DOI:
10.1039/D0RA00176G
(Paper)
RSC Adv., 2020,
10, 7927-7932
Aerobic photooxidative hydroxylation of boronic acids catalyzed by anthraquinone-containing polymeric photosensitizer†
Received
8th January 2020
, Accepted 17th February 2020
First published on 24th February 2020
Abstract
We report herein the synthesis of a polymeric photosensitizer and its application in aerobic photooxidative hydroxylation of boronic acids. The polymeric photosensitizer was synthesized by the condensation of anthraquinone-2-carbonyl chloride (AQ-2-COCl) with poly (2-hydroxyethyl methacrylate) (PHEMA). The photo-oxidative hydroxylation of boronic acids using anthraquinone-containing-poly (2-hydroxyethyl methacrylate) (AQ-PHEMA) was then explored and shown to exhibit high efficiency and broad scope. Moreover, AQ-PHEMA could be easily recovered and reused for more than 20 times without significant loss of the catalytic activity.
Introduction
Oxidative hydroxylation of boronic acids is one of the most important methods for the synthesis of phenols or alcohols.1–7 Strong oxidants such as ozone,8 high-valence halogen compounds,9–11 peroxides and N-oxides12 are efficient reagents for such transformations (Scheme 1a). However, these methods generate large amounts of wastes detrimental to the environment. Oxygen in air is a green oxidant with environment-friendly feature. The methods using oxygen have been well studied.13,14 Some Ru,15 Pd,16 Fe,17 Cu,18–20 complexes have been reported to be effective catalysts in this reaction (Scheme 1b). However, the catalysts are expensive, hard to synthesis, and not environmentally friendly. The metal-free catalyst processes provide a promising alternative (Scheme 1c)21 wherein good catalytic activity could be envisioned.
 |
| | Scheme 1 Oxidative hydroxylation of boronic acids with small molecular catalysts. | |
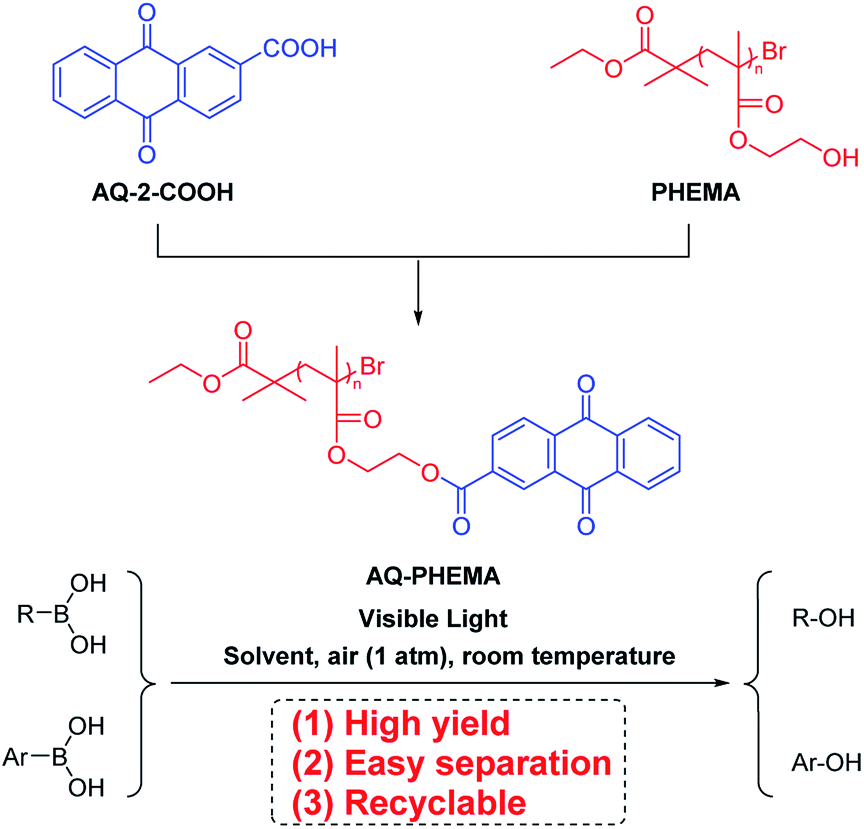
Visible light is an inexhaustible green energy.22–24 Photosensitizer-catalyzed transformations have shown very important applications in photochemistry.25–27 Numerous achievements have been reported in this field. For example, Guo group reported selective cleavage and formation of Csp2–I bond, using carefully-designed thioxanthone derivative.28 This was the first example of Csp2–I bond cleavage by means of photosensitizing, which highlights the potential of small molecular photosensitizer. Visible light photosensitizer-catalyzed oxidative hydroxylation of boronic acids is another hot topic, which has received much attention in the past decade.29–32 In this transformation, the superoxide radical anion generated by single electron transfer could coordinate with the boron atom, then the reaction undergoes rearrangement and hydrolysis to produce the final phenol or alcohol.33,34 Small molecule photosensitizers has the significant disadvantage as a homogenous catalyst, as they are difficult to be recycled or reused. With our continuous interest in the synthesis and application of PHEMA,35 we proposed to synthesize a novel macromolecular photocatalyst by introducing anthraquinone (AQ) group of anthraquinone-2-carboxylic acid (AQ-2-COOH)36–38 into polymeric chains of PHEMA (Scheme 2). This polymeric catalytic material can be easily recovered and reused after catalyzing photoreaction. Herein, we wish to report our recent research in aerobic oxidative hydroxylation of boronic acids employing AQ-PHEMA (for synthesis and characterization of AQ-PHEMA, see ESI†) as catalyst.
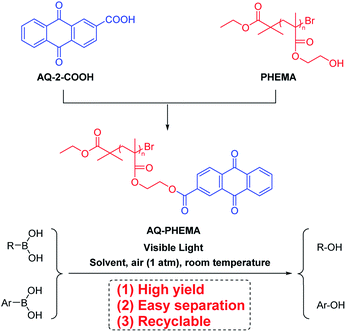 |
| | Scheme 2 The synthetic procedure of AQ-PHEMA and its application in photooxidative hydroxylation of boronic acids. | |
Results and discussion
Optimization and scope investigation
In the beginning, (4-methoxyphenyl)boronic acid (1a) was chosen as the model substrate to optimize the AQ-PHEMA-catalyzed photooxidative hydroxylation of boronic acids. The initial attempt was carried out using i-Pr2NEt as the electron donor, CH3CN as the solvent at rt under air atmosphere. An 86% NMR yield of desired product 4-methoxyphenol (2a) was formed after irradiation under a purple LED for 35 hours (entry 1, Table 1). With this result in hand, a survey of solvents was carried out (entries 2–16, Table 1). Reactions in ether, toluene, or dichloromethane gave lower yield (entries 2–4, Table 1). While methyl tert-butyl ether (MTBE), acetone, CH3NO2, or THF resulted in slightly higher yield (entries 5–8, Table 1). When methyl acetate, EtOH or CH3OH was chosen as the solvent, the yield of 2a was remarkably increased (entries 9–11, Table 1). Reactions in propyl acetate or isopropyl acetate resulted in similar excellent yields (entries 12 and 13, Table 1). When ethyl acetate, dimethyl carbonate (DMC) or 1,4-dioxane was employed as the solvent, the NMR yield of 2a was increased to 99% (entries 14–16, Table 1). Considering the reaction time, 1,4-dioxane was chosen as the optimal solvent. Notably, AQ-PHEMA is insoluble in all the above tested solvents and can only be dispersed in the solvent, so it can be easily recovered by simple filtration. Next, the screening of electron donors was carried out. Ammonia gave a poor yield and the DBU did not work very well (entries 17 and 18, Table 1). Secondary amine (dicyclohexylamine) and tertiary amine (NEt3) gave decreased yields (entries 19 and 20, Table 1). Decreasing the amount of i-Pr2NEt to 1 equivalent resulted in a decreased in yield (entry 21, Table 1). Next, optimization of catalyst loading were conducted (entries 22 and 23, Table 1). The results showed that 3 mol% catalytic is suitable (entry 22, Table 1). Control experiments were also performed which demonstrated the necessity of both the catalyst and light (entries 24 and 25, Table 1). Furthermore, a reaction carried out at 80 °C without light did not lead to any conversion, eliminating the possibility of the thermal effect of the LED light (entry 26, Table 1). Next, the oxidation using AQ as catalyst was conducted as comparasion. The reaction was completed in 12 hours in an excellent yield (entry 27, Table 1). However, AQ was difficult to recover in 1,4-dioxane. Finally, the oxidation using PHEMA as catalyst was investigated and no reaction took place (entry 28, Table 1). The above results clearly demonstrated that AQ was the key catalyst in this transformation. We also tried other different wavelengths for LED lights (Table S1†). The results showed that reactions at other wavelengths were similar to background reactions and purple LED is the only working light source in this reaction. Thus, Condition A (3 mol% of AQ-PHEMA, i-Pr2NEt (2 equiv.), 1,4-dioxane, purple LED, air (1 atm), and room temperature) was considered as the optimized conditions for further studies.
Table 1 Optimization of the reaction conditionsa

|
| Entry |
Solvent |
Catalyst (mol%) |
e-Donor (equiv.) |
Time (h) |
Yieldb (%) |
| The reaction were carried out using 1a (1 mmol) in solvent (5 mL), irradiated by purple LED under air atmosphere at rt. (Based on AQ anchored on PHEMA, the mass of 5 mol% AQ-PHEMA is 17 mg; the mass of 3 mol% AQ-PHEMA is 10 mg; the mass of 1 mol% AQ-PHEMA is 3 mg). Yield determined by 1H NMR analysis using CH2Br2 (1 mmol) as internal standard. Recovered yield of 1a determined by 1H NMR analysis using CH2Br2 (1 mmol) as internal standard. Isolated yield of 2a. The reaction was carried out without light. The reaction was carried out at 80 °C. The reaction was carried out using 3 mol% AQ as catalyst. The reaction was carried out in the presence of 4 mg of PHEMA. |
| 1 |
CH3CN |
5 |
i-Pr2NEt (2) |
35 |
86 |
| 2 |
Et2O |
5 |
i-Pr2NEt (2) |
48 |
34 |
| 3 |
Toluene |
5 |
i-Pr2NEt (2) |
48 |
46 |
| 4 |
DCM |
5 |
i-Pr2NEt (2) |
48 |
51 |
| 5 |
MTBE |
5 |
i-Pr2NEt (2) |
48 |
67 |
| 6 |
Acetone |
5 |
i-Pr2NEt (2) |
48 |
68 |
| 7 |
CH3NO2 |
5 |
i-Pr2NEt (2) |
37 |
74 |
| 8 |
THF |
5 |
i-Pr2NEt (2) |
42 |
78 |
| 9 |
Methyl acetate |
5 |
i-Pr2NEt (2) |
38 |
84 |
| 10 |
EtOH |
5 |
i-Pr2NEt (2) |
33 |
89 |
| 11 |
MeOH |
5 |
i-Pr2NEt (2) |
32 |
92 |
| 12 |
Propyl acetate |
5 |
i-Pr2NEt (2) |
36 |
95 |
| 13 |
Ispropyl acetate |
5 |
i-Pr2NEt (2) |
36 |
96 |
| 14 |
Ethyl acetate |
5 |
i-Pr2NEt (2) |
29 |
99 |
| 15 |
DMC |
5 |
i-Pr2NEt (2) |
34 |
99 |
| 16 |
1,4-Dioxane |
5 |
i-Pr2NEt (2) |
27 |
99 |
| 17 |
1,4-Dioxane |
5 |
NH3·H2O (4 mL) |
27 |
45 (34)c |
| 18 |
1,4-Dioxane |
5 |
DBU |
27 |
69 |
| 19 |
1,4-Dioxane |
5 |
Dicyclohexylamine |
27 |
82 |
| 20 |
1,4-Dioxane |
5 |
NEt3 |
27 |
84 |
| 21 |
1,4-Dioxane |
5 |
i-Pr2NEt (1) |
27 |
81 (14)c |
| 22 |
1,4-Dioxane |
3 |
i-Pr2NEt (2) |
27 |
99 (97)d |
| 23 |
1,4-Dioxane |
1 |
i-Pr2NEt (2) |
27 |
83 (11)c |
| 24 |
1,4-Dioxane |
— |
i-Pr2NEt (2) |
27 |
11 (89)c |
| 25e |
1,4-Dioxane |
3 |
i-Pr2NEt (2) |
27 |
0 (99)c |
| 26e,f |
1,4-Dioxane |
3 |
i-Pr2NEt (2) |
27 |
0 (99)c |
| 27g |
1,4-Dioxane |
3 |
i-Pr2NEt (2) |
12 |
99 |
| 28h |
1,4-Dioxane |
— |
i-Pr2NEt (2) |
27 |
10 (90)c |
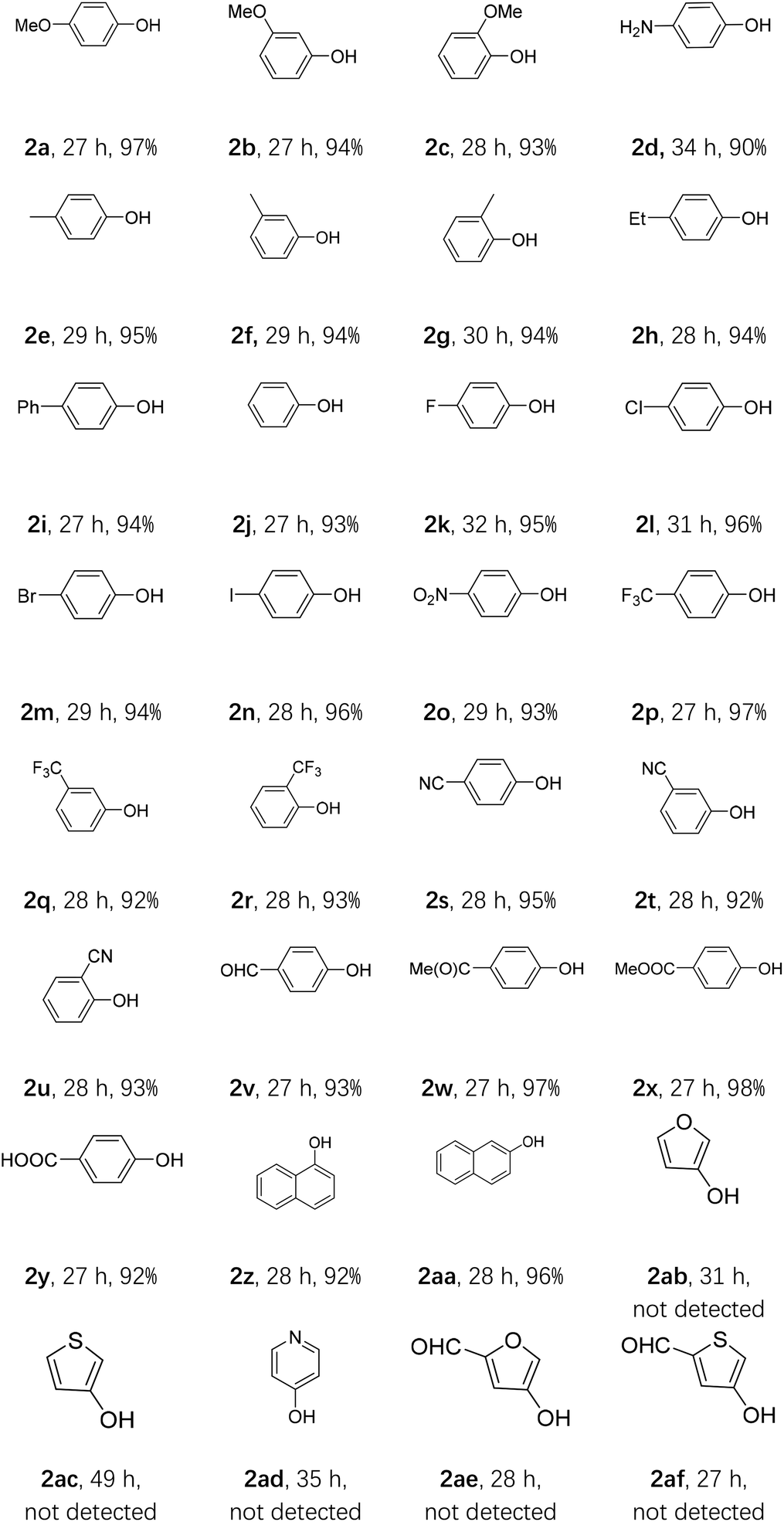
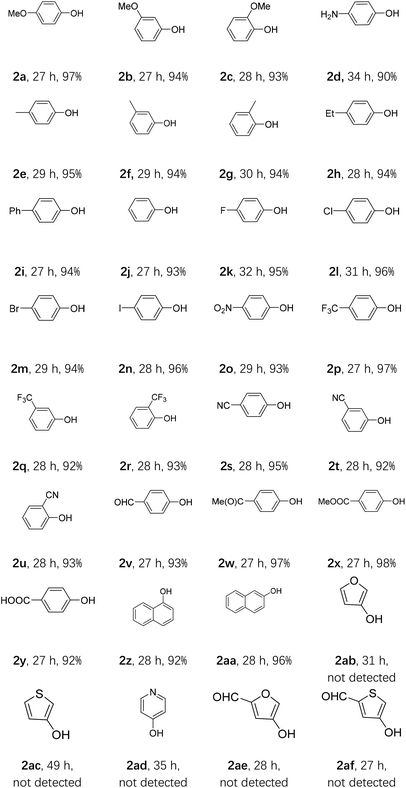
With the optimal conditions in hand, we explored the scope of this oxidative hydroxylation (Table 2). Firstly, the electronic effect of the arylboronic acids was examined. Substrates with strong electron donating group, like ortho-, meta- and para-methoxy, showed nice reactivity with excellent yields (2a–2c). Para-aminophenol was also obtained in an excellent yield (2d). Weak electron donating group such as ortho-, meta- and para-methyl (2e–2g), ethyl (2h), and phenyl (2i) were also conducted and generated in similar excellent yields. Phenol could be obtained from phenylboronic acid with an excellent yield (2j). Nice reactivity was also acquired with halogen bearing arylboronic acids (2k–2n). Substrates with strong electron withdrawing groups (EWGs), including nitro (2o), ortho-, meta- and para-(trifluoromethyl) (2p–2r), and ortho-, meta- and para-nitrile (2s–2u) also showed high reactivities as well as excellent yields. Reactions with other electron withdrawing groups, like formyl (2v), acetyl (2w), methoxy carbonyl (2x) and carboxyl (2y) were also showed excellent reactivities in excellent yields. Further naphthyl groups were employed, and both α- and β-naphthol (2z and 2aa) could obtain excellent yields. Next, we tested some heteroaromatic boronic acids. Unfortunately, the resulting reaction mixtures were complicated (2ab–2af).
Table 2 The oxidative hydroxylation of arylboronic acid under condition Aa
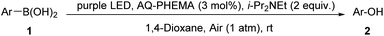
|
| All reactions were carried out using 1 (1 mmol), AQ-PHEMA (3 mol%), i-Pr2NEt (2 equiv.), in 1,4-dioxane (5 mL) irradiated by a purple LED light at rt under air atmosphere. Isolated yield was reported. |
 |
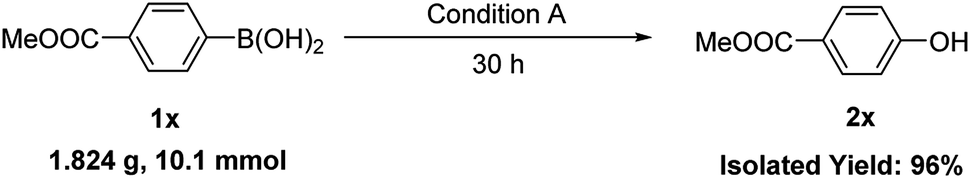
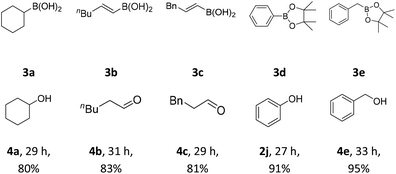
Furthermore, some alkyl and alkenyl boronic acid, as well as aryl boronic acid esters were studied (Table 3). Under condition A, cyclohexylboronic acid (3a) gave a good yield of cyclohexanol (4a). Alkenyl boronic (3b and 3c) gave similar good yields of 4b and 4c. In this case, aldehyde products were formed. Aryl boronic acid ester (3d and 3e) also gave excellent yields of phenol (2j) and phenylmethanol (4e). The above results showed high functional group tolerance and very good substrate scope. Finally, a gram–scale reaction using 1x under condition A afforded 2x in 96% (Scheme 3), demonstrating the scalability and practicality of the current reaction.
Table 3 The oxidative hydroxylation of 3a–e under condition Aa
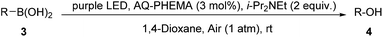
|
| All reactions were carried out using 1 (1 mmol), AQ-PHEMA (3 mol%), i-Pr2NEt (2 equiv.), in 1,4-dioxane (5 mL) irradiated by a purple LED light at rt under air atomsphere. Isolated yield was reported. |
 |
 |
| | Scheme 3 Gram scale reaction. | |
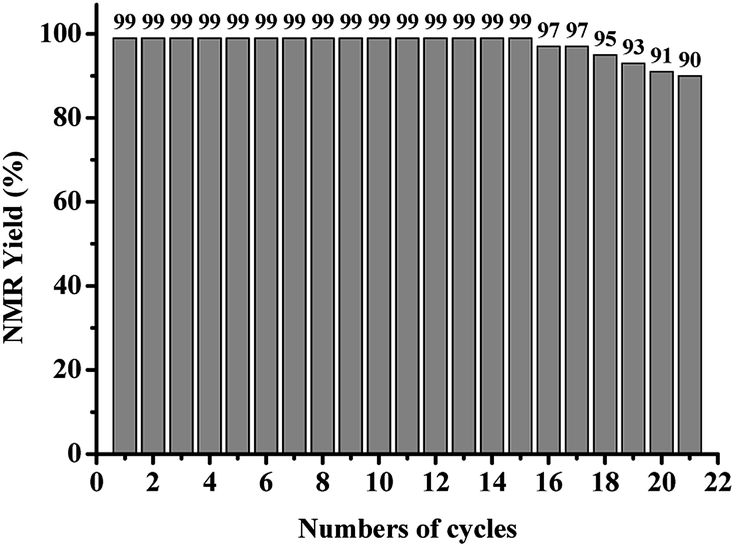
Recycling experiments
To investigate the recyclability of AQ-PHEMA in photooxidative hydroxylation of boronic acids. 4-Methoxycarbonylbenzeneboronic acid (1x) was selected as the model substrate for the AQ-PHEMA recycling experiments under condition A. 1x was completely consumed after 27 hours and the NMR yield of the product 4-methoxycarbonylphenol (2x) was 99% in the first cycle of the photocayalytic reaction. After the first cycle, the catalyst AQ-PHEMA could be easily separated and recovered by simple filtration and directly used for the next cycle under the same procedure. Finally, we found that AQ-PHEMA could be reused more than 20 times without significant loss of the catalytic activity (Fig. 1).
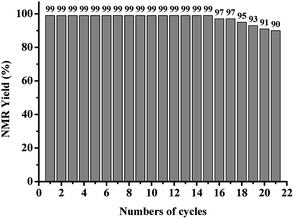 |
| | Fig. 1 Recycling experiments of the photocatalytic reaction of 1x. | |
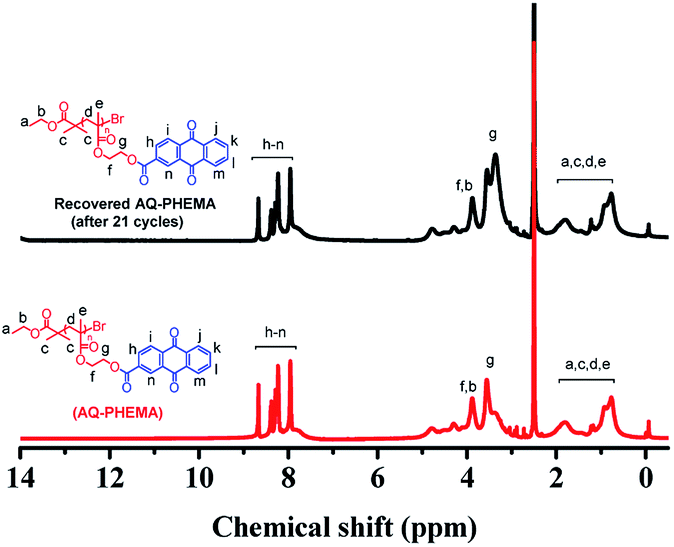
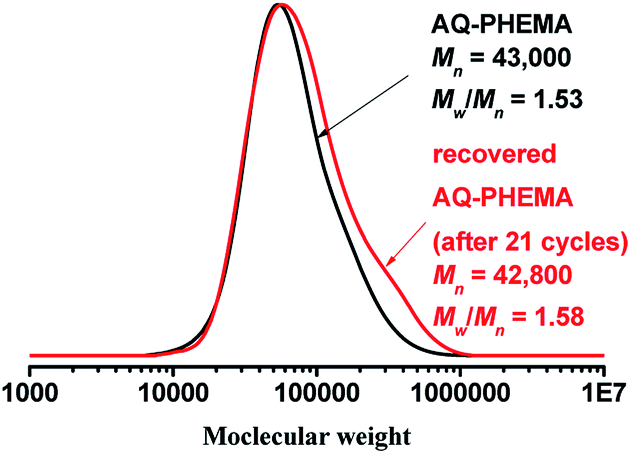
After the recycling experiments (21 cycles), the recovered AQ-PHEMA was characterized by 1H NMR and GPC. As shown in Fig. 2, no significant changes were observed on the 1H NMR spectra compared to the original AQ-PHEMA. The molecular weight Mn,GPC and molecular distribution Mw/Mn of the recovered AQ-PHEMA (after 21 cycles) were very close to the original AQ-PHEMA (Fig. 3). These results clearly indicated that the structure of AQ-PHEMA did not change during the whole catalytic procedure.
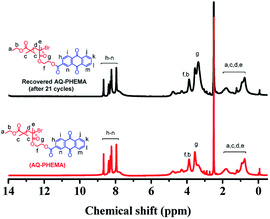 |
| | Fig. 2 1H NMR spectra for the recovered AQ-PHEMA (after 21 cycles) and the original AQ-PHEMA in d6-DMSO. | |
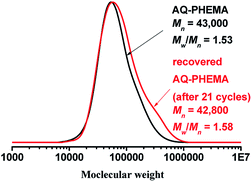 |
| | Fig. 3 The GPC traces of the original AQ-PHEMA and the recovered AQ-PHEMA (after 21 cycles). | |
Mechanism studies
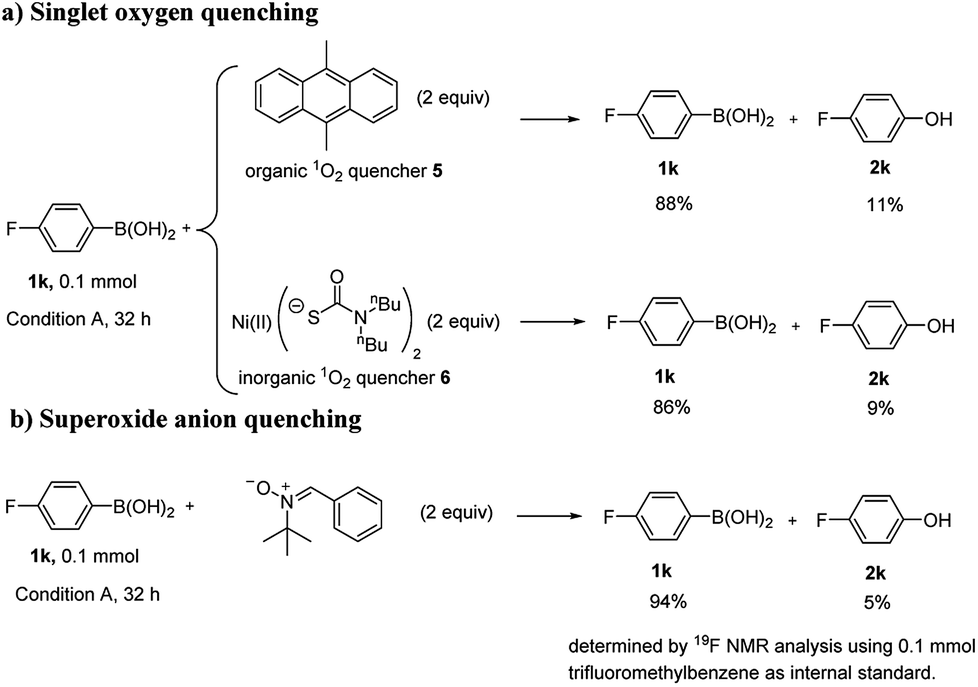
To gain insights into the reaction mechanism, some control experiments were carried out (Scheme 4). When 2 equivalents of 9,10-dimethylanthracene (5) was added to the reaction system as an organic singlet oxygen quencher,39 severe inhibition was observed. Bis((dibutylcarbamothioyl)th-io) nickel(II) (6) was also tried as an inorganic singlet oxygen quencher.40 Similarly, severe inhibition was also observed with 2 equivalents of 6 (Scheme 4a). N-tert-Butyl-1-phenylmethanimine oxide (7), a superoxide radical anion quencher,41–43 was used, the reaction was efficiently inhibited when 2 equivalents of 7 was added (Scheme 4b). The results above suggested that both singlet oxygen and superoxide radical anion were likely involved in this reaction. Cyclic voltammogram of AQ-PHEMA was also recorded under irradiation. (see Fig. S2†) The redox potential validated the single electron oxidation of AQ-PHEMA by oxygen might occur under irradiation.
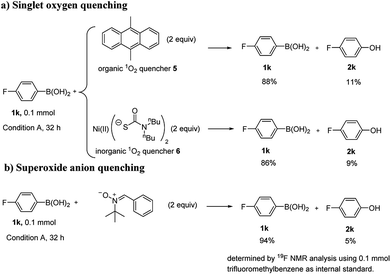 |
| | Scheme 4 Mechanism studies. | |
Proposed mechanism
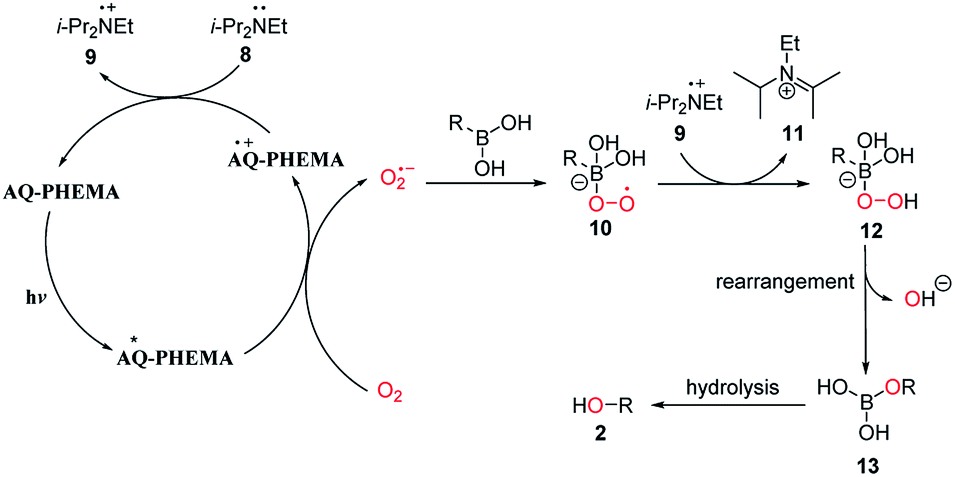
Based on the above control experiments and literature reports,36–38,44–48 a possible reaction mechanism was described as shown in Fig. 4. Firstly, AQ-PHEMA was excited under photo irradiation reacts with molecular oxygen via SET to generate the superoxide radical anion, which further reacted with the boronic acid to form intermediate 10. Intermediate 10 then abstracts a hydrogen atom from 9 to generate intermediate 12. 12 then undergoes rearrangement to form 13 and then hydrolysis to produce the final product 2.
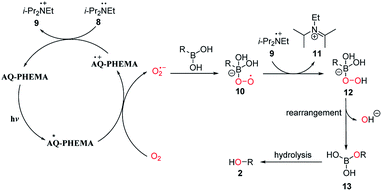 |
| | Fig. 4 Proposed mechanism. | |
Conclusions
In conclusion, we developed a photochemically aerobic photooxidation hydroxylation of boronic acids employing the polymeric photosensitizer AQ-PHEMA as the catalyst. The reaction is excellent yielding and highly efficient and demonstrates broad functional group tolerance. The AQ-PHEMA catalyst could be easily recovered by simple filtration and reused for more than 20 times. The excellent photocatalytic property, easy separation, and recyclability meet the requirements of green chemistry and showed great practical potentials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We greatly acknowledge the financial support from Shanghai Scientific and Technological Innovation Project (18YF1428800).
References
- A. Ding, Y. Zhang, Y. Chen, R. Rios, J. Hu and H. Guo, Tetrahedron Lett., 2019, 60, 660–663 CrossRef CAS
 .
. - H. Kotoucova, I. Strnadova, M. Kovandova, J. Chudoba, H. Dvorakova and R. Cibulka, Org. Biomol. Chem., 2014, 12, 2137–2142 RSC .
- W. Ding, J.-R. Chen, Y.-Q. Zou, S.-W. Duan, L.-Q. Lu and W.-J. Xiao, Org. Chem. Front., 2014, 1, 151–154 RSC .
- H. L. Qi, D. S. Chen, J. S. Ye and J. M. Huang, J. Org. Chem., 2013, 78, 7482–7487 CrossRef CAS PubMed .
- H. Jiang, L. Lykke, S. U. Pedersen, W. J. Xiao and K. A. Jorgensen, Chem. Commun., 2012, 48, 7203–7205 RSC .
- K. Hosoi, Y. Kuriyama, S. Inagi and T. Fuchigami, Chem. Commun., 2010, 46, 1284–1286 RSC .
- E. Kianmehr, M. Yahyaee and K. Tabatabai, Tetrahedron Lett., 2007, 48, 2713–2715 CrossRef CAS .
- Y. K. Bommegowda, N. Mallesha, A. C. Vinayaka and M. P. Sadashiva, Chem. Lett., 2016, 45, 268–270 CrossRef CAS .
- J. Gatenyo, I. Vints and S. Rozen, Chem. Commun., 2013, 49, 7379–7381 RSC .
- U. Bora and A. Gogoi, Synlett, 2012, 23, 1079–1081 CrossRef .
- P. Gogoi, P. Bezboruah, J. Gogoi and R. C. Boruah, Eur. J. Org. Chem., 2013, 2013, 7291–7294 CrossRef CAS .
- R. W. Chen Zhu and J. R. Falck, Org. Lett., 2012, 14, 3494–3497 CrossRef PubMed .
- W. Yin, X. Pan, W. Leng, J. Chen and H. He, Green Chem., 2019, 21, 4614–4618 RSC .
- W. Z. Weng, H. Liang and B. Zhang, Org. Lett., 2018, 20, 4979–4983 CrossRef CAS PubMed .
- N. Gogoi, P. K. Gogoi, G. Borah and U. Bora, Tetrahedron Lett., 2016, 57, 4050–4052 CrossRef CAS .
- A. D. Chowdhury, S. M. Mobin, S. Mukherjee, S. Bhaduri and G. K. Lahiri, Eur. J. Inorg. Chem., 2011, 2011, 3232–3239 CrossRef CAS .
- S. Chatterjee and T. K. Paine, Inorg. Chem., 2015, 54, 9727–9732 CrossRef CAS .
- H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5652–5660 CrossRef CAS .
- X. W. Jimin Xu, C. Shao, D. Su, G. Cheng and Y. Hu, Org. Lett., 2010, 12, 1964–1967 CrossRef .
- K. Inamoto, K. Nozawa, M. Yonemoto and Y. Kondo, Chem. Commun., 2011, 47, 11775–11777 RSC .
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC .
- T. Bach, Angew. Chem., Int. Ed., 2015, 54, 11294–11295 CrossRef CAS PubMed .
- Y. Liu, Y. Song, Y. You, X. Fu, J. Wen and X. Zheng, J. Saudi Chem. Soc., 2018, 22, 439–448 CrossRef CAS .
- S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS .
- E. R. Shilpa and V. Gayathri, J. Saudi Chem. Soc., 2018, 22, 678–691 CrossRef CAS .
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS .
- A. M. Asiri, M. S. Al-Amoudi, S. A. Bazaid, A. A. Adam, K. A. Alamry and S. Anandan, J. Saudi Chem. Soc., 2014, 18, 155–163 CrossRef .
- Y. Zhang, J. Xu and H. Guo, Org. Lett., 2019, 21, 9133–9137 CrossRef CAS .
- M. Jiang, H. Yang and H. Fu, Org. Lett., 2016, 18, 5248–5251 CrossRef CAS .
- I. Kumar, R. Sharma, R. Kumar, R. Kumar and U. Sharma, Adv. Synth. Catal., 2018, 360, 2013–2019 CrossRef CAS .
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed .
- T. Toyao, N. Ueno, K. Miyahara, Y. Matsui, T. H. Kim, Y. Horiuchi, H. Ikeda and M. Matsuoka, Chem. Commun., 2015, 51, 16103–16106 RSC .
- Y.-T. Xu, C.-Y. Li, X.-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Green Chem., 2019, 21, 4971–4975 RSC .
- Y. Q. Zou, J. R. Chen, X. P. Liu, L. Q. Lu, R. L. Davis, K. A. Jorgensen and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS .
- A. Ding, Y. Chen, G. Wang, Y. Zhang, J. Hu and H. Guo, Polymer, 2019, 174, 101–108 CrossRef CAS .
- L. Cui, Y. Matusaki, N. Tada, T. Miura, B. Uno and A. Itoh, Adv. Synth. Catal., 2013, 355, 2203–2207 CrossRef CAS .
- M. Taguchi, Y. Nagasawa, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2016, 57, 230–232 CrossRef CAS .
- Q. Zhou, S. Xu and R. Zhang, Tetrahedron Lett., 2019, 60, 734–738 CrossRef CAS .
- J. M. Carney, R. J. Hammer, M. Hulce, C. M. Lomas and D. Miyashiro, Synthesis, 2012, 44, 2560–2566 CrossRef CAS .
- D. J. Carlsson, T. Suprunchuk and D. M. Wiles, Can. J. Chem., 1974, 52, 3728–3737 CrossRef CAS .
- F. Chalier, M. Hardy, O. Ouari, A. Rockenbauer and P. Tordo, J. Org. Chem., 2007, 72, 7886–7892 CrossRef CAS .
- Y. Liu, L. Wang, Z. Nie, Y. Ji, Y. Liu, K. Liu and Q. Tian, J. Org. Chem., 2006, 71, 7753–7762 CrossRef CAS PubMed .
- S. Kim, Y. Liu, K. M. Nash, J. L. Zweier, A. Rockenbauer and F. A. Villamena, J. Am. Chem. Soc., 2010, 132, 17157–17173 CrossRef CAS .
- L. Fan, J. Jia, H. Hou, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 16437–16440 CrossRef CAS .
- L. Huang, X. Cui, B. Therrien and J. Zhao, Chem.–Eur. J., 2013, 19, 17472–17482 CrossRef CAS .
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS .
- I. G. T. M. Penders, Z. Amara, R. Horvath, K. Rossen, M. Poliakoff and M. W. George, RSC Adv., 2015, 5, 6501–6504 RSC .
- S. D. Sawant, A. D. Hudwekar, K. A. Aravinda Kumar, V. Venkateswarlu, P. P. Singh and R. A. Vishwakarma, Tetrahedron Lett., 2014, 55, 811–814 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization data for all compounds. See DOI: 10.1039/d0ra00176g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b