
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimerization of pentacyclopentacorannulene C30H10 as a strategy to produce C60H20 as a precursor for C60†
Arlette Richaudabc,
María J. López d,
Martha Mojicaa,
Julio A. Alonsode and
Francisco Méndez*a
d,
Martha Mojicaa,
Julio A. Alonsode and
Francisco Méndez*a
aDivisión de Ciencias Básicas e Ingeniería, Departamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, A.P. 55-534, México D. F. 09340, Mexico. E-mail: fm@xanum.uam.mx
bLE STUDIUM RESEARCH FELLOW, Loire Valley Institute for Advanced Studies, Orléans & Tours, France
cConditions Extrêmes et Matériaux: Haute Température et Irradiation (CEMHTI), UPR 3079 CNRS, CEMHTI 1, Avenue de la Recherche Scientifique, 45071 Orléans, France
dDepartamento de Física Teórica, Atómica y Óptica, Universidad de Valladolid, 47011 Valladolid, Spain
eDonostia International Physics Center, 20018 San Sebastián, Spain
First published on 22nd January 2020
Abstract
The chemical synthesis of C60 fullerene in the laboratory is still a challenge. In order to achieve this goal, we propose a synthetic route based on the dimerization between two pentacyclopentacorannulene (C30H10) fragments employing the Diels–Alder cycloaddition reaction. Density functional calculations indicate that a step wise non-concerted dimerization mechanism of C30H10 is favored over a one stage dimerization. The step wise dimerization implies the sequential formation of 2, 4, 6, and 10 new C–C bonds between the two fragments. This leads to the formation of the Diels–Alder cycloadduct C60H20. The results then suggest the synthesis of C60H20 as a precursor for C60. The synthesis of the analogue C60F20 has already been reported.
Introduction
Regarding the utopian idea that assures that “chemists must be able of synthesize everything imaginable”,1 there are a lot of complex molecules that resist the attempts of synthesis, becoming challenging targets for organic chemists. A clear example of this is the controlled bottom-up synthesis of carbon fullerenes.2–5 Even when some successful attempts have been made in the synthesis of prototypes for these systems,6–10 an improvement of the known synthetic methods, as well as the development of new techniques, are still needed.11 The present status of the synthesis of fullerenes has been reviewed previously by three of the authors.5Traditionally, fullerenes are synthesized by a collection of vaporization methods,12 which lead to the production of fullerenes on a commercial scale. However, those methods imply a great environmental and energetic cost, and they are not very efficient, with yields lower than 1%. Additionally, the uncontrollable character of the reaction makes impossible to select a single fullerene or a specific isomer. Anctil et al.13 pointed out that the energy used in the synthesis and the separation of C60 can reach up to 106.9 GJ kg−1 (for comparison, the average annual electricity consumption for a US home in 2010 was about 41 GJ),14 and if we add the energy needed for purification and functionalization, the total energy can suffer a threefold increase, making fullerenes expensive.
In the pursuing of an efficient method for the synthesis of C60, Sygula et al.15,16 have suggested the dimerization of two identical hemispherical C30 fragments, as an “intriguing possibility” to synthesize fullerenes. Scott17 also labelled the dimerization of hemifullerenes as a “particularly seductive strategy”, and highlighted its resemblance with the dimerization of triquinacene to produce dodecahedrane, as was proposed by Woodward et al.18 The work of McElvany et al.19 showed that it is possible to obtain C60 by the coalescence of large monocyclic C30 rings obtained by laser induced fragmentation of carbon oxide precursors. In this case, the high energy annealing conditions provided by the laser allow for the dimerization and reorganization of the molecular cyclic precursors to form C60.
In the synthesis of geodesic polyarenes, all the efforts have been directed to the insertion of the needed curvature in the fragments. An advantage of the dimerization scheme is that the curvature is already present in the starting material, and the main issue, which forms the topic of the present work, is to develop a way to stitch those fragments together. Another advantage is that a successful dimerization will provide a direct method to synthesize the fullerene, consequently increasing the efficiency. Previous works have demonstrated the applicability of the Diels–Alder (DA) cycloaddition reaction in the growing of polycyclic aromatic hydrocarbons (PAHs),20,21 as well as a way to put two PAHs together.22,23 This reaction proceeds in solution and under mild conditions, and could be easily controlled and manipulated by the use of different substituents.24–26
The progress in the synthesis of fullerene fragments has been steady. Scott1 discovered an efficient method to obtain the fragment C20H10 (corannulene; a carbon pentagon surrounded by five hexagons) in high yield. The method combines chemical synthesis with flash vacuum pyrolysis. Many larger curved polycyclic aromatic hydrocarbons, some of them fullerene fragments have also been synthesized by similar methods,4 and Mehta and Panda27 have reviewed the work on the synthesis of bucky-bowl fragments as a previous step in the synthesis of C60. Geneste and coworkers28 have shown that between the known isometric hemifullerenes C30Hx derived from C60, the pentacyclopentacorannulene C30H10 (labelled 1 from now on), is one of the most similar to C60 in terms of curvature. Cyclopentacorannulene C22H12, which can be viewed as having only one five-membered ring at the boundary, instead of the five pentagons of C30H10 (1), has been synthesized by Abdouzarak et al.29 using the Scott method.1 The synthetic methods that use flash vacuum pyrolysis or other high-energy gas phase methods have been scarcely employed for the simple annulation of fully unsaturated five-membered rings onto the perimeter of PAHs.1 However, Scott et al., using microwave-assisted intramolecular arylations, included fused networks of five- and six-membered rings into the corannulene to obtain indenocorannulenes (ICs) and the complete family of all seven ICs (mono-IC, ortho-di-IC, para-di-IC, 1,2,3-tri-IC, 1,2,4-tri-IC, penta-IC and tetra-IC).30 Although the full C30H10 (1) has not been synthesized yet, one can expect that similar methods will allow for the synthesis of this fragment. On the basis of ab initio calculations Mojica et al.31,32 suggested that C30H10 (1) is a good dienophile towards DA cycloaddition reaction with butadiene and possesses high symmetry, which makes it an ideal fragment to produce C60H20 and the C60 fullerene (Fig. 1).
 | ||
| Fig. 1 Schematic view of the dimerization of pentacyclopentacorannulene C30H10 (1). | ||
In this work we have studied, using the density functional theory (DFT)33 and the nudge elastic band (NEB) method,34 the DA dimerization of two C30H10 fragments (Fig. 1). The resulting C60H20 molecule is a C60 fullerene wrapped by a double belt of twenty H atoms forming C–H bonds around the equatorial plane of C60 (the poles are occupied by the central pentagons of the two fragments). Although the C60H20 molecule with the belt of twenty H atoms, has not been obtained experimentally, its structure has been described theoretically by Keppert and Clare,35,36 and it is isostructural with the one that we obtained. Our results expand the work of Sygula et al.15,16 and Scott17 and support the DA dimerization of C30H10 (1) as a viable synthetic strategy to produce C60H20 as a precursor of C60.
Computational methods
Density functional calculations33 have been performed using the supercell method with a basis set of plane waves, as implemented in the DACAPO code.37 The ionic cores are represented by ultrasoft pseudopotentials.38 An energy cutoff of 350 eV was taken for the plane wave expansion of the wave functions, and a cutoff of 1000 eV for the electron density. Electronic exchange and correlation effects are treated by the generalized gradient approximation (GGA) of Perdew and Wang (PW91).39 We considered a supercell of size 25 × 15 × 15 Å and a single k point [1,1,1] of the Monkhorst–Pack type.40 The selected supercell size is sufficiently large to minimize the interaction between molecules in different cells. The activation barriers occurring in the dimerization process have been calculated by using the nudge elastic band method.34 Calculations made with the Gaussian 09 (G09) program41 package at the B3LYP/631-G(d,p) level of theory42,43 for selected structures along the dimerization path fully confirm the results obtained with the DACAPO code.Results and discussion
The DA dimerization of two C30H10 fragments 1 + 1 could occur through a one stage mechanism or a stepwise mechanism. In the one stage mechanism all the ten new C–C bonds will be formed in a single step DA reaction, whereas in the stepwise mechanism two new bonds will first join both fragments together to form an open C60H20, and the eight remaining bonds will be formed subsequently, due to the proximity of the edges of the fragments, leading to the C60H20 with the C60 cage structure. The two dimerization mechanisms of C30H10 have been investigated.(1) One stage dimerization
In this case ten C–C bonds are formed simultaneously between the C atoms of the edges of the two fragments (those C atoms of the fragments saturated with H atoms). To calculate the minimum energy path (MEP) for the one stage dimerization we consider three parts of the process: the first part starts with the two fragments at a large distance, facing each other and properly oriented in order to facilitate the formation of the desired C–C bonds between the diene moiety (blue color) of one fragment and the dienophile moiety (red color) of the other (Fig. 2). The two fragments are stepwise approached one to the other till the distance between the central pentagons of the fragments is 7.5 Å (inset labelled (b) in Fig. 3). At this distance, the two fragments are separated but begin to deform (the corannulene rings are slightly flattened) due to the fragment–fragment interaction. During the approaching path just described, a constrained minimization of the structure is performed at each step, keeping fixed the distance between the central pentagons of the two fragments. The energies along this path, shown in the left panel of Fig. 3, indicate that the energy increases as the two fragments approach each other because of the deformation induced by the interaction between the fragments. The rigidity of C30H10 against flattening deformations was noticed by Liu et al.44 in their theoretical study of the bowl-to-bowl inversion of this molecule. The last part of the dimerization process is investigated going backwards from the final configuration of the C60H20 molecule. The two C30H10 fragments are stepwise separated along their common axis till the distance between the two opposite polar pentagons is of 7.5 Å (inset labelled (d) in Fig. 3). At this distance, the cage structure is still preserved although somewhat deformed. At each step a constrained minimization is performed, similarly as it was explained above. The energies along this path are shown in the right panel of Fig. 3. | ||
| Fig. 2 The reactive sites of the fragments considered for the Diels–Alder reaction are shown in the red and blue circles for the five- and six-membered rings, respectively. | ||
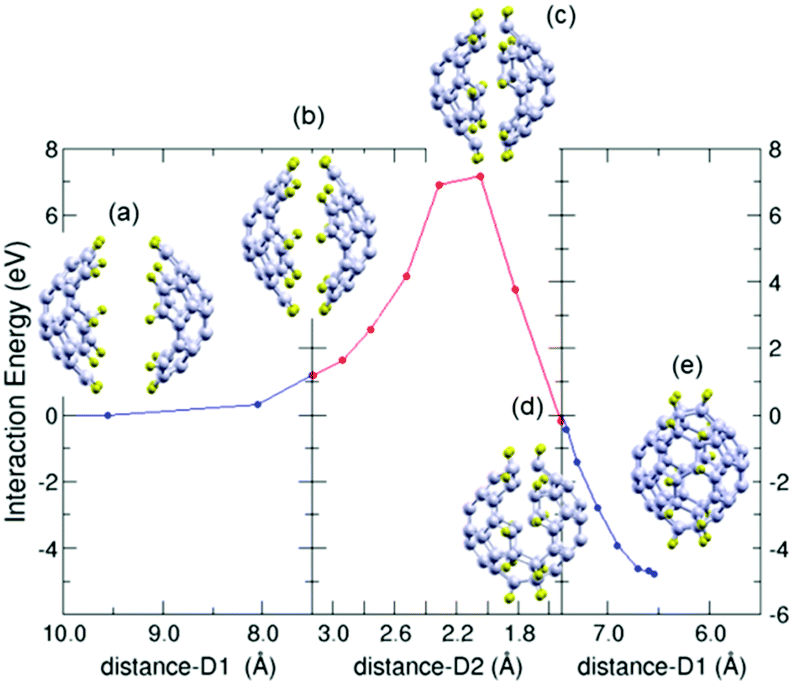 | ||
| Fig. 3 Minimum energy path for the one-stage dimerization of two pentacyclopentacorannulene fragments (1 + 1). The dimerization is decomposed in three stages (panels) explained in the text. The energies are given with respect to the energy of the separated fragments as a function of the distance D1 between the two fragments (distance between the opposite apex pentagons) for the first and third panels, and as a function of distance D2 (average distance between the ten pairs of carbon atoms forming the ten bonds that unite the two fragments) for the intermediate panel. The initial and final structures of the three panels and the transition state for dimerization (intermediate panel) are shown as insets labelled (a)–(e). The coordinates of the selected structures shown in the figure are provided in the ESI.† Carbon and hydrogen atoms are represented in grey and yellow colours, respectively. | ||
Then the minimum energy path (MEP) joining the two previously obtained structures (the structures shown in the insets (b) and (d) of Fig. 3, having, in both cases, a distance of 7.5 Å between the polar pentagons) is calculated by applying the NEB method. A barrier of nearly 7 eV has to be overcome to fuse the two fragments into the C60H20 molecule. The distance between the polar pentagons remains almost constant along this path (although it is allowed to vary). The main structural change is carried out by the C–H bonds. In the separated fragments the C–H bonds prolong the surface of the carbon frame. In contrast, in the cage structure, the C–H bonds are almost perpendicular to the surface of the cage. The hybridization of the C atoms that form the bonds between the two fragments changes from sp2 in the separated fragments to sp3 in the cage structure. Thus, the DA reactions on this preferred orientation will end up forming the C60H20 molecule.
(2) Stepwise dimerization
In this mechanism, two, four, six and ten C–C bonds are formed sequentially between the two fragments (see Fig. 4). A four-stages process is devised starting from the separated fragments, going successively through three intermediate (metastable) structures (2, 3 and 4 in Fig. 4) having, respectively, two, four, and six C–C bonds between the two fragments, and ending up in the C60H20 cage having ten C–C bonds between the two fragments. Notice that a metastable structure having eight C–C bonds between the two fragments does not appear and therefore the system evolves from structure 4 (having six C–C bonds between the two fragments) to the C60H20 cage (having ten C–C bonds between the two fragments) in a single stage. The three intermediate structures (2, 3 and 4) have been fully optimized. Then the minimum energy path between every two successive structures is calculated using the NEB procedure. The MEP energies and some representative structures are shown in Fig. 4, and the coordinates of those representative structures are provided in the ESI.† | ||
| Fig. 4 Minimum energy path for the stepwise dimerization of two pentacyclopentacorannulene fragments (1 + 1). The energies are given with respect to the energy of the separated fragments as a function of the distance D2 (average distance between the ten pairs of carbon atoms forming the ten final bonds that unite the two fragments). Each panel corresponds to the formation of two additional C–C bonds between the two fragments, except for the final panel that corresponds to the formation of four C–C bonds. The initial, final and transition state structures for the processes represented in each panel are shown as insets labelled (a)–(i). The coordinates of the selected structures shown in the figure are provided in the ESI.† Carbon and hydrogen atoms are represented in grey and yellow colours, respectively. | ||
The formation of the first two C–C bonds between the fragments is an endothermic process, a DA cycloaddition reaction31 costing about 2 eV, with an activation barrier of 4 eV (1 + 1 → 2). Once the two fragments have formed the first two C–C bonds, the formation of two additional C–C bonds is exothermic with respect to the structure with only two C–C bonds, releasing an energy of 1.2 eV (2 → 3), although this configuration (3) is still 0.8 eV higher in energy than the separated fragments. The activation barrier for the formation of the third and fourth C–C bonds is 1.1 eV (2 → 3), substantially smaller than the activation barrier in the first step. The formation of the next two C–C bonds occurs by an exothermic process (3 → 4), releasing 1.1 eV after overcoming an activation barrier of 1.1 eV. This is the first intermediate structure (4) in the path of dimerization which is more stable than the separated fragments (by 0.3 eV). Finally, the cage structure is closed with the formation of four more C–C bonds (4 → C60H20). This step is highly exothermic (4.5 eV) with an activation barrier of only 0.5 eV. It is noticeable that the activation barriers decrease and the exothermicity increases along the successive steps leading to the closing of the cage. The stepwise dimerization is more probable than the one-stage process of Fig. 3 because it involves significantly smaller activation barriers.
Producing a C60 fullerene from the C60H20 molecule requires removing the belt of H atoms (see Fig. 5). However, we find that the system formed by C60 and ten free H2 molecules is less stable (about 10 eV higher in energy) than the C60H20 molecule.
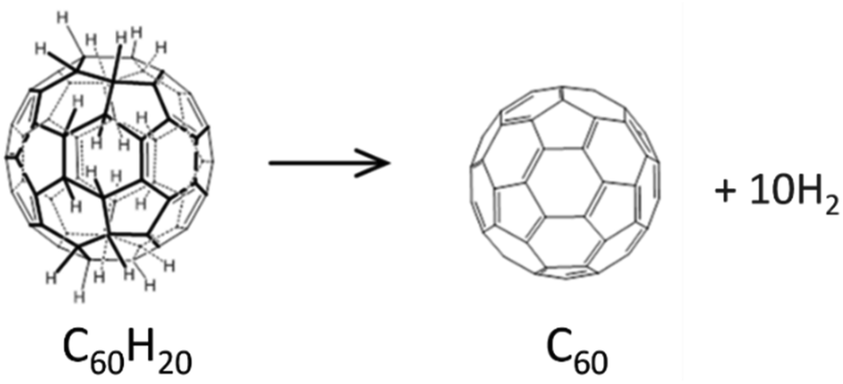 | ||
| Fig. 5 The aromatization of the Diels–Alder cycloadduct produces the C60 fullerene. | ||
The removal of hydrogen in the form of molecular H2 from C60H20 would cost, in average, about 1 eV per H2 molecule to arrive to the clean C60 fullerene. Therefore, the dehydrogenation of C60H20 is an endothermic process that would require high temperatures or appropriate reactants. Other possibility would be to use ultraviolet (UV) radiation. This has been proposed to be the key to the formation of Co60 from large polycyclic aromatic hydrocarbons like C66H20 in the interstellar medium.45,46
Fort and Scott20,21 have shown that the use of nitro-derivatives of ethylene in DA reactions provides an efficient way to increase the number of benzene rings in polycyclic aromatic hydrocarbons. Therefore, a promising way of controlling and manipulating the energy of the C30H10 DA cycloaddition and C60H20 aromatization reactions to obtain C60 could be the use of electron-withdrawing and electron-donating groups47,48 in the two pentacyclopentacorannulene fragments. Our previous studies of the substituent effect in the reactivity of C30H10 suggested that it is possible to increase its reactivity in the DA reactions.31,32
The results presented support the possibility to dimerize C30H10 and the existence of C60H20. In fact, the fullerene hydride C60H20 has been detected in the mass spectra of C60 hydrides synthesized by condensation of benzene molecules,49 although, the structure of C60H20 formed in these experiments is not necessarily the belt-like structure discussed here, which is closely connected to the structure of the precursor C30H10. Boltalina et al.50 synthesized and characterized spectroscopically the analogue molecule C60F20. The C60F20 was obtained through fluorination of fullerene C60 with Cs2PbF6 or with KF/MnF3 mixtures under vacuum at temperatures of 580 °C and 480 °C, respectively. The structure of C60F20 comprises two dehydrocorannulene caps held together by a (CF)20 chain. This suggests that the experimental chemical synthesis of C60H20 should be an achievable challenge.
Conclusions
Using the density functional formalism, we have performed calculations to investigate the dimerization of two identical hemispherical C30H10 fragments to produce C60H20, as a way to synthesize the fullerene C60 in a direct and efficient way in the laboratory. A stepwise mechanism is favoured over the one stage mechanism because the energy barriers involved are lower. The activation barriers decrease and the exothermicity increases along the successive steps leading to the closing of the cage. The results support the possibility to dimerize C30H10 and the existence of C60H20. Even when C30H10 has not been synthetized, Scott et al.30 have pointed out that the syntheses of the indenocorannulenes constitute the first demonstration that the stepwise introduction of curvature can be pushed all the way to produce a level of curvature equal to that of C60 and even beyond. We hope that our results will motivate synthetic chemists to synthesize the fragment C30H10 and attempt its dimerization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work supported by MINECO of Spain and the European Regional Development Fund (grant MAT 2014-54378-R), Junta de Castilla y León (grant VA021G18), and CONACyT-México (project grants 180523 and 163234). M. M. acknowledges CONACyT for a predoctoral fellowship (CB-2012/180523). FM thanks to Programa Especial de Apoyo a la investigación básica UAM-2019, CONACYT. AR acknowledges a LE STUDIUM Research Fellowship, cofunded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 665790. The authors thankfully acknowledge the facilities provided by Centro de Proceso de Datos-Parque Científico (University of Valladolid).Notes and references
- L. T. Scott, Polycyclic Aromat. Compd., 2010, 30, 247 CrossRef CAS.
- F. Diederich and Y. Rubin, Angew. Chem., Int. Ed., 1992, 31, 1101 CrossRef.
- L. T. Scott, Angew. Chem., Int. Ed., 2004, 43, 4994 CrossRef CAS PubMed.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868 CrossRef CAS PubMed.
- M. Mojica, J. A. Alonso and F. Méndez, J. Phys. Org. Chem., 2013, 26, 526 CrossRef CAS.
- L. T. Scott, M. M. Boorum, B. J. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner and A. de Meijere, Science, 2002, 295, 1500 CrossRef CAS.
- G. Otero, G. Biddau, C. Sánchez-Sánchez, R. Caillard, M. F. López, C. Rogero, F. J. Palomares, N. Cabello, M. A. Basanta, J. Ortega, J. Méndez, A. M. Echavarren, R. Pérez, B. Gómez-Lor and J. A. Martín-Gago, Nature, 2008, 454, 865 CrossRef CAS PubMed.
- R. B. M. Ansems and L. T. Scott, J. Am. Chem. Soc., 2000, 122, 2719 CrossRef CAS.
- U. H. F. Bunz, Y. Rubin and Y. Tobe, Chem. Soc. Rev., 1999, 28, 107 RSC.
- O. L. Chapman, M. R. Engel, J. P. Springer and J. C. Clardy, J. Am. Chem. Soc., 1971, 93, 6696 CrossRef CAS.
- L. T. Scott and M. A. Petrukhina, Preface in Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions and Coordination Chemistry, ed. L. T. Scott and M. A. Petrukhina, John Wiley & Sons Inc., Hoboken, New Jersey, 2012, pp. vii–x Search PubMed.
- N. I. Alekseev, B. M. Filippov, I. V. Basargin and A. I. Sedov, J. Eng. Phys. Thermophys., 2011, 84, 1087 CrossRef CAS.
- A. Anctil, C. W. Babbitt, R. P. Raffaelle and B. Landi, J. Environ. Sci. Technol., 2011, 45, 2353 CrossRef CAS PubMed.
- U. S. Energy Information Administration, http://www.eia.gov/tools/faqs/faq.cfm?id=97%26t=3.
- P. Rabideau and A. Sygula, Polynuclear Aromatic Hydrocarbons with Curved Surfaces, in Advances in Theoretically Interesting Molecules, ed. R. P. Thummel, JAI Press Inc., Greenwich, Connecticut, 1995, ch. 1, vol. 3, pp. 1–36 Search PubMed.
- A. H. Abdourazak, Z. Marcinow, A. Sygula, R. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1994, 117, 6410 CrossRef.
- L. T. Scott, Angew. Chem., Int. Ed., 2004, 43, 4994 CrossRef CAS PubMed.
- R. B. Woodward, T. Fukunaga and R. C. Kelly, J. Am. Chem. Soc., 1964, 86, 3162 CrossRef CAS.
- S. W. McElvany, M. M. Ross and N. S. Gorof, Science, 1993, 259, 1594 CrossRef CAS.
- E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006 CrossRef CAS PubMed.
- E. H. Fort and L. T. Scott, Angew. Chem., Int. Ed., 2010, 49, 6626 CrossRef CAS PubMed.
- J. Plater, H. S. Rzepa, F. Stoppa and S. Stossel, J. Chem. Soc., Perkin Trans. 2, 1994, 2, 399 RSC.
- A. Sygula, Eur. J. Org. Chem., 2011, 9, 1611 CrossRef.
- F. Fringuelli and A. Taticchi, The Diels-Alder Reaction: Selected Practical Methods, John Wiley & Sons Ltd., West Sussex, England, 2002, pp. 1–25 Search PubMed.
- B. R. Bear, S. M. Sparks and K. J. Shea, Angew. Chem., Int. Ed., 2001, 40, 820 CrossRef CAS PubMed.
- K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558 CrossRef.
- G. Mehta and G. Panda, PINSA-A: Proc. Indian Natl. Sci. Acad., Part A, 1998, 64, 587 CAS.
- F. Geneste, A. Moradpour, G. Dive, D. Peeters, J. Malthête and J. F. Sadoc, J. Org. Chem., 2002, 67, 605 CrossRef CAS.
- A. H. Abdourazak, A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1993, 115, 3010 CrossRef CAS.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537 CrossRef CAS PubMed.
- M. Mojica, F. Méndez and J. A. Alonso, Molecules, 2013, 18, 2243 CrossRef CAS PubMed.
- M. Mojica, F. Méndez and J. A. Alonso, Molecules, 2016, 21(1), 200 CrossRef PubMed.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- B. W. Clare and D. L. Keppert, THEOCHEM, 1994, 315, 71 CrossRef.
- D. L. Keppert and B. W. Clare, Coord. Chem. Rev., 1996, 155, 1 CrossRef.
- https://wiki.fysik.dtu.dk/dacapo.
- D. Vanderbilt, Phys. Rev. B, 1990, 41, 7892 CrossRef PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671 CrossRef CAS PubMed.
- H. Monkhorst and J. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, 2010 Search PubMed.
- A. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 36, 785 CrossRef PubMed.
- R. F. Liu, X. F. Zhou and N. L. Allinger, J. Phys. Org. Chem., 1994, 7, 551 CrossRef CAS.
- J. Montillaud, C. Joblin and D. Toublanc, Astron. Astrophys., 2013, 552, A15 CrossRef.
- O. Berné, J. Montillaud and C. Joblin, Astron. Astrophys., 2015, 577, A133 CrossRef PubMed.
- K. N. Houk, Y.-T. Lin and F. K. Brown, J. Am. Chem. Soc., 1986, 108, 554 CrossRef CAS PubMed.
- K. N. Houk, R. J. Loncharich, J. F. Blake and W. L. Jorgensen, J. Am. Chem. Soc., 1989, 111, 9172 CrossRef CAS.
- A. Kharlamov, G. Kharlamova, O. Khyzhun and N. Kirillova, in Carbon Nanomaterials in Clean Energy Hydrogen Systems - II. NATO Science for Peace and Security Series C: Environmental Security 2, ed. S. Y. Zaginaichenko et al., Springer, 2011, pp. 287–298 Search PubMed.
- O. V. Boltalina, V. Yu. Markov, P. A. Troshin, A. D. Darwish, J. M. Street and R. Taylor, Angew. Chem., Int. Ed., 2001, 40, 787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09804f |
| This journal is © The Royal Society of Chemistry 2020 |