
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic structure of iron dinitrogen complex [(TPB)FeN2]2−/1−/0: correlation to Mössbauer parameters†
Nidhi Vyas *a,
Aditya Kumarb,
Animesh K. Ojhab and
Abhinav Grover*a
*a,
Aditya Kumarb,
Animesh K. Ojhab and
Abhinav Grover*a
aSchool of Biotechnology, Jawaharlal Nehru University, New Delhi-110067, India. E-mail: nidhivyas12@gmail.com; abhinavgr@gmail.com
bDepartment of Physics, Motilal Nehru National Institute of Technology, Allahabad-211004, India
First published on 25th February 2020
Abstract
Low-valent species of iron are key intermediates in many important biological processes such as the nitrogenase enzymatic catalytic reaction. These species play a major role in activating highly stable N2 molecules. Thus, there is a clear need to establish the factors which are responsible for the reactivity of the metal–dinitrogen moiety. In this regard, we have investigated the electronic structure of low-valent iron (2−/1−/0) in a [(TPB)FeN2]2−/1−/0 complex using density functional theory (DFT). The variation in the oxidation states of iron in the nitrogenase enzyme cycle is associated with the flexibility of Fe→B bonding. Therefore, the flexibility of Fe→B bonding acts as an electron source that sustains the formation of various oxidation states, which is necessary for the key species in dinitrogen activation. AIM calculations are also performed to understand the strength of Fe→B and Fe–N2 bonds. A detailed interpretation of the contributions to the isomer shift (IS) and quadrupole splitting (ΔEQ) are discussed. The major contribution to IS comes mainly from the 3s-contribution, which differs depending on the d orbital population due to different shielding. The valence shell contribution also comes from the 4s-orbital. The Fe–N2 bond distance has a great influence on the Mössbauer parameters, which are associated with the radial distribution, i.e. the shape of the 4s-orbital and the charge density at the nucleus. A linear relationship between IS with Fe–N2 and ΔEQ with Fe–N2 is observed.
Introduction
The study of nitrogen fixation has long been a challenge, as the generation of NH3 from N2 requires high pressure and temperature to stimulate this highly stable molecule.1,2 For the industrial production of ammonia, the Haber–Bosch process also needs a very reactive Fe-based catalyst.3 In contrast, nature facilitates this transformation at ambient temperature and pressure using the nitrogenase enzyme.4–6 Decades of intensive research have been devoted to understanding the exact active site as well as details of the mechanism of the nitrogenase enzyme in the N2 reduction pathway.7–9 A range of complexes have been synthesized based on Mo–N2 and Fe–N2 in previous years to replicate and examine the roles of Fe and Mo in the FeMo cofactor of the nitrogenase enzyme as the N2-activation site.5,9,10 Thus understanding the electronic structure and reactivity of terminal Fe–N2 species is highly relevant to understanding the complex mechanism of nitrogenase. Of particular interest, considerable efforts have been made by Peters and co-workers in the generation of low-valent iron Fe–N2 complexes; they mainly elaborated on studies of iron coordinated tris(phosphine)borate (TPB) ligands.11–14 This specific choice of ligand exhibits the unique characteristic for binding of both terminal N2 as a π-acidic ligand and terminal imido as a π-basic ligand due to the flaccidity of the Fe–B bond.15–18 In one of their studies, the iron nitride [(PB3)Fe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)]+ was synthesized and reported as one of the intermediates generated from [(PB3)Fe–N2)]2−, where the iron centre can have six formal oxidation states, from d10 Fe(−2) to d4 Fe(+4). This [(PB3)Fe–N2)]2− species was generated in situ and used instantly. It was well characterized by Mössbauer and extended X-ray absorption fine structure (EXAFS) spectroscopy.14 In the area of low-valent Fe–N2 species, considerable efforts have been made by Frank Neese and his coworkers.19–21 In other reports, low-spin Fe–N2 complexes with three formal oxidation states +II, +I and 0 were studied with the anionic SiPiPr3 (SiPiPr3 = [Si(o-C6H4PiPr2)3]−) tetradendate tris(phosphino)silyl ligand.19 The group elaborated the studies on iron chemistry with tripodal ligands and thoroughly characterized several species using X-ray, Mössbauer (MB) and EPR spectroscopy.19 The electronic structures of low-valent [Fe(N2)(SiPiPr3)]+/0/− iron–dinitrogen complexes were analyzed using Mössbauer (MB) spectroscopy. They concluded that the metal–ligand bond length has a stronger effect on the Mössbauer (IS) isomer shift relative to the iron physical oxidation state.19 This can be associated with the compression of the Fe–N2 bond distance having fixedly magnified π-backdonation in the consecutive reduction. A large body of literature over the last 15 years has reported DFT calculations to measure the 57Fe Mössbauer parameters in various bioinorganic and inorganic systems.20–27 In order to obtain precious knowledge of the local electronic structure, symmetry and oxidation states, MB spectroscopy has become one of the most important analytic tools.28 The interpretation of MB spectra is not clear, and the support of computation is necessary to obtain all the relevant chemical/physical knowledge from the experimentally measured data.29,30 The two primary parameters that can be obtained from a MB spectrum of a given iron center are the isomer shift (IS) and the quadrupole splitting. These parameters are nicely related to the electron density distribution at the iron nucleus. Computational studies are extensively used in this area to gain insight into the structure, bonding and reactivity of these low-valent Fe–N2 species,31–34 though there are numerous reports available in the literature on the electronic structure, as well as on the mechanistic exploration of mononuclear low-valent iron–dinitrogen species.35–39 In this direction, Phukan et al. examined the mechanistic details (symmetric, asymmetric and hybrid) for ammonia production using the tripodal iron complexes [(SiPMe3)FeI] ([FeSi]) and [(BPMe3)Fe0] ([FeB]).35 The role of [FeB] and [FeSi] was demonstrated and the energetics for the crucial steps of N2 reduction were calculated. De Visser and his co-workers also explored DFT studies of the [(TPB)FeN2]− complex and its conversion to ammonia.37 They demonstrated feasible mechanistic pathways with the addition of protons and electrons. Recently, our group has studied the [(TPB)FeN2] complex with both symmetric and asymmetric pathways and explored its reactivity using molecular orbitals.40 Besides the mechanisms, a comparison of the iron–dinitrogen complexes Fe–N2 across three different formal oxidation states, 2−, 1− and 0, using TPB (tris(phosphine)borane ligand, has not been explored so far. Thus, there is a clear need to establish the factors which control the reactivity of the metal–dinitrogen moiety. In this regard, the model complex [(TPB)FeN2]2−/1−/0 is considered as it possesses various oxidation states which are believed to have varied reactivity patterns. In this work our motive is to perform density functional theory calculations on [(TPB)FeN2] with varying oxidation states, 2−, 1− and 0, and explore its reactivity. With this study, we aim to answer the following captivating questions: (1) What is the nature of the specific bonding (Fe→B and Fe–N2) in the [(TPB)FeN2]2−/1−/0 species which is responsible for this reaction and how does it differ? (2) What is the role of the low-valent oxidation state of iron that is crucial for dinitrogen activation? (3) How do the Mössbauer parameters relate the low-valent oxidation states of these species, and how do these parameters contribute to the various orbitals associated with these complexes? A combination of the Mössbauer parameters with electronic structure studies provides deep insight into the overall catalytic system.
N)]+ was synthesized and reported as one of the intermediates generated from [(PB3)Fe–N2)]2−, where the iron centre can have six formal oxidation states, from d10 Fe(−2) to d4 Fe(+4). This [(PB3)Fe–N2)]2− species was generated in situ and used instantly. It was well characterized by Mössbauer and extended X-ray absorption fine structure (EXAFS) spectroscopy.14 In the area of low-valent Fe–N2 species, considerable efforts have been made by Frank Neese and his coworkers.19–21 In other reports, low-spin Fe–N2 complexes with three formal oxidation states +II, +I and 0 were studied with the anionic SiPiPr3 (SiPiPr3 = [Si(o-C6H4PiPr2)3]−) tetradendate tris(phosphino)silyl ligand.19 The group elaborated the studies on iron chemistry with tripodal ligands and thoroughly characterized several species using X-ray, Mössbauer (MB) and EPR spectroscopy.19 The electronic structures of low-valent [Fe(N2)(SiPiPr3)]+/0/− iron–dinitrogen complexes were analyzed using Mössbauer (MB) spectroscopy. They concluded that the metal–ligand bond length has a stronger effect on the Mössbauer (IS) isomer shift relative to the iron physical oxidation state.19 This can be associated with the compression of the Fe–N2 bond distance having fixedly magnified π-backdonation in the consecutive reduction. A large body of literature over the last 15 years has reported DFT calculations to measure the 57Fe Mössbauer parameters in various bioinorganic and inorganic systems.20–27 In order to obtain precious knowledge of the local electronic structure, symmetry and oxidation states, MB spectroscopy has become one of the most important analytic tools.28 The interpretation of MB spectra is not clear, and the support of computation is necessary to obtain all the relevant chemical/physical knowledge from the experimentally measured data.29,30 The two primary parameters that can be obtained from a MB spectrum of a given iron center are the isomer shift (IS) and the quadrupole splitting. These parameters are nicely related to the electron density distribution at the iron nucleus. Computational studies are extensively used in this area to gain insight into the structure, bonding and reactivity of these low-valent Fe–N2 species,31–34 though there are numerous reports available in the literature on the electronic structure, as well as on the mechanistic exploration of mononuclear low-valent iron–dinitrogen species.35–39 In this direction, Phukan et al. examined the mechanistic details (symmetric, asymmetric and hybrid) for ammonia production using the tripodal iron complexes [(SiPMe3)FeI] ([FeSi]) and [(BPMe3)Fe0] ([FeB]).35 The role of [FeB] and [FeSi] was demonstrated and the energetics for the crucial steps of N2 reduction were calculated. De Visser and his co-workers also explored DFT studies of the [(TPB)FeN2]− complex and its conversion to ammonia.37 They demonstrated feasible mechanistic pathways with the addition of protons and electrons. Recently, our group has studied the [(TPB)FeN2] complex with both symmetric and asymmetric pathways and explored its reactivity using molecular orbitals.40 Besides the mechanisms, a comparison of the iron–dinitrogen complexes Fe–N2 across three different formal oxidation states, 2−, 1− and 0, using TPB (tris(phosphine)borane ligand, has not been explored so far. Thus, there is a clear need to establish the factors which control the reactivity of the metal–dinitrogen moiety. In this regard, the model complex [(TPB)FeN2]2−/1−/0 is considered as it possesses various oxidation states which are believed to have varied reactivity patterns. In this work our motive is to perform density functional theory calculations on [(TPB)FeN2] with varying oxidation states, 2−, 1− and 0, and explore its reactivity. With this study, we aim to answer the following captivating questions: (1) What is the nature of the specific bonding (Fe→B and Fe–N2) in the [(TPB)FeN2]2−/1−/0 species which is responsible for this reaction and how does it differ? (2) What is the role of the low-valent oxidation state of iron that is crucial for dinitrogen activation? (3) How do the Mössbauer parameters relate the low-valent oxidation states of these species, and how do these parameters contribute to the various orbitals associated with these complexes? A combination of the Mössbauer parameters with electronic structure studies provides deep insight into the overall catalytic system.
Computational method
In the current article the Gaussian 16 suite of programs has been used for all calculations.41 The B3LYP density functional was employed for geometry optimization of all the complexes.42–44 For Fe, LanL2DZ was used, which has a double-quality basis set with the Los Alamos effective core potential; for the other atoms (C, H, N, O, P) a 6–31G* basis set was used.45–47 Single point energy calculations were computed using TZVP basis set for all atoms.47 The minimum energy structure was taken and verified by frequency calculations on the optimized structures. The quoted DFT energies are free-energy corrections and zero-point from the frequency computation at a temperature of 298.15 K, unless otherwise mentioned. The polarizable continuum model was used to account for solvent effects.48 THF (tetrahydrofuran) was used as a solvent and visualization was done using Chemcraft software.49 ORCA software was utilized for spectroscopic properties, while the solvation effects were measured using COSMO methodology.50 All spectroscopic parameter calculations incorporated relativistic effects via ZORA (zeroth-order regular approximation) method, as invoked in ORCA software.50 The isomer shifts (IS) were calculated using ρ0, the electron densities at the Fe centre by employing the following linear regression:| δ = α(ρ0 − C) + β |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 au−3. Quadrupole splitting ΔEQ was computed from the electric field gradient Vij (i = x, y, z; Vij are the eigenvalues of the electric field gradient tensor) by using the nuclear quadruple moment Q(57Fe) = 0.16, based on the calibration constants reported by Römelt et al.51,52 Natural bond orbital (NBO) and molecular orbital (MO) studies were executed by G0953 software and Chemcraft software was used for visualization.49,53 Mulliken population analysis was used to determine atomic populations from the single-point energy calculations. We have only focused on the main components of a given molecular orbital because most of the orbitals involved in forming these complexes contain significant contributions from several atomic orbitals. The bond strength was computed using AIM analysis enacted by Bader et al. known as Quantum Theory of Atoms in Molecules (QTAIM), which provides information about electron density at the bond critical point (BCP) (3, −1) in all the species.54,55 We shorten the experimentally reported [(TPB)Fe] complex by placing methyl groups (Me) instead of isopropyl (iPr) groups on the phosphorus end; this reduces the computational cost.
770 au−3. Quadrupole splitting ΔEQ was computed from the electric field gradient Vij (i = x, y, z; Vij are the eigenvalues of the electric field gradient tensor) by using the nuclear quadruple moment Q(57Fe) = 0.16, based on the calibration constants reported by Römelt et al.51,52 Natural bond orbital (NBO) and molecular orbital (MO) studies were executed by G0953 software and Chemcraft software was used for visualization.49,53 Mulliken population analysis was used to determine atomic populations from the single-point energy calculations. We have only focused on the main components of a given molecular orbital because most of the orbitals involved in forming these complexes contain significant contributions from several atomic orbitals. The bond strength was computed using AIM analysis enacted by Bader et al. known as Quantum Theory of Atoms in Molecules (QTAIM), which provides information about electron density at the bond critical point (BCP) (3, −1) in all the species.54,55 We shorten the experimentally reported [(TPB)Fe] complex by placing methyl groups (Me) instead of isopropyl (iPr) groups on the phosphorus end; this reduces the computational cost.
Results and discussions
The electronic structures and reactivity were broadly explored at the B3LYP level of theory. Our modeled structure for study is shown in Fig. 1. This biomimetic species provides a basis to examine the complex process of nitrogen fixation at a molecular level. | ||
| Fig. 1 DFT model structure of iron dinitrogen complexes. | ||
Electronic structure of iron dinitrogen complexes [(TPB)FeN2]2−/1−/0
To better understand the reactivity between different oxidation states of the iron [(TPB)FeN2]2−/1−/0 complexes, we first compared their distinct bonding situations. In the nitrogen fixation mechanism, the N2 molecule is first activated at the iron centre by giving electrons from its π-bonding and σ-orbitals to iron, while simultaneously gaining back-donating electrons from iron with its antibonding π-orbitals (π*). The beauty of the TPB iron-based complex is that the sp2 hybridized boron atom behaves as a Lewis acid, providing an unoccupied pz orbital to create a reverse-dative bond. The optimized parameters and energies of the [(TPB)FeN2]2−/1−/0 complexes are tabulated in Table S1.†The Fe→B bond has a redox-flexible environment. Flexibility in the ligand environment apparently plays a key role in allowing a single iron site to activate N2 reduction for this reaction to occur. This reverse dative bond acts as an electron source that supplies donating bonding electrons from iron to enlarge the bond length the schematic process is shown in Fig. 2. Here the specific nature of this bulky [P]3 ligand, which is electron-rich, thrusts the iron (Fe) 3d energy level to a higher one through orbital interaction; this in turn enhances the probability of forming better backbonding with the π-antibonding orbitals of N2. The previously reported value for the Fe→B bond length of [(TPB)-FeCl] is 2.531 Å, which is greater than the sum of Pyykkö’s single-bond radius of 2.010 Å for Fe–B, illustrating Fe→B dative bonding.36,56,57 Further, according to the mechanism, the complex undergoes reduction to an unusual Fe(1−) and Fe(2−) oxidation state in [(TPB)-FeN2]− and [(TPB)-FeN2]2−, respectively. These species play a major role in successive N2 activation. In [(TPB)FeN2]2−/1−/0, the Fe→B and Fe–N bond distances are generally shorter than in the [(TPB)-FeCl] complex. The Fe→B bond lengths are observed to be 2.369 Å, 2.345 Å and 2.280 Å for the [(TPB)FeN2]2−/1−/0 complexes, respectively (Table S1†). It can be seen that as reduction at the iron centre takes place, the bond length increases (Fig. 3). Also, charge transfer from iron to boron takes place as we go from the Fe2− to the Fe0 oxidation state (see Table S2†). The iron dinitrogen bond distance is shortened in the reduced state. The N2 complexes are always linear end-on at the proximal nitrogen atom, allowing π-back bonding interactions from the metal d orbitals to the π* orbitals of N2. In Fe–N2 bonding the three orbitals are involved. The iron (Fe) dxz and (Fe) dyz orbitals participate in the bonding of N2 via the πy* and πz* orbitals (Fig. S2†). Most of this charge transfer takes place through π-backdonation from the Fe dxz and dyz orbitals to the N2 πy* and πz* orbitals, respectively. Also, negative charge is transferred to the terminal nitrogen atom, N, indicating that Nβ is more receptive to protonation than Nα. Iron complexes featuring strongly activated N2 ligands can exhibit N–N bond elongation (Fig. 3, Table S1†) from Fe0 to Fe2−. The computed values of the NN stretching frequencies are 2246 cm−1, 2142 cm−1 and 2079 cm−1 for the Fe0, Fe1− and Fe2− complexes, which is in agreement with experimental observations.14,15 This shows that there is a considerable red shift in the NN vibration frequencies and the N–N bond distance is eventually lengthened as reduction takes place. As far as the gap between the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) is concerned, its value is 4.43 eV, 3.59 eV and 2.76 eV for Fe0, Fe1− and Fe2− species, respectively (Table S2† and Fig S3†). It decreases as the oxidation state is decreased, which means that iron will be more reactive in the Fe2− oxidation state. It is notable that the HOMO is basically based on the metal d orbital and the LUMO is related to the antibonding orbitals of the ligand. The Fe→B and Fe–N bonds in [(TPB)FeN2]2−/1−/0 complexes were also analyzed through natural bond orbitals (NBO) and the details are given in Table S2.† The Fe→B σ bond was formed with 53.79% (Fe), 46.21% (B), and has a larger contribution from boron than the other Fe1− and Fe0 complexes. This suggests that the boron atom will have the largest capacity to act as an electron reservoir, which enhances the reactivity in the reduced state. From the QTAIM analysis, the BCPs provide useful knowledge about the nature of different chemical interactions or chemical bonds, for example covalent bonds, metal–ligand interactions, hydrogen bonds and weak orbital interactions. The BCP is a saddle point on the electron density curvature, having the smallest value in the path of atomic interaction direction and the largest value in the perpendicular direction to this interaction. All the topological (interaction) parameters such as electron density ρ(rc), Laplacian of the charge density (∇2ρ), kinetic energy density G(rc), potential energy density V(rc), and total energy density E(rc) corresponding to the Fe→B and Fe–N bonds are discussed here (see Table S3†). The electron density ρ at the BCP corresponds to the Fe→B bond ranges from 0.063 e Å−3 to 0.061 e Å−3 and 0.059 e Å−3 with the characteristics of “closed-shell” interactions; also for this ∇2ρ > 0 and the kinetic and potential energies are nearly equal in magnitude,58 which represents the dative bond between Fe and B. As the reduction of Fe takes place from Fe0 to Fe2−, the charge density distribution between Fe and B is decreased, which implies that the strength of bonding is reduced with increasing reduction. As far as the Fe–N bond length is concerned, it decreases as the iron goes from oxidation state 0 to 2− (Fig. 1). For this bond, the electron density ρ values are 0.081(0) e Å−3, 0.124(1−) e Å−3 and 0.133(2−) e Å−3; also for this ∇2ρ > 0, which again represents a closed shell type interaction. The measured parameters suggest the bond has a metallic nature but with slight variation towards ionic character, and has a tendency to participate in donor–acceptor interactions.
 | ||
| Fig. 2 Model structure of [(TPB)FeN2], along with illustration of Fe→B, Fe–N2 and NN bonding. | ||
 | ||
| Fig. 3 Variation of selected bond lengths (Å) versus oxidation states. | ||
Here the detailed analysis of the contributions to the isomer shift and quadrupole splitting is elaborated. Theoretically computed isomer shifts for the Fe2− to Fe0 complexes are 0.26 mm s−1, 0.40 mm s−1 and 0.56 mm s−1, respectively, which correspond to the reported experimental values.14,59 These values are directly related to the electron density at the nucleus. In the isomer shift the major contributions include (a) variation in the 3s-orbital, which is notably due to alterations in the shielding of the d-orbital population of iron; and (b) changes in the valence shell part. The computed theoretical electron densities of the [(TPB)FeN2]2−/1−/0 complexes are mentioned in Table 1.
| Contribution | Fe2− | Fe1− | Fe0 |
|---|---|---|---|
| 4s population | 0.35 | 0.36 | 0.37 |
| 3d population | 6.97 | 6.90 | 6.70 |
| ρ0(1s) (au−3) | 12454.47 |
12454.17 |
12454.28 |
| ρ0(2s) (au−3) | 1157.72 | 1157.70 | 1157.71 |
| ρ0(3s) (au−3) | 165.16 | 164.97 | 164.70 |
| ρ0(4s) (au−3) | 1.89 | 1.86 | 1.61 |
| ρ0 (au−3) | 13782.17 |
13782.17 |
13781.58 |
| δexpt (mm s−1) | 0.2614 |
0.4059 |
0.5659 |
| δcalcd (mm s−1) | 0.30 | 0.32 | 0.55 |
It is apparent from Table 1 that the contributions from 1s and 2s orbitals are insignificant and can be ignored for qualitative discussion. Furthermore, the contributions from 3s and 4s orbitals influence the electron density at the Fe nucleus (Table 1). As reduction at the metal centre takes place, it leads to a decrease in the isomer shift. This is due to the change in the 4s-contribution to the electron density ρ0 at the Fe nucleus. Also the 3d-population increases, which enhances shielding effect of the extra 3d charge and pushes the 4s electrons further away from the nucleus. Besides the effective nuclear change being weakened, the energy separation is broadened among the iron 4s orbital and ligand σ-donating orbitals with totally symmetric combinations; therefore the total 4s population decreases throughout the series. The computed Fe–N2 bond distances for [(TPB)FeN2]2−/1−/0 complexes are 1.779 Å, 1.807 Å and 1.973 Å, respectively. It is noted that the reduction of the iron centre species leads to the reduction of this bond distance. A linear relationship between the calculated theoretical electron densities and Fe–N2 bond distance is obtained (Fig. 4). Since the Fe–N2 bond distance is essential parameters which affects the architecture of the 4s-orbital and therefore tune the isomer shift (Fig. 5).
 | ||
| Fig. 4 Variation of isomer shift versus Fe–N2 bond distance of [(TPB)FeN2]2−/1−/0 complex. | ||
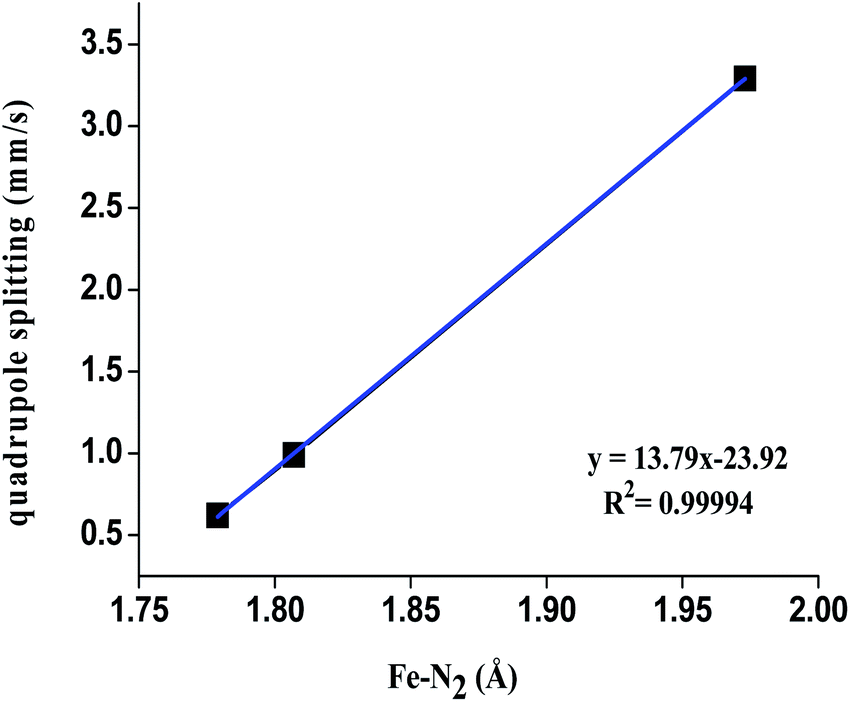 | ||
| Fig. 5 Variation of quadrupole splitting versus Fe–N2 bond distance of [(TPB)FeN2]2−/1−/0 complex. | ||
This is directly related to the radial extension of the 4s-orbital, which in turn affects the electron density at the nucleus. Since a decrease in the 4s-orbital population in the reduced state leads to a contraction of the Fe–N2 bond length, which eventually tends to attract the 4s-orbital towards the core, this causes an enhancement in the electron density at the iron nucleus ρ0(4s). Therefore the values of the isomer shift will be lower as reduction at the iron centre takes place.
Another significant parameter which provides information about the electronic structure of the environment of a Mössbauer nucleus is the quadrupole splitting, which is directly related to the electric field gradient tensor (efg) (the second derivative of the potential produced by the surrounding charges). Therefore, it important to know the effects of geometrical distortion on electronic structure. Basically, the efg involves two parts: the valence term, associated with the anisotropy of the 3d orbital of Fe; and the lattice term, related to the charge distribution of the surrounding ions in the point charge approximation. Therefore, the anisotropy needs non-zero efg and local distortion of coordination at the Fe centre. In Table 2, the contribution to the electric field gradient tensor is given for all three complexes. Significant variation was observed for the quadrupole splitting for all these species. The values of the quadrupole splitting parameters are 0.82 mm s−1, 1.01 mm s−1 and 3.34 mm s−1 for the [(TPB)FeN2]2−/1−/0 complexes. Large values are noted for the [(TPB)FeN2]0 complex. As shown in Table 2, there is a considerable change in the electric field gradient, particularly the Vzz component. The largest contribution arises from the d orbital valence and its ligand-centered bonding orbitals. For the [(TPB)FeN2]2− complex, these values are 0.41 and 0.052 au−3, respectively. As the core electrons are closed shell, they do not contribute to the efg and their effect is very small; however, it cannot be neglected. Here we see that the lattice contribution is small over a two-centre bond contribution. The computed asymmetry parameter (η) for the Fe2− species is 0.15; this small value is dependent on effective C3v symmetry. This is reflected in the computed values of ∠P–Fe–P ≈ 116.3°, 116.2°, 116.2°. However, a large value of η is observed for the Fe1− species, i.e. 0.45, which has angles ∠P–Fe–P ≈ 106.8°, 107.1°, 134.0°. For complex [(TPB)FeN2]0 there is large quadrupole splitting, i.e. 3.30 mm s−1. This is because the species likely undergoes a (pseudo) Jahn–Teller distortion. This distortion reduces the axial symmetry of the complex, which leads to a finite asymmetry parameter of the electric field gradient at the Fe centre.
| Contribution | Fe2− | Fe1− | Fe0 |
|---|---|---|---|
| Local valence | 0.41 | 45.8 | −1.87 |
| Core polarization (Fe-2p, 3p) (au−3) | −0.01 | −46.22 | 0.066 |
| Two-center point charge (lattice) (au−3) | −0.014 | −0.05 | −0.047 |
| Two-center bond (au−3) | 0.052 | −0.20 | −0.209 |
| Total (au−3) | −0.37 | −0.59 | −2.01 |
| ΔEQ expt (mm s−1) | 0.8214 |
1.0159 |
3.3659 |
| ΔEQ calcd (mm s−1) | 0.62 | 0.99 | 3.30 |
| η calcd | 0.15 | 0.45 | 0.24 |
Orbital analysis and assignment of redox states
The molecular orbital representation of the [(TPB)FeN2]2− complex is shown in Fig. 6. The occupation number can be understood as Fe-(dxz/yz)4, (dz2)2 and (dxy, dx2−y2)4 due to its trigonal bipyramidal coordination geometry. From the MO diagram of this complex, two Fe-dxz/yz orbitals are doubly occupied and are π-bonding; there are corresponding π* antibonding orbitals (N2), which will be vacant. These orbitals are responsible for π-backdonation from Fe to the N2 centre. The molecular orbitals Fe-dxy/dx2−y2 strongly interact with the lone pair of the P atom and are doubly occupied; therefore the Fe–P bond distances are stronger for the Fe2− oxidation state than for the Fe1− and Fe0 complexes, i.e. 2.137 Å, 2.137 Å, 2.137 Å; 2.254 Å, 2.224 Å, 2.242 Å; and 2.404 Å, 2.404 Å, 2.404 Å, respectively (Table S1†). The metal centre reduction of the [(TPB)FeN2]2−/1−/0 complexes can be considered as electron transfer series in the reaction pathways. For these complexes the electron accepting orbitals are dxy and dx2−y2. As seen in Fig. 6, 7 and 8, in the reduction process the nitrogen antibonding orbitals become close to the Fexz/yz orbitals, and the noted values for these orbitals are 5.53 eV, 6.02 eV and 6.76 eV for the Fe2−, Fe1− and Fe0 complexes. This is explained by the contraction in the nuclear charge at the Fe centre.
orbitals become close to the Fexz/yz orbitals, and the noted values for these orbitals are 5.53 eV, 6.02 eV and 6.76 eV for the Fe2−, Fe1− and Fe0 complexes. This is explained by the contraction in the nuclear charge at the Fe centre.
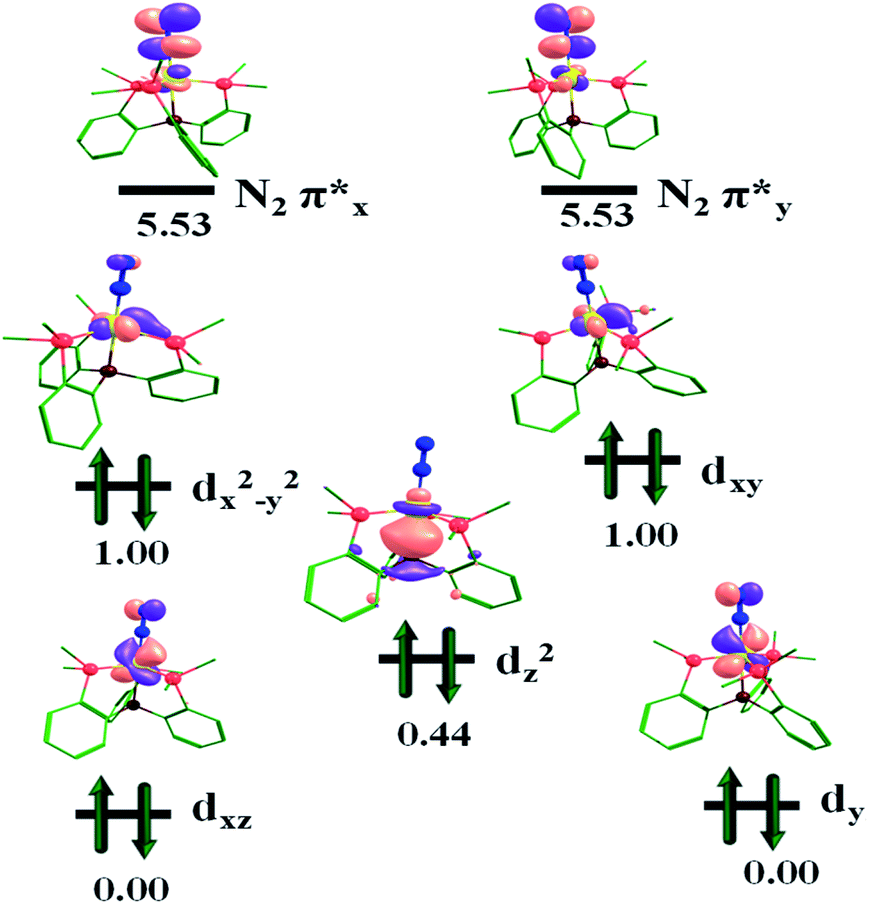 | ||
| Fig. 6 Frontier molecular orbital (MO) diagram for complex [(TPB)FeN2]2−. The orbital energy values are given in eV. | ||
 | ||
| Fig. 7 Frontier molecular orbital (MO) diagram for complex [(TPB)FeN2]1−. The orbital energy values are given in eV. | ||
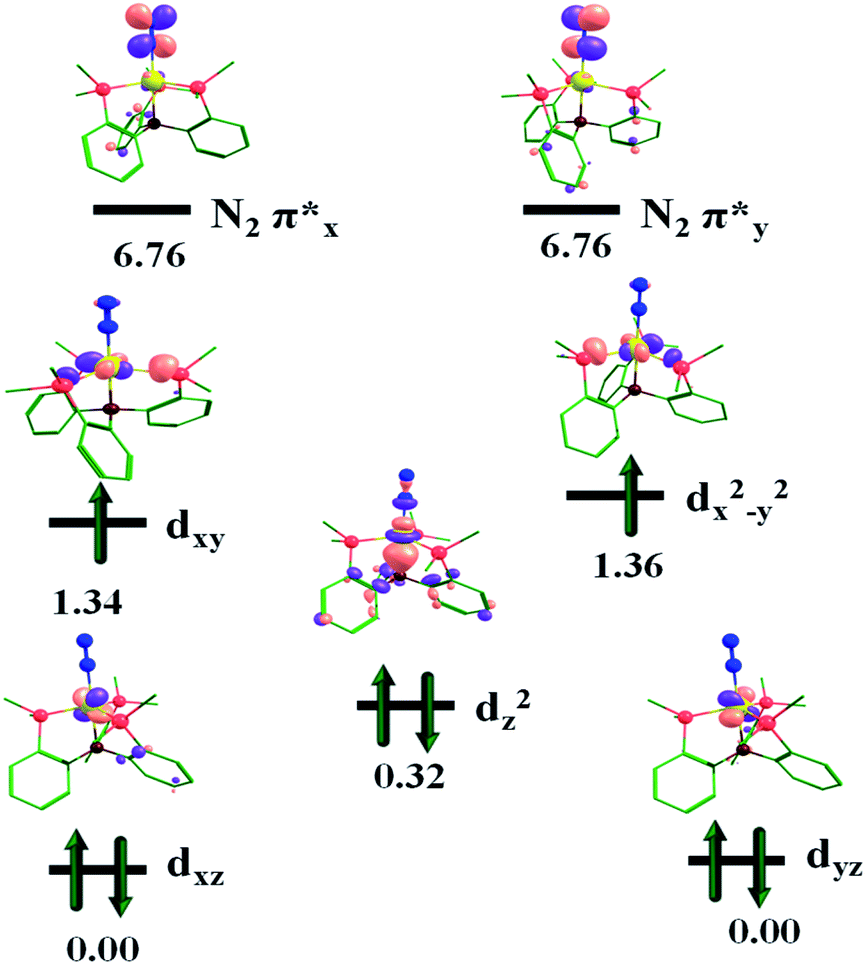 | ||
| Fig. 8 Frontier molecular orbital (MO) diagram for complex [(TPB)FeN2]0. The orbital energy values are given in eV. | ||
In the reduced state, iron to N2 π-backdonation increases; this makes the Fe–N2 bond stronger and weakens the N–N bond distance. Therefore the low-valent iron strongly activates the dinitrogen molecule and makes the process more feasible.
Conclusions
Activation of the highly non-polar N2 molecule by the nitrogenase enzyme is an emerging topic in the field of catalysis. Despite the fact that several transition metal-based complexes readily perform this transformation, biomimetic model compounds which perform this reaction efficiently are limited. Studies of low-valent [(TPB)Fe–N2]2−/1−/0 complexes to understand their behaviour in activating dinitrogen are a significant breakthrough in this area. In the present study we have explored the electronic structure of iron–dinitrogen complexes [(TPB)Fe–N2]2−/1−/0 and discuss the correlation of the electronic structure with reactivity. The main factors for analysis are electronic structure changes that happen during the metal-centred reduction of the nitrogenase enzyme by [(TPB)Fe–N2]2−/1−/0 species. The main point in discussing the electronic structure of this complex is the allocation of a physical oxidation state at Fe center, i.e. the effective electronic configuration of the mainly metal-based d orbitals. Based on this, conclusions derived from our work are summarized below.(1) From the optimized structures of [(TPB)Fe–N2]2−/1−/0 complexes, the lowest energy was obtained for the [(TPB)Fe–N2]2− complex. From the bonding analysis it is established that the Fe→B bond acts an electron source due to its flexible nature in the course of the nitrogenase enzyme cycle.
(2) Mössbauer spectroscopic parameters computed for the [(TPB)Fe–N2]2−/1−/0 species generally agree with the experimental values. The computed values of the isomer shift decrease as the iron goes from Fe0 to the Fe2− state. The main contribution to the isomer shift is a 3s-orbital contribution, which differs depending on the d orbital population due to different shielding. The valence shell contribution is also due to the 4s-orbital.
(3) The large value of quadrupole splitting ΔEQ in the case of the [(TPB)Fe–N2]0 complex is associated with the Jahn–Teller distortion, which decreases the axial symmetry of the species and eventually generates a finite asymmetry parameter and thus an electric field gradient at the Fe centre.
(4) Our calculations establish that the iron–dinitrogen bond length is interrelated with the iron oxidation state, which has a considerable effect on the Mössbauer parameters. A linear correlation of the iron oxidation state with the isomer shift and quadrupole splitting was obtained. This can be regarded as shortening the iron–dinitrogen bond length as it enhances π-backdonation by successive metal-centred reductions.
(5) From the molecular orbital analysis, it is demonstrated that as the reduction at the iron centre takes place, the electron accepting orbitals dxy and dx2−y2 become closer to the  antibonding orbital. This in turn increases the π-backdonation and thus reduces the Fe–N2 bond length, which apparently weakens the NN bond. Therefore, the complex [(TPB)Fe–N2]2− will be more reactive than the other two complexes.
antibonding orbital. This in turn increases the π-backdonation and thus reduces the Fe–N2 bond length, which apparently weakens the NN bond. Therefore, the complex [(TPB)Fe–N2]2− will be more reactive than the other two complexes.
In the above report, presented here for the first time, we have analyzed and compared the subtle electronic structures of low-valent [(TPB)Fe–N2]2−/1−/0 complexes using DFT and unravelled the factors that play key roles in the high reactivity of the metal–dinitrogen moiety. The concept of electronic cooperativity studied here has a wider scope for other dinitrogen models/enzymes.
Note
The references for the experimental values of isomer shift and quadrupole moments are mentioned in the tables.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Nidhi Vyas is thankful to the DST Inspire fellowship for the research grant. A. Kumar is thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi, for the award of the junior research fellowship (JRF).References
- T. Bazhenova and A. Shilov, Coord. Chem. Rev., 1995, 144, 69–145 CrossRef CAS.
- J. Postgate, Nitrogen fixation, Cambridge University Press, Cambridge, UK, 3rd edn, 1998 Search PubMed.
- J. R. Jennings, Catalytic Ammonia Synthesis, Plenum Press, New York, 1st edn, 1991, ch. 2, pp. 19–108 Search PubMed.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS PubMed.
- L. M. Rubio and P. W. Ludden, J. Bacteriol., 2005, 187, 405–414 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- K. Rajagopalan and J. Johnson, J. Biol. Chem., 1992, 267, 10199–10202 CAS.
- C. E. MacBeth, S. B. Harkins and J. C. Peters, Can. J. Chem., 2005, 83, 332–340 CrossRef CAS.
- N. P. Mankad, M. T. Whited and J. C. Peters, Angew. Chem., 2007, 46, 5768–5771 CrossRef CAS PubMed.
- B. D. Matson and J. C. Peters, ACS Catal., 2018, 8, 1448–1455 CrossRef CAS PubMed.
- N. B. Thompson, M. T. Green and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 15312–15315 CrossRef CAS PubMed.
- M. E. Moret and J. C. Peters, Angew. Chem., 2011, 50, 2063–2067 CrossRef CAS PubMed.
- T. M. Buscagan, P. H. Oyala and J. C. Peters, Angew. Chem., 2017, 56, 6921–6926 CrossRef CAS PubMed.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105–1115 CrossRef CAS PubMed.
- M. A. Nesbit, P. H. Oyala and J. C. Peters, J. Am. Chem. Soc., 2019, 141, 8116–8127 CAS.
- S. Ye, E. Bill and F. Neese, Inorg. Chem., 2016, 55, 3468–3474 CrossRef CAS PubMed.
- J. F. Berry, E. Bill, E. Bothe, S. D. George, B. Mienert, F. Neese and K. Wieghardt, Science, 2006, 312, 1937–1941 CrossRef CAS PubMed.
- S. Ye, C.-Y. Geng, S. Shaik and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 8017–8030 RSC.
- S. Sinnecker, L. D. Slep, E. Bill and F. Neese, Inorg. Chem., 2005, 44, 2245–2254 CrossRef CAS PubMed.
- F. Neese, J. Am. Chem. Soc., 2006, 128, 10213–10222 CrossRef CAS PubMed.
- C. Geng, S. Ye and F. Neese, Dalton Trans., 2014, 43, 6079–6086 RSC.
- M. Ansari, N. Vyas, A. Ansari and G. Rajaraman, Dalton Trans., 2015, 44, 15232–15243 RSC.
- M. Jaccob and G. Rajaraman, Dalton Trans., 2012, 41, 10430–10439 RSC.
- T. Chachiyo and J. H. Rodriguez, Dalton Trans., 2012, 41, 995–1003 RSC.
- S. J. Clark, J. D. Danaldson and S. M. Grimes, Spectroscopic Properties of Inorganic and Organometallic Compounds, TheRoyal Society of Chemistry, 1997, pp. 380–453 Search PubMed.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341–5350 CrossRef CAS PubMed.
- L. Walker, G. K. Wertheim and V. Jaccarino, Phys. Rev. Lett., 1961, 6, 98 CrossRef CAS.
- R. B. Yelle, J. L. Crossland, N. K. Szymczak and D. R. Tyler, Inorg. Chem., 2009, 48, 861–871 CrossRef CAS PubMed.
- S. Chen, M. H. Chisholm, E. R. Davidson, J. B. English and D. L. Lichtenberger, Inorg. Chem., 2009, 48, 828–837 CrossRef CAS PubMed.
- M. Reiher, B. Le Guennic and B. Kirchner, Inorg. Chem., 2005, 44, 9640–9642 CrossRef CAS PubMed.
- Z. Benedek, M. Papp, J. Oláh and T. Szilvási, Inorg. Chem., 2018, 57, 8499–8508 CrossRef CAS PubMed.
- U. Gogoi, A. K. Guha and A. K. Phukan, Chem.–Eur. J., 2013, 19, 11077–11089 CrossRef CAS PubMed.
- J.-B. Lu, X.-L. Ma, J.-Q. Wang, J.-C. Liu, H. Xiao and J. Li, J. Phys. Chem. A, 2018, 122, 4530–4537 CrossRef CAS PubMed.
- M. A. Kaczmarek, A. Malhotra, G. A. Balan, A. Timmins and S. P. de Visser, Chem.–Eur. J., 2018, 24, 5293–5302 CrossRef CAS PubMed.
- J. Varley, Y. Wang, K. Chan, F. Studt and J. Nørskov, Phys. Chem. Chem. Phys., 2015, 17, 29541–29547 RSC.
- N. B. Thompson, P. H. Oyala, H. T. Dong, M. J. Chalkley, J. Zhao, E. E. Alp, M. Hu, N. Lehnert and J. C. Peters, Inorg. Chem., 2019, 58, 3535–3549 CrossRef CAS PubMed.
- N. Vyas, B. Pandey, A. Ojha and A. Grover, Int. J. Quantum Chem., 2019, e26025 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- B. Mennucci, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 CAS.
- G. A. Zhurko, Chemcraft version 16, 2014 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- M. Roemelt, S. Ye and F. Neese, Inorg. Chem., 2009, 48, 784–785 CrossRef CAS PubMed.
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef.
- F. Weinhold, J. Comp. Chem., 2012, 33, 2363–2379 CrossRef CAS PubMed.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- R. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549–558 RSC.
- A. F. Hill, G. R. Owen, A. J. White and D. J. Williams, Angew. Chem., 1999, 38, 2759–2761 CrossRef CAS.
- R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360–2366 CrossRef CAS PubMed.
- J. S. Anderson, G. E. Cutsail III, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7803–7809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables for optimized parameters, NBO along with charge, topological parameters, molecular orbitals, HOMO–LUMO plot. See DOI: 10.1039/c9ra10481j |
| This journal is © The Royal Society of Chemistry 2020 |