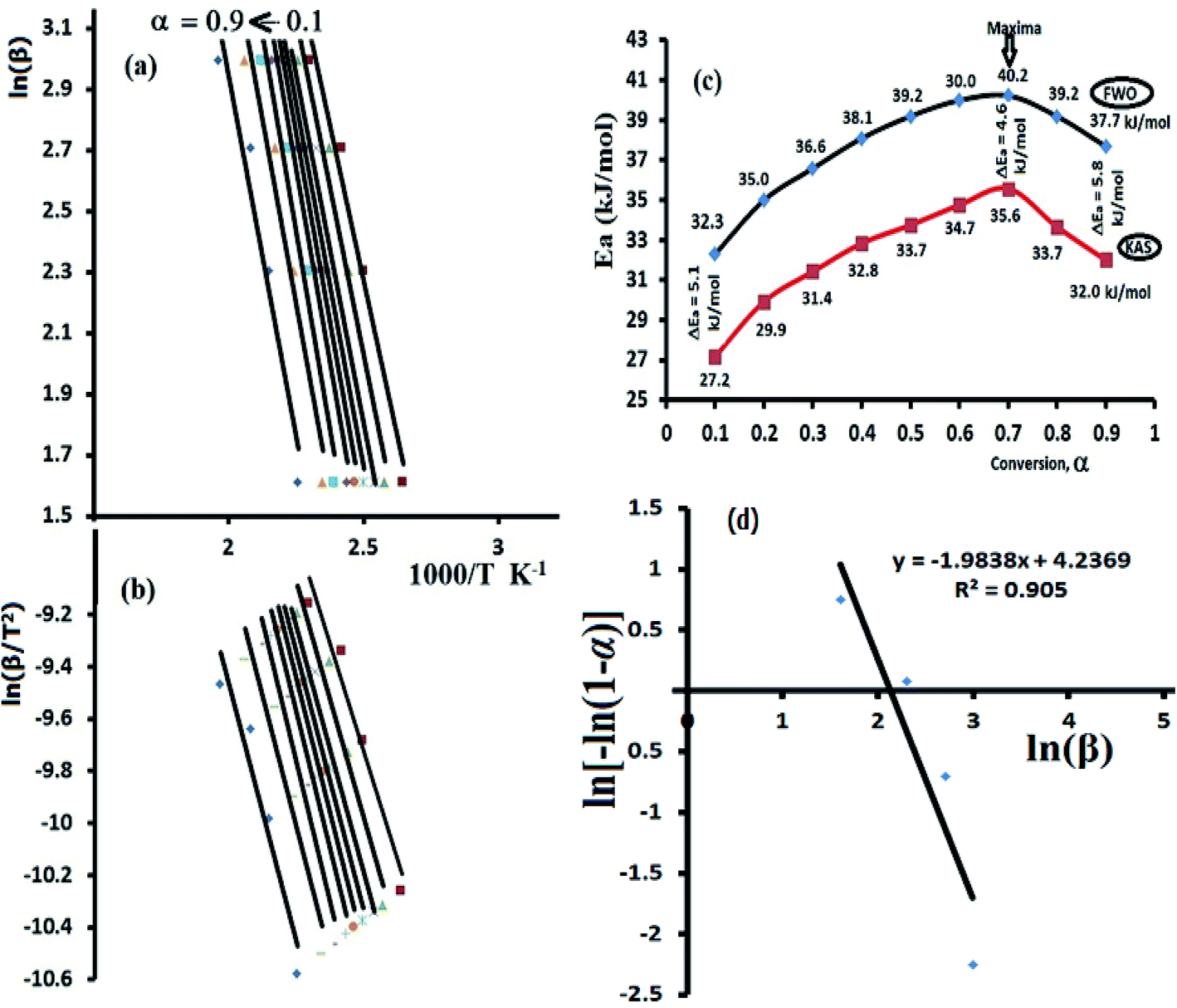 | ||
| Fig. 9 Ea's calculations via FWO (a), KAS (b), α–Ea's relation (c) and Avrami's plot (d). | ||
The activation energies were changed by rising α from 0.1 to 0.9 (Fig. 9c), at α = 0.1 FWO model revealed Ea with 32.3 kJ mol−1, while KAS gave it 27.2 kJ mol−1. Both models reached up to the maxima at α = 0.7 with 40.2 kJ mol−1 (FWO) and 35.6 kJ mol−1 (KSA) then decreased back with the extent of conversion to 37.7 kJ mol−1 at α = 0.9 (FWO) and 32.0 kJ mol−1 by KSA method. In general, a high degree of matching between the two models was recorded with about ΔEa ∼ 5 kJ mol−1 in all the trials. Moreover, the significant variation in Ea values indicated a complex kinetics stimulated decomposition process, the non-linear relationship between Ea and α provided collected via both models indicated that the ligand is prospective to be decomposed via more than one steps reaction (n > 1) and not through one broad step as thermal analysis reflected.37–39 Therefore, Avrami kinetic equation39 was applied at fixed suitable temperature 160 °C and in α = 0.1–0.9 range to support the complexity (multi-step) of thiophene-2-carbohydrazide thermal degradation reaction as in eqn (11).
ln[−ln(1 − α)] = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) χ(T) − nlnβ χ(T) − nlnβ
| (11) |
Plotting of ln[−ln(1 − α)] vs. logβ at 160 °C reflected a straight line with slope = −1.98 (Fig. 9d), therefore, the kinetic order n of the process is ∼2 (second order reaction), Avrami theory plot supported the non-simplicity decomposition of thiophene-2-carbohydrazide as well as the FWO/KAS models did.
4. Conclusion
The small-molecule endo-thiophene-2-carbohydrazide isomer was confirmed by XRD-crystal structure (CCDC 936463); the DFT/XRD-structure parameters reflected a semi-unity graphical correlations. An excellent matching in the hydrogen bonds computed by (HSA and MEP) was recorded compared to the experimental XRD-packing result. Both nucleophilic and electrophilic sites on the molecule surface were detected by Mulliken and NPA population charge analysis. A high degree of matching between the theoretical HOMO/LUMO, DOS, GRD and TD-SCF/DFT/B3LYP compared to the experimental spectral results.The DFT-computation of amide  imidic prototropic tautomerization through the QST2 method provided the possibility of such process via two steps reaction, simple flip-rotation with T.S1 and single proton intramigration with T.S2 (pseudo-four-membered-heterocyclic ring) formation.
imidic prototropic tautomerization through the QST2 method provided the possibility of such process via two steps reaction, simple flip-rotation with T.S1 and single proton intramigration with T.S2 (pseudo-four-membered-heterocyclic ring) formation.
The dependence of Ea's on α was confirmed by applying both FWO and KAS isoconversional thermal kinetic methods, the kinetic decomposition process is likely to be multistep reaction, Avrami's kinetic plot supported the second order decomposition reaction.
(E)/(Z)-thiophene-2-carbohydrazonic acid isomers reflected an excellent DNA-docking with cisplatin binding mode, moreover, Z-isomer is consider to be more active since more shorter H-bonds in [drug:DNA] complex were detected, meanwhile, no docking effect was detected in both endo and exo-thiophene-2-carbohydrazide isomers.
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no (RG-1440-141).References
- Y. L. Niu and T. Li, Anal. Chim. Acta, 2019, 1049, 196–212 CrossRef PubMed.
- G. Kumar, V. S. Krishna, D. Sriram and S. M. Jachak, Eur. J. Med. Chem., 2018, 156, 871–884 CrossRef CAS PubMed.
- W. Pan, J. Liu and X. Wang, Tetrahedron, 2018, 17, 1468–1475 CrossRef.
- L. K. Kummari, M. S. Butler, E. Furlong, R. Blundell and A. Robertson, Bioorg. Med. Chem., 2018, 26, 5408–5419 CrossRef CAS PubMed.
- S. Tolosa, N. Mora-Diez, A. Hidalgo and J. A. Sanson, RSC Adv., 2014, 4, 44757–44768 RSC.
- M. Boobalan, S. Ramalingam, M. Amaladasan, D. Tamilvendan, G. Venkatesa Prabhu and M. Bououdina, J. Mol. Struct., 2014, 1072, 153–172 CrossRef CAS.
- B. Parrino, S. Ullo, A. Attanzio, S. Cascioferro and P. Diana, Eur. J. Med. Chem., 2018, 158, 236–246 CrossRef CAS PubMed.
- Z. Wojnarowska, P. Wlodarczyk, K. Kaminski, K. Grzybowska, L. Hawelek and M. Paluch, J. Chem. Phys., 2010, 133, 94507–94515 CrossRef CAS PubMed.
- Z. Wojnarowska, K. Grzybowska, L. Hawelek, M. Dulski, R. Wrzalik, I. Gruszka and M. Paluch Drug, Mol. Pharmaceutics, 2013, 10, 3612–3627 CrossRef CAS PubMed.
- V. Balachandran, A. Janaki and A. Nataraj, Spectrochim. Acta, Part A, 2014, 118, 321–330 CrossRef CAS PubMed.
- E. Mateo-Marti and C. M. Pradier, Spectrochim. Acta, Part A, 2013, 109, 247–252 CrossRef CAS PubMed.
- R. Arora, U. Issar and R. Kakkar, Comput. Theor. Chem., 2017, 1105, 18–26 CrossRef CAS.
- A. Rajavel, A. Aditya Prasad and T. Jeyakumar, J. Mol. Struct., 2017, 1130, 138–149 CrossRef CAS.
- A. Yildırım, M. Yıldırım and Ç. Kastas, J. Mol. Struct., 2017, 1127, 275–282 CrossRef.
- A. K. Srivastava, A. Kumar, N. Misra, P. S. Manjula, B. K. Sarojini and B. Narayana, J. Mol. Struct., 2016, 1107, 137–144 CrossRef CAS.
- B. Bax, C. Chung and C. Edge, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 131–140 CrossRef CAS PubMed.
- M. R. Aouad, M. Messali, N. Rezki, N. Al-Zaqri and I. Warad, J. Mol. Liq., 2018, 264, 621–630 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, A. Lyčka and G. Gilli, J. Am. Chem. Soc., 2002, 124, 13554–13567 CrossRef CAS PubMed.
- B. Bandyopadhyay and P. Biswas, RSC Adv., 2015, 5, 34588–34593 RSC.
- H. Tavakol, J. Mol. Struct.: THEOCHEM, 2010, 956, 97–102 CrossRef CAS.
- H. Tavakol, Int. J. Quantum Chem., 2012, 112, 1215–1224 CrossRef CAS.
- H. Tavakol, Struct. Chem., 2011, 22, 1165–1177 CrossRef CAS.
- H. Tavakol, Int. J. Quantum Chem., 2011, 111, 3717–3724 CAS.
- H. Tavakol, T. Hadadi and H. Roohi, J. Mol. Struct., 2012, 53, 649–658 CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka, and M. A. Spackman, Crystal Explorer 2.1, University of Western Australia, Perth, 2007 Search PubMed.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- A. Barakat, M. Islam, A. Al-Majid, H. A. Ghabbour, S. Atef, A. Zarrouk and I. Warad, J. Theor. Comput. Chem., 2018, 17, 1–23 CrossRef.
- M. R. Aouad, M. Messali, N. Rezki, M. A. Said, D. Lentz, L. Zubaydi and I. Warad, J. Mol. Struct., 2019, 1180, 455–465 CrossRef CAS.
- A. Titi, T. Shiga, H. Oshio, R. Touzani, B. Hammouti, M. Mouslim and I. Warad, J. Mol. Struct., 2020, 1199, 126995–127003 CrossRef CAS.
- I. Warad, F. F. Awwadi, B. Abd Al-Ghani, A. Sawafta, N. Shivalingegowda, N. K. Lokanath, M. S. Mubarak, T. Ben Hadda, A. Zarrouk, F. Al-Rimawi, A. B. Odeh and S. A. Barghouthi, Ultrason. Sonochem., 2018, 48, 1–9 CrossRef CAS PubMed.
- I. Warad, Y. Al-Demeri, M. Al-Nuri, S. Shahwan, M. Abdoh, S. Naveen, N. K. Lokanath, M. S. Mubarak, T. B. Hadda and Y. N. Mabkhot, J. Mol. Struct., 2017, 1142, 217–227 CrossRef CAS.
- F. A. Saleem, S. Musameh, A. Sawafta, P. Brandao, C. J. Tavares, S. Ferdov, A. Barakat, A. Al Ali, M. Al-Noaimi and I. Warad, Arabian J. Chem., 2017, 10, 845–855 CrossRef.
- M. V. P. Ravi and S. P. Tewari, J. Phys. Chem. A, 2013, 117, 10162–10169 CrossRef PubMed.
- J. Shi, Y. Lou, K. Zhou and D. Pan, Spectrochim. Acta, Part A, 2018, 204, 209–216 CrossRef CAS PubMed.
- I. Warad, S. Musameh, A. Sawafta, P. Brandão, C. J. Tavares, A. Zarrouk, S. Amereih, A. Al Ali and R. Shariah, Ultrason. Sonochem., 2019, 52, 428–436 CrossRef CAS PubMed.
- N. Koga and J. Sestak, J. Therm. Anal., 1991, 37, 1103–1114 CrossRef.
Footnote |
| † CCDC 936463. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09831c |
| This journal is © The Royal Society of Chemistry 2020 |