
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light driven photo-reduction of Cu2+ to Cu2O to Cu in water for photocatalytic hydrogen production†
Shuang Cao‡
ab,
Chuan-Jun Wang‡ *c,
Guo-Qiang Wangc,
Yong Chenab,
Xiao-Jun Lvab and
Wen-Fu Fu*abd
*c,
Guo-Qiang Wangc,
Yong Chenab,
Xiao-Jun Lvab and
Wen-Fu Fu*abd
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, CAS-HKU Joint Laboratory on New Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: fuwf@mail.ipc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cCollege of Chemistry and Material Science, Shandong Agricultural University, Tai'an 271018, P. R. China. E-mail: wangchuanjun@sdau.edu.cn
dCollege of Chemistry and Engineering, Yunnan Normal University, Kunming 650092, P. R. China
First published on 5th February 2020
Abstract
Metal nanoparticles are synthesized via various methods and have found many applications in areas such as sensing, electronics and catalysis. Light induced formation of noble metal nanoparticles, especially platinum, in solution or loaded on semiconductor surfaces, is an established practice in photocatalysis. Nevertheless, preparation of catalytically-active non-precious metal nanoparticles via photo-reduction still have room to be further explored. Here, we report a visible light driven system that can coordinate photo-reduction of CuSO4 to selectively prepare Cu2O or Cu nanoparticles, while at the same time, mediating efficient hydrogen production with in situ generating Cu catalyst without further need to add any components. The Cu2O and Cu nanoparticles in situ generated are crystalline in nature and can perform as pre-catalyst (Cu2O) or catalyst (Cu) to catalyze hydrogen production when reincorporated into the same photo-reduction system with organic photosensitizers. Our work offers an exploratory pathway to prepare target metal nanoparticles while provides some insight into harnessing solar energy for multi-functional purposes.
Introduction
The worldwide fossil-fuel-depletion propels urgent quests for alternative solutions regarding renewable energy resource supplies. Solar energy provides our planet with a vast amount of energy that possess great potential to solve energy problems. Major approaches concerning utilization and conversion of solar energy like the photosynthesis of green plants in nature and the solar cells for electricity generation are versatile but also are limited.1 Developing means of artificial photosynthesis for effectively harnessing solar energy is crucial for the sustainable development of human society.2–5 Photocatalytic hydrogen production has been regarded as a promising solution to relieve the crisis triggered by depletion of fossil fuels.6 Multi-component systems with metal complex mimics as light sensitizer or catalyst have been extensively explored during the past few decades.6–9 Many research studies in this area concern the development of non-precious metal based molecular catalysts to replace noble metal Pt nanoparticles for efficient hydrogen production.10–17 Pt is rare and expensive, which limits its general application. A substantial amount of active molecular photocatalysts based on active metal centers like Fe,8,18 Co,10 Ni8,11–13 have been developed. However, one side effect for most homogeneous molecular catalysts is their degradation when either light intensity or irradiation time is increased.18 Heterogeneous nanoparticle catalysts possess advantages such as easy preparation, good stability, excellent recyclability and reusability. The development of hybrid systems where metal nanoparticles could act as efficient photocatalysts with organic photosensitizers could combine the versatility of homogeneous catalysis with the advantages of heterogeneous catalysts. Copper, one of the most widely used metal in human society, has received increasing attention in photocatalytic hydrogen production. For instance, there have been previous reports on Cu loaded on semiconductors for water reduction under light irradiation.19–23 Copper based molecular catalysts are rare, and there have been literature reported the pre-synthesis of PVP stabilized Cu nanoparticles with 2-phenyl-4-(1-naphthyl)quinolinium as photosensitizer and NADH as sacrificial electron donor in organic water mixed solution under acidic conditions for water reduction, but the hydrogen evolution efficiency is low compared with Ni nanoparticles.24 To make further contribution in this area of study, it is highly desirable to explore systems where stable Cu nanoparticles can combine with cheap organic sensitizers for water reduction in pure neutral or alkaline water solution. The research would hold prospect for coupling to water oxidation reaction, which is thermodynamically possible in basic solution.On the other hand, preparation of metal or metal oxide nanoparticles is crucial for the subsequent use in catalysis. Currently, it has been mostly conducted via chemical methods25 with thermal decomposition, phase-transfer method, use of ionic liquids and microwave synthesis. Some of these processes require coating of the nanoparticles with stabilizing agent, which is often unfavorable for catalytic reactions to take place on active metal surface sites.26,27 Light induced in situ generation of nanoparticles from metal salt precursors holds significance in water splitting reactions.28–32 There have been reports on photo-generation of Cu in Cu(OH)2/TiO2 (ref. 33) and Cu2O/TiO2 (ref. 34) photocatalysts under UV light irradiation. But as far as we know, there has been few work on photo-generation of Cu based nanoparticles in a homogeneous system incorporating organic dyes as reduction agent and photosensitizer, the investigation of such systems may provide further details of mechanistic insight.
In this work, we demonstrate a bi-functional visible light driven system incorporating fluorescein as photosensitizer and reducing agent, and CuSO4 salt as precursor in alkaline TEA or TEOA aqueous solution, for photo-generation of Cu2O and Cu nanoparticles and simultaneous photocatalytic hydrogen production. Tunable preparation of Cu2O or Cu particles is realized by adjusting the choice of TEA and TEOA sacrificial electron donors and exerting control over photo-irradiation time. A stepwise light driven photochemical process from Cu(OH)2 to Cu2O to Cu, which was the real photocatalyst, was proposed based on real-time X-ray diffraction (XRD) monitoring and corroborated hydrogen production experiments. When the in situ generated Cu2O and Cu were incorporated into the same photocatalytic system in place of CuSO4, robust hydrogen production were observed, the system lasted for more than 140 hours with no significant decrease in efficiency and large amounts of hydrogen evolved. A single crystal of a Cu(TEOA)(H2O)2 complex was also cultivated and analyzed, together with a series of dynamic light scattering (DLS), XRD and induction period measurements, it greatly contributes to the clarification of mechanism. It is intriguing that by incorporating Fl and CuSO4 in an alkaline TEOA or TEA solution, and presiding control over photo-irradiation time, we have successfully acquired Cu2O and Cu nanocrystals while simultaneously achieved hydrogen evolution in one system. Our work is expected to provide some new insight on harnessing solar energy for applications in photocatalysis.
Experimental
Assembly of photocatalytic system and hydrogen measurement
In a typical reaction, Fl (10−3 M) was added into a 60 mL quartz tube containing CuSO4·5H2O (1.0 × 10−5 M) and TEA or TEOA (5%, v/v) aqueous solution (30 mL) at pH 11 or 10 (adjusted with 0.1 M HCl). The solution was deoxygenized with N2 for 30 minutes and then the tube was sealed with a rubber cap. The 10 samples (10 tubes containing the same components for parallel experiment) were subjected to irradiation apparatus comprising LED light source (30 × 3 W, λ > 420 nm) and magnetic stirrer. The focused intensity on the flask was about 16 mW cm−2. The number of incident photons was 3.35 × 1017 photons per s as measured by using an irradiance meter. The generated hydrogen from the systems was measured at different time intervals by GC-14C (Shimadzu) which was equipped with a 5 Å molecular sieve column (3 m × 2 mm), thermal conductivity detector and N2 carrier gas. The amount of hydrogen was quantified by an external standard method. The turnovers were calculated based on the mole amount of added CuSO4.Characterization methods
The prepared Cu2O and Cu nanoparticles collected at different time intervals was washed several times with deoxygenated ethanol, centrifuged and dried under an inert atmosphere. The dried nanoparticles were directly used for XRD measurement. For TEM measurement, the nanoparticles were dispersed in deoxygenated ethanol solution. High-resolution transmission electron microscopy (HRTEM) was performed on JEM 2100F which is operated at an accelerating voltage of 200 kV. The SEM images were taken on a Hitachi S-4800 field emission scanning electron microscope operating at 5.0 kV. SEM samples were prepared by drop-casting suspensions onto silicon wafers. X-ray powder diffraction (XRD) spectra were collected on a Bruker D8 Focus under Cu-Kα radiation at (λ = 1.54056 Å). The pH values were adjusted with a Model pH S-3C meter (Mettler Toledo FE20). Dynamic light scattering (DLS) measurements were performed with a Dynapro NanoStar instrument (Wyatt Technology). The DLS instrument used in this study can detect particle sizes ranging from 0.5 to 2000 nm. The light source for the scattering experiments is a He–Ne gas laser (100 mW, λ = 658 nm). Data were obtained using a scattering angle of 90° at 25 °C.Results and discussion
Hydrogen production and tunable preparation of nanoparticles
The photocatalytic system was assembled by adding a certain amount of catalyst precursor CuSO4 (10−4 M) into 30 mL 5% TEA or TEOA sacrificial electron donor aqueous solution, fluorescein (1 mM) was introduced as light sensitizer and reduction agent. Under visible light irradiation, synchronous production of nanoparticles and hydrogen could be observed and determined. Moreover, control experiments showed that when either light irradiation is absent or any component is missing from the system, neither particle formation nor any significant amount of hydrogen evolution can be detected. Optimization of photocatalytic parameters includes different sacrificial donors, pH, photosensitizers, the concentration of Fl and precursor Cu2+ ions.A screen of sacrificial reagents used, from TEA, TEOA to methanol to acetic acid and ascorbic acid revealed that only in the presence of basic TEA and TEOA, can the photocatalytic system show photo-reduction and hydrogen evolution activity. The fact that ascorbic acid, a common effective electron donor13,16 with smaller oxidation potential and is more easily oxidized,35 renders as ineffective sacrificial agent, indicates the strong influence of pH value on the photocatalytic activity of the system. The reason the system is functional in alkaline solution could be associated with the formation of Cu(OH)2 pre-catalyst, fulfilling the photo-reduction process of Cu(OH)2 to Cu2O (−0.08 V vs. NHE) and Cu2O to Cu (−0.358 V vs. NHE). This supposition is in accordance with the photocatalytic phenomenon and will be further discussed in mechanistic part. pH often has strong influence on the excited states of photocatalytic components.12 The influence of pH values on H2 production is scrutinized, the rate of H2 production is highest at pH 11 and 10 in TEA and TEOA solutions respectively (ESI Fig. S1 and S2†). A scrutiny of the photosensitizers used in TEOA and TEA systems revealed that Fl is the most effective one compared with other rhodamine dyes like Erythrosin B, Eosin Yellowish and Rose Bengal (ESI Fig. S3 and S4†). In addition, the concentration of Fl was also tested, the highest hydrogen production activity in TEOA and TEA solutions were found with Fl concentration of 1 mM (ESI Fig. S5 and S6†). At low concentrations of Fl (0.2, 0.5 mM) the system quickly lost hydrogen evolution activity within 12 hours of light irradiation due to the bleach of sensitizers to generate excited states. However, hydrogen production rates are also low at high Fl concentrations, (0.2, 0.5 mM) which may be ascribed to the inefficient input of light and self-quenching of excited state molecules caused by aggregation of dyes in high concentration.36
When varying CuSO4 concentrations for hydrogen production, interesting experimental phenomenons were observed which are crucial to the selective preparation of Cu2O and Cu nanoparticles. The influence of Cu2+ concentration was examined with Fl (1.0 × 10−3 M) and in 30 mL 5% TEA or TEOA aqueous solution adjusted to the optimized pH value (Fig. 1a and b).
 | ||
| Fig. 1 Time course of H2 evolution from systems containing Fl (1.0 mM) and (5% v/v) TEA (a) or TEOA (b) in 30 mL H2O at pH 11 (a) or 10 (b) in a 60 mL volume quartz tube upon irradiation with λ > 420 nm LED light with various concentrations of CuSO4·5H2O. Induction period experiments of H2 evolution in the first 2 hours from system containing Fl (1.0 mM), Cu2+ (1.0 × 10−3 M) in (5% v/v) TEA (c) or TEOA (d) aqueous solution at optimized pH values. (e) XRD patterns of the precipitates collected at different irradiation time from a system have the same experimental conditions as (c). (f) XRD patterns of the precipitates collected at different irradiation time from a system have the same experimental conditions as (d). | ||
In the TEA system, the hydrogen production rate enhanced dramatically with increasing amounts of added CuSO4. At 10−3 M Cu2+ concentration, particle formation was quickly observable to eye upon light irradiation and 4.6 mL H2 was generated within 4 hours of irradiation, corresponding to an initial rate of 1.15 mL h−1. However, the system had shorter lifetime at higher Cu2+ concentration, which is probably due to the fast consummation of Fl and TEA for photo-reduction of particles and photo-evolution of hydrogen. At 10−6 M, the hydrogen production system was low in efficiency but with longer lifetimes, hydrogen production was almost linear for the first 10 hours of irradiation with a turnover frequency (TOF) value of 173 h−1 and a turnover number of 2633 was achieved based on added CuSO4 concentration after 22 hours of irradiation. By contrast, the TEOA system exhibited different experimental results. The initial hydrogen production rate was low but the system displayed longer lifetime compared with those in TEA solution. For instance, at 10−4 M Cu2+ concentration, hydrogen production rate of the first 6 hours is only 0.45 mL h−1 with TEOA compared with 0.71 mL h−1 with TEA as sacrificial reagent, but hydrogen evolution was almost linear for an irradiation period of 25 hours in TEOA aqueous solution, but ceased activity within 12 hours of illumination in TEA system. The improvement in hydrogen evolution rate was also observed in TEOA solution with increasing of Cu2+ from 10−6 M to 10−4 M. However, when the catalyst precursor concentration was further increased to 10−3 M, no hydrogen evolution activity was detected for the first 30 min of light irradiation and hydrogen evolution rate was all the way lower than when Cu2+ concentration was 10−4 M.
Accordingly, we performed some induction-period-measurements together with real-irradiation-time XRD experiments to shed light on the situation. Induction period experiments with Cu2+ concentration of 10−3 M in 5% TEOA and TEA solution was conducted (Fig. 1c and d). It revealed that no hydrogen evolution occurred within the first 40 min in TEOA solution, but for system with TEA, hydrogen evolution happens after 10 min of light irradiation. On examination of the photocatalytic solution, we observed the solution become turbid with particles. Therefore, the precipitates at different irradiation times were collected and characterized by XRD measurements to clarify the reason behind the different hydrogen production results in TEOA and TEA solution, and to identify the changes of added Cu2+ going through during photocatalytic process. For the TEA system, blue precipitate was immediately formed when CuSO4 (10−3 M) was added into the basic TEA aqueous solution. From the XRD patterns (Fig. 1e), characteristic diffraction peaks of Cu(OH)2 were observed. There were six main diffraction peaks near or at 2θ = 16.7, 23.8, 34.1, 35.9, 39.8, 53.2 and 63.2°, corresponding to (020), (021), (002), (111), (130), (150) and (043) diffraction planes of Cu(OH)2 (JCPDS 12-420),31 respectively. After 30 minutes irradiation, black particles were obtained and proved to be mainly Cu(0), with the reflection peaks corresponding to the Cu diffraction plane of (111), (200) and (220).37 At the same time, only a small amount of Cu2O was also detected with the diffraction plane of (111).38 Under the same experimental conditions, particles collected after 2 h of irradiation exhibited the similar XRD patterns with that irradiated for 30 minutes, but with decrease in intensity for Cu2O (111) reflection peak, indicating a further diminish in Cu2O amount. Samples tested after 4 h of light illumination displayed only the Cu(0) reflection peaks and a complete absence of Cu2O. Collectively, these XRD measurements provide us with three items of important information. First, XRD measurements at different hours revealed a fast photochemical process from Cu(OH)2 to Cu2O and Cu, and the gradual transformation of Cu2O to Cu under visible light irradiation in the TEA solution. Second, when coordinating these XRD data with hydrogen production results (Fig. 1a and c), we can identify Cu(0) as an effective photocatalyst and Cu(OH)2 is not. As in the first five minutes when the system is overall with the presence of Cu(OH)2, but no hydrogen is detected and hydrogen evolution is low within the first 20 minutes. In addition, the fact that Cu2O was fully transformed into Cu(0) after 4 h of irradiation while hydrogen production was active for more than 10 hours, is proof that Cu(0) can act as effective catalyst. Third, we have made clear the irradiation time necessary for the selective preparation of pure Cu nanoparticles in the TEA system is irradiation after 4 hours.
For the TEOA system, when Cu2+ (10−3 M) was added into the solution, no precipitation but an indigo blue clear solution was observed. Single crystals were cultivated and we found Cu(TEOA)(H2O)2 complex was formed in the solution based on X-ray single crystal diffraction method. However, the clear solution quickly turned turbid upon light irradiation. As irradiation for 40 min produced no H2 (Fig. 1d), the particles formed at 40 min were collected and were found to be dark red in color. XRD measurement showed that these nanoparticles were pure Cu2O in form (Fig. 1f).35 However, samples measured at 70 min showed that Cu(0) is predominant in these particles. A further diminish in Cu2O amount is observed for samples irradiated 2 h, and complete transformation of Cu2O to Cu was also observed for samples measured after 4 h of irradiation (Fig. 1f). Three important pieces of information can also be extracted from these experiments. First, Cu(TEOA)(H2O)2 complex is not photocatalyst for hydrogen production in our system. As no hydrogen production activity is observed in the first 40 min of irradiation (Fig. 1d). Second, Cu is the sole efficient catalyst for hydrogen evolution in our system. There has been previous report of using Cu2O for overall water splitting under visible light irradiation with a comparatively large amount of Cu2O under prolonged irradiation time (1900 h). The amount of Cu2+ within our system is much less (10−3 M), irradiation of the system for 40 min produced pure Cu2O but no accountable amount H2 proved that Cu2O is not an active photocatalyst to accept excited state electrons from Fl for hydrogen evolution. Only when Cu was generated, can efficient hydrogen evolution occur. Third, by adopting TEOA as the sacrificial agent and exerting precise control over the irradiation time, we have successfully realized the tunable-phase selective preparation of pure Cu2O and Cu nanoparticles in one photocatalytic system while at the same time achieved substantial hydrogen evolution. In addition, when the concentration of Cu2+ is lowered, although evolution of hydrogen could be detected earlier, it was difficult to collect the precipitate for XRD measurement as the few particles in situ generated were well dispersed in the solution. However, we could perorate that Cu2+ was reduced to Cu2O and then to Cu(0) as in 1 mM Cu2+ concentration systems. Lower the concentration of Cu2+ would result in faster photo-reduction processes from Cu2+ to Cu2O to Cu(0), and therefore the earlier evolution of hydrogen gas.
Characterization of Cu2O and Cu nanoparticles
The identity of the tunably prepared Cu2O and Cu nanoparticles was initially characterized by XRD methods. We then utilized a couple of other techniques to acquire more information regarding the morphology and nano-scale interaction of these in situ generated particles, which we hope will provide some mechanistic insights regarding the photocatalytic process. The isolated Cu2O and Cu nanoparticles were examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. 2). It was found that most of the Cu2O (Fig. 2a and b) and Cu (Fig. 2d and e) particles were spherical in form and the majority of them were in the state of agglomeration. Some of the Cu2O nanoparticles are 200 nm to 600 nm in size (Fig. 2a), HRTEM image of their edges showed that these particles were composed of numerous tiny highly crystalline nanoparticles with the size around 2–5 nm (Fig. 2a, inset). The selected area electron diffraction (SAED) pattern showed that both the Cu2O (Fig. 2d inset) and Cu (Fig. 2e inset) nanoparticles had diffraction rings of single crystal. The nanoparticles were found to have good crystalline behaviour, with clear single crystal lattices observed. The spacing of 0.21 nm between adjacent lattice planes was measured for Cu2O, which corresponds to the distance between two (200) crystal planes of Cu2O (Fig. 2d).38 Lattice crystal plane spacing of 0.208 nm was also measured for Cu nanoparticles and can be ascribed to the diffraction of (111) Cu planes.37 In addition, the XPS results were also conducted on Cu nanoparticles collected after 4 h of irradiation in both systems, and on Cu2O nanoparticles collected after 40 min of irradiation in TEOA system. The results showed that compared to that of Cu, The Cu 2p2/3 and Cu 2p1/2 peaks of Cu2O shifted to lower binding energy. The same trend was reported in previous literature (Fig. S7†).20 | ||
| Fig. 2 TEM (a), SEM (b) and HR-TEM (c) images of the tunably prepared Cu2O nanoparticles. TEM (d), SEM (e) and HR-TEM (f) images of the Cu nanoparticles formed in situ under visible light irradiation. Inset of (a): HRTEM image of the edges of the nanoparticles. | ||
Robust hydrogen production with isolated Cu2O and Cu nanoparticles
The tunably prepared Cu2O and Cu nanoparticles were reincorporated into the photocatalytic system as precatalyst or catalyst for photocatalytic hydrogen evolution. When Cu2O (30 μM, 4.4 mg) or Cu (30 μM, 2 mg) was added into systems containing 2 mM Fl photosensitizer in 30 mL 5% TEA or 5% TEOA aqueous solution (Fig. 3c and d), different experimental results were obtained. In the first place, for systems incorporating Cu(0) nanoparticles, instantaneous evolution of hydrogen could be detected upon visible light irradiation. The in situ generated Cu(0) could perform as stable and robust photocatalyst for hydrogen production in the alkaline aqueous solution. For system incorporating TEA as sacrificial electron donor, a total amount of 7.7 mL H2 could be obtained after 31 hours of irradiation (Fig. 3a); for systems with TEOA as sacrificial agent, the system exhibited extraordinarily stable activity, photocatalytic reaction steadily went on for more than 140 h with no significant decrease in hydrogen production rate, producing 35 mL H2 in a single run without extra adding of components (Fig. 3b). In the second place, when Cu2O pre-catalyst was added into the systems, hydrogen production was found to experience an induction period (Fig. S8 and S9†), due to the photo-reduction of Cu2O to Cu(0) photocatalyst (Fig. 3c–f). In TEA solution, the induction period was found to be short with hydrogen evolution happened after 10 min of irradiation (Fig. S8†), and hydrogen production continued for more than 20 h, with more than 9.7 mL hydrogen generated (Fig. 3a). In contrast, the induction period took almost 40 min before hydrogen evolution occurred in TEOA solution (Fig. S9†). Similar as when using Cu as photocatalyst, hydrogen production was more robust in TEOA solution, with more than 44 mL H2 produced after about 140 h of photocatalytic reaction (Fig. 3b). Previous reports showed that TEOA could prevent back-reaction between PS+ and PS− and stabilize the radicals, thereby prolonging the lifetime of singlet excited state of PS. On the contrary, a higher concentration of TEA led to decrease in photocatalytic efficiency because of back-reaction between Fl* and TEA+.39–41 Therefore, the sharp decrease of activity after 20 h in TEA was due to the consumption of Fl. In the third place, we again employed XRD methods to shed light on the time-course transformation of Cu2O to Cu in the photocatalytic system with either TEA or TEOA sacrificial agents (Fig. 3c and d). Most of the Cu2O was found to be transformed into Cu(0) after 40 min of light irradiation in TEA solution while in TEOA solution only trace amounts of Cu(0) was generated at that time. In accordance with XRD measurements in Fig. 1, these results proved that Cu(0) is the active catalyst in our system and also demonstrated a faster photo-reduction rate of Cu2O to Cu when TEA was adopted as the sacrificial electron donor. In the fourth place, we found that for both Cu2O and Cu incorporated, when use TEA as sacrificial agent, the system had a higher H2 evolution rate than when TEOA was used before 10 h of irradiation (Fig. S10 and S11†). After 10 h, hydrogen evolution of both Cu2O and Cu in TEA experienced sharp decrease in efficiency and nearly lost activity after 20 h (Fig. S10†). However, hydrogen evolution of Cu2O and Cu in TEOA increased almost linearly even after 10 hours of irradiation and continually produced hydrogen for more than 120 hours (Fig. S10†). In the last place, hydrogen evolution rate with Cu2O (30 μM, 4.4 mg) was lower than with Cu (30 μM, 2 mg) in the first few hours, which is due to the lower amount of Cu catalyst in the system during the Cu2O transformation process (Fig. 3a and b). However, for systems incorporate Cu2O (30 μM, 4.4 mg), hydrogen production surpassed both in rate and amount after a few hours of irradiation. This can be attributed to the completion of induction period and fully transformation of Cu2O (30 μM, 4.4 mg) to Cu catalyst (theoretically 60 μM, 3.9 mg), which has a larger value than systems with added Cu (30 μM, 4.4 mg). In short, we have proved that the in situ generated Cu2O and Cu nanoparticles could serve as effective pre-catalyst or catalyst for photocatalytic hydrogen production when incorporated into the same system containing Fl and TEA or TEOA. Transformation of Cu2O to Cu, which is the real efficient catalyst in our system, was established and the high photocatalytic activity may be in part attributed to the active facets of the highly crystalline nanoparticles. | ||
| Fig. 3 Hydrogen production from systems incorporating in situ generated Cu2O (black line) (30 μM, 4.4 mg) or Cu (red line) nanoparticles (30 μM, 2 mg) with Fl (2.0 mM) in 30 mL (5% v/v) pH = 11 TEA (a) or (5% v/v) pH = 10 TEOA (b) aqueous solution. (c) and (d) XRD patterns measured at different irradiation times. Sample precipitates were collected from systems initially containing Cu2O nanoparticles (30 μM, 4.4 mg) with Fl (2.0 mM) in 30 mL pH = 11 (5% v/v) TEA (c) or 30 mL pH = 10 (5% v/v) TEOA (d) aqueous solution. | ||
Furthermore, as can be seen from the microscopic images (Fig. 2), the generated Cu nanoparticles showed agglomerated behaviour, which self-assembly into larger nanoparticles. To improve the catalytic efficiency, it is highly necessary to adopt methods that can prevent the overgrowth of nanoparticles,42 such as carbon layer protection,43 introducing graphene as support to form composite material,30,31 use of growth directing agents44 et al. In a following work, we managed to prepare dendritic copper based nanoparticles with uniform size around 20 nm in a photocatalytic system with PVP as directing agent, which we would hopefully to demonstrate in further work.
Mechanistic insights
The photochemical processes governing the photo-reduction of Cu2+ in solution is further investigated to provide some mechanistic insights into the time-coursed in situ phase formation of Cu2O and Cu nanoparticles and the disparity of hydrogen evolution in TEOA or TEA aqueous solution (Fig. 4). As we have discussed above, when CuSO4 (10−3 M) was added into TEA solution, instantaneous formation of blue precipitate was observed and prove to be Cu(OH)2 by XRD measurements (Fig. 1e). Subsequent fast transformation of Cu(OH)2 to Cu2O and then to Cu was happening within 10 minutes and was also monitored by real-time XRD experiments (Fig. 1e and 3c). Bernhard's group reported the photo-reduction of Zn2+ to Zn metal with molecular Ir complex as photosensitizer and TEA as electron donor under visible light irradiation.45 For our system, excited state Fl* and Fl− generated upon light irradiation have reduction potentials of −1.7 and −1.3 V,12 which possess sufficient driving force to provide electrons for reduction of Cu(OH)2 → Cu2O (−0.08 V vs. NHE) and Cu2O → Cu (−0.358 V vs. NHE). Upon generation of Cu, the excited state fluorescein further provide electrons to Cu(0) for photocatalytic hydrogen production. As has been discussed in literature,31 when the concentration of TEA is high (5% v/v) in the system, the excited state Fl* would be first quenched by TEA through a reductive quenching mechanism to form Fl−, which then transfers electron to the precatalyst for reduction process. Therefore, we may safely conclude that the photochemical process in TEA solution is the photo-induced electron transfer from Fl* to Cu(OH)2 through reductive quenching process.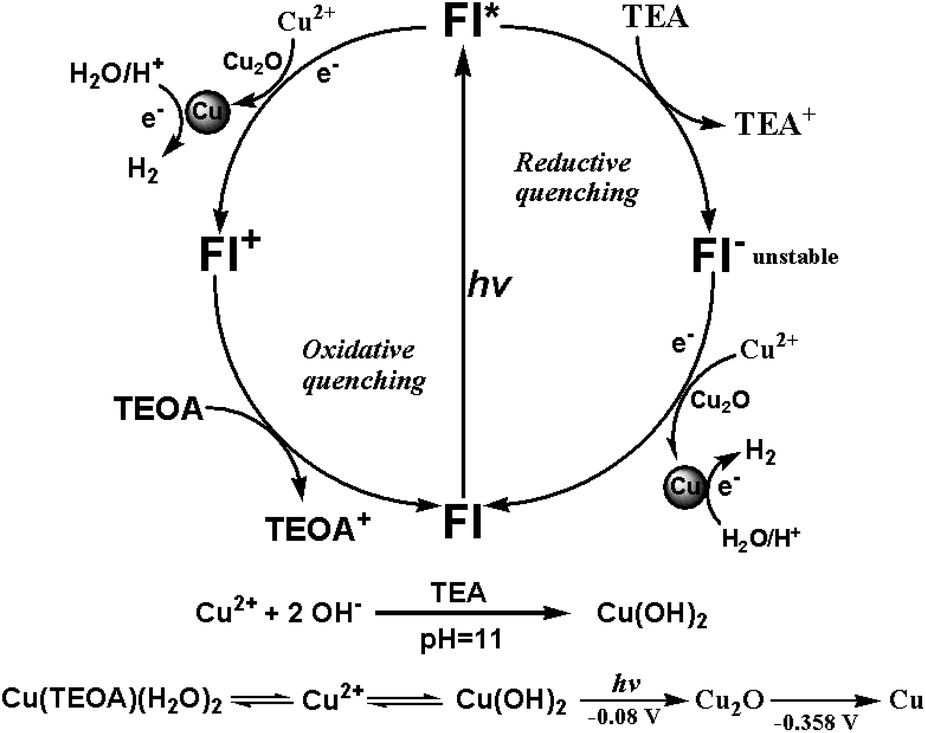 | ||
| Fig. 4 Schematic illustration of the photocatalytic process. | ||
In TEOA solution, however, no sign of the formation of Cu(OH)2 was observed but an indigo blue clear solution when CuSO4 was added. It is reasonable to assume that Cu2+ would coordinate with TEOA to form molecular complex which is soluble in the solution as previously reported for Ni2+ and TEOA.46 Fortunately, single crystals were cultivated by adding CuSO4 in aqueous solution in the presence of excess TEOA, the crystal structure determined by X-ray single crystal method revealed the structure of a Cu(TEOA)(H2O)2 complex, with all of the three O and N atoms of one TEOA molecule plus two water molecules coordinated to Cu2+ (Fig. S12†). On the other hand, we have already proved that this Cu(TEOA)(H2O)2 complex is not a molecular catalyst for hydrogen production through a series of hydrogen production induction-period and XRD experiments (Fig. 1d and f). During photocatalytic experiments, the system quickly turned turbid upon light irradiation for a few minutes, continually producing Cu2O within the first 40 minutes of irradiation. Only when Cu nanoparticles were generated after 40 min could evolution of hydrogen occur (Fig. 1d and f). Therefore, it is likely that the same photochemical process from Cu(OH)2 to Cu2O to Cu catalyst was happening in TEOA solution.
For systems incorporating TEOA sacrificial donor, the pH of the solution optimized for hydrogen evolution is at pH = 10 (Fig. S2†), fast formation of turbids in the solution upon light irradiation means that the Cu(TEOA)(H2O)2 complex was unstable and could dissociate Cu2+ upon light irradiation, which would coordinate with OH− in the alkaline condition to form Cu(OH)2 precatalyst to undergo transformation to Cu2O and Cu. To support this supposition, dynamic light scattering experiments were employed to supply additional evidence (Fig. S13†). Before light irradiation, some particles with the size around 200 nm were detected in the photocatalytic system (Fig. S13a†), which indicates the presence of Cu(OH)2 nanoparticles in the system. Photo-irradiation of the TEOA system for 4 min saw the emergence of particles (Fig. S13c†) around 1500 nm in size, which amount increased after 7 min of irradiation (Fig. S13d†). The size of the particles generated corresponds to the Cu2O size in the SEM images (Fig. 2b) and proves the photo-reduction process from Cu(OH)2 to Cu. So formation of Cu(TEOA)(H2O)2 complex in TEOA solution has the effect of controlled-release of Cu2+ to continually form Cu(OH)2 precatalyst for slow transformation to Cu2O and then to Cu. This proposition is in accordance with the longer induction period measured for hydrogen production in TEOA system. As for the longer induction period observed for TEOA system directly incorporating Cu2O as precatalyst, we may also find root in the unique structural property of TEOA with its three coordinating oxygen atoms, which upon dissociation with Cu2+ may have the ability to readily enclose the in situ generated Cu2O nanoparticles for gradual photo-reduction to Cu(0) nanoparticles. Furthermore, it has been reported in literature, TEOA cannot quench the excited state Fl*.12,31 Therefore, when Fl is used as photosensitizer with TEOA as sacrificial electron donor in aqueous solution, the photochemical process is an oxidative quenching one: the electron transfers from Fl* to Cu(OH)2 → Cu2O (−0.08 V vs. NHE) and Cu2O → Cu (−0.358 V vs. NHE).
Conclusions
In conclusion, we have successfully prepared Cu2O and Cu nanoparticles from inorganic CuSO4 precursor in alkaline aqueous solution in one photocatalytic system; while at the same time achieved efficient hydrogen evolution with in situ generated Cu nanoparticles. The photochemical process regarding the evolution of hydrogen and transformation from Cu(OH)2 to Cu2O to Cu was clarified based on XRD and induction period hydrogen evolution experiments. Cu2O nanoparticles were obtained by photo-reduction of CuSO4 (10−3 M) in TEOA solution after irradiation for 30 min, while Cu nanoparticles were obtained after 4 h of irradiation in both TEA and TEOA solutions. When the in situ prepared Cu2O and Cu were isolated and re-added into the same photocatalytic system, the same photo-reduction process from Cu2O to Cu photocatalyst was detected and the system was highly efficient for photocatalytic hydrogen production with TEOA as sacrificial regent. Single crystal of a Cu(TEOA)(H2O)2 complex and DLS experiments helped to reveal the presence of association and dissociation balance between decomposition of Cu2+ from TEOA and formation of Cu(OH)2 in the alkaline solution. Our work was expected to provide some insight into utilization of solar energy for hydrogen evolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Basic Research Program of China (973 Program 2013CB834804) and the Ministry of Science and Technology of China (2012DFH40090). W.-F. F. thanks the National Natural Science Foundation of China (21471155, 21777136) for financial support. C. J. Wang thanks the Incubation Program of Youth Innovation in Shandong Province for financial support.References
- G. W. Crabtree and N. S. Lewis, Phys. Today, 2007, 60, 37–42 CrossRef CAS.
- D. G. Nocera, ChemSusChem, 2009, 2, 387–390 CrossRef CAS PubMed.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- D. G. Nocera, Energy Environ. Sci., 2010, 3, 993–995 RSC.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680–20683 CrossRef CAS.
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667–1670 CrossRef CAS.
- M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989–7991 RSC.
- H.-Y. Wang, G. Si, W.-N. Cao, W.-G. Wang, Z.-J. Li, F. Wang, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2011, 47, 8406–8409 RSC.
- J. Han, W. Zhang, T. Zhou, X. Wang and R. Xu, RSC Adv., 2012, 2, 8293–8296 RSC.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- T. Yu, Y. Zeng, J. Chen, Y.-Y. Li, G. Yang and Y. Li, Angew. Chem., Int. Ed., 2013, 52, 5631–5635 CrossRef CAS PubMed.
- N. Wu, Int. J. Hydrogen Energy, 2004, 29, 1601–1605 CrossRef CAS.
- W. J. Foo, C. Zhang and G. W. Ho, Nanoscale, 2013, 5, 759–764 RSC.
- I. Mondal, S. Gonuguntl and U. Pal, J. Phys. Chem. C, 2019, 123(43), 26073–26081 CrossRef CAS.
- P. DeSario, J. Pietron, T. Brintlinger, M. McEntee, J. Parker, O. Baturina, R. Stroud and D. Rolison, Nanoscale, 2017, 9, 11720–11729 RSC.
- L. Tong, L. Ren, A. Fu, D. Wang, L. Liu and J. Ye, Chem. Commun., 2019, 55, 12900–12903 RSC.
- Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 6111–6118 RSC.
- C. N. R. Rao, H. S. S. Ramakrishna Matte, R. Voggu and A. Govindaraj, Dalton Trans., 2012, 41, 5089–5120 RSC.
- F. Alonso, P. Riente, J. A. Sirvent and M. Yus, Appl. Catal., A, 2010, 378, 42–51 CrossRef CAS.
- P.-Z. Li, A. Aijaz and Q. Xu, Angew. Chem., Int. Ed., 2012, 51, 6753–6756 CrossRef CAS.
- B. F. DiSalle and S. Bernhard, J. Am. Chem. Soc., 2011, 133, 11819–11821 CrossRef CAS.
- P. Jarosz, P. Du, J. Schneider, S.-H. Lee, D. McCamant and R. Eisenberg, Inorg. Chem., 2009, 48, 9653–9663 CrossRef CAS.
- C. J. Wang, S. Cao and W. F. Fu, Chem. Commun., 2013, 49, 11251–11253 RSC.
- C. J. Wang, S. Cao, B. Qin, C. Zhang, T. T. Li and W. F. Fu, ChemSusChem, 2014, 7, 1924–1933 CrossRef CAS.
- C. J. Wang, Y. Chen, X. J. Lv and W. F. Fu, Appl. Catal., B, 2016, 182, 59–67 CrossRef CAS.
- K. Lalitha, G. Sadanandam, V. D. Kumari, M. Subrahmanyam, B. Sreedhar and N. Y. Hebalkar, J. Phys. Chem. C, 2010, 114, 22181–22189 CrossRef CAS.
- J. Yu and J. Ran, Energy Environ. Sci., 2011, 4, 1364–1371 RSC.
- F. Wen, X. Wang, L. Huang, G. Ma, J. Yang and C. Li, ChemSusChem, 2012, 5, 849–853 CrossRef CAS PubMed.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CrossRef CAS.
- Y. Liu, Y. Chu, Y. Zhuo, L. Dong, L. Li and M. Li, Adv. Funct. Mater., 2007, 17, 933–938 CrossRef CAS.
- S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870–5874 RSC.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CrossRef CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35–41 CrossRef CAS.
- X. Zhang, Z. Jin, Y. Li, S. Li and G. Lu, J. Phys. Chem. C, 2009, 113, 2630–2635 CrossRef CAS.
- M. Zhou, M. Lin, Y. Wang, X. Guo, X. Guo, L. Peng and W. Ding, Chem. Commun., 2011, 47, 9268–9274 RSC.
- M. Zhou, M. Li, C. Hou, Z. Li, Y. Wang, K. Xiang and X. Guo, Chin. Chem. Lett., 2018, 29, 787–790 CrossRef CAS.
- K. Koczkur, S. Mourdikoudis, L. Polavarapu and S. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- A. C. Brooks, K. Basore and S. Bernhard, Inorg. Chem., 2013, 52, 5794–5800 CrossRef CAS.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09590j |
| ‡ The authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |