
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient method to obtain anti-inflammatory phenolic derivatives from Scindapsus officinalis (Roxb.) Schott. by a high speed counter-current chromatography coupled with a recycling mode†
Jinqian Yu a,
Xiaowei Suna,
Lei Zhaob,
Xueyong Wang*c and
Xiao Wang*a
a,
Xiaowei Suna,
Lei Zhaob,
Xueyong Wang*c and
Xiao Wang*a
aQilu University of Technology (Shandong Academy of Sciences), Shandong Analysis and Test Center, Shandong Key Laboratory of TCM Quality Control Technology, Jinan, 250014, P. R. China. E-mail: wangx@sdas.org
bReyoung Pharmaceutical Co., Ltd, No. 44 Cultural West Road, Jinan 250012, P. R. China
cCollege of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing, 100102, P. R. China. E-mail: xueyongwang@bucm.edu.cn
First published on 17th March 2020
Abstract
Herein, we provide an effective separation strategy based on liquid–liquid extraction and two different modes of high speed counter-current chromatography (HSCCC) for the rapid enrichment and separation of compounds from n-butanol-partitioned samples of S. officinalis. Liquid–liquid extraction of the crude sample was performed using a two-phase solvent system composed of ethyl acetate–n-butanol–water with volume ratios of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:9, 5:4:9 and 3:6:9 (v/v), which allowed components with lower polarity and higher polarity to be enriched separately with the first ratio and the other two ratios, respectively. For separation, the conventional and recycling mode HSCCC were combined to develop a strategy for the acquisition of eight phenolic derivatives from the enriched samples, including one new compound, 7-O-[β-D-xylopyranosyl-(1–4)-β-D-glucopyranosyl-(1–4)-α-L-rhamnopyranosyl]-5-hydroxy-2-methyl-4H-1-benzopyran-4-one (5), three caffeoylquinic acid isomers, 3-O-caffeoylquinic acid butyl ester (6), 5-O-caffeoylquinic acid butyl ester (7), 4-O-caffeoylquinic acid butyl ester (8), salidroside (1), drynachromoside B (2), 3,4-dihydroxy-benzoic acid (3), and 5,7-dihydroxy-2-methyl chromone (4). Recycling HSCCC separation was successfully applied to separate the three isomers after six cycles. Furthermore, all the isolates were evaluated for their anti-inflammatory activity against nitric oxide (NO) production in vitro, with 6 and 7 showing significant inhibitory effects with IC50 values of 13.8 μM and 17.6 μM, respectively.
:0:9, 5:4:9 and 3:6:9 (v/v), which allowed components with lower polarity and higher polarity to be enriched separately with the first ratio and the other two ratios, respectively. For separation, the conventional and recycling mode HSCCC were combined to develop a strategy for the acquisition of eight phenolic derivatives from the enriched samples, including one new compound, 7-O-[β-D-xylopyranosyl-(1–4)-β-D-glucopyranosyl-(1–4)-α-L-rhamnopyranosyl]-5-hydroxy-2-methyl-4H-1-benzopyran-4-one (5), three caffeoylquinic acid isomers, 3-O-caffeoylquinic acid butyl ester (6), 5-O-caffeoylquinic acid butyl ester (7), 4-O-caffeoylquinic acid butyl ester (8), salidroside (1), drynachromoside B (2), 3,4-dihydroxy-benzoic acid (3), and 5,7-dihydroxy-2-methyl chromone (4). Recycling HSCCC separation was successfully applied to separate the three isomers after six cycles. Furthermore, all the isolates were evaluated for their anti-inflammatory activity against nitric oxide (NO) production in vitro, with 6 and 7 showing significant inhibitory effects with IC50 values of 13.8 μM and 17.6 μM, respectively.
1 Introduction
Currently, natural products are underexplored resources for structurally unique and pharmacologically active compounds.1 Accordingly, various separation methods, such as conventional open column chromatography and preparative column chromatography, have been developed due to the growing demands for pure compounds in clinical experiments to treat corresponding diseases.2,3 Recently, high-speed counter-current chromatography (HSCCC), a liquid-only partition chromatography separation technique, has been broadly used to separate active constituents from natural medicinal herbs not only due to its simple elimination of adsorption loss of constituents onto solid support matrices, but also its high resolution and separation efficiency. In general, a suitable two-phase solvent system is crucial to HSCCC separation, the selection of which relies on the satisfactory partition coefficient (KD) of the target component.4–6 However, not all components in natural medicinal herbs can distribute discriminately in two-phase solvents due to the similar structures and properties of their isomers, which can be co-eluted in one peak in HSCCC separation. Accordingly, for isomers with similar KD values, recycling mode HSCCC (R-HSCCC) separation has been established as a useful method, which is characteristic of recycling effluents in the same column.7–10 R-HSCCC is equipped with a six-way valve to perform the recycling strategy, which can greatly improve the separation potential of compounds with poor resolution. Additionally, R-HSCCC can diminish the sample- and mobile-phase loss during the separation cycles because the solvents in the column are used both as the sample and mobile phases.As is known, the crude extracts of natural medicinal herbs possess numerous compounds, which make it very difficult to obtain compounds with high purity. Therefore, it is highly challenging to pre-dispose crude extracts properly and simply before HSCCC separation. Currently, the pretreatments available, e.g. silica gel column chromatography (CC), polyamide resin CC, and liquid–liquid extraction, serve not only to enrich the target components but also to remove non-targets.11 Among them, liquid–liquid extraction is one of the most widely used pretreatment techniques for crude extracts because there is no adsorption loss of constituents, and it has high efficiency and easy operation.12,13 Usually, the solvent systems used for traditional liquid–liquid extraction only include water and another immiscible solvent, such as petroleum–water, chloroform–water, ethyl acetate–water and n-butanol–water. However, the polarity changes for the above solvent systems are limited due to their simple solvent constituents, which may result in the incomplete extraction of the targets or loss of minor compounds. Nowadays, an increasing number of two-phase solvent systems composed of multiple solvents are being used in liquid–liquid extraction, which can provide more solvent systems with different polarities, resulting in the targets being completely enriched and impurities completely removed.11 Recently, a quaternary system of n-hexane–ethyl acetate–methanol–water was selected for liquid–liquid extraction and HSCCC separation simultaneously,14 which demonstrates that if the polarities of the solvent systems used for liquid–liquid extraction are in sequential order, the components in the treated samples can be fractioned by polarity sequentially. Correspondingly, the solvent systems can be utilized as a two-phase solvent system for HSCCC separation or substituted with similar solvent systems.
Scindapsus officinalis (Roxb.) Schott. (Araceae), is a Miao folk medicine usually distributed in South China and the tropical region of India. In Yunnan Traditional Chinese Medicine, S. officinalis is decocted for the treatment of bronchitis and pertussis. Previous phytochemical investigations have led to the isolation of chromones, alkaloids, and phenolic cyclobutantetraol esters from S. officinalis,15–17 the first of which have been reported to exhibit promising anti-inflammatory activities. Additionally, it was revealed that the n-butanol extract of the whole plant showed significant anti-inflammatory activity against nitric oxide (NO) production in our earlier pharmacological investigation on S. officinalis. Thus, to obtain the significant constituents from S. officinalis for further pharmacological investigation, an efficient method combining liquid–liquid extraction and HSCCC was employed in the present work to purify one new compound, 7-O-[β-D-xylopyranosyl-(1–4)-β-D-glucopyranosyl-(1–4)-α-L-rhamnopyranosyl]-5-hydroxy-2-methyl-4H-1-benzopyran-4-one (5), and seven known compounds salidroside (1), drynachromoside B (2), 3,4-dihydroxy-benzoic acid (3), 5,7-dihydroxy-2-methyl chromone (4), 3-O-caffeoylquinic acid butyl ester (6), 5-O-caffeoylquinic acid butyl ester (7), and 4-O-caffeoylquinic acid butyl ester (8) (Fig. 1), of which three caffeoylquinic acid isomers (6–8) were isolated by R-HSCCC separation. Additionally, all isolates were evaluated for their anti-inflammatory activity against NO production in vitro.
 | ||
| Fig. 1 Structures of compounds 1–8. | ||
2 Materials and methods
2.1 Apparatus
The HSCCC instrument employed was a TBE-300C high-speed counter-current chromatography (Tauto Biotechnique, Shanghai, China) with a polytetrafluoroethylene (PTFE) multilayer coil (1.6 mm internal diameter and 320 mL total volume) and a 20 mL sample loop. A constant temperature circulator (HX series, Zhengzhou, Changcheng Company, China) was used to control the temperature. The solvent was pumped into the column with an NS-1007 constant-flow pump (Beijing Emilion Science & Technology, Beijing, China). The other instruments included an 8823A-UV detector (Beijing Emilion Science & Technology) at 254 nm and a 3507 portable recorder (Yokogawa, Sichuan Instrument Factory, Chongqing, China).2.2 Chemicals and plant material
Organic solvents including ethanol, petroleum ether, ethyl acetate, methanol, n-butanol, and chloroform used for crude extract preparation and HSCCC separation were all of analytical grade (Damao Chemical Factory, Tianjin, China). Chromatographic grade acetonitrile (Tedia Company Inc, Fairfield, USA) was used for HPLC analysis. The water used was pretreated with a Super Pure Water system (TYP10S, Jinan Taipingma Environmental Protection Equipment Co., Ltd.). The plant material of S. officinalis used here was the same as that in our previous study,14,15 as well as the medicine origin.2.3 Preparation of crude sample
Dried and powdered stems of S. officinalis (1.0 kg) were extracted with 95% EtOH (2 h, 1 h, and 1 h) under reflux, and subsequently partitioned with petroleum ether, EtOAc, and n-butanol sequentially. The n-butanol-partitioned fraction was combined and evaporated to recover the solvent. Finally, 75.0 g of n-butyl alcohol-partitioned fraction was obtained.2.4 Preparation of samples I–V by liquid–liquid extraction
The lower phase (50 mL) of ethyl acetate–n-butanol–water (9:0:9, v/v) was used to dissolve a certain amount of n-butanol-partitioned sample (1.5 g) in a separating funnel, after which an equal volume of the upper phase (50 mL) of the same solvent system was added and the mixture in the separating funnel was shaken. After equilibrium of the sample distribution between the two phases was achieved, the upper phase was separated. The lower phase was extracted twice by the upper phase. The upper phase was combined and evaporated to dryness as sample I. Subsequently, the lower phase (50 mL) of ethyl acetate–n-butanol–water (7:2:9, v/v) was used to dissolve the dried sample in the previous lower phase, and then further extracted twice with the upper phase (50 mL) of ethyl acetate–n-butanol–water (7:2:9, v/v). The upper phase was combined and evaporated to dryness as sample II. Samples III–V were produced using the same method described above with the solvent systems of ethyl acetate–n-butanol–water at the volume ratios of 5:4:9, 3:6:9, and 1:8:9, respectively (Fig. 2).
 | ||
| Fig. 2 Schematic diagram of the liquid–liquid extraction procedure for the n-butanol extracts. n-Butanol extracts: 1.5 g; solvent system: ethyl acetate–n-butanol–water; upper phase: 50 mL, and lower phase: 50 mL. | ||
2.5 HSCCC separation procedure
:1:3, v/v) was shaken and mixed in a separating funnel, and then left to settle. Two phases were separated and treated with ultrasonication to remove any air bubbles. The upper phase was used as the stationary phase and the lower phase as the mobile phase. Besides, an equal volume of the upper and lower phases (5 mL for each phase) was added to dissolve samples III and IV for conventional HSCCC separation.The solvent systems and sample preparation for conventional and recycling HSCCC separation of sample I were the same. Specifically, 2 L of the solvent system of chloroform–methanol–water (4:3:2, v/v) was shaken and mixed in a separating funnel, and then left to settle. Two phases were separated and treated with ultrasonication to remove any air bubbles. The upper phase was used as the stationary phase and the lower phase as the mobile phase. Besides, an equal volume of the upper and lower phases (5 mL for each phase) was added to dissolve sample I and its isomers for conventional and recycling HSCCC separation.
2.6 Analysis and identification of HSCCC peak fractions
Each purified fraction from the HSCCC separation was analyzed by HPLC using a Waters C18 column (250 × 4.6 mm, 5 μm) at 210 and 254 nm. The HPLC system was composed of a Waters 2695 pump, Waters 2695 controller and Waters photodiode array detector (Waters, USA). Evaluation and quantification were performed using an Empower Pro Data System (Waters, USA). The mobile phase consisted of acetonitrile (A) and water (B). The flow rate was kept constant at 1.0 mL min−1 for a total run time of 30 min. The gradient elution program was carried out as follows: 0–20 min, 15% A to 25% A; and 20–30 min, 25% A to 35% A.Identification of the peak fractions was carried out via HRESI-MS and NMR spectroscopy on a Bruker Impact II (Bruker Daltonics Inc., USA) and Varian-600 spectrometer (Varian, CA, USA, using tetramethylsilane as the internal standard), respectively.
2.7 Anti-inflammatory activity assay against NO production
The bioassay for anti-inflammatory activity against NO production was carried out mainly based on the previously described method.15,16 Briefly, Raw 264.7 macrophage cells were treated with compounds 1–8 and dexamethasone at five concentrations of 20.0, 10.0, 5.0, 2.5, and 1.25 μM, and then the stimulant of LPS (1.0 μg mL−1) was added. Finally, Griess reagents I and II were mixed with the cell supernatants to check the NO production, with the absorbance measured at 570 nm. NO inhibition was designated as the percentage reduction in LPS stimulation.3 Results and discussion
3.1 Preparation of the crude samples by liquid–liquid extraction
Traditional liquid–liquid extraction usually involves only two types of solvents, water and an immiscible solvent, which results in the variation in the polarity between the solvent systems being large and the selectivity of different compounds poor. Therefore, two-phase solvent systems composed of three or four solvents were used in the liquid–liquid extraction, and thus the components in the treated samples could be fractioned based on different polarities according to their KD values.11 The samples with different polarities could be further separated by HSCCC using the corresponding solvent systems.A series of solvent systems based on ethyl acetate–n-butanol–water mixed at different volume ratios was selected for liquid–liquid extraction pretreatment of the crude samples of S. officinalis because the sample was from the n-butanol partitioning step. Firstly, the lower phase of ethyl acetate–n-butanol–water (9:0:9, v/v) was used to dissolve the n-butanol-partitioned samples of S. officinalis, after which an equal volume of the upper phase of the same solvent system was added and mixed with the lower phase. After extracting the upper phase twice, it was combined and evaporated to dryness as sample I. It was found that most of the compounds with lower polarities (compounds 5–8 with retention times ranging from 20 min to 30 min in the HPLC analysis) were extracted in the upper phase of ethyl acetate–n-butanol–water (9:0:9, v/v). To obtain the target compounds with higher polarities, n-butanol at a volume ratio of 2 was added and the volume ratio of ethyl acetate was decreased to 7 to promote the transfer of the highly polar compounds from the lower phase to the upper phase, and correspondingly, to ensure the equal distribution of the two phases. As shown in Table 1, the compounds with higher polarities (compounds 1–4 with retention times ranging from 10 min to 20 min in the HPLC analysis) were extracted by the upper phase of ethyl acetate–n-butanol–water (7:2:9, v/v); however, the compounds with lower polarities (compounds 5–8) also appeared in the upper phase, which means the selectivity of the extraction by this solvent system was poor. Therefore, the solvent system of ethyl acetate–n-butanol–water (5:4:9, v/v) was used to extract the compounds with higher polarities. It was found that most of the compounds with higher polarities (compounds 1–4) were extracted in the upper phase of ethyl acetate–n-butanol–water (5:4:9, v/v). Liquid–liquid extraction with the solvent system of ethyl acetate–n-butanol–water (3:6:9, v/v) was carried out, providing similar results as the above solvent system. Finally, the solvent system of ethyl acetate–n-butanol–water (1:8:9, v/v) was employed to extract the target compounds, and the results shown in Table 1 indicate that the components with the highest polarity were present in the lower phase (pure compounds appeared before compound 1 with retention times ranging from 0 min to 4 min in the HPLC analysis) and almost no compounds in the upper phase.
| No. | Ratio (v/v) | Amount of extracts (mg) |
|---|---|---|
| I | 9:0:9 |
368 |
| II | 7:2:9 |
258 |
| III | 5:4:9 |
461 |
| IV | 3:6:9 |
280 |
| V | 1:8:9 |
77 |
According to the above experiments, 368 mg sample I, 258 mg sample II, 461 mg sample III, 280 mg sample IV, and 77 mg sample V were obtained by liquid–liquid extraction using solvent systems of ethyl acetate–n-butanol–water of 9:0:9, 7:2:9, 5:4:9, 3:6:9, and 1:8:9 (v/v) from 1.5 g n-butanol-partitioned sample of S. officinalis. HSCCC separation was further carried out for sample I for compounds 5–8, and samples III and IV for compounds 1–4 (Fig. 3).
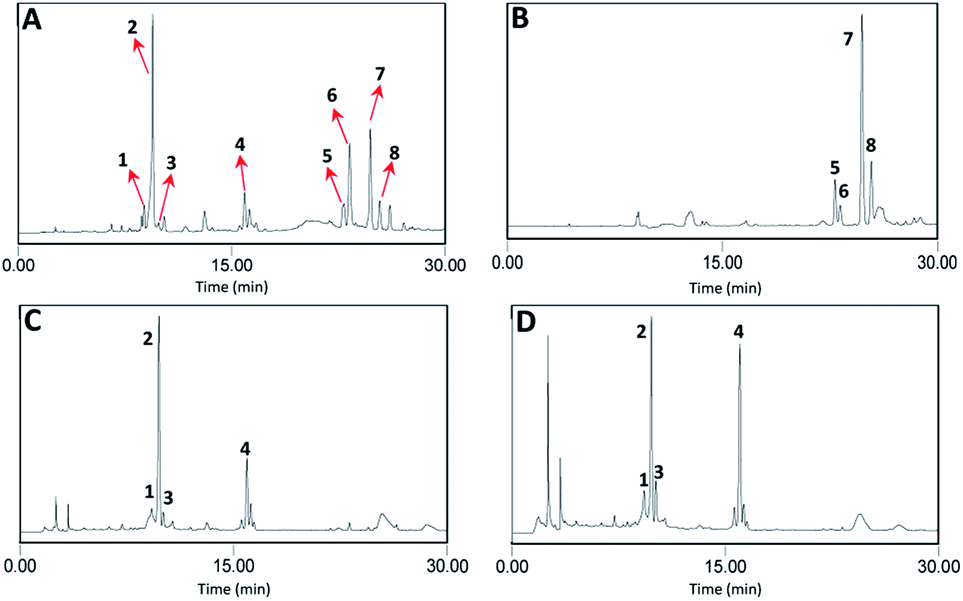 | ||
| Fig. 3 HPLC chromatograms of the n-butanol extract (A), and samples obtained by liquid–liquid extraction of I (B), III (C), and IV (D). Conditions: column, Waters C18 (250 mm × 4.6 mm, 5 μm); mobile phase, acetonitrile (A) and water (B), gradient elution program: 0–20 min, 15% A to 25% A; 20–30 min, 25% A to 35% A; flow rate, 1.0 mL min−1; and detection wavelength, 254 nm. | ||
3.2 Optimization of HSCCC separation conditions for sample I
As one of the most important factors for HSCCC, optimization of the two-phase solvent system can prompt the target compounds to be separated from each other, which can be achieved based on their expected K values ranging from 0.5 to 2.0.18Sample I was obtained by liquid–liquid extraction of the upper phase of ethyl acetate–n-butanol–water (9:0:9, v/v) from the n-butanol-partitioned samples of S. officinalis due to its low polarity. Because sample I was mainly extracted with ethyl acetate, a solvent system containing ethyl acetate was employed to separate the target compounds (5–8), i.e. ethyl acetate–methanol–water with different volume ratios were tested for the KD values of HSCCC separation (Table 2). The results indicate that these solvent systems were too hydrophilic to attract compounds 5–8 in the lower phase. Thus, petroleum ether was added to the solvent system to reduce its hydrophilicity. After employing several solvent systems of petroleum ether–ethyl acetate–methanol–water with different volume ratios, it was found that a suitable KD value for compound 5 could not be obtained. Finally, ethyl acetate in the solvent system of ethyl acetate–methanol–water was changed to chloroform to adjust the polarity of the solvent system. After employing several solvent systems of chloroform–methanol–water with different volume ratios, it was found that chloroform–methanol–water (4:3:2, v/v) gave suitable KD values for 5–8 (Table 2). When HSCCC separation for sample I was carried out with chloroform–methanol–water (4:3:2, v/v), compound 5 was successfully separated; however, compounds 6–8 were eluted together in one peak (Fig. 4). After analysis by HPLC-HRESIMS, compounds 6–8 were determined to possess the same molecular structure and indicated as three isomers, which may be difficult to separate by the conventional HSCCC.
| Solvent system (v/v) | Peak no. | |||
|---|---|---|---|---|
| 5 (KD) | 6 (KD) | 7 (KD) | 8 (KD) | |
| a KD was calculated as the ratio of the sample concentration in the upper (stationary) to lower (mobile) phases.b Compound mainly distributed in the upper phase.c Compound mainly distributed in the lower phase. | ||||
| Ethyl acetate–methanol–water (10:1:10) |
23.5 | 24.3 | 6.4 | 21.8 |
| Ethyl acetate–methanol–water (8:1:7) |
13.8 | 18.9 | 3.2 | 14.2 |
| Ethyl acetate–methanol–water (5:1:5) |
12.1 | 17.4 | 4.1 | 14.9 |
| Petroleum ether–ethyl acetate–methanol–water (1:5:1:5) |
—b | 12.3 | 5.0 | 6.13 |
| Petroleum ether–ethyl acetate–methanol–water (1:2:1:2) |
—b | 3.29 | 0.64 | 0.93 |
| Petroleum ether–ethyl acetate–methanol–water (1:1:1:1) |
—c | 0.71 | 0.02 | 0.05 |
| Chloroform–methanol–water (4:2:3) |
1.17 | 2.49 | 2.87 | 2.66 |
| Chloroform–methanol–water (4:3:2) |
0.74 | 1.02 | 1.10 | 1.21 |
| Chloroform–methanol–water (4:1:4) |
1.18 | 2.75 | 3.53 | 6.10 |
 | ||
| Fig. 4 HSCCC chromatograms of samples III and IV (a), sample I (b), and recycling HSCCC chromatogram of the three isomers (6–8) from sample I (c), obtained from the n-butanol extract of S. officinalis. Solvent system: (a) ethyl acetate–n-butanol–water (2:1:3, v/v); (b) and (c) chloroform–methanol–water (4:3:2, v/v); stationary phase for (a)–(c): upper phase, mobile phase: lower phase; revolution speed for (a)–(c): 800 rpm; sample loading: (a) 200 mg of sample I; (b) 200 mg of sample II; and (c) 80 mg of the three isomers (6–8); detection wavelength for (a)–(c): 254 nm; flow rate for (a)–(c): 2.0 mL min−1; and retention of the stationary phase: (a) 64.2%, (b) 57.4%, and (c) 38.7%. | ||
R-HSCCC is improved HSCCC with a designed six-way valve to form the injecting and recycling positions, and although, it takes a long time, it consumes the same amount of solvent as the conventional HSCCC. After the sample solution was injected through the injection six-way valve, HSCCC was changed to the recycling mode by switching the six-way valve to the recycling position, recycling the effluents in the same column. Thus, for the three isomers, recycling HSCCC was carried out to obtain successful separation. As shown in Fig. 4, one signal peak of the three isomers was separated into three peaks after six cycles of HSCCC with chloroform–methanol–water (4:3:2, v/v). In the second cycle, compound 6 appeared in the slight shoulder peak, with compounds 7 and 8 mixed in the main peak. In the next cycle, the two shoulder peaks were further separated from each other, and after one cycle, compound 8 appeared in the slight shoulder of the second peak. Finally, after six cycles, three peaks were present clearly with the three isomers being separated. Meanwhile, the peak range for the three isomers increased from 56 min to 232 min after six cycles. At the end of the separation, the three isomers were collected separately. As shown in Fig. 4, compounds 5–8 were successfully separated from the combined samples with the flow rate and revolution speed setting as 2.0 mL min−1 and 800 rpm, respectively, and analyzed by HPLC, which indicated that their purities were over 95% (Fig. 5).
 | ||
| Fig. 5 HPLC chromatograms of the n-butanol extract, the three isomers (6–8), and the purified eight peaks. | ||
3.3 Optimization of HSCCC separation conditions for samples III and IV
Samples III and IV were obtained by liquid–liquid extraction of the upper phases of ethyl acetate–n-butanol–water (5:4:9 and 3:6:9, v/v) due to their higher polarity. Because the compositions of samples III and IV were almost the same, these two samples were combined or further HSCCC separation. The two extraction solvent systems were both screened; however, the KD values for compounds 1–4 were larger than 2.0, which would prolong the separation time. Therefore, the volume ratios of ethyl acetate and n-butanol were adjusted, and eventually, the solvent system of ethyl acetate–n-butanol–water (2:1:3, v/v) was employed to separate compounds 1–4, the polarity of which was slightly lower than that of the two extraction solvents. As shown in Fig. 4, compounds 1–4 were successfully separated from the combined samples with the flow rate and revolution speed setting of 2.0 mL min−1 and 800 rpm, respectively, and analyzed by HPLC, which indicated that their purities were over 95% (Fig. 5).
3.4 Identification of the isolated compounds
Identification of the obtained compounds was carried out by spectroscopic analysis and comparison with the literature.19–23 Eventually, one new compound (5) was obtained, together with seven known compounds, including salidroside (1), drynachromoside B (2), n-butyl-O-β-D-fructopyranoside (3), 5,7-dihydroxy-2-methyl chromone (4), 3-O-caffeoylquinic acid butyl ester (6), 5-O-caffeoylquinic acid butyl ester (7), and 4-O-caffeoylquinic acid butyl ester (8).Compound 5 was established to possess a molecular structure of C27H36O17 based on its 13C NMR and HR-ESIMS spectra, which provided an m/z 631.1926 [M − H]−, indicating ten degrees of unsaturation. The characteristic signals for the 5,7-dioxygenated chromone moiety were observed in the 1H NMR spectrum of 5, appearing as an OH group at δH 12.65 (s), two meta-coupled aromatic protons at δH 6.54 (1H, d, J = 2.0 Hz, H-8) and 6.33 (1H, d, J = 2.0 Hz, H-6), an olefinic proton at δH 6.13 (1H, s, H-3), and a tertiary methyl at δH 2.32 (3H, s, Me-11).14 In addition, three anomeric protons at δH 5.44 (1H, d, J = 1.2 Hz, H-1′) and 4.54 (2H, overlapped, J = 6.4 Hz, H-1′′, H-1′′′), which correlated to the corresponding anomeric carbons at δC 98.5 (C-1′), 104.3 (C-1′′), 102.8 (C-1′′′) in the HSQC spectrum, and one secondary methyl at δH 1.20 (3H, d, J = 6.0 Hz, H-6′) were observed in the 1H NMR spectrum. The NMR spectroscopic data of 5 was compared in detail with that of drynachromoside A,15 which indicated that they are superimposable with each other except for the existence of one additional sugar moiety linked at C-4′′. This exception was further verified by the HMBC correlations of 4.54 (H-1′′′) with δC 82.3 (C-4′′). For the sugar moiety, 2D NMR and acid hydrolysis analyses were employed to assign the three sugars as one α-L-rhamnopyranose, one β-D-glucopyranose, and one β-D-xylopyranose. Therefore, compound 5 was elucidated to be 7-O-[β-D-xylopyranosyl-(1–4)-β-D-glucopyranosyl-(1–4)-α-L-rhamnopyranosyl]-5-hydroxy-2-methyl-4H-1-benzopyran-4-one.
7-O-[β-D-xylopyranosyl-(1–4)-β-D-glucopyranosyl-(1–4)-α-L-rhamnopyranosyl]-5-hydroxy-2-methyl-4H-1-benzopyran-4-one (5): ESI-MS, m/z 631.1926 [M − H]−. 1H (DMSO-d6, 400 MHz): 1.20 (3H, d, J = 6.0 Hz, H-6′), 2.32 (3H, s, H-11), 3.13 (2H, overlapped, H-3′, H-3′′), 3.20 (1H, t, J = 8.0 Hz, H-4′′), 3.49 (5H, overlapped, H-2′, H-2′′, Hb-6′′, H-2′′′, Hb-5′′′), 3.53 (1H, t, J = 9.6 Hz, H-4′), 3.66–3.91 (5H, overlapped, H-5′, H-5′′, Ha-6′′, H-4′′′, Ha-5′′′), 4.54 (2H, overlapped, J = 6.4 Hz, H-1′′, H-1′′′), 5.44 (1H, J = 1.2 Hz, H-1′), 6.13 (1H, s, H-3), 6.33 (1H, s, H-6), 6.54 (1H, s, H-8), 12.65 (1H, s, OH-5); 13C NMR (DMSO-d6, 100 MHz): 18.4 (C-6′), 20.3 (C-11), 61.4 (C-6′′), 65.1 (C-5′′′), 67.2 (C-5′), 68.9 (C-2′), 70.0 (C-4′′′), 70.2 (C-3′), 70.3 (C-5′′), 71.8 (C-2′′′), 72.3 (C-2′′), 76.9 (C-3′′, 3′′′), 81.5 (C-4′), 82.3 (C-4′′), 94.9 (C-8), 98.5 (C-1′), 99.9 (C-6), 102.8 (C-1′′′), 104.3 (C-1′′), 105.4 (C-10), 108.6 (C-3), 157.7 (C-9), 161.5 (C-5), 161.8 (C-7), 168.8 (C-2), 182.4 (C-4).
3.5 Anti-inflammatory activity assay against NO production
Since our earlier pharmacological investigation on S. officinalis revealed that the n-butanol extract of the whole plant showed significant anti-inflammatory activity against NO production, the obtained eight compounds (1–8) were evaluated for their anti-inflammatory activity against NO production in vitro using dexamethasone (IC50 value of 2.1 μM) as a positive control. The results (Fig. 6) indicate that all eight compounds show dose-dependent inhibitory effects, i.e. higher inhibitory rates at high concentrations, and lower inhibitory rates at low concentrations, among of which, only compound 1 exhibited no inhibitory effect at the concentration of 1.25 μM, and compounds 6 and 7 showed significant inhibitory effects with IC50 values of 13.8 μM and 17.6 μM, respectively. The pharmacological investigation on the isolated compounds was in accordance with the earlier pharmacological investigation on the n-butanol extract of S. officinalis, offering further research prospects.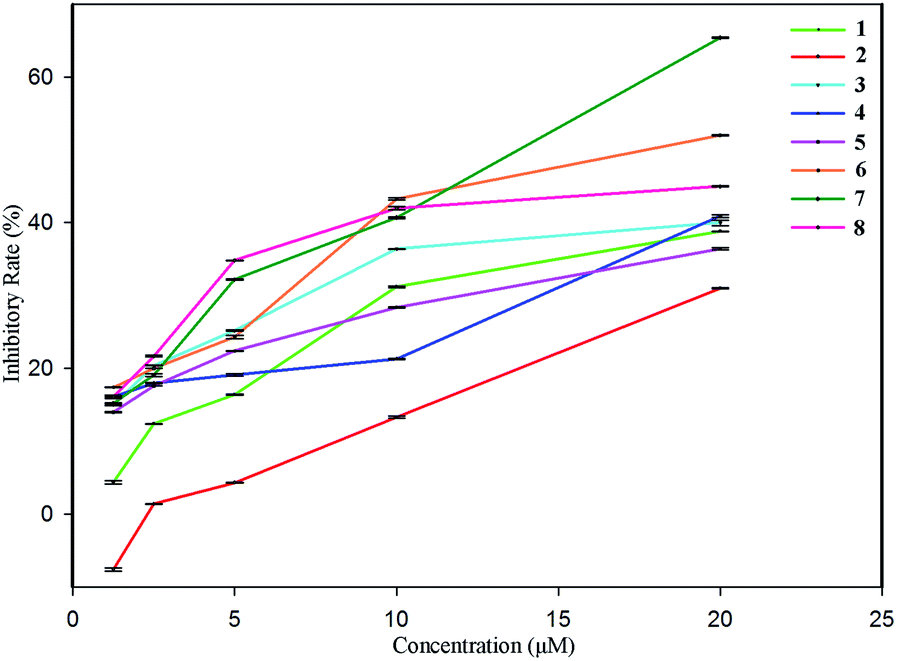 | ||
| Fig. 6 Inhibitory activities of compounds 1–8 against NO production. | ||
4 Conclusions
Limited by the complexity of natural medicines and similarity of their isomers, effective pretreatment and separation methods are necessary. In this investigation, a sample pretreatment method assisted by modified liquid–liquid extraction using two-phase solvent systems of ethyl acetate–n-butanol–water with five different volume ratios was selected to enrich the compounds from the n-butanol-partitioned samples of S. officinalis based on polarity, which allowed the complex sample to be separated into two parts with different polarities. For the separation, conventional and recycling HSCCC were combined to develop a strategy to obtain eight compounds, including three isomers. Accordingly, the results indicate that the modified liquid–liquid extraction process using two-phase solvent systems is an effective pretreatment method for complex samples, which can enrich compounds according to their polarity. It is unquestionable that the recycling mode HSCCC separation is a powerful method to obtain compounds with similar structures and partition coefficient values, which can improve the poor peak resolution of isomers compared with the conventional HSCCC separation. Finally, the obtained compounds were evaluated for their anti-inflammatory activity against NO production according to our earlier pharmacological investigation on the n-butanol extract of S. officinalis, which revealed that nearly all the obtained compounds show certain inhibitory effects. In conclusion, the pretreatment and separation strategy reported herein is efficient for the comprehensive separation and rapid discovery of the main active compounds from complicated natural medicinal plants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation (ZR2018QH006), Shandong Provincial Key Research and Development Plan (2019GSF108004), the National Key R&D Program of China (2017YFC1700703, 2017YFC1702701) and the Priority Research Program of the Shandong Academy of Sciences.References
- A. Stierle, J. Nat. Prod., 2018, 81, 1125–1126 CrossRef CAS.
- S. T. Xu, H. Yao, M. Hu, D. H. Li, Z. Y. Zhu, W. J. Xie, H. Q. Yao, L. Wu, Z. S. Chen and J. Y. Xu, J. Nat. Prod., 2017, 80, 2391–2398 CrossRef CAS PubMed.
- H. Song, J. Lin, X. Zhu and Q. Chen, J. Sep. Sci., 2016, 39, 1574–1591 CrossRef CAS PubMed.
- C. L. Sun, J. Li, D. J. Wang, J. Q. Yu, X. Wang and L. Q. Huang, RSC Adv., 2015, 92, 75831–75837 RSC.
- Q. Liu, H. L. Zeng, S. J. Jiang, L. Zhang, F. Z. Yang, X. Q. Chen and H. Yang, Food Chem., 2015, 186, 139–145 CrossRef CAS PubMed.
- D. J. Wang, H. Zhu, M. Li, X. Y. Song, H. J. Yan, J. Q. Yu and X. Wang, Ind. Crops Prod., 2018, 126, 69–75 CrossRef CAS.
- H. J. Yan, L. Chen, A. Y. Bai, H. Q. Zhao, M. Kong and L. Cui, Ind. Eng. Chem. Res., 2018, 57, 15840–15845 CrossRef CAS.
- D. J. Wang, H. Q. Zhao, H. Zhu, L. Wen, J. Q. Yu, L. L. Li and Y. L. Geng, Ind. Crops Prod., 2019, 129, 224–230 CrossRef CAS.
- T. Y. Ma, H. J. Dong, H. Lu, H. Q. Zhao, L. P. Guo and X. Wang, J. Chromatogr. A, 2018, 1578, 61–66 CrossRef CAS PubMed.
- T. Chen, X. Yang, N. N. Wang, H. M. Li, J. Y. Zhao and Y. L. Li, J. Sep. Sci., 2018, 41, 3660–3668 CrossRef CAS PubMed.
- J. J. Wang, D. Y. Gu, M. Wang, X. F. Guo, H. Q. Li, Y. Dong, H. Guo, Y. Wang, M. Q. Fan and Y. Yang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1053, 16–19 CrossRef CAS PubMed.
- D. L. Zou, X. L. Zhu, F. Zhang, Y. R. Du, J. B. Ma and R. W. Jiang, J. Agric. Food Chem., 2018, 66, 1008–1014 CrossRef CAS PubMed.
- L. H. Wang, Y. H. Mei, F. Wang, X. S. Liu and Y. Chen, Sep. Purif. Technol., 2011, 77, 397–401 CrossRef CAS.
- J. Yang, D. Y. Gu, Z. N. Ji, C. Fang, F. Xu and Y. Yang, Ind. Crops Prod., 2018, 124, 363–368 CrossRef CAS.
- J. Q. Yu, X. Y. Song, D. J. Wang, X. Y. Wang and X. Wang, Fitoterapia, 2017, 122, 101–106 CrossRef CAS PubMed.
- J. Q. Yu, X. Y. Song, P. Yang, X. Y. Wang and X. Wang, Fitoterapia, 2018, 129, 54–61 CrossRef CAS PubMed.
- J. Q. Yu, X. W. Sun, Y. Mu, X. G. Shi, X. Y. Wang and X. Wang, Fitoterapia, 2019, 137, 104244 CrossRef CAS PubMed.
- Y. C. Zhang, C. M. Liu, Y. J. Qi, Y. C. Li and S. N. Li, Ind. Eng. Chem. Res., 2015, 54, 3009–3017 CrossRef CAS.
- T. Y. Shi, H. Chen, L. L. Jing, X. Y. Liu, X. L. Sun and R. Jiang, Synth. Commun., 2011, 41, 2594–2600 CrossRef CAS.
- L. F. Han, F. Zheng, Y. Zhang, E. W. Liu, W. Li, M. H. Xia, T. Wang and X. M. Gao, Nat. Prod. Res., 2015, 29, 1703–1710 CrossRef CAS PubMed.
- Z. P. Shang, J. J. Meng, Q. C. Zhao, M. Z. Su, Z. Luo, L. Yang and J. J. Tan, Fitoterapia, 2013, 84, 130–134 CrossRef CAS PubMed.
- Y. L. Lin, C. K. Lu, Y. J. Huang and H. J. Chen, J. Agric. Food Chem., 2011, 59, 8789–8795 CrossRef CAS PubMed.
- N. Tanaka, Y. Kashiwada, T. Nakano, H. Shibata, T. Higuchi, M. Sekiya and Y. Takaishi, Phytochemistry, 2009, 70, 141–146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for the isolated compounds. See DOI: 10.1039/c9ra09453a |
| This journal is © The Royal Society of Chemistry 2020 |