
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new oxadiazol-phthalazinone derivatives with anti-proliferative activity; molecular docking, pro-apoptotic, and enzyme inhibition profile†
Mohamed H. Hekal a,
Abeer M. El-Naggar*a,
Fatma S. M. Abu El-Azma and
Wael M. El-Sayed*b
a,
Abeer M. El-Naggar*a,
Fatma S. M. Abu El-Azma and
Wael M. El-Sayed*b
aDepartment of Chemistry, Faculty of Science, Ain Shams University, Abbassia 11566, Cairo, Egypt. E-mail: elsayedam@sci.asu.edu.eg
bDepartment of Zoology, Faculty of Science, Ain Shams University, Abbassia 11566, Cairo, Egypt. E-mail: waelelhalawany@hotmail.com; wael_farag@sci.asu.edu.eg; Fax: +202/2684-2123; Tel: +202/2482-1633
First published on 22nd January 2020
Abstract
Background and aim: The current study reports the synthesis and biological evaluation of two novel series of 4-(5-mercapto-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one derivatives. Methods: The synthetic reactions were carried out under both conventional and ultrasonic irradiation conditions. The anti-proliferative activity of the newly synthesized compounds against two human epithelial cell lines; liver (HepG2) and breast (MCF-7) in addition to normal fibroblasts (WI-38) was investigated. In addition to molecular docking studies, the possible mechanism(s) of action were also explored. Results: In general, an improvement in synthetic rates and yields was observed when reactions were carried out under sonication compared with classical conditions. The structures of the products were established based on analytical and spectral data. Derivatives 2e and 7d, in addition to compound 1, had significant and selective anti-proliferative activity against liver and breast cancer cell lines without harming normal fibroblasts. These derivatives arrested the cell cycle progression and/or induced apoptosis. This has been manifested by the elevation in the expression of p53 and caspase 3, down-regulation of cdk1, and a reduction in the concentrations of MAPK and Topo II at submicromolar concentrations. The latter results confirmed the molecular docking study. Conclusions: Compound 1 had the best profile on the gene and protein levels (arresting cell cycle and inducing apoptosis). The ability of compounds 1 and 2e to inhibit both MAPK and Topo II nominates these derivatives as potential candidates for further anticancer and antitumor studies.
1. Introduction
Phthalazin-1(2H)-ones and (alkyl or aryl)thio-1,3,4-oxadiazoles serve as important building blocks for biologically active molecules. The structures of these two important biologically active pharmacophore component systems are shown in Fig. 1. | ||
| Fig. 1 Phthalazinone-oxadiazoles scaffold. | ||
The phthalazin-1(2H)-one nucleus has emerged as a promising and attractive scaffold in the preparation of promising antitumor agents. For example, phthalazin-1,4-diones have been reported as potent type II IMP dehydrogenase inhibitors1 and as active anti-proliferative agents against various human and murine tumor cells,2,3 particularly hepatocellular carcinoma.4 In addition, a series of 4-substituted-2H-phthalazin-1-ones has been studied as potent orally bioavailable poly ADP ribose polymerase (PARP) inhibitors.5–7 Olaparib (1), MRU-868 (2) and KU0058958 (3) are the most interesting PARP inhibitors based on the 4-substituted-2H-phthalazin-1-one scaffold (Fig. 2).
 | ||
| Fig. 2 Structure of the lead anticancer phthalazin-1(2H)-one derivatives 1–3. | ||
The second pharmacophore component, 1,3,4-oxadiazoles, is an important category of heterocyclic compounds with a wide range of biological activities such as antimicrobial,8 antiviral,9 fungicidal,10 antineoplastic,11 inhibition of tyrosinase,12 anticancer13–16 and inhibition of cathepsin K.17 They are also useful intermediates in organic synthesis18 and they are widely utilized as electron transporting and hole-blocking materials.19 Further, 1,3,4-oxadiazole heterocycles are recognized as bioisosteres of esters and amides that can contribute interesting pharmacokinetic properties due to the presence of N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O linkage in the oxadiazole nucleus, which increases lipophilicity and affects the ability of the drugs to reach molecular targets by transmembrane diffusion. These molecules are often utilized as pharmacophores due to their ability to bind target proteins through hydrogen bond formation.20
O linkage in the oxadiazole nucleus, which increases lipophilicity and affects the ability of the drugs to reach molecular targets by transmembrane diffusion. These molecules are often utilized as pharmacophores due to their ability to bind target proteins through hydrogen bond formation.20
The biological activities of these pharmacophores, phthalazin-1(2H)-one and 1,3,4-oxadiazole, led us to design and synthesize novel lead compounds containing the two pharmacophores in a single molecule with the goal of increasing pharmacological activities. The synthetic reactions were carried out under both conventional and ultrasonic irradiation conditions. Ultrasound has been increasingly used in organic synthesis in recent years. A large number of organic reactions can be carried out in higher yield, shorter reaction time and milder conditions under ultrasonic irradiation.21,22 The phenomenon observed is known as “acoustic cavitation” and generates high temperatures and pressures in localized zones of the liquid, creating rate acceleration in many chemical reactions.23 In addition, multicomponent reactions have received considerable attention, since they show good atom economy; they are highly convergent and require minimal time and effort to obtain complex molecules.24
A series of 18 new compounds was synthesized and examined for anti-proliferative activity against two human epithelial breast and liver cancer cell lines, as well as normal fibroblasts. The lead compounds showing significant anti-proliferative activity were further investigated on the expression of crucial genes involved in cell cycle progression and apoptosis. Finally, these lead compounds were evaluated as inhibitors of key enzymes involved in cell cycle and cellular proliferation: p38 mitogen-activated protein kinase (MAPK) and topoisomerase II. The ability of molecular docking to predict the inhibitory effects of the lead derivatives against topoisomerase II and serine/threonine-protein kinase (PIM1) was also examined. The parent compound 1 was docked against these two key enzymes because the two pharmacophores used in the current study were separately demonstrated to possess significant anticancer activity.
2. Results and discussion
2.1. Chemistry
As a part of our interest in the synthesis of a wide range of heterocyclic systems with biological applications,25–29 and in a continuation of using green chemistry tools in heterocyclic synthesis,30–33 our current research is concerned with the utility of 4-(5-mercapto-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (1)34 in the synthesis of new substituted 4-(1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one derivatives under both conventional and ultrasonic irradiation conditions.In this study, we report an efficient method for the synthesis of a series of 4-(1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one derivatives via alkylation of compound 1 with the appropriate alkyl and acyl halides namely, ethyl iodide, benzyl chloride, phenacyl bromide, m-nitrophenacyl bromide and chloroacetyl chloride in ethanol and in the presence of catalytic amount of triethylamine to afford the target compounds 2a–e (Scheme 1). The structures of compounds 2a–e were elucidated from their spectroscopic data. Their IR spectra exhibited absorption bands corresponding to NH group in the range 3247–3158 cm−1 as well as bands for CO group at 1657–1688 cm−1.
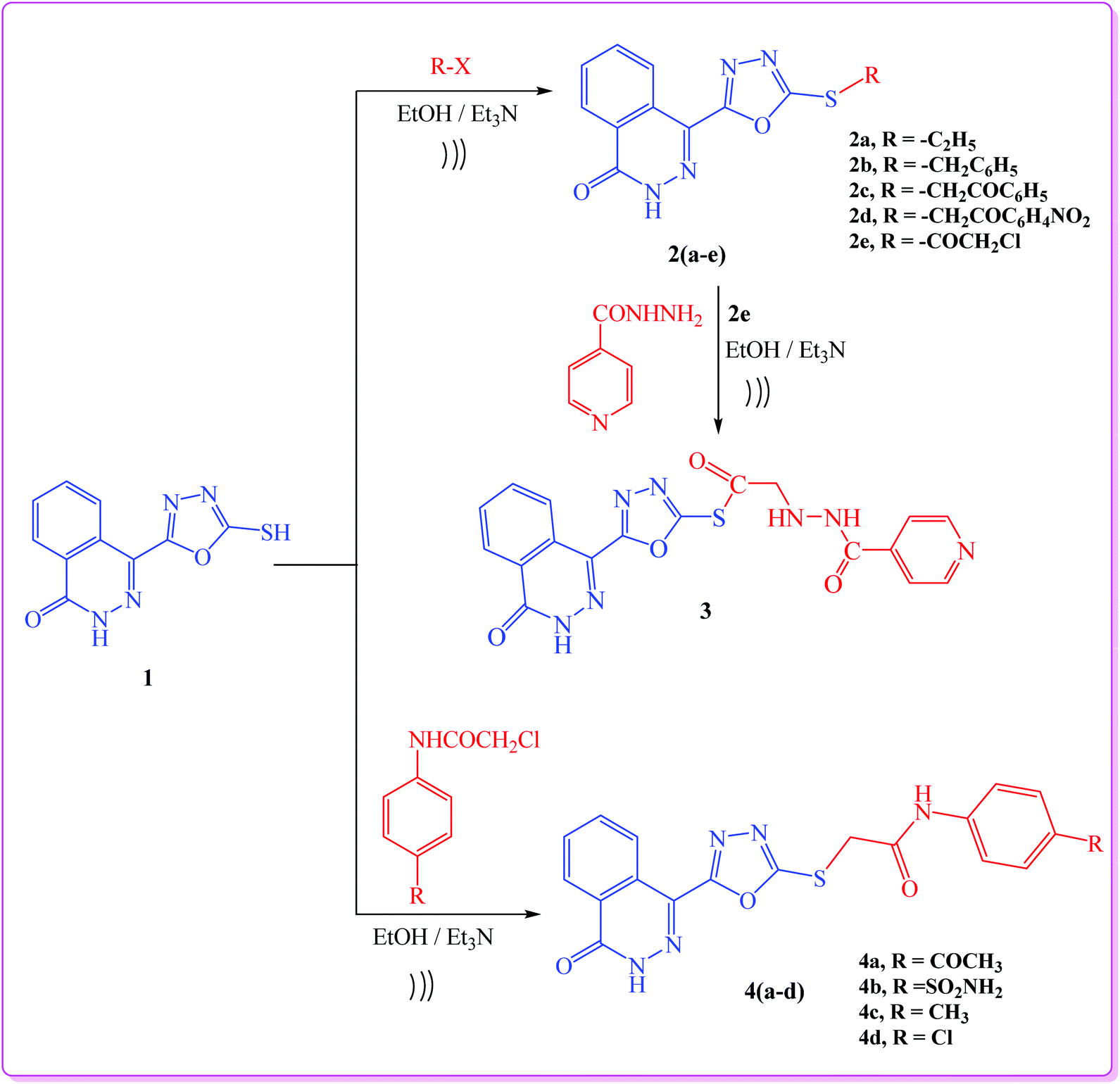 | ||
| Scheme 1 General procedure for synthesis of target compounds (2a–e, 3, 4a–d). | ||
When compound 2e was allowed to react with isonicotinohydrazide, it yielded the hydrazide derivative 3 (Scheme 1). Finally, reaction of compound 1 with the appropriate 2-chloro-N-substituted phenylacetamide derivatives afforded 4-(1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one derivatives 4a–d (Scheme 1). The 1H NMR (DMSO) spectra of compounds 4a–d revealed the presence of broad signals for (NH) protons at 10.77–13.38 ppm, and a singlet signal characteristic for SCH2 protons at 4.45 ppm.
In addition, a Mannich reaction was performed through a one-pot three-component reaction. An ultrasound-assisted reaction between 4-(5-mercapto-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (1), formaldehyde (5), and primary amines (6) namely, p-toluidine, p-chloroaniline, o-chloroaniline, p-anisidine, p-nitroaniline, 3-aminopyridine and cyclohexyl amine, afforded 4-(1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one derivatives (7a–g), respectively, in good yield (Scheme 2).
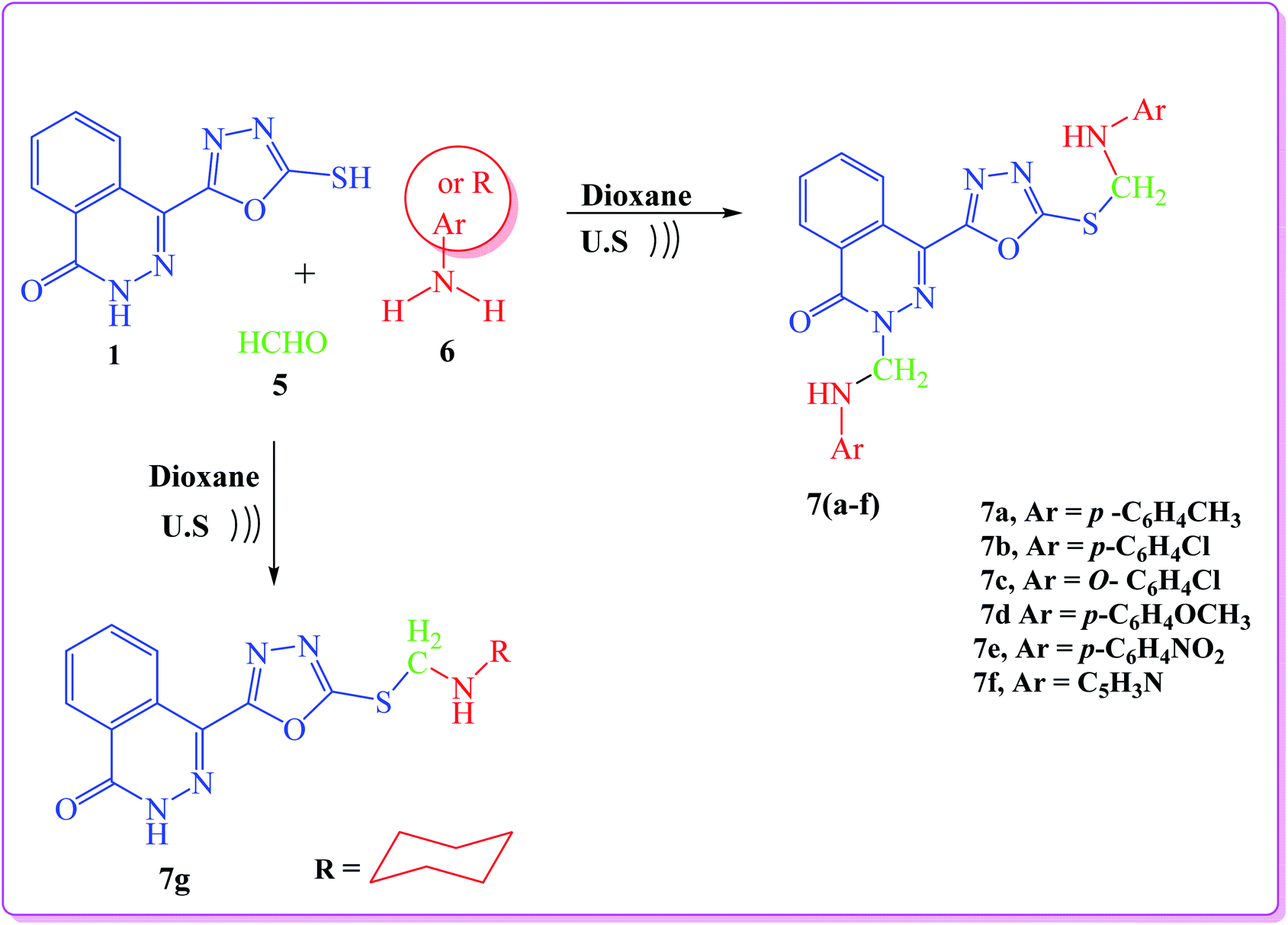 | ||
| Scheme 2 General procedure for synthesis of target compounds (7a–g). | ||
The IR spectra of the products lacked the SH absorption peak and showed the CO peak at 1666–1691 cm−1. The 1H NMR spectra of the products (7a–g) showed that the methylene groups (SCH2N and NCH2N) attached to asymmetric nitrogen are magnetically non-equivalent (diastereotopic proton) and they appear as dd (quartet) signals in the range of δ 5.39–5.56 and 5.50–5.99, respectively. The NH proton appears as multiplet in the range 12.99–13.45 due to the quadrapole moment of nitrogen atom. The 1H NMR spectra of compound 7f showed two multiplet signals at 4.57–5.08 and 5.12–5.49 (high field) attributed to the two NH groups (SCH2NH and NCH2NH), respectively. This is due to the presence of NH group in position three of pyridine moiety.
The methodology presented is easy, atomically economical, fast, and environmentally benign. The synthesis of the target compounds was carried out as outlined in Schemes 1 and 2. It was observed that the ultrasonic approach proved to be extremely fast providing good to excellent yields (85–94%) as compared with the conventional method (62–77%). The results are summarized in Table 1. The structures of all newly prepared compounds were confirmed by spectral and elemental analyses, which were in full agreement with the proposed structures.
| Entry | Comp. no. | Ultrasonic irradiation | Conventional | ||
|---|---|---|---|---|---|
| Time (min) | Yield (%) | Time (min) | Yield (%) | ||
| 1 | 2a | 5 | 92 | 55 | 70 |
| 2 | 2b | 7 | 90 | 65 | 71 |
| 3 | 2c | 5 | 87 | 65 | 67 |
| 4 | 2d | 5 | 85 | 50 | 62 |
| 5 | 2e | 4 | 86 | 50 | 66 |
| 6 | 3 | 8 | 93 | 55 | 75 |
| 7 | 4a | 10 | 92 | 75 | 73 |
| 8 | 4b | 9 | 94 | 70 | 77 |
| 9 | 4c | 7 | 90 | 65 | 74 |
| 10 | 4d | 10 | 91 | 75 | 70 |
| 11 | 7a | 5 | 94 | 75 | 68 |
| 12 | 7b | 7 | 93 | 80 | 76 |
| 13 | 7c | 7 | 90 | 72 | 74 |
| 14 | 7d | 5 | 91 | 80 | 67 |
| 15 | 7e | 7 | 92 | 83 | 77 |
| 16 | 7f | 8 | 88 | 71 | 69 |
| 17 | 7g | 10 | 95 | 80 | 75 |
2.2. Biology
| Cpd | IC50 (μM) | ||
|---|---|---|---|
| HepG2 | MCF7 | WI-38 | |
| DOX | 4.5 ± 0.3 | 4.2 ± 0.2 | 6.7 ± 0.5 |
| 1 | 4.5 ± 1.1 | 4.9 ± 0.8 | 74.9 ± 2.4 |
| 2a | 61.3 ± 4.2 | 55.8 ± 4.0 | 39.1 ± 2.9 |
| 2b | 58.9 ± 4.0 | 68.2 ± 4.4 | 42.2 ± 3.3 |
| 2c | 66.1 ± 4.5 | 57.3 ± 4.2 | 23.5 ± 1.9 |
| 2d | >100 | >100 | 29.5 ± 2.2 |
| 2e | 6.9 ± 1.3 | 5.0 ± 1.5 | 72.4 ± 4.8 |
| 3 | 38.4 ± 3.5 | 40.0 ± 3.6 | 44.6 ± 4.2 |
| 4a | 72.6 ± 4.8 | 76.2 ± 4.7 | 35.4 ± 2.7 |
| 4b | 83.9 ± 5.2 | >100 | 32.9 ± 2.5 |
| 4c | 28.1 ± 2.9 | 36.3 ± 3.2 | 66.0 ± 4.7 |
| 4d | 53.9 ± 3.8 | 49.5 ± 3.9 | 45.7 ± 3.5 |
| 7a | 23.5 ± 2.8 | 26.2 ± 2.5 | 62.8 ± 4.5 |
| 7b | 33.3 ± 3.4 | 31.3 ± 2.8 | 51.4 ± 4.1 |
| 7c | 17.2 ± 1.9 | 15.4 ± 1.2 | 23.2 ± 1.2 |
| 7d | 6.3 ± 0.7 | 5.5 ± 0.4 | 68.6 ± 2.1 |
| 7e | 7.0 ± 1.5 | 8.4 ± 0.9 | 21.6 ± 1.8 |
| 7f | 21.8 ± 2.3 | 20.8 ± 1.9 | 58.1 ± 4.3 |
| 7g | 42.9 ± 3.7 | 45.5 ± 3.8 | 49.2 ± 3.9 |
The viability of cells was measured after 48 h of incubation with 5 different concentrations (three wells per every concentration) of the investigated compounds by MTT assay. The IC50 was determined from the dose–response curves as the mean of two parallel experiments. Doxorubicin (Dox) was used as a positive control. The data are expressed as mean ± SEM.
| Treatment | Fold induction from untreated cells | |||
|---|---|---|---|---|
| p53 | Cdk1 | Caspase 3 | Txnrd1 | |
| a Significant difference (p < 0.05) as compared to untreated cells. The data are expressed as mean ± SEM for two independent experiments. | ||||
| DOX | 2.15 ± 0.41a | 0.25 ± 0.06a | 4.10 ± 0.35a | 0.44 ± 0.11a |
| 1 | 3.83 ± 0.11a | 0.22 ± 0.03a | 3.10 ± 0.21a | 0.67 ± 0.03 |
| 2e | 1.63 ± 0.18 | 1.04 ± 0.25 | 0.98 ± 0.12 | 0.93 ± 0.09 |
| 7c | 1.37 ± 0.05 | 1.21 ± 0.14 | 1.19 ± 0.13 | 0.96 ± 0.14 |
| 7d | 2.16 ± 0.12a | 0.98 ± 0.11 | 1.96 ± 0.09a | 1.02 ± 0.16 |
| 7e | 2.02 ± 0.08a | 1.04 ± 0.09 | 2.22 ± 0.14a | 1.11 ± 0.10 |
p53 is a tumor suppressor gene and widely known as the guardian of the genome. It is mutated in more than 50% of human cancer types.38 It plays a central role in the regulation of cell cycle progression, apoptosis, and DNA stability. DNA damage signals the increase in p53 expression, which arrests the cell cycle at the G1/S checkpoint, activates DNA repair mechanisms, and if the damage is severe, p53 initiates apoptosis.39 The elevation in p53 in the current study could be responsible for the parallel elevation in the expression of caspase 3. Caspase 3 is the main execution member responsible for apoptosis induced by both extrinsic and intrinsic pathways.40 However, the elevation in the expression level of caspase 3 should result in the elevation of active cleaved enzyme to indicate the occurrence of apoptosis. The ability of the cell to progress to S phase depends on cdk1.41 The downregulation of cdk1 by doxorubicin and compound 1, along with the elevation in p53 expression, suggests cell cycle arrest. Derivative 1 was the only investigated compound that downregulated the expression of cdk1. This could be attributed to the 4-fold elevation in p53 versus the 2-fold increase by derivatives 7d and 7e. However, this assumption of the p53 threshold elevation is not supported by the doxorubicin results. Doxorubicin also caused only a 2-fold increase in p53 and, at the same time, downregulated cdk1 expression. Therefore, we believe that derivative 1 directly acted on the cdk1 gene in addition to its effects on p53, but this assumption needs further investigation. The last gene investigated, txnrd1, is the only known enzyme that reduces thioredoxin.42 Thioredoxin in turn reduces several proteins and forms the disulfide bonds crucial for the activity of these proteins. Thioredoxin and txnrd1 are essential for cell growth and survival. Therefore, this enzyme is upregulated in many types of cancer.43 Inhibition of txnrd1 leads to cell death.44 Unfortunately, none of oxadiazol-phthalazinone derivatives significantly inhibited the expression of this enzyme. The 33% inhibition by compound 1 did not achieve statistical significance.
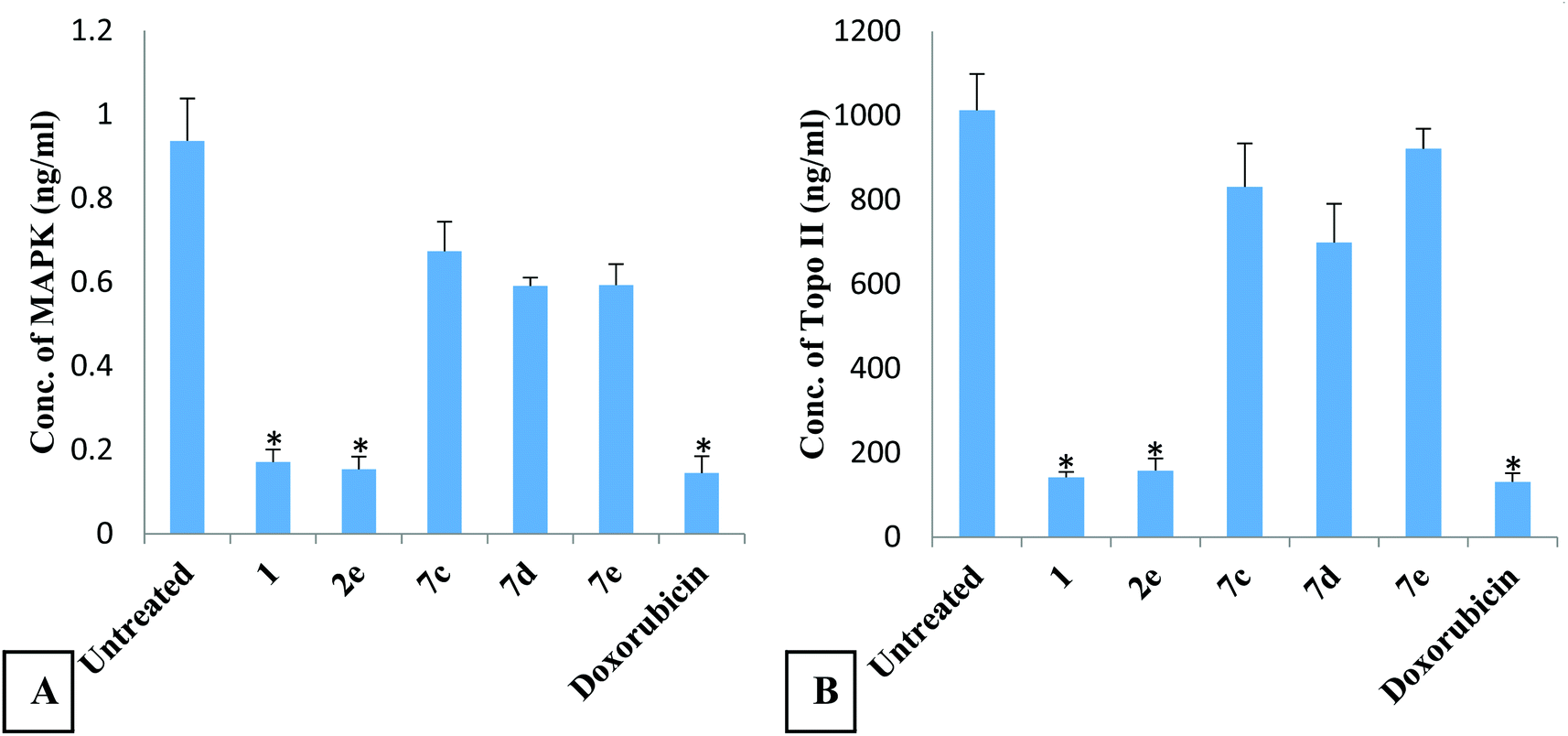 | ||
| Fig. 3 The effect of the novel oxadiazol-phthalazinone derivatives on MAPK (A) and topoisomerase II (Topo II) (B) concentrations in HepG2 cells. Each compound has been assigned three wells and the experiment was performed twice. Data are expressed as mean ± SEM. * Significant inhibition compared to untreated cells. There was no significant difference between derivatives 1 and 2e and doxorubicin. | ||
 | ||
| Fig. 4 The effect of derivatives 1 and 2e on cleaved caspase 3 activity in HepG2 cells. Data are expressed as mean ± SEM. * Significant inhibition compared to untreated cells. | ||
| Comp. no. | PIM1 | Topoisomerase II | ||||
|---|---|---|---|---|---|---|
| ΔG kcal mol−1 | No. of H bonds | No. of π bonds | ΔG kcal mol−1 | No. of H bonds | No. of π bonds | |
| 1 | −5.69 | 2 (Asp168, Met109) | 1 (Ile84) | −6.06 | 2 (Asp78, Asn51) | 2 (Met83 and Met83) |
| 2e | −5.93 | 3 (Thr106, Met109 and Lys53) | (Val38, Leu108) | −6.04 | Ser124 | — |
| Ligand | −6.61 | 3 (Asp112, Thr106 and Met109) | — | −6.44 | 3 (Asp78, Arg140 and Arg 140) | — |
The interaction of etoposide with PIM1 (Fig. 5A) illustrates three hydrogen bonding interactions between important amino acid residues of the active site: Asp112, Thr106 and Met109. Additionally, it shows a good interaction CDOCKER score (−6.61 kcal mol−1). The proposed binding mode of compound 1 with PIM1 showed an affinity value of −5.69 kcal mol−1 (Table 4). The interaction demonstrated two hydrogen bond interactions; one between the S atom of the phthalazinonethione moiety and Met109, and a second between the nitrogen atom of position 1 of the phthalazinone moiety and Asp 168. The aromatic moiety was also involved in aromatic stacking interactions with Ile84 (Fig. 5B). Scrutinizing the binding pattern between derivative 2e and PIM1 showed an affinity value of −5.93 kcal mol−1 (Table 4). Several interactions with the protein were observed including the formation of three hydrophobic interactions with the sulfur atom on the phthalazinone moiety and Thr106, Met109 and Lys53. Moreover, the phthalazinone moiety was involved in aromatic stacking interactions with Val38 and Leu108 (Fig. 5C).
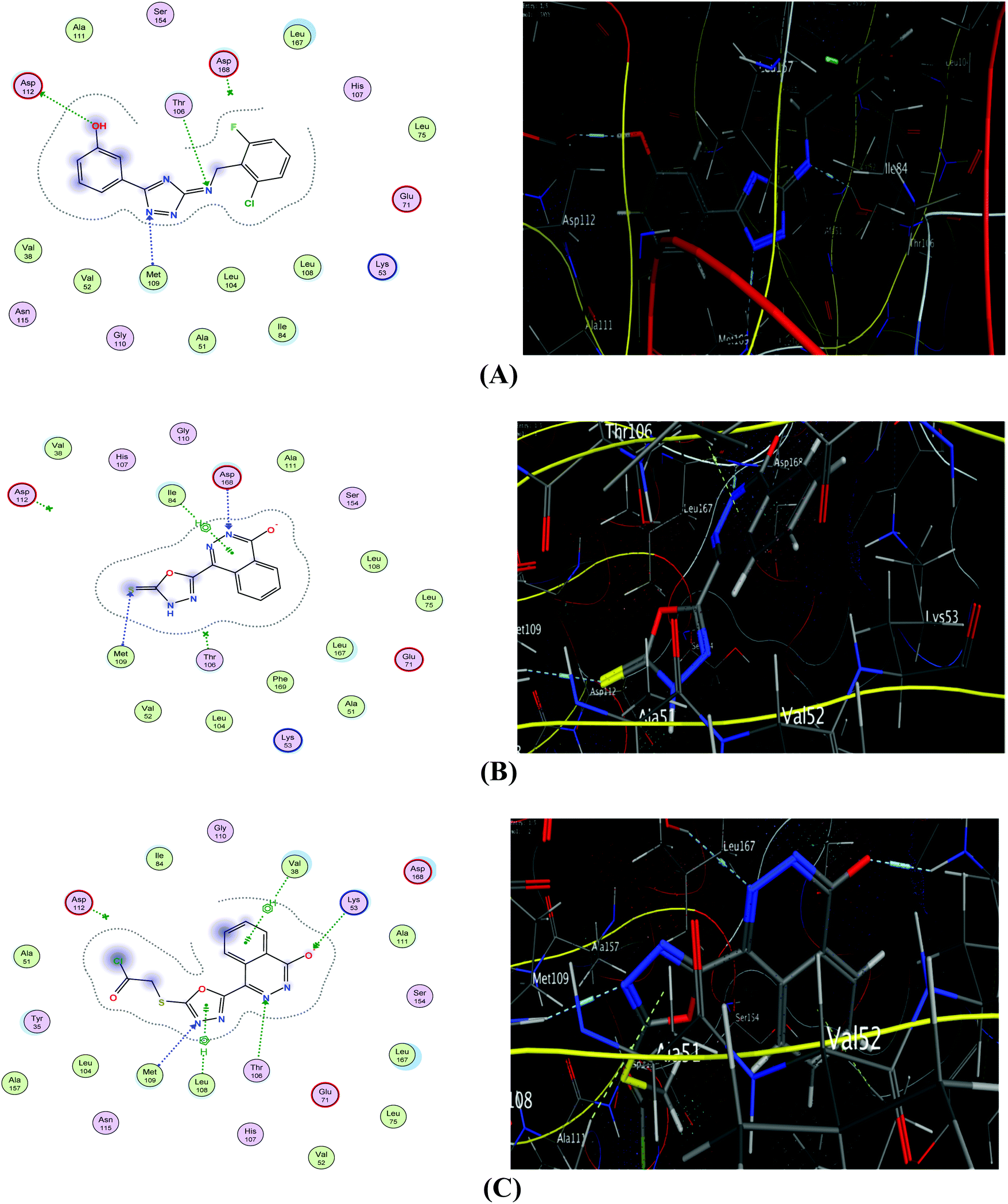 | ||
| Fig. 5 2D (left) and 3D (right) diagrams of binding interactions of ligand (A), compound 1 (B), and derivative 2e (C) with PIM1. | ||
The interaction of the ligand with DNA-topoisomerase II illustrated three hydrogen bonds between important amino acid residues of the active site, one with Asp78 and a second with Arg140 with an interaction CDOCKER score −6.44 kcal mol−1 (Table 4 and Fig. 6A). The proposed binding mode of compound 1 showed an affinity value of −6.06 kcal mol−1. It showed two hydrogen bond interactions; the first between the sulphur atom of the phthalazinone moiety and Asn51 and the second between the nitrogen atom and the amino acid Asp78, in addition to two π bonds with Met83 (Fig. 6B). The binding interaction of derivative 2e showed an affinity value of −6.04 kcal mol−1 with one hydrogen bonding interaction between the oxygen atom of the phthalazinone moiety atom and Ser124 (Fig. 6C).
 | ||
| Fig. 6 2D (left) and 3D (right) diagrams of binding interactions of ligand (A), compound 1 (B), and derivative 2e (C) with Topo II. | ||
The results of the docking study with PIM-1 kinase and Topo II are in good agreement with the inhibition profile of derivatives 1 and 2e on MAPK and Topo II reported in this study.
3. Materials and methods
3.1. Chemistry
Method A: ultrasonic conditions. A mixture of compound 1 (10 mmol) and alkyl or acyl halides, namely, ethyl iodide, benzyl chloride, phenacyl bromide, m-nitrophenacyl bromide and chloroacetyl chloride (10 mmol) in ethanol (20 mL) and catalytic amount of triethylamine (0.5 mL) was placed in an Erlenmeyer flask (50 mL) and subjected to ultrasound waves at room temperature for 5–10 min (controlled by TLC). The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent to afford the pure products 2a–e.
Method B: conventional conditions. A mixture of compound 1 (10 mmol) and alkyl halides or acyl halides, namely, ethyl iodide, benzyl chloride, phenacyl bromide, m-nitrophenacyl bromide and chloroacetyl chloride (10 mmol) in ethanol (20 mL) and a catalytic amount of triethylamine (0.5 mL) was heated under reflux for 50–65 min. The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent. All structures were confirmed by spectral data.
4-(5-(Ethylthio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (2a). Pale yellow crystals (92, 70%), mp 262–264 °C (dioxane). IR (ν/cm−1): 3214, 3158 (NH), 3002 (CH aromatic), 2937, 2895 (CH aliphatic), 1667 (C
O). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.43 (t, 3H, CH2CH3), 3.33 (q, 2H, CH2CH3), 7.91–8.06 (m, 2H, Ar–Hphthalazinone), 8.32 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.94 (d, 1H, Ar–Hphthalazinone, J = 8.4 Hz), 13.34 (br.s, 1H, NH, exchangeable with D2O). Anal. calcd for C12H10N4O2S (274.30): C, 52.55; H, 3.67; N, 20.43. Found C, 52.46; H, 3.71; N, 20.51.
4-(5-(Benzylthio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (2b). White crystals (90, 71%), mp 224–226 °C (ethanol/dioxane). IR (ν/cm−1): 3223, 3167 (NH), 3051 (CH aromatic), 2925, 2894 (CH aliphatic), 1668 (C
O). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 4.60 (s, 2H, SCH2), 7.25–7.50 (m, 5H, Ar–HC6H5), 7.91–7.95 (dd, 1H, Ar–Hphthalazinone, J = 8.4 Hz), 8.02–8.06 (dd, 1H, Ar–Hphthalazinone, J = 7.9 Hz), 8.33 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.91 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 13.41 (br.s, 1H, NH, exchangeable with D2O). Anal. calcd for C17H12N4O2S (336.37): C, 60.70; H, 3.60; N, 16.66. Found: C, 60.59; H, 3.54; N, 16.73.
4-(5-((2-Oxo-2-phenylethyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (2c). White crystals (87, 67%), mp 260–262 °C (dioxane). IR (ν/cm−1): 3247, 3201 (NH), 3063 (CH aromatic), 2976 (CH aliphatic), 1680, 1675 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.20 (s, 2H, SCH2), 7.56–7.60 (m, 2H, Ar–HC6H5), 7.69–7.72 (m, 1H, Ar–Hphthalazinone), 7.91–8.07 (m, 3H, Ar–HC6H5 + 1H, Ar–Hphthalazinone), 8.32 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.88 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 13.36 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 41.1, 126.6 (2), 127.4, 127.8, 128.9 (2), 129.4 (2), 130.8, 132.8, 134.5, 134.8, 135.4, 159.6, 162.3, 164.7, 192.9. Anal. calcd for C18H12N4O3S (364.38): C, 59.33; H, 3.32; N, 15.38. Found: C, 59.47; H, 3.40; N, 15.29.
4-(5-((2-(3-Nitrophenyl)-2-oxoethyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (2d). White crystals (85, 62%), mp 276–278 °C (dioxane). IR (ν/cm−1), 3210 (NH), 3056 (CH aromatic), 2966, 2938 (CH aliphatic), 1687, 1676 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.28 (s, 2H, SCH2), 7.86–7.94 (m, 2H, Ar–H), 8.00–8.04 (m, 1H, Ar–Hphthalazinone), 8.31 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.47–8.53 (m, 2H, Ar–H Ar–Hphthalazinone), 8.74 (s, 1H, Ar–HC6H4NO2), 8.87 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 13.36 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 41.0, 123.2, 126.5, 126.6, 127.3, 127.8, 128.5, 130.8, 131.2, 132.8, 134.8, 135.0, 136.7, 148.5, 159.6, 162.4 (2), 191.8. Anal. calcd for C18H11N5O5S (409.38): C, 52.81; H, 2.71; N, 17.11. Found: C, 52.99; H, 2.80; N, 17.22.
S-(5-(4-Oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)2-chloroethanethioate (2e). Pale yellow crystals (86, 66%), mp 260–262 °C (dioxane). IR (ν/cm−1): 3161 (NH), 3066 (CH aromatic), 2940, 2894 (CH aliphatic), 1688 (C
O). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.54 (s, 2H, COCH2), 7.91–7.95 (m, 1H, Ar–Hphthalazinone), 8.01–8.05 (m, 1H, Ar–Hphthalazinone), 8.32 (d, 1H, Ar–Hphthalazinone, J = 7.6 Hz), 8.69 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 13.39 (br.s, 1H, NH, exchangeable with D2O). Anal. calcd for C12H7ClN4O3S (322.72): C, 44.66; H, 2.19; N, 17.36. Found: C, 44.84; H, 2.22; N, 17.45.
Method A: ultrasonic conditions. A mixture of compound 2e (10 mmol) and isonicotinohydrazide (10 mmol) in dioxane (20 mL) was placed in an Erlenmeyer flask (50 mL) and subjected to ultrasound waves at room temperature for 8 min (controlled by TLC). The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent to afford the pure product 3.
Method B: conventional conditions. A mixture of compound 2e (10 mmol) and isonicotinohydrazide (10 mmol) in dioxane (20 mL) was heated under reflux for 55 min. The precipitated solid was filtered off, dried, and recrystallized from dioxane giving orange crystals of compound 3.
S-(5-(4-Oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)2-(2-isonicotinoylhydrazinyl)ethanethioate (3). Orange crystals (93, 75%), mp > 300 °C (dioxane). IR (ν/cm−1): 3430, 3176 (NH), 3070 (CH aromatic), 2937, 2890 (CH aliphatic), 1724, 1663 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 4.03 (s, 2H, SCH2), 7.84–8.94 (m, 8H, Ar–H + 2H, NH, exchangeable with D2O), 13.00 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 46.0, 121.9, 126.1, 126.3, 126.5, 126.6, 127.4, 127.8, 131.0, 132.4, 132.6, 132.7, 134.5, 134.7, 159.6, 161.7, 166.3, 169.2. Anal. calcd for C18H13N7O4S (423.4): C, 51.06; H, 3.09; N, 23.16. Found: C, 52.22; H, 3.18; N, 23.26.
Method A: ultrasonic conditions. A mixture of compound 1 (10 mmol) and 2-chloro-N-phenylacetamide derivatives (10 mmol) in dioxane (20 mL) was placed in an Erlenmeyer flask (50 mL) and subjected to ultrasound waves at room temperature for 10 min (controlled by TLC). The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent to afford the pure products 4a–d.
Method B: conventional conditions. A mixture of compound 1 (10 mmol), and 2-chloro-N-phenylacetamide derivatives (10 mmol) in dioxane (20 mL) was heated under reflux for 65–75 min. The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent. All structures were confirmed by spectral data as discussed below.
N-(4-Acetylphenyl)-2-((5-(4-oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)thio)acetamide (4a). White crystals (92, 73%), mp > 300 °C (dioxane). IR (ν/cm−1): 3322, 3215 (NH), 3045 (CH aromatic), 2969, 2895 (CH aliphatic), 1691, 1668 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 2.49 (s, 3H, COCH3), 4.44 (s, 2H, SCH2), 7.70 (d, 2H, Ar–HC6H4OCH3, J = 8.4 Hz), 7.92–7.95 (m, 2H, Ar–H + 1H, Ar–Hphthalazinone), 8.00–8.03 (m, 1H, Ar–Hphthalazinone), 8.31 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.90 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 10.78 (br.s, 1H, NH, exchangeable with D2O), 13.37 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 26.6, 37.4, 118.9 (2), 126.6 (2), 127.4, 127.8, 130.0 (2), 130.8, 132.5, 132.8, 134.8, 143.3, 159.6, 162.3, 164.7, 165.7, 196.9. Anal. calcd for C20H15N5O4S (421.43): C, 57.00; H, 3.59; N, 16.62. Found: C, 57.22; H, 3.64; N, 16.71.
2-((5-(4-Oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (4b). White crystals (94, 77%), mp > 300 °C (dioxane). IR (ν/cm−1): 3311, 3268, 3163 (NH, NH2), 3065 (CH aromatic), 2934, 2895 (CH aliphatic), 1691, 1663 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 4.43 (s, 2H, SCH2), 7.25 (br.s, 2H, NH2, exchangeable with D2O), 7.71–7.78 (m, 4H, Ar–H), 7.91–8.05 (m, 2H, Ar–Hphthalazinone), 8.32 (d, 1H, Ar–Hphthalazinone, J = 8.8 Hz), 8.90 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 10.79 (br.s, 1H, NH, exchangeable with D2O), 13.37 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (400 MHz, DMSO-d6) δ (ppm): 37.4, 119.2 (2), 126.6, 127.2, 127.4 (3), 127.8, 130.8, 132.8, 134.8, 139.2, 141.9, 159.6, 162.3, 164.7, 165.7. Anal. calcd for C18H14N6O5S2 (485.47): C, 47.16; H, 3.08; N, 18.33. Found: C, 47.33; H, 3.18; N, 18.44.
2-((5-(4-Oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)thio)-N-(p-tolyl)acetamide (4c). Grey crystals (90, 74%), mp 265–267 °C (dioxane). IR (ν/cm−1): 3308, 3215 (NH), 3074 (CH aromatic), 2934, 2894 (CH aliphatic), 1689, 1657 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 2.29 (s, 3H, CH3), 4.40 (s, 2H, SCH2), 7.01 (d, 2H, Ar–H, J = 8.4 Hz), 7.46 (d, 2H, Ar–H, J = 8.4 Hz), 7.89–8.04 (m, 2H, Ar–Hphthalazinone), 8.31 (d, 1H, Ar–Hphthalazinone, J = 8.1 Hz), 8.90 (d, 1H, Ar–Hphthalazinone, J = 8.1 Hz), 10.41 (br.s, 1H, NH, exchangeable with D2O), 13.36 (br.s, 1H, NH, exchangeable with D2O). Anal. calcd for C19H15N5O3S (393.42): C, 58.01; H, 3.84; N, 17.80. Found C, 58.19; H, 3.92; N, 17.91.
N-(4-Chlorophenyl)-2-((5-(4-oxo-3,4-dihydrophthalazin-1-yl)-1,3,4-oxadiazol-2-yl)thio)acetamide (4d). White crystals (91, 70%), mp 206–208 °C (ethanol). IR (ν/cm−1): 3305, 3163 (NH), 3044 (CH aromatic), 2978, 2941 (CH aliphatic), 1691, 1657 (2C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 4.40 (s, 2H, SCH2), 7.35–7.38 (dd, 2H, Ar–HC6H4Cl, J = 8 Hz), 7.60–7.63 (d, 2H, Ar–HC6H4Cl, J = 8 Hz), 7.92–8.05 (m, 2H, Ar–Hphthalazinone), 8.32 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 8.91 (d, 1H, Ar–Hphthalazinone, J = 8 Hz), 10.76 (br.s, 1H, NH, exchangeable with D2O), 13.42 (br.s, 1H, NH, exchangeable with D2O). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 37.3, 121.1, 121.2, 126.6, 127.4 (2), 127.8 (2), 129.2, 130.9, 131.8, 134.8 (2), 138.0, 159.6, 162.3, 164.7, 165.3. Anal. calcd for C18H12ClN5O3S (413.84): C, 52.24; H, 2.92; N, 16.92. Found: C, 52.16; H, 2.83; N, 17.09.
Method A: ultrasonic conditions. A mixture of compound 1 (10 mmol), formaldehyde (20 mmol) and primary amines namely, p-toluidine, p-chloroaniline, o-chloroaniline, p-anisidine, p-nitroaniline, 3-aminopyridine and cyclohexyl amine (20 mmol) in dioxane (20 mL) were placed in an Erlenmeyer flask (50 mL) and subjected to ultrasound waves at room temperature for 5–10 min (controlled by TLC). The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent to afford the pure products 7a–g.
Method B: conventional conditions. A mixture of compound 1 (10 mmol), formaldehyde (20 mmol) and primary amines (20 mmol) (Table 1) in dioxane (20 mL) was stirred at room temperature for 72–83 min. The precipitated solid was filtered off, dried, and recrystallized from the appropriate solvent.
2-((p-Tolylamino)methyl)-4-(5-(((p-tolylamino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7a). Buff crystals (94, 68%), mp 175–177 °C (ethanol). IR (ν/cm−1): 3180 (NH), 3078 (CH aromatic), 2920, 2892 (CH aliphatic), 1669 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 2.28 (s, 3H, CH3), 2.50 (s, 3H, CH3), 5.42–5.52 (dd, 2H, SCH2N, J = 9 Hz), 5.90–6.08 (dd, 2H, NCH2N, J = 9 Hz), 6.45–7.80 (m, 4H, Ar–H), 7.92–8.05 (m, 4H, Ar–H), 8.31–8.34 (m, 2H, Ar–Hphthalazinone), 8.66–8.71 (m, 2H, Ar–Hphthalazinone), 13.38–13.44 (m, 2H, 2NH, exchangeable with D2O). Calcd for C26H24N6O2S (484.6): C, 64.44; H, 4.99; N, 17.34. Found: C, 64.25; H, 4.89; N, 17.27.
2-(((4-Chlorophenyl)amino)methyl)-4-(5-((((4-chlorophenyl)amino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7b). Buff crystals (93, 76%), mp 138–140 °C (ethanol). IR (ν/cm−1): 3356 (NH), 3079 (CH aromatic), 2961, 2889 (CH aliphatic), 1667 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.39–5.48 (dd, 2H, SCH2N, J = 16 Hz), 5.50–5.77 (dd, 2H, NCH2N, J = 16 Hz), 6.62–7.69 (m, 8H, Ar–H), 7.85–8.74 (m, 4H, Ar–Hphthalazinone), 13.00–13.45 (m, 2H, 2NH, exchangeable with D2O). 13C NMR (400 MHz, DMSO-d6) δ (ppm): 57.8, 73.9, 113.6 (2), 115.6, 119.6 (2), 120.8, 125.8, 127.04, 127.5, 128.4, 128.6, 130.1, 132.9, 133.2, 133.3, 134.6, 141.7 (2), 155.3, 158.2, 159.8, 175.7. Anal. calcd for C24H18Cl2N6O2S (525.4): C, 54.86; H, 3.45; N, 16.00. Found: C, 54.67; H, 3.35; N, 16.11.
2-(((2-Chlorophenyl)amino)methyl)-4-(5-((((2-chlorophenyl)amino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7c). White crystals (90, 74%), mp 240–242 °C (ethanol). IR (ν/cm−1): 3171, 3111 (NH), 3071 (CH aromatic), 2956, 2893 (CH aliphatic), 1669 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.51–5.81 (dd, 2H, SCH2N, J = 11.6 Hz), 5.90–5.99 (dd, 2H, NCH2N, J = 11.6 Hz), 6.16–7.92 (m, 8H, Ar–H), 7.94–8.41 (m, 4H, Ar–Hphthalazinone), 13.00–13.48 (m, 2H, 2NH exchangeable with D2O). Anal. calcd for C24H18Cl2N6O2S (525.4): C, 54.86; H, 3.45; N, 16.00. Found: C, 54.65; H, 3.37; N, 15.90.
2-(((4-Methoxyphenyl)amino)methyl)-4-(5-((((4-methoxyphenyl)amino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7d). Brown crystals (91, 67%), mp 158–161 °C (ethanol). IR (ν/cm−1): 3203 (NH), 3074 (CH aromatic), 2955 (CH aliphatic), 1666 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 3.73 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 5.51–5.89 (dd, 2H, SCH2N, J = 12 Hz), 5.75–5.89 (dd, 2H, NCH2N, J = 10.4 Hz), 6.68–6.95 (m, 4H, Ar–H), 7.04–7.37 (m, 4H, Ar–H), 7.43–8.75 (m, 4H, Ar–Hphthalazinone), 12.99–13.40 (m, 2H, 2-NH, exchangeable with D2O). Anal. calcd for C26H24N6O4S (516.6): C, 60.45; H, 4.68; N, 16.27. Found: C, 60.23; H, 4.57; N, 16.19.
2-(((4-Nitrophenyl)amino)methyl)-4-(5-((((4-nitrophenyl)amino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7e). Yellow crystals (92, 77%), mp 213–215 °C (ethanol/dioxane). IR (ν/cm−1): 3393 (NH), 3079 (CH aromatic), 2924, 2895 (CH aliphatic), 1671 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.50–5.52 (dd, 2H, SCH2N, J = 6.9 Hz), 5.68–5.70 (dd, 2H, NCH2N, J = 6.9 Hz), 6.78–6.80 (d, 2H, Ar–Hnitrophenyl, J = 6 Hz), 7.07–7.10 (d, 2H, Ar–Hnitrophenyl, J = 6 Hz), 7.84–8.61 (m, 8H, Ar–H), 13.41–13.42 (m, 2H, 2NH exchangeable with D2O). Anal. calcd for C24H18N8O6S (546.5): C, 52.75; H, 3.32; N, 20.50. Found: C, 52.56; H, 3.24; N, 20.38.
2-((Pyridin-3-ylamino)methyl)-4-(5-(((pyridin-3-ylamino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7f). White crystals (88, 69%), mp 190–192 °C (ethanol). IR (ν/cm−1): 3395 (NH), 3085 (CH aromatic), 2925 (CH aliphatic), 1691 (C
O), 1650 (CN). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 4.57–5.08 (m, 1H, NH, exchangeable with D2O), 5.12–5.49 (m, 1H, NH, exchangeable with D2O), 5.49–5.65 (dd, 2H, SCH2N, J = 12 Hz), 5.75–5.82 (dd, 2H, NCH2N, J = 12 Hz), 7.08–7.50 (m, 4H, Ar–Hpyridyl), 7.78–7.96 (m, 4H, Ar–Hpyridyl), 8.18–8.34 (m, 2H, Ar–Hphthalazinone), 8.49 (d, 1H, Ar–Hphthalazinone, J = 8.4 Hz), 8.59 (d, 1H, Ar–Hphthalazinone, J = 8.4 Hz). 13C NMR (400 MHz, DMSO-d6) δ (ppm): 59.3, 82.3, 119.3 (2), 120.8, 124.1, 126.2 (2), 126.8 (2), 127.6, 127.7, 132.7 (2), 134.5 (2), 136.1, 138.7, 143.1 (2), 159.1, 164.9. Anal. calcd for C22H18N8O2S (458.50): C, 57.63; H, 3.96; N, 24.44. Found C, 57.79; H, 4.01; N, 24.52.
4-(5-(((Cyclohexylamino)methyl)thio)-1,3,4-oxadiazol-2-yl)phthalazin-1(2H)-one (7g). Pale yellow crystals (95, 75%), mp 240–242 °C (ethanol/dioxane). IR (ν/cm−1): 3306 (NH), 3112 (CH aromatic), 2971, 2894 (CH aliphatic), 1672 (C
O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 1.18–1.85 (m, 11Hcyclohexyl), 5.38–5.48 (dd, 2H, CH–Ncyclohexyl), 7.86–8.06 (m, 2H, Ar–Hphthalazinone), 8.25–8.74 (m, 2H, Ar–Hphthalazinone), 12.99–13.04 (m, 2H, 2NH, exchangeable with D2O). Anal. calcd for C17H19N5O2S (357.4): C, 57.13; H, 5.36; N, 19.59. Found C, 56.98; H, 5.27; N, 19.48.
3.2. Biology
| Gene | Forward primer | Reverse primer |
|---|---|---|
| p53 | CTGCCCTCAACAAGATGTTTTG | CTATCTGAGCAGCGCTCATGG |
| Cdk1 | CCGAAATCTGCCAGTTTGAT | CTGGCCAGTTCATGGATTCT |
| Caspase-3 | TTAATAAAGGTATCCATGGAGAACACT | TAGTGATAAAAATAGAGTTCTTTTGTGAG |
| Txnrd1 | GTTACTTGGGCATCCCTGGTGA | CGCACTCCAAAGCGACATAGGA |
| GAPDH | TCAAGAAGGTGGTGAAGCAG | AGGTGGAAGAATGGGAGTTG |
3.3. Statistical analysis of data
Statistical analysis was performed using One Way ANOVA for comparing different groups. The distribution of data was first tested using the Kolmogorov–Sminrov test. Results were expressed as mean ± SEM. Paired comparisons between groups were performed using Tukey-HSD test. The confidence limit was set at 95% and the difference at p < 0.05 was considered significant.Conflicts of interest
The authors declare no conflict of interest, financial or otherwise.Acknowledgements
The authors sincerely acknowledge Dr Jeanette Roberts (Professor of Medicinal Chemistry and Dean Emerita, University of Wisconsin-Madison) for critically reading the manuscript.References
- I. H. Hall, B. J. Barnes, E. S. Ward, J. R. Wheaton, K. A. Shaffer, S. E. Cho and A. E. Warren, Arch. Pharm., 2001, 334, 229–234 CrossRef CAS PubMed
.
- I. H. Hall, E. S. Hall and O. T. Wong, Anti-Cancer Drugs, 1992, 3, 55–62 CrossRef CAS PubMed
- I. H. Hall, D. W. Covington, J. R. Wheaton, R. A. Izydore and X. Zhou, Pharmazie, 2001, 56, 168–174 CAS
- M. C. Cardia, S. Distinto, E. Maccioni, A. Plumitallo, L. Sanna, M. L. Sanna and S. Vigo, J. Heterocycl. Chem., 2009, 46, 674–679 CrossRef CAS
- V. M. Loh, X.-l. Cockcroft, K. J. Dillon, L. Dixon, J. Drzewiecki, P. J. Eversley, S. Gomez, J. Hoare, F. Kerrigan, I. T. W. Matthews, K. A. Menear, N. M. B. Martin, R. F. Newton, J. Paul, G. C. M. Smith, J. Vilec and A. J. Whittlec, Bioorg. Med. Chem. Lett., 2005, 15, 2235–2238 CrossRef CAS PubMed
- X. L. Cockcroft, K. J. Dillon, L. Dixon, J. Drzewiecki, F. Kerrigan, V. M. Loh, N. M. B. Martin, K. A. Menear and G. C. M. Smith, Bioorg. Med. Chem. Lett., 2006, 16, 1040–1044 CrossRef CAS PubMed
- K. A. Menear, C. Adcock, R. Boulter, X. Cockcroft, L. Copsey, A. Cranston, K. J. Dillon, J. Drzewiecki, S. Garman, S. Gomez, H. Javaid, F. Kerrigan, C. Knights, A. Lau, V. M. Loh, I. T. W. Matthews, S. Moore, M. J. O'Connor, G. C. M. Smith and N. M. B. Martin, J. Med. Chem., 2008, 51, 6581–6591 CrossRef CAS
- S. L. Gaonkar, K. M. L. Rai and B. Prabhuswamy, Eur. J. Med. Chem., 2006, 41, 841–846 CrossRef CAS
- T. M. C. Tan, Y. Chen, K. H. Kong, J. Bai, Y. Li, S. G. Lim, T. H. Ang and Y. Lam, Antiviral Res., 2006, 71, 7–14 CrossRef CAS PubMed
- Y. Li, J. Liu, H. Zhang, X. Yang and Z. Liu, Bioorg. Med. Chem. Lett., 2006, 16, 2278–2282 CrossRef CAS PubMed
- A. S. Aborai, H. M. Abdel-Rahman, N. M. Mahfouz and M. A. Gendy, Bioorg. Med. Chem., 2006, 14, 1236–1246 CrossRef PubMed
- M. T. Khan, M. I. Choudhary, K. M. Khan, M. Rani and A. U. Rahman, Bioorg. Med. Chem., 2005, 13, 3385–3395 CrossRef CAS PubMed
- C. Loetchutinat, F. Chau and S. Mankhetkorn, Chem. Pharm. Bull., 2003, 51, 728–730 CrossRef CAS PubMed
- A. H. Abadi, A. A. H. Eissa and G. S. Hassan, Chem. Pharm. Bull., 2003, 51, 838–844 CrossRef CAS
- B. G. Szczepankiewicz, G. Liu, H. S. Jae, A. S. Tasker, I. W. Gunawardana, T. W. v. Geldern, S. L. Gwaltney, J. R. Wu-Wong, L. Gehrke, W. J. Chiou, R. B. Credo, J. D. Alder, M. A. Nukkala, N. A. Zielinski, K. Jarvis, K. W. Mollison, D. J. Frost, J. L. Bauch, Y.
H. Hui, A. K. Claiborne, Q. Li and S. H. Rosenberg, J. Med. Chem., 2001, 44, 4416–4430 CrossRef CAS
- D. Kumar, S. Sundaree, E. O. Johnson and K. Shah, Bioorg. Med. Chem. Lett., 2009, 19, 4492–4494 CrossRef CAS
- J. T. Palmer, B. L. Hirschbein, H. Cheung, J. McCarter, J. W. Janc, W. Z. Yu and G. Wesolowski, Bioorg. Med. Chem. Lett., 2006, 16, 2909–2914 CrossRef CAS
- R. N. Warrener, Eur. J. Org. Chem., 2000, 3363–3380 CrossRef CAS
- M. Guan, Z. Q. Bian, Y. F. Zhou, F. Y. Li, Z. J. Li and C. H. Huang, Chem. Commun., 2003, 2708–2709 RSC
- H. Lai, D. Dou, S. Aravapalli, T. Teramoto, G. H. Lushington, T. M. Mwania, K. R. Alliston, D. M. Eichhorn, R. Padmanabhan and W. C. Groutas, Bioorg. Med. Chem., 2013, 21, 102–113 CrossRef CAS PubMed
- T. J. Mason and J. P. Lorimer, J. Chem. Technol. Biotechnol., 2004, 79, 207–208 Search PubMed
- M. R. Shaaban, T. S. Saleh, F. H. Osman and A. M. Farag, J. Heterocycl. Chem., 2007, 44, 177–181 CrossRef CAS
- S. Dandapani and L. A. Marcaurelle, Curr. Opin. Chem. Biol., 2010, 14, 362–370 CrossRef CAS PubMed
- T. J. Mason and D. Peters, Angew. Chem., Int. Ed., 2003, 42, 2331–2335 CrossRef PubMed
- M. A. EL-Hashash, E. A. El-Bordany, M. I. Marzouk, A. M. El-Naggar, T. M. S. Nawar and W. M. El-Sayed, Anti Cancer Agents Med. Chem., 2018, 18, 1589–1598 CrossRef CAS
- I. H. Eissa, A. M. El-Naggar and M. A. El-Hashash, Bioorg. Chem., 2016, 67, 43–56 CrossRef CAS
- I. H. Eissaa, A. M. El-Naggar, N. E. A. Abd El-Sattar and A. S. A. Youssef, Anti Cancer Agents Med. Chem., 2018, 18, 195–209 CrossRef PubMed
- M. A. Ismail, A. Negm, R. K. Arafa, E. Abdel-Latif and W. M. El-Sayed, Eur. J. Med. Chem., 2019, 169, 76–88 CrossRef CAS
- M. A. Ismail, M. M. Youssef, R. K. Arafa, S. S. Al-Shihry and W. M. El-Sayed, Eur. J. Med. Chem., 2017, 126, 789–798 CrossRef CAS
- k. A. M. Abouzid, G. H. Al-Ansary and A. M. El-Naggar, Eur. J. Med. Chem., 2017, 134, 357–365 CrossRef CAS
- A. M. El-Naggar, M. M. Hemdan and S. R. Atta-Allah, J. Heterocycl. Chem., 2017, 54, 3519 CrossRef CAS
- A. M. El-Naggar, A. K. Khalil, H. M. Zeidan and W. M. El-Sayed, Anti Cancer Agents Med. Chem., 2017, 17, 1644–1651 CAS
- M. H. Hekal and F. S. M. Abu El-Azm, J. Heterocycl. Chem., 2017, 54, 3056–3064 CrossRef CAS
- M. R. Mahmoud, W. S. I. Abou-Elmagd, H. A. Derbala and M. H. Hekal, Chin. J. Chem., 2011, 29, 1446–1450 CrossRef CAS
- Y. Sun, P. Xia, H. Zhang, B. Liu and Y. Shi, Am. J. Cancer Res., 2016, 6, 114–125 CAS
- J. Herrero-Ruiz, M. Mora-Santos, S. Giráldez, C. Sáez, M. Á. Japón, M. Tortolero and F. Romero, Oncotarget, 2014, 5, 7563–7574 CrossRef
- J. J. Liu, Q. Liu, H. L. Wei, J. Yi, H. S. Zhao and L. P. Gao, Pharmazie, 2011, 66, 440–444 CAS
- T. Ozaki and A. Nakagawara, Cancers, 2011, 3, 994–1013 CrossRef CAS
- E. Senturk and J. J. Manfredi, Methods Mol. Biol., 2013, 962, 49–61 CrossRef PubMed
- M. Olsson and B. Zhivotovsky, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS PubMed
- J. M. Enserink and R. D. Kolodner, Cell Div., 2010, 5, 1–41 CrossRef PubMed
- E. S. Arnér and A. Holmgren, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef PubMed
- P. Nguyen, R. T. Awwad, D. D. Smart, D. R. Spitz and D. Gius, Cancer Lett., 2006, 2361, 64–74 Search PubMed
- K. F. Tonissen and G. Di Trapani, Mol. Nutr. Food Res., 2009, 53, 87–103 CrossRef CAS
- G. Ambrosini, H. S. Cheema, S. Seelman, E. B. Sambol, S. Singer and G. K. Schwartz, Mol. Cancer Ther., 2008, 7, 890–896 CrossRef CAS PubMed
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 327–337 CrossRef CAS
- E. Willmore, S. de Caux and N. J. Sunter, Blood, 2004, 103, 4659–4665 CrossRef CAS
- X. Zhang, M. Song, J. K. Kundu, M. H. Lee and Z. Z. Liu, Journal of Cancer Prevention, 2018, 23, 109–116 CrossRef
- B. T. Le, M. Kumarasiri, J. R. Adams, M. Yu, R. Milne, M. J. Sykes and S. Wang, Future Med. Chem., 2015, 7, 35–53 CrossRef CAS
- Y. Tursynbay, J. Zhang, Z. Li, T. Tokay, Z. Zhumadilov, D. Wu and Y. Xie, Biomed. Rep., 2016, 4, 140–146 CrossRef CAS
- I. Malík, J. Csöllei, J. Jampílek, L. Stanzel, I. Zadražilová, J. Hošek, Š. Pospíšilová, A. Čížek, A. Coffey and J. O' Mahony, Molecules, 2016, 21, 1274 CrossRef
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS
- R. A. Hussein, E. A. El-Husseiny, L. A. Hassanin and W. M. El-Sayed, Int. J. Clin. Pharmacol. Toxicol., 2017, 6, 270–279 Search PubMed
- S. A. El-Metwally, A. K. Khalil, A. M. El-Naggar and W. M. El-Sayed, Anti-Cancer Agents Med. Chem., 2018, 18, 1761–1769 CrossRef CAS PubMed
- S. Kamada, M. Washida, J. I. Hasegawa, H. Kusano, Y. Funahashi and Y. Tsujimoto, Oncogene, 1997, 15, 285–290 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09016a |
| This journal is © The Royal Society of Chemistry 2020 |