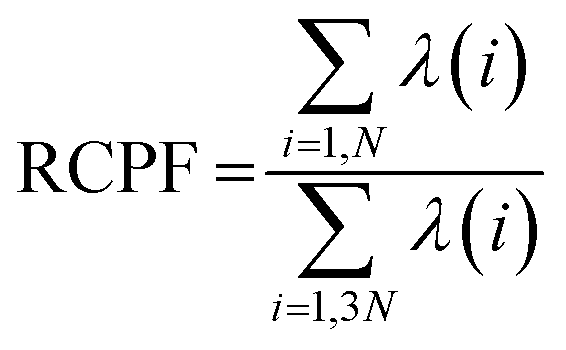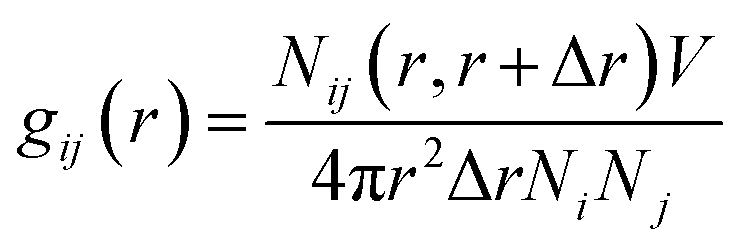DOI:
10.1039/C9RA09612D
(Paper)
RSC Adv., 2020,
10, 7933-7947
Structure and dynamics of ionic liquid tolerant hyperthermophilic endoglucanase Cel12A from Rhodothermus marinus†
Received
18th November 2019
, Accepted 4th February 2020
First published on 24th February 2020
Abstract
Economic deconstruction of lignocellulose remains a challenge due to the complex architecture of cellulose, hemicellulose, and lignin. Advancements in pretreatment processes have introduced ionic liquids (ILs) as promising non-derivatizing solvents for reducing biomass recalcitrance and for promoting enzymatic hydrolysis. However, available commercial cellulases are destabilized or inactivated even in low concentration of residual ILs. Thus, a molecular understanding of IL-enzyme interactions is crucial for developing IL-tolerant enzymes with high catalytic activity. In this study, molecular insight behind the IL tolerance of hyperthermophilic endoglucanase Cel12A from Rhodothermus marinus (RmCel12A) has been investigated in 20%, 40%, and 60% 1-ethyl-3-methylimidazolium acetate (EmimAc) through molecular dynamic simulations at 368 K. Though the enzyme retained its stability in all EmimAc concentrations, the activity was affected due to the loss of essential dynamic motions. A protein structure network was constructed using the snapshots of protein structures from the simulation trajectories and the hub properties of residues R20, Y59, W68, W197, E203, and F220 were found to be lost in 60% EmimAc. Emim cations were observed to intrude the active site tunnel and interact with more number of catalytic residues with higher cumulative fractional occupancy in 60% EmimAc than in 20% or 40% EmimAc. Some non-catalytic residues have also been identified at the active site, which can be probable mutation targets for improving the IL tolerance. Our findings reveal the molecular understanding behind the origin of activity loss of RmCel12A and proposed insights for the further improvement of IL sensitivity.
1. Introduction
Recent globalization, overconsumption of fossil fuels, and its detrimental consequences on the environment have raised the demand for sustainable energy resources.1–3 Biomass-derived fuels are a promising alternative to fossil fuels along with the production of numerous value-added chemicals in biorefineries analogous to petroleum industries.4 A lignocellulosic feedstock is the most abundant organic material on earth and currently, research is underway for developing sustainable lignocellulosic biorefineries.5 Both the cellulosic and hemicellulosic parts of lignocellulose are reservoirs of enormous fermentable sugars for biorefinery products, including bioethanol.6 However, the highly recalcitrant properties of lignocellulose require extensive physical, chemical, or biological pretreatment to extract the polysaccharide chains, which can be further hydrolyzed and depolymerized into monosaccharides.7–9 Several physical, chemical, and biological pretreatment methods have been developed to enhance the accessibility of biomass polysaccharides to hydrolytic enzymes.10 Currently, industrial pretreatment technologies mostly involve acid–alkali treatments that require huge amounts of water for washing purposes, thus generating chemical hazards and environmental concerns.11 Moreover, due to degradation of the cellulose backbone, a significant amount of cellulose is lost in such processing techniques. In the past few years, ionic liquids (IL) have shown promises to be greener alternatives and more effective non-derivatizing solvents for cellulose dissolution.11 ILs are salts with a melting point of around or below 100 °C, having low vapor pressure, high thermal stability, and non-volatile properties, offering great opportunity for replacing traditional organic solvents in several industrial processes.12,13 ILs can dissolve lignocellulosic biomass and effectively separate the complex mixture of cellulose, hemicellulose, and lignin for further processing.14 The ability of ILs as cellulose solvents was pioneered in early 2000s.15 Since then, significant advancements have been made in ionic liquid research in carbohydrate chemistry among different ILs and 1-ethyl-3-methylimidazolium acetate (EmimAc) was found to be one of the most effective pretreatment agents for cellulose dissolution.16 Moreover, the ILs can be recycled for further pretreatment cycles. After the IL pretreatment, the regenerated cellulose is carried for enzymatic hydrolysis for generating fermentable sugars. Pretreatment with IL accelerates the hydrolysis step by exposing the cellulose surface to cellulolytic enzymes and resulting in a remarkably improved glucose yield.17–22 Besides, IL/water mixed solvents have also been proposed to enhance the process efficiency and the cost-effectiveness, and provide an aqueous environment in the hydrolysis step.23,24 In addition, simultaneous pretreatment and saccharification strategies have been attempted by combining IL pretreatment and IL tolerant cellulases.25–28 This combined technology for the ‘one-pot’ process, if it becomes a reality in the near future, may drastically cut down the cost of the IL pretreatment technology. However, a critical issue is that commercial cellulases or hemicellulases are either inactivated or the activity is drastically reduced in the presence of low concentration of ILs.29,30 Thus, in an ideal scenario, the residual ILs in the biomass should be completely removed from the pretreated biomass before moving onto the hydrolysis step so that the cellulolytic activities of the enzymes remain unaffected. However, some amount of ILs always remain in the biomass even after processing. Additional washing of the residual IL increases the overall processing cost, which restricts the implementation of the IL pretreatment at a commercial scale. In order to overcome this challenge, researchers have focused either on discovering new cellulases or engineering the existing cellulases with higher IL tolerance. For example, endoglucanase from Thermotoga maritima and Pyrococcus horikoshii were found to have higher IL tolerance than the industrial benchmark Trichoderma viride cellulase, and retained 50% and 100% of their specific activities in 15% (v/v) and 20% (v/v) EmimAc,31 respectively. A cocktail of cellulases and β-glucosidase from Trichoderma reesei and Aspergillus niger showed 77% and 65% activities in 15% and 20% (w/v) EmimAc,32 respectively. A halophilic cellobiohydrolase from Halorhabdus utahensis was observed to tolerate upto 20% (w/w) of ILs such as EmimAc, 1-ethyl-3-methylimidazolium chloride (EmimCl), 1-butyl-3-methylimidazolium chloride (BmimCl), and 1-allyl-3-methylimidazolium chloride (AmimCl).33 In another study, endoglucanases and xylanases from Aspergillus fumigatus were shown to retain enzymatic activity in 20% EmimAc.34 Cellulase cocktail from microbial consortia was also developed and it showed 54% activity in 20% EmimAc.35 Cellulase from Aspergillus fumigatus was isolated from the chemically polluted microhabitat with the maximum activity of 127%, 111%, and 109% in 30% (v/v) of 1-ethyl-3-methylimidazolium dimethyl phosphate, AmimCl, and 1-ethyl-3-methylimidazolium methylammonium-methylsulfate respectively.36 Furthermore, the study involving the isolation and characterization of cellulases from switchgrass-adapted microbial community have led to the discovery of high IL tolerant cellulases that exhibited considerable enzymatic activities from 10% (v/v) EmimAc to 40% (v/v) EmimAc.37 Though such extensive studies may lead to discoveries of novel cellulases with high IL tolerance, understanding the molecular basis behind the high stability and activity will provide important insights for the further improvement of enzyme activity in the presence of IL. Molecular dynamic (MD) simulations have been extensively used to investigate the cellulase-IL interaction and generate hypotheses for the probable activity loss, as found in experiments. For example, the process of competitive inactivation of Trichoderma reesei cellobiohydrolase I (CBHI) by 1-butyl-3-methylimidazolium (Bmim) cations was studied through MD simulations and in silico mutagenesis of H228R was also carried out for enhancing the IL tolerance by reducing the binding affinity of Bmim towards the enzyme.38 In another study, NMR experiments and MD simulations were combined to elucidate the denaturation of local structures and cation induced competitive inhibition of Acidothermus cellulolyticus endoglucanase in BmimCl.39 The MD simulations of three glycoside hydrolase (GH) family 5 cellulases from T. viride, T. maritima, and P. horikoshii revealed the molecular details behind the loss of activities of the T. viride and T. maritima cellulase and stability of the P. horikoshii in increasing the EmimAc concentration.31,40 Different surface charge variants of endoglucanase homologs from Acidothermus cellulolyticus have revealed both denaturing and inhibitory effects of EmimCl.41 Apart from cellulases, dynamics of many other proteins have been investigated in IL solutions including GH 11 xylanase,42 lytic polysaccharide monooxygenases,43 lipase,44 protease,45 and lysozyme.46 In the very first MD simulation study of an enzyme in ionic liquids, the structure and dynamics of the serine protease cutinase from Fusarium solani pisi in 1-butyl-3-methylimidazolium hexafluorophosphate ([Bmim][PF6]) and 1-butyl-3-methylimidazolium nitrate ([Bmim][NO3]) was analyzed.47 This study revealed that the enzyme was less stable in [Bmim][NO3] compared to that in [Bmim][PF6], providing an insight that the properties of the IL solvents could be altered by varying the anion species for designing an optimum IL for bimolecular applications. Furthermore, recent reports have shed light on the role of MD simulations to understand the structure and dynamics of enzymes in various non-aqueous solvents.48,49
Different families of cellulases have been well explored in search of IL tolerant GH enzymes.50,51 GH Family 12 endoglucanase from Rhodothermus marinus (RmCel12A) exhibits both hyperthermophilic and high IL tolerant properties. It is an aerobic, halophile, and thermophile bacterium found in extreme environments.52 Based on a previous experimental study, RmCel12A has been observed to be one of the highest IL tolerant enzymes with 100% activity in the presence of 40% (v/v) EmimAc with an optimum temperature of 95 °C. Thus, RmCel12A is an attractive hyperthermophilic cellulase to study the IL tolerance behavior. However, the structural and dynamic behavior of RmCel12A behind such high IL tolerance is still unclear. The elucidation of molecular-level details will provide new insights for rational design and engineering of cellulases for industrial applications. In this study, the theoretical understanding behind the high IL tolerance of endoglucanase RmCel12A has been investigated by atomistic molecular dynamic simulation in different concentrations (20%, 40%, and 60%) of EmimAc and water mixtures. As RmCel12A was found to exhibit an optimum temperature of 95 °C in the experiments,37 all the simulations were performed at 368 K. It was observed that the enzyme retained a stable structure in all IL strengths. With increasing IL concentration, the essential dynamics of the protein, especially the opening and the closing motions, were found to be altered and the dynamically stable hub residues were lost. Moreover, the Emim cations populated the active site cleft of the enzyme with higher cumulative fractional occupancy with the catalytic residues, which suggests that competitive inhibition causes the loss of enzymatic activity in high IL concentration.
2. Methods
2.1 Preparation of simulation systems
All MD simulations were performed in AMBER 18 (ref. 53) at elevated temperatures (368 K). X-ray crystallographic structure for RmCel12A endoglucanase was obtained from the Protein Data Bank (PDB ID: 2BWA;54 resolution: 1.68 Å). The AMBERff14SB55 force field was used to describe the topology of the protein. The force field parameters for EmimAc were the same as that used in our previous study.24 A total of three EmimAc/water mixed solvent systems were prepared: S1 (20% EmimAc and 80% water), S2 (40% EmimAc and 60% water), and S3 (60% EmimAc and 40% water), each containing the RmCel12A enzyme (Table 1). The standard TIP3P56 water model was used for the water molecules. Periodic boundary conditions were applied to mimic the properties of the bulk system. Particle Mesh Ewald (PME)57 summation method was implemented for the calculation of electrostatic interaction with a non-bonded cut-off distance of 10 Å. The temperature (368 K) was controlled by Langevin thermostat and the SHAKE algorithm58 was used for constraining the bonds involving hydrogen atoms. Monte Carlo barostat was implemented for pressure regulation. Energy minimization was carried out for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of steepest descent algorithm on the initial geometries of all the systems. The MD production run was performed under the NPT ensemble with a time step of 2 femtoseconds (fs). Three sets of simulations were performed (denoted by set a: S1a, S2a, S3a; set b: S1b, S2b, S3b, and set c: S1c, S2c, S3c) for 300 ns each. All the three sets of simulations were started from the initial coordinates of the individual systems and had the same simulation conditions. Trajectory data were collected at an interval of 1 picosecond (ps) and analysis was performed by excluding first 100 ns for the equilibration stage. CPPTRAJ59 module was used for the post-processing of the MD simulation trajectories and visual molecular dynamics (VMD)60 was used as a visualization tool.
000 steps of steepest descent algorithm on the initial geometries of all the systems. The MD production run was performed under the NPT ensemble with a time step of 2 femtoseconds (fs). Three sets of simulations were performed (denoted by set a: S1a, S2a, S3a; set b: S1b, S2b, S3b, and set c: S1c, S2c, S3c) for 300 ns each. All the three sets of simulations were started from the initial coordinates of the individual systems and had the same simulation conditions. Trajectory data were collected at an interval of 1 picosecond (ps) and analysis was performed by excluding first 100 ns for the equilibration stage. CPPTRAJ59 module was used for the post-processing of the MD simulation trajectories and visual molecular dynamics (VMD)60 was used as a visualization tool.
Table 1 Details of the simulated systems
| Serial no. |
System names |
System property |
Number of IL ions (Emim, Ac) |
Number of water molecules |
Temperature (K) |
Total simulation time (ns) |
| 1 |
S1a |
S1b |
S1c |
20% EmimAc |
248, 243 |
8233 |
368 |
300 |
| 2 |
S2a |
S2b |
S2c |
40% EmimAc |
491, 486 |
6175 |
368 |
300 |
| 3 |
S3a |
S3b |
S3c |
60% EmimAc |
734, 729 |
4116 |
368 |
300 |
2.2 Principal component analysis (PCA)
Principal Component Analysis (PCA) can reveal the collective motions in the protein structure during the simulation process. PCA was carried out with one frame for each 100 ps interval from the simulation trajectory. First, an average structure (Cα atoms) was generated by aligning all the structural ensembles to the starting structure for removing the global rotational and translational motions. A covariance matrix (Cij) was constructed between the position vector (r) of the atom i and j, as given below:| | |
Covij = 〈(ri − 〈ri〉)(rj − 〈rj〉)〉
| (1) |
where the angle brackets denote an average over the simulation trajectory. The covariance matrix was diagonalized to get a set of orthogonal eigenvectors. The eigenvalues associated with each eigenvector signify the contribution of that eigenvector in the overall dynamic motions of the protein. Finally, the displacement vectors of each atom were projected along the eigenvectors to obtain the principal components (PCs). To investigate how much each of the PCs contribute in the total protein dynamics, Relative Cumulative Positional Fluctuation (RCPF) of the eigenvectors for each system was calculated using the following formula,61| |
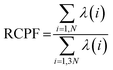 | (2) |
where λ(i) is the ith eigenvalue and N is the number of atoms considered for the construction of the covariance matrix.
2.3 Construction of the protein structure network (PSN)
The Protein Structure Network (PSN) was constructed from the atomic coordinates of amino acids in the protein structure snapshots from the simulation trajectories. Each amino acid was represented as a node in the network, where the nodes were connected by forming edges. The strength of the edges are defined based on the non-covalent interactions between a pair of interacting nodes. The interaction strength between ith and jth residues was calculated by62| |
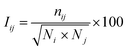 | (3) |
where Iij and nij denote the percentage interaction and the number of atom–atom pairs within a distance cut-off of 4.5 Å between the ith and jth residues, respectively; Ni and Nj are the normalization factors that consider the type of ith and jth residues, their side chain length, and their propensities to form maximum number of contacts with other amino acids in the protein. A cutoff (Imin = 4.0) for effective interaction was set and for any selected residue pairs, if Iij > Imin, the residues were considered to be interacting and connected in the protein structure network. Besides, a hub node is defined if a node contains four or more number of edges.
2.4 Radial distribution function (RDF) analysis
Radial Distribution Function (RDF) provides insights into the distribution of the solute–solvent at the molecular level in different systems. It describes how a given particle is organized around a reference particle, leading to a change in the density as a function of radial distance. The RDF was calculated around the nitrogen atom of residue D106, which resides at the center of mass (COM) of the active site of RmCel12A (Fig. 1). In case of the solvent molecules, atoms having varying chemical environments were chosen. For example, N2 atom in Emim was considered as the positively charged region, whereas C2 atom of the acetate was selected to represent the polar part of the anion (Fig. S1, ESI†). The oxygen atom (O) of water was selected to represent the water molecule. Finally, RDF, i.e., g(r) was calculated using the formula as given below.24| |
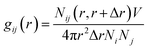 | (4) |
where Nij (r, r + ∇r) is the number of j particles around i within a shell radius of r to (r + ∇r), which is normalized by the actual number of Ni and Nj atoms at that distance. V denotes the total volume of the system, whereas 4πr2dr is the variable spherical volume as a function of radial distance.
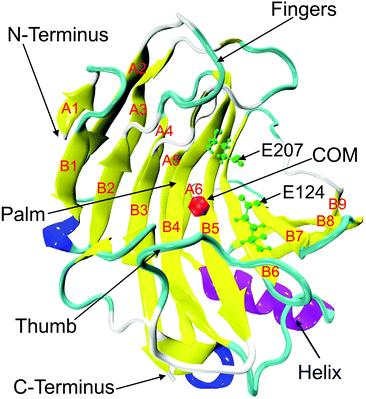 |
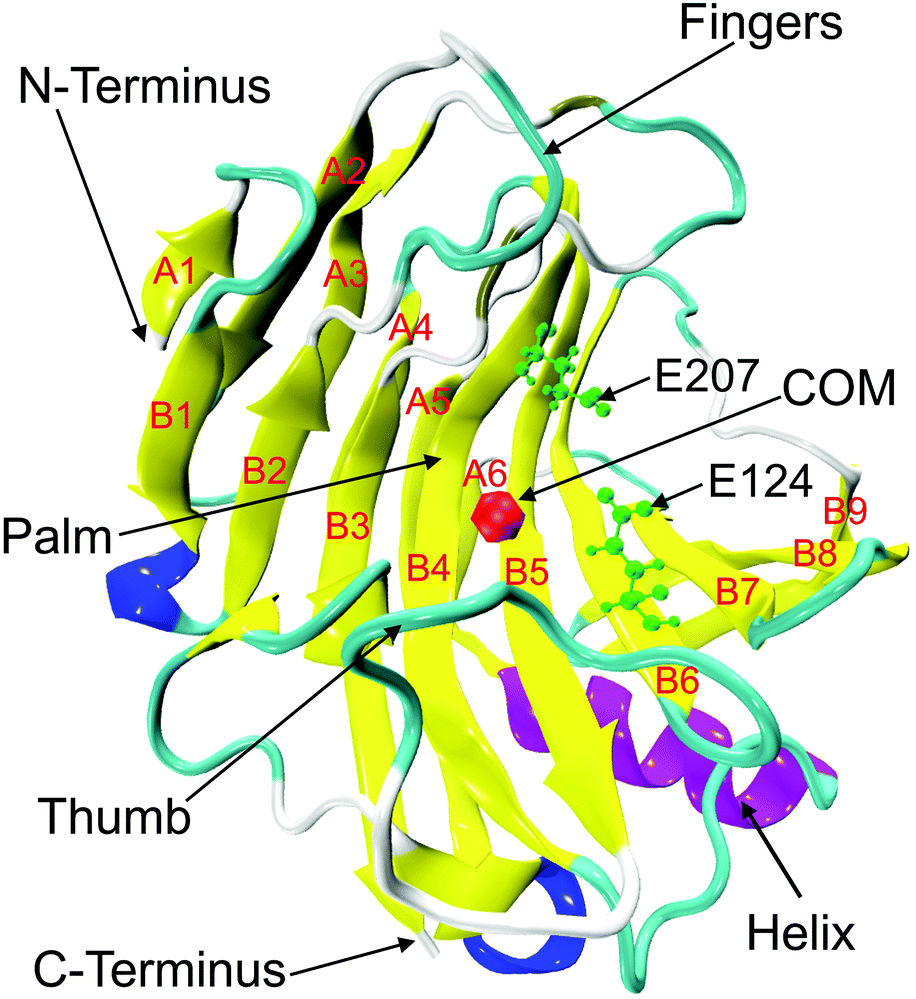
| | Fig. 1 Crystal structure of endoglucanase Cel12A from Rhodothermus marinus (PDB ID: 2BWA, chain A) with the β-jelly roll structure consisting of β-sheets (yellow), A (strands A1 to A6), and B (strands B1–B9). The alpha and 310 helices are colored in purple and blue, respectively. Two important catalytic residues (E124 and E207) are represented in green color using ball and stick model in the active site tunnel. The red colored bead close to the residue D106 on B5 strand indicates the center of mass (COM) of the protein. | |
3. Results and discussion
To have the theoretical understanding behind the high ionic liquid tolerance of hyperthermophilic endoglucanase Cel12A from Rhodothermus marinus (RmCel12A), all-atom MD simulation was performed in varying concentrations of EmimAc and water mixtures (20%, 40%, and 60% EmimAc) in this study. The 20% and 40% EmimAc simulation systems can provide insights into the native behavior of the enzyme where the enzyme does not lose activity experimentally. Besides, 60% EmimAc with much higher Emim concentration has been designed, which might help to the predict loss in enzymatic activity due to the structural or dynamical changes compared to the 20% and 40% EmimAc systems. Three replicate sets of simulations were carried out to confirm the statistical significance of the results.
3.1 Structural stability of RmCel12A in different IL/water mixed solvent systems
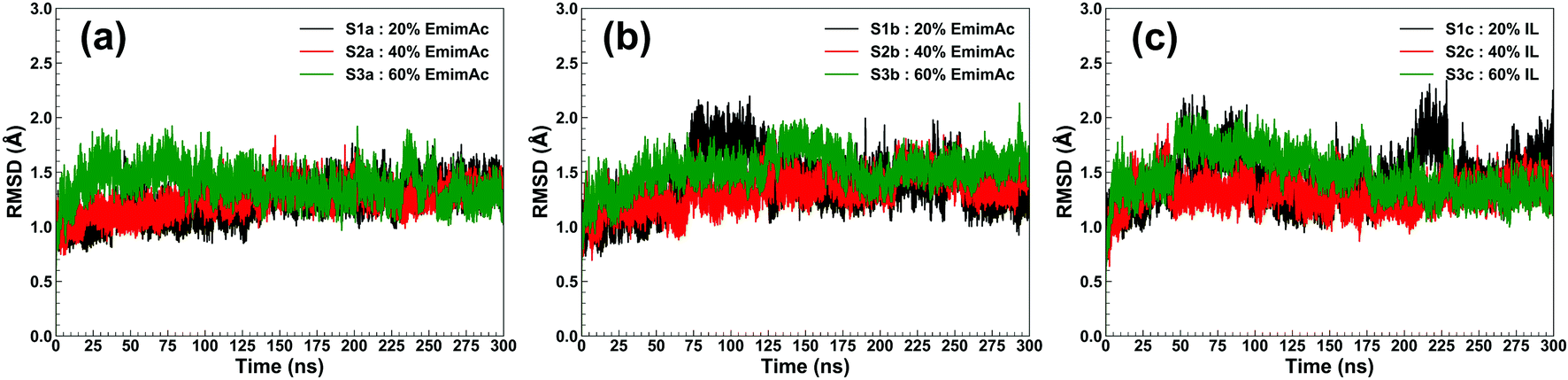
RmCel12A contains a β-jelly roll structural fold with two β-sheets and an alpha-helix at the bottom (Fig. 1). The outer and inner sheets consist of six (A1 to A6) and nine (B1 to B9) antiparallel β-strands, respectively. To evaluate the structural stability of the protein, the root-mean-square deviation (RMSD) of RmCel12A was calculated with reference to the crystal structure. The average Cα-RMSD values were observed to be (1.29 ± 0.12 Å, 1.41 ± 0.18 Å, and 1.44 ± 0.20 Å), (1.30 ± 0.09 Å, 1.39 ± 0.11 Å, and 1.29 ± 0.11 Å), (1.35 ± 0.12 Å, 1.56 ± 0.13 Å, and 1.43 ± 0.16 Å) in 20% EmimAc (S1a, S1b, and S1c), 40% EmimAc (S2a, S2b, and S2c), and 60% EmimAc (S3a, S3b, and S3c), respectively (Fig. 2). It shows that the average RMSD of the enzyme over three replicates in 60% EmimAc (1.45 ± 0.09 Å) is slightly higher than that of 20% EmimAc (1.38 ± 0.09 Å) and 40% EmimAc (1.32 ± 0.06 Å) systems. Furthermore, the end structure of the enzyme after the 300 ns simulations in each system was superimposed with the initial crystal structure to investigate if there are any small noticeable structural changes (Fig. S2, ESI†). It was observed that the secondary structures almost overlapped with the crystal structure with low RMSD value of 1.35 Å, 1.19 Å, and 1.08 Å in 20% EmimAc (S1a), 40% EmimAc (S2a), and 60% EmimAc (S3a), respectively. The loop regions seem to be more dynamic, giving rise to the RMSD fluctuations. The RMSD values in all the systems at elevated temperature (368 K) suggest that RmCel12A maintains a stable structure in both low (20%) and high (60%) EmimAc concentration in the simulation time scale.
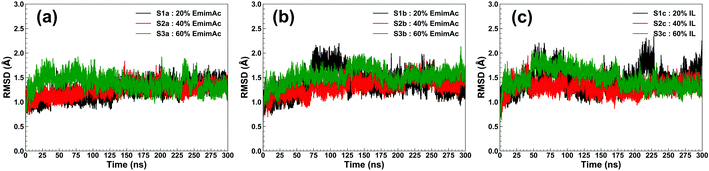 |
| | Fig. 2 RMSD of RmCel12A for MD trajectories in 20% EmimAc (black), 40% EmimAc (red), and 60% EmimAc (green) for (a) replicate set a, (b) replicate set b, and (c) replicate set c at 368 K. | |
3.2 Dynamic motion of RmCel12A in different IL/water mixed solvent systems
The enzymatic activity of RmCel12A depends on its dynamic motion in different EmimAc concentrations. Dynamic motions populating the native conformation in the open and closed form play key roles during the activity of an enzyme.63 Principal component analysis (PCA) has been extensively used to identify essential dynamics of proteins from MD simulation trajectories.42,64–66 Notably, the eigenvectors having larger eigenvalues correspond to the most dominant dynamic motions taking place in the protein. To identify the modes that contribute to major dynamic motions in the enzyme, the relative cumulative positional fluctuation (RCPF) of RmCel12A was evaluated for each system (Fig. S3, ESI†). It is notable that only the first few eigenvectors with the largest eigenvalues contribute the most to system dynamics. The relative cumulative positional fluctuation of principal component 1 (PC1) and principal component 2 (PC2) were observed to be (17.66%, 14.29%), (17.07%, 11.64%), and (22.16%, 6.94%) in 20% EmimAc, 40% EmimAc, and 60% EmimAc, respectively. The two modes were significant as they comprised of nearly ∼30% of the total modes. Thus, PC1 and PC2 were analyzed as representative dominant modes to study the essential dynamics of the protein. To elucidate how these dominant modes influence protein dynamics, residue-specific mobility (square-fluctuations) was calculated (Fig. 3). It was observed that PC1 mode contributed towards higher residue motions across the loop between β-strands B2–A2 (residues 23 to 31) and B5–B6 (residues 111 to 122) of RmCel12A in both 20% EmimAc and 40% EmimAc. On the other hand, the motion of the residues of the loops consisting of strands B2–A2 in 20% EmimAc and B2–A2, A3–B3, B5–B6, and B8–B7 (residues 159 to 161) in 40% EmimAc were due to PC2 mode. The aromatic residues Y119 and (W26, Y119) in the active site tunnel were found to be comparatively more mobile in 20% EmimAc and 40% EmimAc, respectively. In case of 60% EmimAc, the loop between B5–B6 and B8–B7 strands exhibited higher mobility due to PC1 mode. Also, PC2 mode contributed to the mobility of the loops between strands A1–B1 (residues 6 to 10), B2–A2, B5–B6, B8–B7 and B6–B9 (residues 133 to 139) in 60% EmimAc. Some of the active site mobile residues were W9, W26, W68, R100, Y119, E124, Y156, W159, W161, and Y163, which have been experimentally found to play important roles in the function of RmCel12A.54,67 The high mobility of these residues signifies that the native motions related to enzyme function were deviated in 60% EmimAc as compared to 20% EmimAc and 40% EmimAc. Thus, the mobility plot depicted a change in the residue fluctuation pattern of RmCel12A in 60% EmimAc in comparison to 20% EmimAc and 40% EmimAc. Moreover, these dominant modes were projected on the Cα atoms of the average structure of RmCel12A using porcupine plots (Fig. 4). The hand-like structure of RmCel12A exhibits an opening and closing motion at the active site due to the shuffling of the thumb and fingers in a coordinated fashion. These dynamic motions of the enzyme are crucial for substrate binding, catalysis, and product ejection during cellulose hydrolysis. The dominant modes of opening and closing motion at the active site are present in 20% EmimAc (Fig. 4a and d) and 40% EmimAc (Fig. 4b and e). On the other hand, dominant PCA modes are only involved in the opening of the active site in 60% EmimAc (Fig. 4c and f). Thus, the dynamic motions for enzymatic catalysis involving the opening and closing of active site were preserved in low IL strength, and significantly affected due to the increase in IL concentrations. The loss of these essential motions related to the enzyme function may affect the activity of the enzyme at higher IL strength.
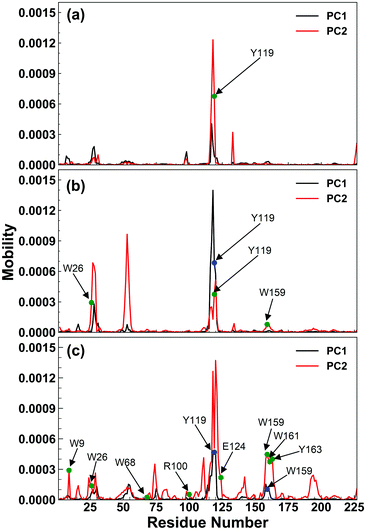 |
| | Fig. 3 Residue based mobility plot of RmCel12A showing only the mobility of different residues due to the dominant modes, i.e., PC1 (black line) and PC2 (red line) in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc systems. The blue and green circles indicate the highly mobile active site residues due to PC1 and PC2 modes, respectively. | |
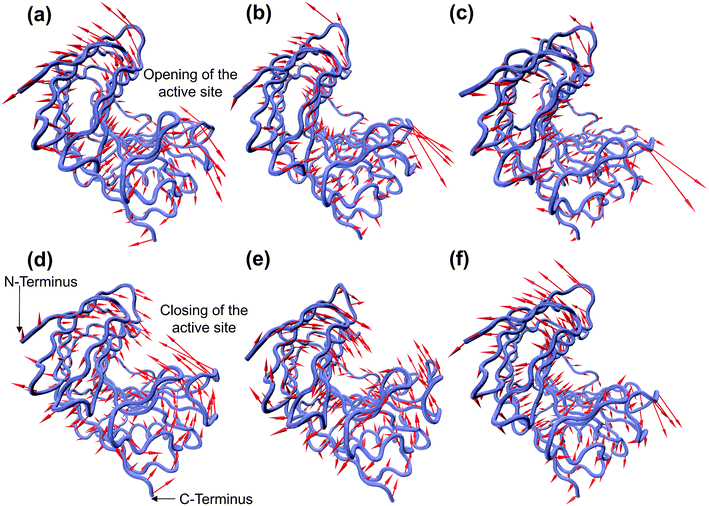 |
| | Fig. 4 Porcupine plot of the dominant modes (PC1 and PC2) involved in opening and closing motion of the hand-like structure of RmCel12A. PC1 corresponds to the opening of the active site in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc systems, whereas PC2 contributes to the closing of the active site in (d) 20% EmimAc and (e) 40% EmimAc. However, in the case of 60% EmimAc, PC2 exhibits the opening motion (f) only, suggesting the loss of the essential dynamic motions of the enzyme in high EmimAc concentration. | |
3.3 Insight into the conformational changes of RmCel12A using protein structure network
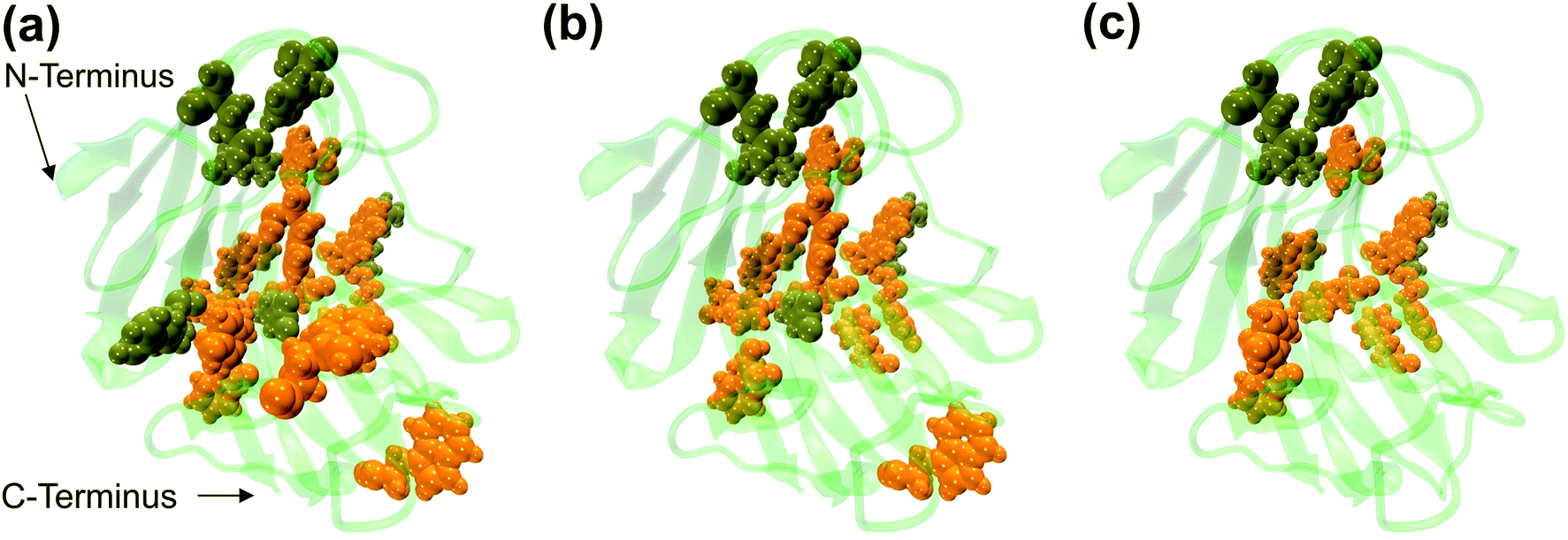
The dynamic behavior of RmCel12A in the presence of varying IL concentrations was investigated by constructing the Protein Structure Network (PSN) from each snapshot in the MD simulation trajectories. PSN has been extensively applied to analyze protein folding/unfolding and protein–tRNA communication pathways in MD trajectories earlier.62,68,69 To understand the conformational change of RmCel12A, hub residues were examined in the PSN. The residues that retained the hub properties for 25% of the simulation snapshots were considered as dynamically stable hubs. These dynamically stable hubs are crucial for maintaining network stability and thereby influence the dynamics and activity of the protein. The total number of dynamically stable hub residues was found to be (18, 18, 17), (15, 15, 15), and (12, 13, 13) in 20% EmimAc (S1a, S1b, S1c), 40% EmimAc (S2a, S2b, S2c), and 60% EmimAc (S3a, S3b, S3c), respectively (Table 2). Out of the 18 hub residues from 20% EmimAc, hub residues R20, Y63, and W68 were lost in 40% EmimAc simulation. In case of 60% EmimAc, the hub properties of residue R20, Y59, W68, W197, E203, and F220 were lost in the network. However hub residues Y59, W197, and E203 appeared as hubs in one of the three replicate simulations. Among the lost hub residues, W68 and Y59 were found to be involved in aromatic stacking interaction during enzyme–substrate (cellulose) binding.67 On the other hand, residue Y59 is conserved in all GH 12 families and resides near the catalytic base (E207) in the active site cleft in RmCel12A. Thus, it is suggested that the loss of these two catalytically critical hubs residues (Y59 and W68) affect the network integrity and thereby the activity of RmCel12A in 60% EmimAc. Moreover, some non-catalytic hub residues (R20, W197, E203, and F220) were also lost in high IL concentration. These residues are predicted to be important for maintaining stable dynamics for catalysis in RmCel12A. On the other hand, it is notable that all the hub residues were mostly concentrated over the surface of the active site tunnel in all the simulated systems. The number of aromatic residues in the hub were (12, 12, 12), (10, 10, 10), and (8, 8, 9) in 20% EmimAc (S1a, S1b, S1c), 40% EmimAc (S2a, S2b, S2c), and 60% EmimAc (S3a, S3b, S3c) respectively. These dynamically stable hubs form a stable aromatic network cluster (Fig. 5). These aromatic residues provide a hydrophobic core with increased stacking interactions, leading to the stability and hyperthermophilic properties of the enzyme. Previously, similar aromatic network clusters were found in multiple thermophilic proteins and have been reported to be absent in their mesophilic homologs.70 To summarize, PSN provides insights at the level of amino acid side-chain interactions that lead to changes in RMCel12A with increasing IL concentration.
Table 2 List of the dynamically stable hubs of RmCel12A in 20% EmimAc, 40% EmimAc, and 60% EmimAc systems
| 20% EmimAc |
40% EmimAc |
60% EmimAc |
| S1a |
S1b |
S1c |
S2a |
S2b |
S2c |
S3a |
S3b |
S3c |
| R20 |
R20 |
R20 |
— |
— |
— |
— |
— |
— |
| Q32 |
Q32 |
Q32 |
Q32 |
Q32 |
Q32 |
Q32 |
Q32 |
Q32 |
| H50 |
H50 |
H50 |
H50 |
H50 |
H50 |
H50 |
H50 |
H50 |
| Y59 |
Y59 |
Y59 |
Y59 |
Y59 |
Y59 |
— |
— |
Y59 |
| Y63 |
Y63 |
Y63 |
— |
— |
— |
Y63 |
Y63 |
Y63 |
| F64 |
F64 |
F64 |
F64 |
F64 |
F64 |
— |
F64 |
F64 |
| W68 |
W68 |
W68 |
— |
— |
— |
— |
— |
— |
| W91 |
W91 |
W91 |
W91 |
W91 |
W91 |
W91 |
W91 |
W91 |
| W101 |
W101 |
W101 |
W101 |
W101 |
W101 |
W101 |
W101 |
W101 |
| Y105 |
Y105 |
Y105 |
Y105 |
Y105 |
Y105 |
Y105 |
Y105 |
Y105 |
| W128 |
W128 |
W128 |
W128 |
W128 |
W128 |
W128 |
W128 |
W128 |
| Y166 |
Y166 |
Y166 |
Y166 |
Y166 |
Y166 |
Y166 |
Y166 |
Y166 |
| R167 |
R167 |
R167 |
R167 |
R167 |
R167 |
R167 |
R167 |
R167 |
| F183 |
F183 |
F183 |
F183 |
F183 |
F183 |
F183 |
F183 |
F183 |
| W197 |
W197 |
W197 |
W197 |
W197 |
W197 |
W197 |
— |
— |
| E203 |
E203 |
E203 |
E203 |
E203 |
E203 |
— |
E203 |
— |
| L215 |
L215 |
— |
L215 |
L215 |
L215 |
L215 |
L215 |
L215 |
| F220 |
F220 |
F220 |
F220 |
F220 |
F220 |
— |
— |
— |
| Total hubs = 18 |
Total hubs = 18 |
Total hubs = 17 |
Total hubs = 15 |
Total hubs = 15 |
Total hubs = 15 |
Total hubs = 12 |
Total hubs = 13 |
Total hubs = 13 |
| Aromatic hubs = 12 |
Aromatic hubs = 12 |
Aromatic hubs = 12 |
Aromatic hubs = 10 |
Aromatic hubs = 10 |
Aromatic hubs = 10 |
Aromatic hubs = 8 |
Aromatic hubs = 8 |
Aromatic hubs = 9 |
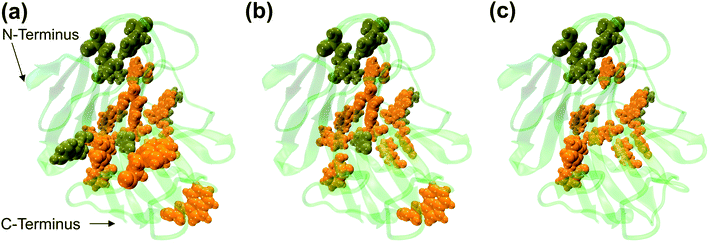 |
| | Fig. 5 Representation of the dynamically stable hubs in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc. The aromatic and non-aromatic hubs are colored in orange and tan green, respectively. With increasing EmimAc concentration, the number of hub residues decreases in RmCel12A. | |
3.4 Interaction of RmCel12A with the IL/water mixed solvent systems
Radial distribution function describes the organization of the solvent molecules around the enzyme and helps to trace the specific interaction between the enzyme and IL/water molecules. The RDF profile shows an interesting distribution of the solvent molecules around the active site of the enzyme in different IL/water solvent systems. The Emim cations were found to have g(r) of (3.515, 2.786, 2.649), (2.783, 1.257, 1.669), and (1.072, 0.974, 0.544) at a radial distance of (5.75 Å, 5.75 Å, 5.75 Å), (5.65 Å, 5.55 Å, 5.65 Å), and (5.45 Å, 5.35 Å, 5.35 Å) in 20% EmimAc (S1a, S1b, S1c), 40% EmimAc (S2a, S2b, S2c), and 60% EmimAc (S3a, S3b, S3c), respectively (Fig. 6a and S4, ESI†). It appears that Emim cations exist very close to the active site of the enzyme and the RDF peaks decrease with increasing EmimAc concentration. It is noticeable that with increasing EmimAc concentration, there is a left-shift in the RDF peaks, which signifies that the RmCel12A–Emim interaction increases from 20% EmimAc to 60% EmimAc system. On the contrary, the radial distance of the acetate (Ac) anions were (11.15 Å, 11.35 Å, and 11.35 Å), (11.35 Å, 11.15 Å, and 11.15 Å) and (12.45 Å, 11.45 Å, and 13.35 Å) with very low RDF peak values of (0.610, 0.208, and 0.800), (0.475, 0.211, and 0.407) and (0.174, 0.141, and 0.212) in 20% EmimAc (S1a, S1b, and S1c), 40% EmimAc (S2a, S2b, and S2c), and 60% EmimAc (S3a, S3b, and S3c), respectively (Fig. 6b and S4, ESI†). This signifies that Ac anions were distributed far away (≥11.15 Å) from the active site in comparison to the Emim cations (≤5.75 Å). The RmCel12A–Ac RDF peaks are shifted to the right from 20% EmimAc (11.15 Å) to 60% EmimAc (13.35 Å). This suggests that RmCel12A–Ac interactions are reduced with increasing EmimAc concentration. The RDF peaks of RmCel12A–water first interaction shell were found at about (5.55 Å, 5.65 Å, and 5.55 Å), (5.55 Å, 5.65 Å, and 5.55 Å), and (5.55 Å, 5.55 Å and 5.55 Å) with very low RDF peak values of (0.267, 0.280, and 0.242), (0.337, 0.333, and 0.398), and (0.155, 0.431, and 0.220) in 20% EmimAc (S1a, S1b, and S1c), 40% EmimAc (S2a, S2b, and S2c), and 60% EmimAc (S3a, S3b, and S3c), respectively (Fig. 6c and S4, ESI†). The water molecules also reside close (≤5.65 Å) to the active site of the enzyme; as it has been described previously, these water molecules are required during the hydrolysis of cellulose.71 From the RDF profile, it is clear that the population of the Emim cations is significantly higher than that of water or Ac anions at the active site of RmCel12A. However, it is interesting to observe that the intensity of the Emim peaks was reduced with the increase in EmimAc strength. This phenomenon is due to the micro-architecture of EmimAc and water mixed solvent systems. With the addition of water, the colloquial attraction between Emim and Ac anions is perturbed, as reported earlier.24 In low EmimAc concentration, e.g., 20% EmimAc, acetate ions form small network clusters with the water molecules and subsequently, the Emim cations become comparatively free to interact with the enzyme, leading to higher RDF peaks. However, in high EmimAc concentration, e.g., 40% EmimAc and 60% EmimAc, as the attraction between Emim and Ac anions is retained, the distribution of Emim cations around the COM of RmCel12A is relatively less in these systems than that in the 20% EmimAc system. Due to the same reason, the height of the Emim peaks are slightly higher in the RDF profile in 20% EmimAc and gradually decrease in 40% and 60% EmimAc. In contrast, the Ac anions interact with RmCel12A in a different manner. The RDF for Ac ions starts to rise at a distance of ∼11.15–13.35 Å and reaches a g(r) value of 1 at about ∼25 Å, signifying very low distribution of Ac at the active site of the enzyme. Thus, it is predicted that only Emim cations effectively interact with the active site, whereas the Ac ions do not have specific binding interactions with RmCel12A. However, water molecules are found in a very low concentration at the active site, which is very reasonable as water molecules are crucial for enzymatic catalysis.
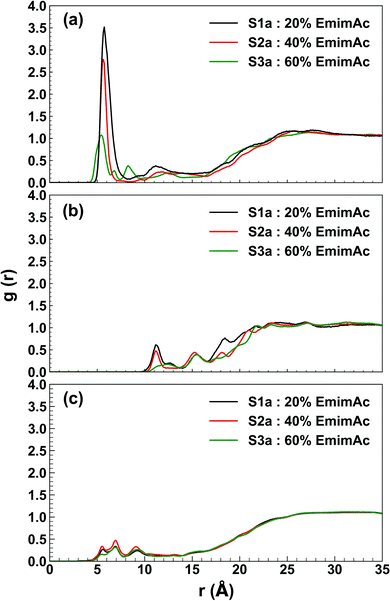 |
| | Fig. 6 Plot of radial distribution function (RDF) of the solvent molecules with respect to the center of mass (COM) of RmCel12A in 20% EmimAc (black line), 40% EmimAc (red line), and 60% EmimAc (green line) for the interactions of (a) RmCel12A–Emim, (b) RmCel12A–Ac, and (c) RmCel12A–water. | |
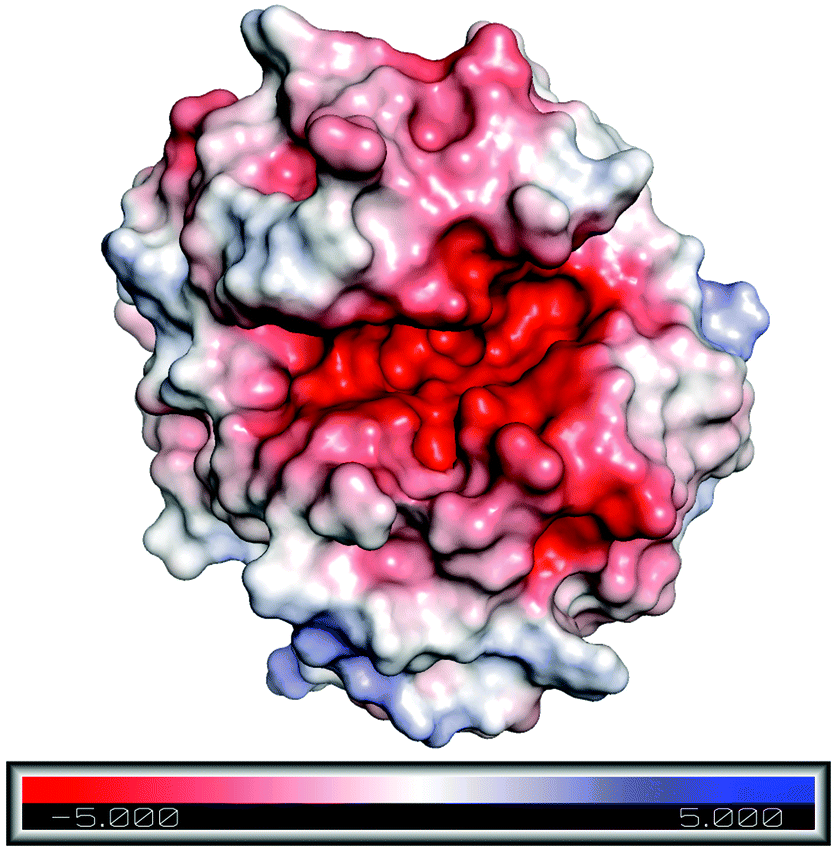
To understand the reason behind the high concentration of Emim at the active site, as observed in the RDF analysis, electrostatic surface charge distribution was calculated on the crystal structure and the end structures of the enzyme from each simulation trajectories by using Adaptive Poisson–Boltzmann Solver (APBS).72 It was noticed that the active site of RmCel12A exhibits a comparatively negative charge density with respect to the rest of the enzyme surface and can be visualized from the charge distribution scale from −5.0 (red) to +5.0 (blue) (Fig. 7 and S5, ESI†). This indicates the fact that the negatively charged active site is the primary reason for the attraction of Emim, resulting in a higher population of Emim cations, as found in the RDF analysis. On the other hand, it also explains that there lies repulsion between the Ac anions and the active site, which resulted in the distribution of Ac at a significant distance. Thus, it is suggested that the interaction between Emim and RmCel12A could affect the enzymatic activity at higher concentration of EmimAc, i.e., 60% EmimAc. However, it is noticeable that the highest peak of RDF corresponds to 20% EmimAc, where RmCel12A is found to be active experimentally. On the contrary, RmCel12A is supposed to have reduced activity in 60% EmimAc, where it shows the lowest RDF peak, meaning comparatively less distribution of Emim cations than that in the 20% EmimAc system. Thus, analysis of the Emim binding pattern and its occupancy at the active site of RmCel12A in both low and high concentration of EmimAc are crucial to identify the probable reasons for the loss of enzyme activity.
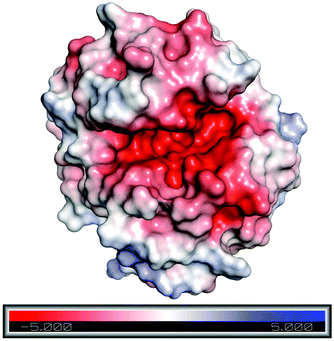 |
| | Fig. 7 Electrostatic surface charge distribution of the RmCel12A crystal structure as obtained from Adaptive Poisson–Boltzmann Solver (APBS) electrostatics calculation. Comparatively negative charge distribution can be observed at the active site cleft with respect to the rest of the enzyme surface. | |
3.5 Binding pattern of Emim at the active site of RmCel12A
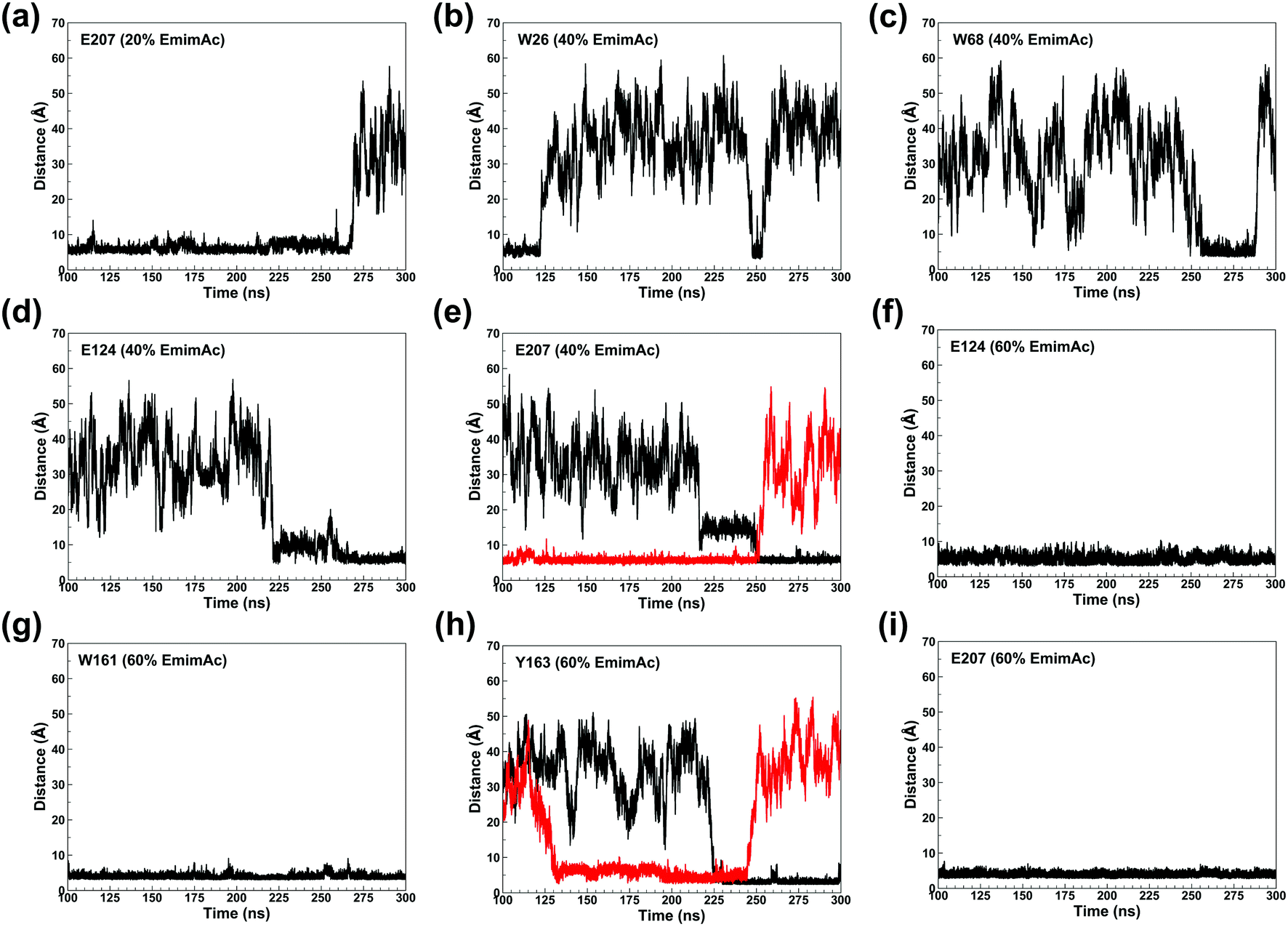
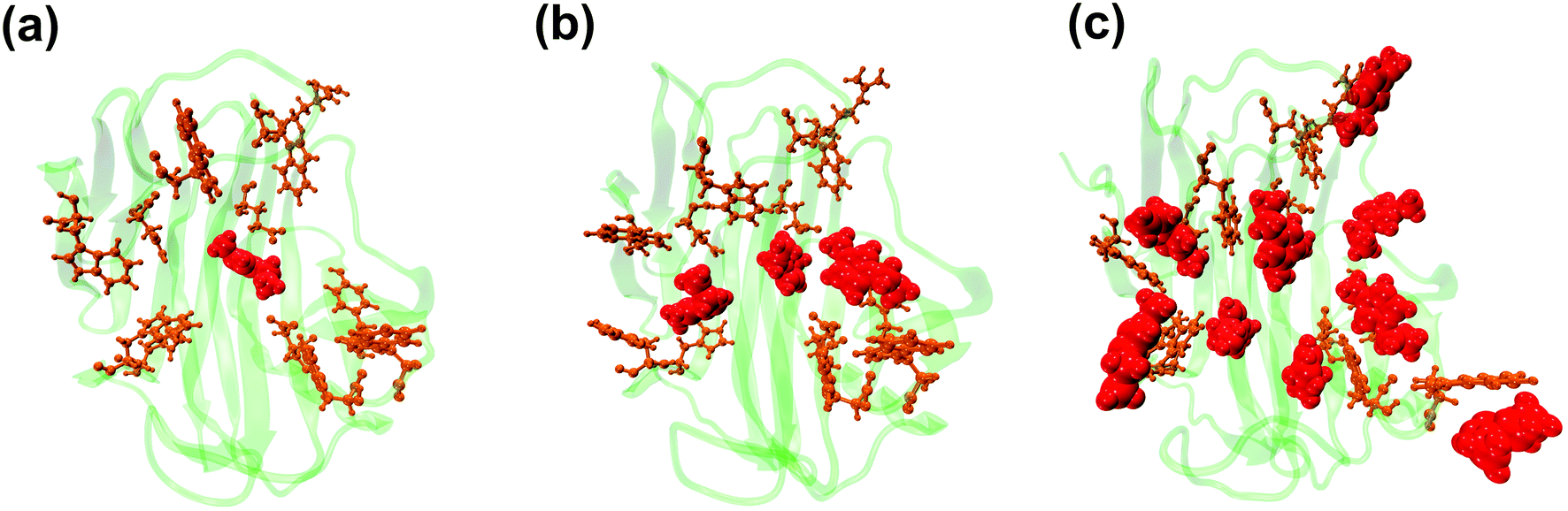
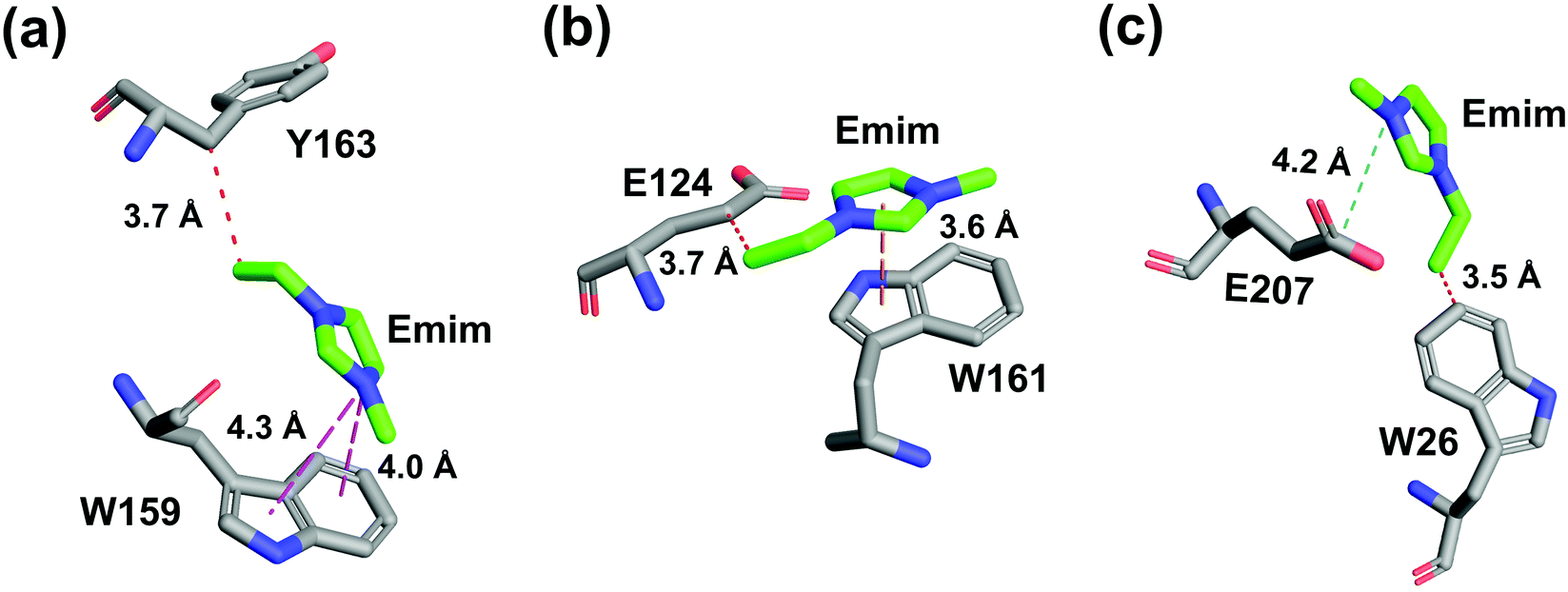
The binding of Emim cations with RmCel12A can explain the mode of Emim interaction in the simulation trajectory. The binding interaction of Emim cations with RmCel12A was calculated by considering a distance cut-off of 4.5 Å. In order to describe the binding statistics, the cumulative binding time (t), for which an Emim cation was in contact with the protein, was calculated and is presented as histogram plot (Fig. 8). Notably, binding time greater than 10%, i.e., 20 ns of the analyzed snapshots (200 ns) were considered as effective binding in every system. Three types of interactions were observed from the binding pattern of Emim cations with RmCel12A, i.e., weakly interacting (t < 50 ns), moderately interacting (50 ns > t < 100 ns), and strongly interacting (t > 100 ns). The weakly and moderately interacting Emim cations were freely diffusing near the protein surface in all the systems. However, the number of strongly bound Emim cations were 2, 3, and 9 in 20% EmimAc, 40% EmimAc, and 60% EmimAc, respectively. This suggests that the Emim–RmCel12A interaction increases with the rising concentration of EmimAc. Moreover, to have residue specific interaction profile with the Emim cations, the cumulative fractional occupancy of Emim binding was analyzed by combining all the snapshots, for which any Emim cation was interacting with a particular amino acid residue, and have been averaged over three replicate simulation trajectories (Fig. 9). It is interesting to observe that the distribution of Emim population with high occupancy of the enzyme was very low in 20% EmimAc. On the other hand, the Emim population with significantly high cumulative fractional occupancy was observed in 60% EmimAc. This revealed that the interaction between RmCel12A and Emim is low in 20% and 40% EmimAc, which does not affect the activity of RmCel12A, as observed in the experiments.37 A total of 12, 25, and 52 residues in the enzyme exhibit high occupancy (>0.1) in 20% EmimAc, 40% EmimAc, and 60% EmimAc, respectively. This suggests that interaction of Emim and RmCel12A increases with increasing IL concentration. Besides, the number of active site catalytic residues with effective Emim binding was found to be 1 (E207), 6 (W9, W26, H67, W68, E124, E207), and 10 (W9, N24, W26, H67, W68, E124, W159, W161, Y163, E207) in 20% EmimAc, 40% EmimAc, and 60% EmimAc, respectively (ESI Table S1†). Notably, in case of 40% EmimAc, most of the active site interacting residues have very low cumulative fractional occupancy (∼0.103–0.162) except that of E207 (0.621). On the other hand, occupancy is much higher for most of the residues in 60% EmimAc system, ranging from 0.179 to 0.630. Moreover, to investigate whether these binding interactions are stable or transient, the distances between the center of mass of the active site residues and particular Emim cations with high occupancy were examined (Fig. 10 and S6, ESI†). Using the contact distance cut-off (≤4.5 Å), the Emim cations' interaction with E207 was present in 3% and 2.5% of the total analyzed snapshots in 20% EmimAc and 40% EmimAc systems, respectively (Fig. 10a and e). However, the Emim cations were within the contact distance cut-off of residues H67, W68, E124, W161, Y163, and E207 for 60.04%, 29.69%, 49.29%, 87.97%, 52.50%, and 88.40% of the total analyzed snapshots, respectively, in 60% EmimAc system (Fig. 10f–i and S6c–g, ESI†). This reveals that the Emim cations interact with the active site residues for longer periods of time in 60% EmimAc than that in 20% or 40% EmimAc system. It is suggested that with increasing EmimAc concentration, Emim cations intrude into the active site tunnel and strongly bind to the catalytic residues to block the enzyme–substrate interaction (Fig. 11). Protein–ligand interaction profiler73 has been used on the representative snapshots obtained from the simulation trajectories in 60% EmimAc system to obtain a more detailed view of the enzyme–Emim interaction. Emim cations form multiple non-bonding interactions including hydrophobic, π-cation, π–π stacking, and salt-bridges with the amino acid residues (Fig. 12). The hydrophobic interactions take place between carbon atoms of the amino acids (e.g., W26, E124, and Y163) and the ethyl (–CH2–CH3) tail of the Emim cations. Besides, the π-cation interactions are found between the pair of aromatic rings of the amino acids (e.g., W159) and a positive charge center in the Emim cation (Fig. 12a). Moreover, the π–π stacking was observed between the aromatic rings of the amino acids (e.g., W161) and the imidazolium ring of Emim (Fig. 12b). Salt-bridge interactions are present between two opposite charge centers from the amino acids (e.g., E207) and the Emim cations, respectively (Fig. 12c). Thus, with increasing EmimAc concentration, the Emim cations strongly bind to more number of catalytic residues as in 60% EmimAc system compared to 40% and 20% EmimAc systems. This is in agreement with the RDF analysis where the left-shift of the peaks with increasing IL concentration suggested an increase in the interaction strength of RmCel12A–Emim. Though there was a higher RDF peak in 20% EmimAc, the cumulative fractional occupancy of Emim at the active site of RmCel12A was very low compared to 60% EmimAc system. In contrast, there were low RDF peaks in the 60% EmimAc system, indicating fewer Emim cations. But the binding became stronger due to higher fractional occupancy of Emim at the active site. Moreover, experimental findings have revealed that these strongly interacting residues in case of 60% EmimAc were also found to be crucial for cellulose binding, catalysis, and product ejection.54,67 Besides, some non-catalytic residues (D13, R18, N55, Y119, and E203) around the active site also exhibited considerable fractional occupancy with the Emim cations. Moreover, the energetics behind the interaction between Emim and the active site catalytic residues were evaluated by calculating the interaction energy (IE = electrostatic energy + van der Waals energy) in different systems. The average IE over three replicate simulations were found to be −105.69 ± 4.72 kcal mol−1, −119.923 ± 1.83 kcal mol−1, and −130.52 ± 13.93 kcal mol−1 in 20% EmimAc, 40% EmimAc, and 60% EmimAc systems, respectively. This shows tighter binding of the Emim cations at the active site of RmCel12A in 60% EmimAc than in 40% and 20% EmimAc. Thus, with rising IL concentration, there exists a strong affinity of the Emim cations towards the cellulose-binding tunnel of RmCel12A. This attraction of the Emim cations can be attributed to the high negative charge distribution at the active site, as elucidated by the APBS electrostatics calculation. Moreover, this strong binding of Emim with the cellulase leads to a competition between Emim and the cello-oligosaccharides to occupy the binding pocket. In case of 20% and 40% EmimAc, the competition is optimal and transient, as revealed by the cumulative fractional occupancy, leading to no considerable loss of enzyme activity, as found in the experiments.37 Moreover, with increasing EmimAc concentration, as in the case of 60% EmimAc, the Emim cations compete and block the cello-oligosaccharide binding and affect the enzymatic hydrolysis. Though no cellulose ligand was considered in the simulation framework, the competitive inhibitory effect of Emim has been well captured, similar to earlier studies.38,39,42 Thus, the cumulative binding statistics of Emim cations indicate probable competitive inhibition of RmCel12A at higher concentration of IL.
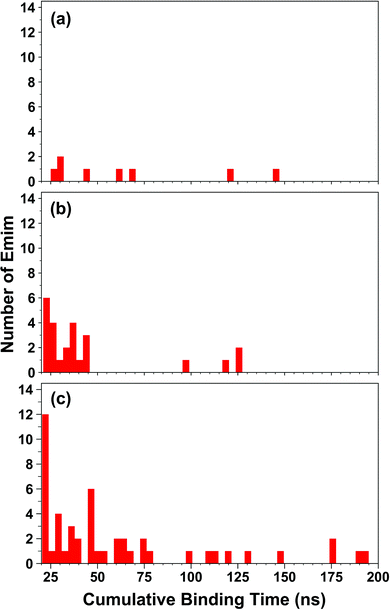 |
| | Fig. 8 Histogram plot of the cumulative binding time of Emim cations interacting with the RmCel12A in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc systems. | |
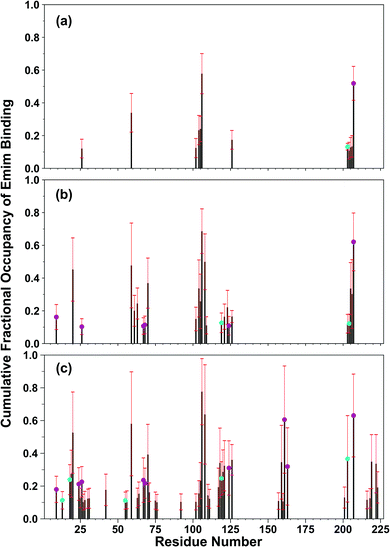 |
| | Fig. 9 Residue-wise cumulative fractional occupancy of Emim cations averaged over the three replicate simulations in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc systems. The black bars represent the average of the fractional occupancy, whereas the red bars show the respective standard deviation. The plot indicates that RmCel12A interacts strongly with increasing concentration of EmimAc. The magenta circles over the bars indicate the catalytic residues. The cyan circles are the predicted non-catalytic residues having high fractional occupancy of Emim at the active site of the enzyme. | |
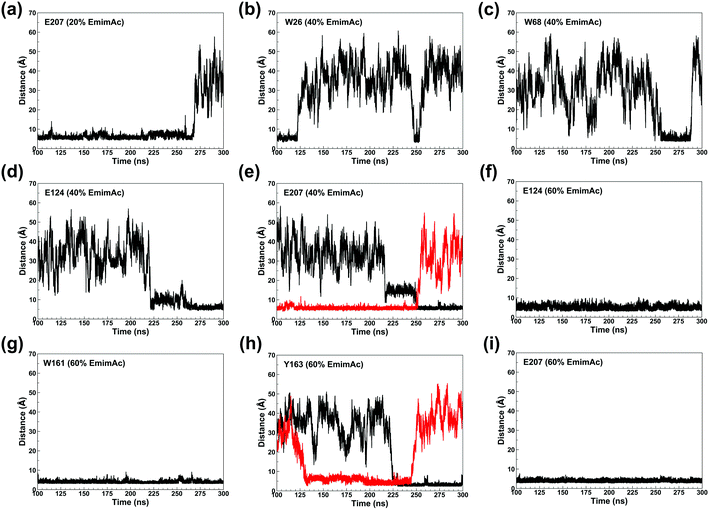 |
| | Fig. 10 Plot of the time dependent contact distances of representative active site residues and the interacting Emim cations in 20% EmimAc: (a) E207; 40% EmimAc: (b) W26, (c) W68, (d) E124, (e) E207; and 60% EmimAc: (f) E124, (g) W161, (h) Y163, (i) E207. The black and the red colors indicate different Emim cations interacting with a particular amino acid residue. | |
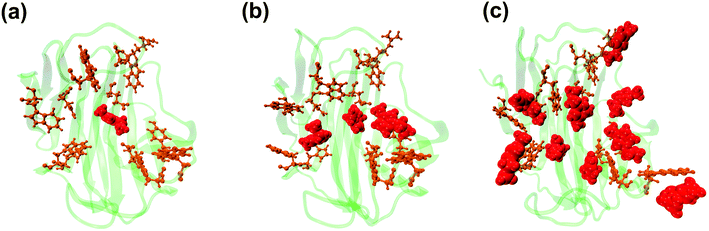 |
| | Fig. 11 Representative snapshots from the simulation trajectory depicting the interaction of Emim cations at the active site of RmCel12A in (a) 20% EmimAc, (b) 40% EmimAc, and (c) 60% EmimAc. The Emim cations and the active site catalytic residues are shown in solid van der Waals' spheres (red) and licorice (orange) representation, respectively. Notably, with increasing concentration of EmimAc, more number of Emim cations with higher fractional occupancy are trapped at the active site of RmCel12A. | |
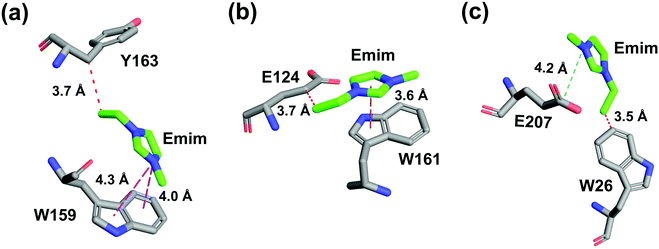 |
| | Fig. 12 Atomic level interactions between the Emim cations and the active site residues are represented from the simulation in 60% EmimAc system, which includes (a) hydrophobic and π-cation interaction, (b) hydrophobic and π–π stacking interaction, and (c) hydrophobic and salt-bridge interaction. Hydrophobic, π-cation, π–π stacking, and salt-bridge interactions are shown by red, magenta, yellow, and cyan dashed lines, respectively. | |
4. Conclusion
Endoglucanase Cel12A from R. marinus is one of the highest IL tolerant (upto 40% (v/v) EmimAc) cellulases reported so far. In order to have theoretical insights into such high IL tolerance of the enzyme, atomistic MD simulations were carried out in 20%, 40%, and 60% EmimAc systems. The RMSD evolution shows the structural stability of RmCel12A in all the IL concentrations at 368 K for all the three replicate simulations during the 300 ns simulation time scale. The principal component analysis revealed that the essential dynamic motions of RmCel12A in 20% and 40% EmimAc systems were severely dampened in 60% EmimAc system. Notably, the opening and closing motion of RmCel12A in the 60% EmimAc was the most affected and was predicted to reduce the enzyme activity. Besides, protein structure network (PSN) was constructed for every snapshot in the simulation trajectory to examine any change in the tertiary conformation of RmCe112A. From the PSN analysis, few dynamically stable native hubs in 20% EmimAc were found to be lost in 40% EmimAc (R20, Y63, and W68) and 60% EmimAc (R20, Y59, W68, W197, E203, and F220), suggesting the loss of network integrity with rising IL concentration. Also, stable dynamic hubs were found to form an aromatic network cluster, which is hypothesized to play a crucial role in hyperthermophilicity in the protein. The RDF analysis suggested that the Emim cations were majorly populated near (average radial distance ∼5.62 Å) the active site of the enzyme, whereas the Ac anions were present at a significant distance (average radial distance ∼11.58 Å). This behavior was due to the negatively charged active site tunnel. The cumulative fractional occupancy of Emim indicated that with increasing IL concentration, the cumulative retention time increased with the active site catalytic residues. The interactions were observed to be mostly transient in both 20% EmimAc and 40% EmimAc. However, the RmCel12A–Emim interactions were stable in 60% EmimAc. The Emim cation forms multiple non-bonded interactions, e.g., hydrophobic, π-cation, π–π stacking, and salt-bridges with the active site residues in 60% EmimAc, leading to competitive inhibition. Some non-catalytic residues, e.g., D13, R18, N55, Y119, and E203 with significant cumulative fractional occupancy with the Emim cations were also identified. These can be probable mutation targets for weakening the binding affinity of Emim at the active site of the enzyme. Furthermore, this study provides an understanding of the high IL tolerant behavior of a hyperthermophilic representative of GH family 12 and paves the way for engineering this enzyme to improve its activity for industrial application.
Conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgements
This work was supported by a research grant from the Science and Engineering Board (SERB), DST, Govt. of India [No. ECR/2016/001096], Scheme for Promotion of Academic and Research Collaboration (SPARC), MHRD, Govt. of India [No. SPARC/2018-2019/P265/SL] and DBT-Ramalingaswami fellowship [No. BT/RLF/Re-entry/06/2013] to A. G. The authors would like to acknowledge Mr Gourab Bhattacharje for proofreading the manuscript.
References
- C. Withagen, Pollution and exhaustibility of fossil fuels, Resour. Energy Econ., 1994, 16, 235–242 CrossRef.
- K. Mukhopadhyay and O. Forssell, An empirical investigation of air pollution from fossil fuel combustion and its impact on health in India during 1973-1974 to 1996-1997, Ecol. Econ., 2005, 55, 235–250 CrossRef.
- I. Dincer and M. A. Rosen, Energy, environment and sustainable development, Appl. Energy, 1999, 64, 427–440 CrossRef CAS.
- S. Octave and D. Thomas, Biochimie, 2009, 91, 659–664 CrossRef CAS PubMed.
- M. FitzPatrick, P. Champagne, M. F. Cunningham and R. A. Whitney, Bioresour. Technol., 2010, 101, 8915–8922 CrossRef CAS PubMed.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- M. Rastogi and S. Shrivastava, Renew. Sustain. Energy Rev., 2017, 80, 330–340 CrossRef.
- A. W. Bhutto, K. Qureshi, K. Harijan, R. Abro, T. Abbas, A. A. Bazmi, S. Karim and G. Yu, Energy, 2017, 122, 724–745 CrossRef CAS.
- H. Chen, J. Liu, X. Chang, D. Chen, Y. Xue, P. Liu, H. Lin and S. Han, Fuel Process. Technol., 2017, 160, 196–206 CrossRef CAS.
- M. Balat, Production of bioethanol from lignocellulosic materials via the biochemical pathway: a review, Energy Convers. Manag., 2011, 52, 858–875 CrossRef CAS.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. R. Seddon, Biocatalysis in ionic liquids, Green Chem., 2002, 4, 147–151 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, Dissolution of cellulose with ionic liquids, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodríguez and R. D. Rogers, Complete dissolution and partial delignification of wood in the ionic liquid 1-ethyl-3-methylimidazolium acetate, Green Chem., 2009, 11, 646 RSC.
- A. P. Dadi, S. Varanasi and C. A. Schall, Enhancement of cellulose saccharification kinetics using an ionic liquid pretreatment step, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS PubMed.
- H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, Regenerating cellulose from ionic liquids for an accelerated enzymatic hydrolysis, J. Biotechnol., 2009, 139, 47–54 CrossRef CAS PubMed.
- A. A. Elgharbawy, M. Z. Alam, M. Moniruzzaman and M. Goto, Biochem. Eng. J., 2016, 109, 252–267 CrossRef CAS.
- H. T. Tan and K. T. Lee, Understanding the impact of ionic liquid pretreatment on biomass and enzymatic hydrolysis, Chem. Eng. J., 2012, 183, 448–458 CrossRef CAS.
- W. Xiao, W. Yin, S. Xia and P. Ma, The study of factors affecting the enzymatic hydrolysis of cellulose after ionic liquid pretreatment, Carbohydr. Polym., 2012, 87, 2019–2023 CrossRef CAS.
- Z. Qiu, G. M. Aita and M. S. Walker, Effect of ionic liquid pretreatment on the chemical composition, structure and enzymatic hydrolysis of energy cane bagasse, Bioresour. Technol., 2012, 117, 251–256 CrossRef CAS PubMed.
- J. Shi, K. Balamurugan, R. Parthasarathi, N. Sathitsuksanoh, S. Zhang, V. Stavila, V. Subramanian, B. A. Simmons and S. Singh, Understanding the role of water during ionic liquid pretreatment of lignocellulose: co-solvent or anti-solvent?, Green Chem., 2014, 16, 3830–3840 RSC.
- B. Manna and A. Ghosh, Dissolution of cellulose in ionic liquid and water mixtures as revealed by molecular dynamics simulations, J. Biomol. Struct. Dyn., 2019, 37, 3987–4005 CrossRef CAS PubMed.
- E. Husson, T. Auxenfans, M. Herbaut, M. Baralle, V. Lambertyn, H. Rakotoarivonina, C. Rémond and C. Sarazin, Sequential and simultaneous strategies for biorefining of wheat straw using room temperature ionic liquids, xylanases and cellulases, Bioresour. Technol., 2018, 251, 280–287 CrossRef CAS PubMed.
- T. Auxenfans, E. Husson and C. Sarazin, Simultaneous pretreatment and enzymatic saccharification of (ligno) celluloses in aqueous-ionic liquid media: a compromise, Biochem. Eng. J., 2017, 117, 77–86 CrossRef CAS.
- J. Sun, N. V. S. N. M. Konda, R. Parthasarathi, T. Dutta, M. Valiev, F. Xu, B. A. Simmons and S. Singh, One-pot integrated biofuel production using low-cost biocompatible protic ionic liquids, Green Chem., 2017, 19, 3152–3163 RSC.
- J. Grewal, R. Ahmad and S. K. Khare, Development of cellulase-nanoconjugates with enhanced ionic liquid and thermal stability for in situ lignocellulose saccharification, Bioresour. Technol., 2017, 242, 236–243 CrossRef CAS PubMed.
- J. M. Gladden, M. Allgaier, C. S. Miller, T. C. Hazen, J. S. VanderGheynst, P. Hugenholtz, B. A. Simmons and S. W. Singer, Glycoside hydrolase activities of thermophilic bacterial consortia adapted to switchgrass, Appl. Environ. Microbiol., 2011, 77, 5804–5812 CrossRef CAS PubMed.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Ionic liquid salt-induced inactivation and unfolding of cellulase from Trichoderma reesei, Green Chem., 2003, 5, 443–447 RSC.
- S. Datta, B. Holmes, J. I. Park, Z. Chen, D. C. Dibble, M. Hadi, H. W. Blanch, B. A. Simmons and R. Sapra, Ionic liquid tolerant hyperthermophilic cellulases for biomass pretreatment and hydrolysis, Green Chem., 2010, 12, 338–345 RSC.
- Y. Wang, M. Radosevich, D. Hayes and N. Labbé, Compatible Ionic liquid-cellulases system for hydrolysis of lignocellulosic biomass, Biotechnol. Bioeng., 2011, 108, 1042–1048 CrossRef CAS PubMed.
- T. Zhang, S. Datta, J. Eichler, N. Ivanova, S. D. Axen, C. A. Kerfeld, F. Chen, N. Kyrpides, P. Hugenholtz, J. F. Cheng, K. L. Sale, B. Simmons and E. Rubin, Identification of a haloalkaliphilic and thermostable cellulase with improved ionic liquid tolerance, Green Chem., 2011, 13, 2083–2090 RSC.
- S. W. Singer, A. P. Reddy, J. M. Gladden, H. Guo, T. C. Hazen, B. A. Simmons and J. S. Vandergheynst, Enrichment, isolation and characterization of fungi tolerant to 1-ethyl-3-methylimidazolium acetate, J. Appl. Microbiol., 2011, 110, 1023–1031 CrossRef CAS PubMed.
- J. I. Park, E. J. Steen, H. Burd, S. S. Evans, A. M. Redding-Johnson, T. Batth, P. I. Benke, P. D’haeseleer, N. Sun, K. L. Sale, J. D. Keasling, T. S. Lee, C. J. Petzold, A. Mukhopadhyay, S. W. Singer, B. A. Simmons and J. M. Gladden, A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels, PLoS One, 2012, 7(5), e37010 CrossRef CAS PubMed.
- J. Xu, B. He, B. Wu, B. Wang, C. Wang and L. Hu, An ionic liquid tolerant cellulase derived from chemically polluted microhabitats and its application in in situ saccharification of rice straw, Bioresour. Technol., 2014, 157, 166–173 CrossRef CAS PubMed.
- J. M. Gladden, J. I. Park, J. Bergmann, V. Reyes-Ortiz, P. D’Haeseleer, B. F. Quirino, K. L. Sale, B. A. Simmons and S. W. Singer, Discovery and characterization of ionic liquid-tolerant thermophilic cellulases from a switchgrass-adapted microbial community, Biotechnol. Biofuels, 2014, 7(1), 15 CrossRef PubMed.
- W. Li, L. Wang, R. Zhou and Y. Mu, Ionic liquid induced inactivation of cellobiohydrolase i from Trichoderma reesei, Green Chem., 2015, 17, 1618–1625 RSC.
- S. R. Summers, K. G. Sprenger, J. Pfaendtner, J. Marchant, M. F. Summers and J. L. Kaar, Mechanism of Competitive Inhibition and Destabilization of Acidothermus cellulolyticus Endoglucanase 1 by Ionic Liquids, J. Phys. Chem. B, 2017, 121, 10793–10803 CrossRef CAS PubMed.
- V. Jaeger, P. Burney and J. Pfaendtner, Comparison of three ionic liquid-tolerant cellulases by molecular dynamics, Biophys. J., 2015, 108, 880–892 CrossRef CAS PubMed.
- L. B. Johnson and C. D. Snow, Molecular dynamics simulations of cellulase homologs in aqueous 1-ethyl-3-methylimidazolium chloride, J. Biomol. Struct. Dyn., 2017, 35, 1990–2002 CrossRef CAS PubMed.
- V. W. Jaeger and J. Pfaendtner, Structure, dynamics, and activity of xylanase solvated in binary mixtures of ionic liquid and water, ACS Chem. Biol., 2013, 8, 1179–1186 CrossRef CAS PubMed.
- K. G. Sprenger, A. Choudhury, J. L. Kaar and J. Pfaendtner, Lytic Polysaccharide Monooxygenases ScLPMO10B and ScLPMO10C Are Stable in Ionic Liquids As Determined by Molecular Simulations, J. Phys. Chem. B, 2016, 120, 3863–3872 CrossRef CAS PubMed.
- P. R. Burney, E. M. Nordwald, K. Hickman, J. L. Kaar and J. Pfaendtner, Molecular dynamics investigation of the ionic liquid/enzyme interface: application to engineering enzyme surface charge, Proteins: Struct., Funct., Bioinf., 2015, 83, 670–680 CrossRef CAS PubMed.
- M. A. M. Latif, B. A. Tejo, R. Abedikargiban, M. B. Abdul Rahman and N. M. Micaêlo, Modeling stability and flexibility of Chymotrypsin in room temperature ionic liquids, J. Biomol. Struct. Dyn., 2014, 32, 1263–1273 CrossRef CAS PubMed.
- S. Ghosh, S. Parui, B. Jana and K. Bhattacharyya, Ionic liquid induced dehydration and domain closure in lysozyme: FCS and MD simulation, J. Chem. Phys., 2015, 143(12), 125103 CrossRef PubMed.
- N. M. Micaêlo and C. M. Soares, Protein structure and dynamics in ionic liquids. Insights from molecular dynamics simulation studies, J. Phys. Chem. B, 2008, 112, 2566–2572 CrossRef PubMed.
- D. Lousa, A. M. Baptista and C. M. Soares, Phys. Chem. Chem. Phys., 2013, 15, 13723–13736 RSC.
- S. Wang, X. Meng, H. Zhou, Y. Liu, F. Secundo and Y. Liu, Enzyme stability and activity in non-aqueous reaction systems: a mini review, Catalysts, 2016, 6, 32 CrossRef.
- S. Badieyan, D. R. Bevan and C. Zhang, Study and design of stability in GH5 cellulases, Biotechnol. Bioeng., 2012, 109, 31–44 CrossRef CAS PubMed.
- A. S. Borisova, E. V. Eneyskaya, S. Jana, S. F. Badino, J. Kari, A. Amore, M. Karlsson, H. Hansson, M. Sandgren, M. E. Himmel, P. Westh, C. M. Payne, A. A. Kulminskaya and J. Ståhlberg, Correlation of structure, function and protein dynamics in GH7 cellobiohydrolases from Trichoderma atroviride, T. reesei and T. harzianum, Biotechnol. Biofuels, 2018, 11(1) CAS.
- G. A. Alfredsson, J. K. Kristjansson, S. Hjorleifsdottir and K. O. Stetter, Rhodothermus marinus,
gen. nov., sp. nov., a Thermophilic, Halophilic Bacterium from Submarine Hot Springs in Iceland, Microbiology, 1988, 134, 299–306 CAS.
- D. A. Case, S. R. Brozell, D. S. Cerutti, T. E. I. Cheatham, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Lu, D. M. York and P. A. Kollman, Amber 18, Univ. California, San Fr., p. 2734527 Search PubMed.
- S. J. Crennell, D. Cook, A. Minns, D. Svergun, R. L. Andersen and E. Nordberg Karlsson, Dimerisation and an increase in active site aromatic groups as adaptations to high temperatures: X-ray solution scattering and substrate-bound crystal structures of Rhodothermus marinus endoglucanase Cel12A, J. Mol. Biol., 2006, 356, 57–71 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, L. Perera, L. Li and P. Lee, New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations, Structure, 1999, 7(3), R55 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. R. Roe and T. E. Cheatham, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Bhattacharyya, A. Ghosh, P. Hansia and S. Vishveshwara, Allostery and conformational free energy changes in human tryptophanyl-tRNA synthetase from essential dynamics and structure networks, Proteins: Struct., Funct., Bioinf., 2010, 78, 506–517 CrossRef CAS PubMed.
- A. Ghosh, K. V Brinda and S. Vishveshwara, Dynamics of Lysozyme Structure Network: Probing the Process of Unfolding, Biophys. J., 2007, 92, 2523–2535 CrossRef CAS PubMed.
- K. A. Henzler-Wildman, V. Thai, M. Lei, M. Ott, M. Wolf-Watz, T. Fenn, E. Pozharski, M. A. Wilson, G. A. Petsko, M. Karplus, C. G. Hübner and D. Kern, Intrinsic motions along an enzymatic reaction trajectory, Nature, 2007, 450, 838–844 CrossRef CAS PubMed.
- L. Casalino, G. Palermo, A. Spinello, U. Rothlisberger and A. Magistrato, All-atom simulations disentangle the functional dynamics underlying gene maturation in the intron lariat spliceosome, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6584–6589 CrossRef CAS PubMed.
- S. Bhakat, A. J. M. Martin and M. E. S. Soliman, An integrated molecular dynamics, principal component analysis and residue interaction network approach reveals the impact of M184V mutation on HIV reverse transcriptase resistance to lamivudine, Mol. Biosyst., 2014, 10, 2215–2228 RSC.
- I. Aier, P. K. Varadwaj and U. Raj, Structural insights into conformational stability of both wild-type and mutant EZH2 receptor, Sci. Rep., 2016, 6(1), 1–10 CrossRef PubMed.
- S. J. Crennell, G. O. Hreggvidsson and E. Nordberg Karlsson, The structure of Rhodothermus marinus Cel12A, a highly thermostable family 12 endoglucanase, at 1.8 Å resolution, J. Mol. Biol., 2002, 320, 883–897 CrossRef CAS PubMed.
- A. Ghosh and S. Vishveshwara, A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis, Proc. Natl. Acad. Sci., 2007, 104, 15711–15716 CrossRef CAS PubMed.
- S. Vishveshwara, A. Ghosh and P. Hansia, Intra and Inter-Molecular Communications Through Protein Structure Network, Curr. Protein Pept. Sci., 2009, 10, 146–160 CrossRef CAS PubMed.
- N. Kannan and S. Vishveshwara, Aromatic clusters: a determinant of thermal stability of thermophilic proteins, Protein Eng. Des. Sel., 2002, 13, 753–761 CrossRef PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Electrostatics of nanosystems: application to microtubules and the ribosome, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, PLIP: fully automated protein-ligand interaction profiler, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09612d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab