DOI:
10.1039/C9RA09429F
(Paper)
RSC Adv., 2020,
10, 4568-4578
Novel ultra-small Pd NPs on SOS spheres: a new catalyst for domino intramolecular Heck and intermolecular Sonogashira couplings†
Received
12th November 2019
, Accepted 21st December 2019
First published on 28th January 2020
Abstract
We report a novel catalyst Pd/SOS that catalyzes the dual C–C bond forming coupling of an iodoarene moiety with an internal alkene and an external alkyne via an intramolecular Heck reaction, followed by an intermolecular Sonogashira reaction, respectively. The catalyst was characterized using XRD, IR, XPS, SEM and TEM analyses. Notably, for the first time, cheap and readily available new silica [nanosilica on microsilica (SOS)] material-supported ultra-small Pd nanoparticles (2.20 nm) are employed for the efficient synthesis of dihydrobenzofuran and oxindole derivatives in a domino one-pot reaction. Significantly, a sub-molar quantity of Pd (0.3 mol%) was found to be sufficient to furnish the products in very good to near quantitative yields. Gratifyingly, the catalyst could be recycled up to five cycles with a marginal loss (∼no loss) of the product.
Introduction
Nowadays, nanomaterials are an integral part of almost all technological developments. Although the field of the synthesis of nanomaterials has well expanded in the last two decades, the innovation of unique nanostructures with scalable and sustainable synthesis remains a critical challenge. In the recent past, supported ultra-small Pd nanoparticles have been the area of intense research.1 The unsupported Pd nanoparticles tend to aggregate and are difficult to handle in catalytic reactions.1 To overcome this aggregation, various supporting materials have been developed including microporous and mesoporous metal oxides,2 carbon,3,4 and polymer materials.5 The recent development of silica-supported metal nanoparticles has further boosted the fundamental research and industrial potential of heterogeneous catalysts because these are inexpensive and eco-friendly materials with reasonable catalytic performance and stability.6
High-surface-area silica has broad applications in many fields such as catalysis, medical imaging and drug delivery.7 Vitally, the natural properties of silica can be tuned by fluctuating various parameters, for example, the size, shape and morphology.7 The interest in silica nanospheres with different sizes, measurements and morphology is persistently increasing since modern industries have a huge preference for such materials.8 The effectiveness of these materials is mostly due to their micro and mesostructures, which enable dynamic atoms to scatter on the extensive inner surface and enhance the activity.9 The availability of active sites inside the nano-silica particles is vital as the poor accessibility of active sites will restrict their applications when significant mass transport is essential.7 Hence, high-surface-area nano-silica with the better availability of active sites is required. Moreover, silica has tunable pore structures with a non-toxic nature. Therefore, due to these reasons, silica is the best solid support for organic transformations. Based on the advantages of silica, we have synthesized novel silica material-supported ultra-fine Pd particles and showed their efficacy for the domino preparation of dihydrobenzofuran and oxindole derivatives.
Particularly, dihydrobenzofurans and oxindoles are ubiquitous scaffolds with interesting biological properties.10,11 Specifically, 3,3′-disubstituted dihydrobenzofurans and oxindoles constitute the main core structures of natural products and pharmaceutical compounds.12,13 Therefore, these core structures (dihydrobenzofurans and oxindoles) have drawn considerable attention from the synthetic community. As a result, notable domino protocols were established for their synthesis via homogeneous catalysis.12,13 We have also established the efficient synthesis of benzofurans, oxindoles, and indolines under homogenous catalysis.14,15 Domino reactions have been proven to be incredible synthetic organic reactions in chemistry as they permit the formation of at least two or more bonds in a single operation. To the best of our knowledge, there are no reports on domino reactions via heterogeneous catalysis using sub-molar quantities of the catalyst. Therefore, the development of novel heterogeneous catalysts has attracted interest from the scientific community. Moreover, heterogeneous catalysis is a powerful tool for environmentally benign processes. For this reason, when compared to traditional methods, heterogeneous catalysis is the new and promising alternative for the formation of 3,3′-disubstituted dihydrobenzofurans and oxindole derivatives.
In this study, for the first time, we synthesized a novel silica material, i.e., nano-silica uniformly distributed on the surface of bulk silica (SOS). Moreover, it was identified that ultra-small particles were dispersed on the surface of the SOS material. As an application in organic transformation, the products were obtained in very good to near quantitative yields in a domino fashion and by using sub-molar quantities of the catalyst. Also, we successfully recycled the novel catalyst without a considerable loss of the product.
Results and discussion
Synthesis of SOS
Polyvinylpyrrolidone (PVP, 10k MW, 1.262 g) and cetyltrimethylammonium bromide (CTAB, 0.0625 g) were dissolved in distilled water (20 mL). To this solution, methanol (40 mL) was added with stirring; then, ammonium hydroxide (2.8%, 10 mL) was added and the solution was stirred for 15 min. Finally, to this stirred solution, 3-mercaptopropyl trimethoxysilane (MPTMS, 2 mL) was added. The resultant solution was stirred overnight at room temperature. The formed SOS spheres were collected by centrifugation and dried. Furthermore, these SOS spheres were calcined at 600 °C for 5 h. The synthetic process of SOS is shown in Scheme 1.
 |
| | Scheme 1 Diagrammatic representation of SOS material synthesis. | |
Synthesis of Pd/SOS catalyst
To a round bottom flask, we added 55 mg (0.31 mmol) of PdCl2, 30 mg of PVP (10k MW) and water (25 mL) at room temperature. The resultant mixture was stirred at 80 °C for 30 min. To the reaction mixture cooled to room temperature, we added 0.126 M of aq. NaBH4 solution and 500 mg of SOS and stirred at 80 °C for 3 h. The resultant Pd/SOS was washed with water (2 × 10 mL) and EtOH (2 × 10 mL) and dried at 110 °C for 12 h under vacuum. The resultant black coloured material was grained and used as such for further analysis and its catalytic applications. The synthetic process of Pd/SOS is shown in Scheme 2.
 |
| | Scheme 2 Diagrammatic representation of Pd/SOS material synthesis. | |
Characterization
XRD analysis. The powder XRD pattern of Pd/SOS is shown in Fig. 1, which confirms the structure and phases of Pd/SOS. For Pd/SOS, the Pd nanoparticles showed diffraction peaks at the 2θ values of 40.13°, 46.64°, 67.96° and 81.97° and the corresponding d-spacing values were 2.246 Å, 1.947 Å, 1.379 Å and 1.175 Å. These 2θ and d-spacing values can be attributed to the (111), (200), (220), and (311) lattice planes of Pd (JCPDS no. 87-0638).16 The broad amorphous peak at the 2θ of 24° indicates the presence of silica (SOS).
 |
| | Fig. 1 The powder XRD pattern of the Pd/SOS catalyst. | |
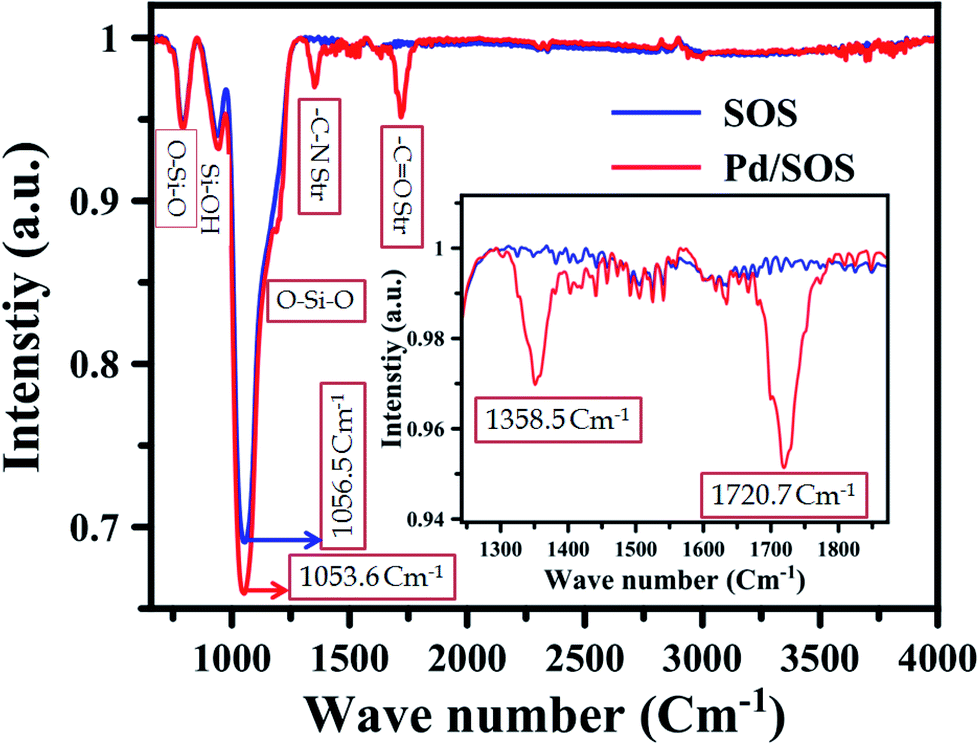
IR spectrum analysis. The FTIR spectra of SOS and Pd on PVP-decorated SOS were obtained in the range of 650–4000 cm−1; the spectral data are shown in Fig. 2. For SOS and Pd/SOS, the observable peaks were found at 3798.2 cm−1, 1630.0 cm−1, 1525.2 cm−1, 1056.5 cm−1, 941.8 cm−1 and 795.9 cm−1, whereas for Pd/SOS, peak shifts were observed. The small broad peak due to the adsorbed water shows an asymmetric broadband range from 3560 to 3640 cm−1. This band is attributed to moisture, which exists everywhere around us.17 The moisture on SOS was mainly trapped as Si–OH by breaking the bonds in the network of the SiO2 nanospheres.18,19 Therefore, the resultant stretching frequency of the –OH bond in Si–OH was observed as a sharp peak at 3710.5 cm−1 and the bending mode of the –OH bond in Si–OH could be seen at around 1630.0 cm−1 but was hindered by the many stretching peaks of Si–O.19 A small broad peak can be seen at 1525.2 cm−1, which indicates the stretching bands of Si–O.19 The main and highest intense band of the SOS nanospheres was observed at 1056.5 cm−1, which indicated the antisymmetric stretching frequency of the Si–O–Si bond.20–22 In contrast, the symmetric stretching frequency of the Si–O–Si bond was observed at 795.9 cm−1.21 For the weakly bonded silanols (Si–OH) on the SOS surface, we observed a vibrational band at 940.3 cm−1.20 When Pd nanoparticles were dispersed on the surface of SOS, the highest intense band for Si–O–Si shifted to the lower frequency side, i.e., 1053.6 cm−1 and the peak for Si–OH shifted to the higher frequency side, i.e., 946.0 cm−1.20 The shifts in the Si–O–Si and Si–OH bands indicate the successful modification of the SOS spheres by Pd nanoparticles.20 In addition, the sharp peaks observed at 1358.5 cm−1 and 1720.7 cm−1 indicate the stretching frequencies of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in polyvinylpyrrolidone (PVP), which is decorated on the surface of SOS.23
O in polyvinylpyrrolidone (PVP), which is decorated on the surface of SOS.23
 |
| | Fig. 2 The FT-IR spectra of SOS and Pd/SOS materials. | |
XPS analysis. The oxidation states and quantifications of the elements in the catalyst were characterized by X-ray photoelectron spectroscopy (XPS). The wide scan XPS spectra of Pd/SOS and the Pd 3d, Si 2p, O 1s and C 1s core-level spectra are shown in Fig. 3. In Fig. 3, the characteristic peaks for 3d3/2 and 3d5/2 are observed at 334.7 eV and 339.7 eV, respectively, and these peaks are assigned to Pd (0).24,25 Moreover, the core-level spectrum of Si 2p shows binding energies at 101.3 eV, 102.2 eV, 103.0 eV, 105.5 eV, 108.3 eV and 109.1 eV. The binding energies of Si 2p observed at 101.3 eV, 102.2 eV and 103.0 eV confirm the presence of silicon suboxide peaks, i.e., Si2+, Si3+ and Si4+, respectively.26 In contrast, the peaks at 105.5 eV, 108.3 eV and 109.1 eV account for the unique chemical state of Si atoms (Si4+ from SiO2).27,28 The oxygen atoms are surrounded by the Si atoms in SOS, which supplies a certain density to the Si atoms. Therefore, the local position of the peak for Si 2p shifts to the higher binding energy side.27–29 Also, the O 1s core-level spectrum shows six components of binding energies at 530.1 eV, 530.9 eV, 532.5 eV and 533.5 eV, which indicate the oxygen atom-surrounded environments, i.e., Si–O–Pd, Si–O–Si, Si–O–H, and adsorbed oxygen, respectively;30 in contrast, the peaks at 534.2 eV and 537.7 eV indicate the oxygen atoms surrounded by Si atoms.28 Also, the C 1s peaks at 285.3 eV, 285.8 eV, 286.9 eV, and 289.5 eV indicate the carbon atoms in CC/C–C, C–N, C–O, and O–CO, respectively.31 The core-level spectrum of N 1s shows a peak at 399.1 eV, which originates from the pyridinic nitrogen (N) in PVP.32
 |
| | Fig. 3 The XPS pattern of Pd/SOS catalyst. | |
Quantification of elements by XPS. The quantification of elements in the Pd/SOS catalyst was determined by XPS. The atomic and mass concentrations of each element in the Pd/SOS catalyst are shown in Table 1.
Table 1 The quantification of elements in Pd/SOS catalyst
| Element |
Atomic conc. [%] |
Error [%] |
Mass conc. [%] |
Error [%] |
| Pd |
0.10 |
0.05 |
0.74 |
0.34 |
| C |
62.75 |
0.50 |
50.95 |
0.62 |
| Si |
0.96 |
0.41 |
18.90 |
0.70 |
| O |
27.19 |
0.39 |
29.41 |
0.44 |
SEM analysis. The FE-SEM images of nanosilica spheres on bulk silica spheres (SOS) are shown in Fig. 4. From the SOS SEM images, we demonstrate that SOS are smooth and spherical in shape. Moreover, Fig. 4 reveals that the nano-silica spheres are uniformly distributed on the surface of each bulk silica sphere. Also, the average size of nano-silica spheres is 250 nm. In contrast, the average size of bulk silica spheres is 3.5 μm. Moreover, the particle size distribution of nano and bulk silica is shown in Fig. 4.
 |
| | Fig. 4 SEM images and average particle distribution of SOS. | |
TEM analysis. For the further confirmation of the morphology of SOS and the particle size of Pd nanoparticles, we used transmission electron microscopy (TEM) (Fig. 5). In this TEM analysis, we observe that the morphology of SOS is spherical and all Pd nanoparticles and nano-silica spheres are distributed on the surface of bulk silica, which is clearly shown in Fig. 5. Interestingly, we found the presence of ultra-small particles on the surface of the SOS spheres. From Fig. 5, the average particle size of ultrafine Pd particles is found to be 2.20 nm. These TEM results support the SEM results.
 |
| | Fig. 5 TEM images of Pd/SOS and particle size distribution of ultrafine Pd nanoparticles. | |
Catalytic performance
Experimental procedure for the synthesis of dihydrobenzofurans 3. In an oven-dried Schlenk tube, 1-iodo-2-((2-methylallyl)oxy)benzene 1a (0.25 mmol), alkyne 2 (0.5 mmol), K2CO3 base (0.5 mmol), Pd/SOS sphere catalyst (0.3 mol% of Pd), and solvent (DMF) (1.0 mL) were added. The resulting reaction mixture was stirred at 100 °C for 1–24 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was diluted with ethyl acetate (5 mL) and neutral NH4Cl solution (5 mL) was added, followed by extraction with ethyl acetate (2 × 4 mL). The organic layers were dried with Na2SO4 and concentrated in reduced vacuum. The purification of the residue by silica gel column chromatography using distilled petroleum ether (hexane) as the eluent furnished the dihydrobenzofurans 3.
Experimental procedure for the synthesis of oxindoles 5. In an oven-dried Schlenk tube, N-(2-iodophenyl)-N-alkylmethacrylamide 4 (0.25 mmol), alkyne 2 (0.5 mmol), K2CO3 base (0.5 mmol), Pd/SOS sphere catalyst (0.3 mol% of Pd), and solvent (DMF) (1.0 mL) were added. The resulting reaction mixture was stirred at 100 °C for 12–24 h. The progress of the reaction was monitored by TLC. After completion of reaction, the reaction mixture was diluted with ethyl acetate (5 mL) and neutral NH4Cl solution (5 mL) was added, followed by extraction with ethyl acetate (2 × 4 mL). The organic layers were dried with Na2SO4 and concentrated in reduced vacuum. The purification of the residue by silica gel column chromatography using distilled petroleum ether/ethyl acetate as the eluent furnished the oxindoles 5.
Optimization studies
To begin with, the optimization for the domino intramolecular Heck cyclization followed by intermolecular Sonogashira coupling (for the synthesis of dihydrobenzofuran derivatives) was planned. In this regard, 1-iodo-2-((2-methylallyl)oxy)benzene 1a and phenylacetylene 2a were chosen as the starting materials. Thus, 1a (0.25 mmol) and 2a (0.5 mmol) were treated in the presence of the Pd/SOS (0.3 mol%) catalyst and K2CO3 (0.5 mmol) base in DMF (0.5 mL) at 100 °C for 1 h. To our delight, the expected dihydrobenzofuran 3aa was isolated in a near quantitative yield (Table 2, entry 1). In contrast, the reaction was found to be inferior with other bases [Cs2CO3 (53%), diazabicycloundecene (DBU) (82%) and NEt3 (44%), (Table 2, entries 2 to 3)]. When water was used as the solvent, 3aa was obtained in poor to moderate yields (Table 2, entries 5 to 8). The addition of 0.25 mmol of phase-transfer catalysts [tetrabutylammonium iodide (TBAI), tetrabutylammonium bromide (TBAB) and benzyl triethylammonium chloride (BTEAC)] to the reaction in water and in the presence of the base K2CO3 improved the yields of 3aa (Table 2, entries 9 to 11). The reaction with 0.25 mmol of BTEAC with the base DBU afforded 3aa in 75% yield (Table 2, entry 12). On the other hand, the product 3aa was obtained in 78% and 90% yields in the polar solvents DMA and DMSO, respectively (Table 2, entries 13 and 14). Only starting materials were recovered back without the base or the catalyst or at room temperature (Table 2, entries 15 to 17). Only trace amounts of the product 3aa were noted when the reaction was performed at 50 °C (Table 2, entry 18). A poor yield of 3aa was obtained when the reaction was conducted under neat conditions (Table 2, entry 19). From the above-mentioned results, the best optimal conditions are those given in Table 1 entry 1 for the formation of the dihydrobenzofuran 3aa.
Table 2 Screening study for the formation of 3aaa,b

|
| Entry |
Base |
Additive (PTC) |
Solvent |
Yield 3aa (%) |
| Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), phenylacetylene 2a (51 mg, 0.5 mmol), catalyst (0.3 mol% of Pd), base (0.5 mmol), additive (0.25 mmol), solvent (0.5 mL). Isolated product yield. With no catalyst. At room temperature. At 50 °C. |
| 1 |
K2CO3 |
— |
DMF |
99 |
| 2 |
Cs2CO3 |
— |
DMF |
53 |
| 3 |
DBU |
— |
DMF |
82 |
| 4 |
NEt3 |
— |
DMF |
44 |
| 5 |
K2CO3 |
— |
Water |
42 |
| 6 |
Cs2CO3 |
— |
Water |
30 |
| 7 |
DBU |
— |
Water |
40 |
| 8 |
NEt3 |
— |
Water |
15 |
| 9 |
K2CO3 |
TBAI |
Water |
60 |
| 10 |
K2CO3 |
TBAB |
Water |
65 |
| 11 |
K2CO3 |
BTEAC |
Water |
77 |
| 12 |
DBU |
BTEAC |
Water |
75 |
| 13 |
K2CO3 |
— |
DMA |
78 |
| 14 |
K2CO3 |
— |
DMSO |
90 |
| 15 |
— |
— |
DMF |
— |
| 16c |
K2CO3 |
— |
DMF |
— |
| 17d |
K2CO3 |
— |
DMF |
— |
| 18e |
K2CO3 |
— |
DMF |
Trace |
| 19 |
K2CO3 |
— |
Neat |
35 |
Effect of no. of equivalent. Furthermore, to optimize the reaction, the reaction was performed with different equivalents of the base K2CO3 and phenylacetylene 2a, as outlined in Fig. 6. Thus, the reaction was carried out using 0.5, 1, 1.5, 2, 2.5 and 4 equivalents of the base K2CO3 relative to 1a under standard conditions; the benzofuran 3aa was isolated in 55%, 77%, 86%, 99%, 99% and 99% yields, respectively. From these results, it is evident that 2 equivalents of K2CO3 is the optimal quantity. Similarly, the reaction was explored with 0.5, 1, 1.5 and 2 equivalents of 2a with respect to 1a and the product 3aa was formed in 62%, 85%, 93%, and 99% yields, respectively. Therefore, it was concluded that 2 equivalents of 2a with respect to 1a is the optimal loading.
 |
| | Fig. 6 Optimization with regards to equivalents of 2a and K2CO3 with respect to 1a. Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), phenylacetylene 2a, Pd/SOS (0.3 mol% of Pd), K2CO3, DMF (0.5 mL). Isolated product yield. | |
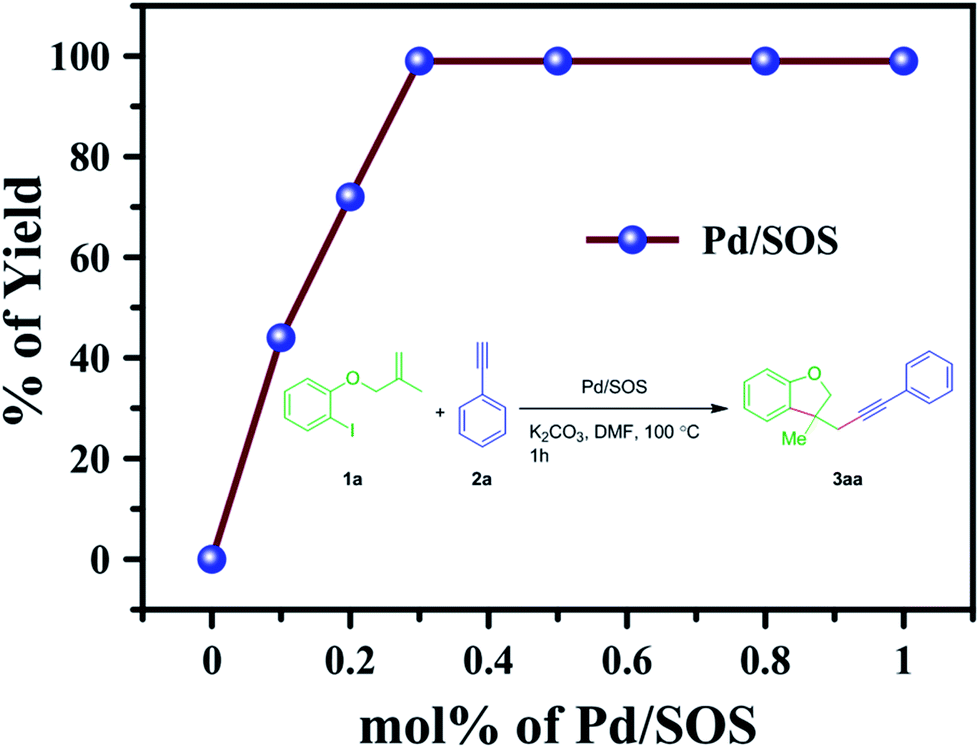
Effect of catalyst concentration. To optimize the amount of the catalyst needed, the reaction was conducted with different amounts of the catalyst (we used 0.0, 0.1, 0.2, 0.3, 0.5, 0.8 and 1 mol% of Pd in Pd/SOS with regards to 1a); the product 3aa was furnished in 0%, 44%, 72%, 99%, 99%, 99% and 99% yields, respectively. From these results, we confirm that 0.3 mol% of Pd in Pd/SOS is the optimal loading of the catalyst, as shown in Fig. 7.
 |
| | Fig. 7 Study with regards to mol% of the catalyst for the formation of 3aa. Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), phenylacetylene 2a (51 mg, 0.5 mmol), Pd/SOS catalyst, K2CO3(0.5 mmol), DMF (0.5 mL). Isolated product yield. | |
Effect of reaction time. In this study, we also tested the effect of reaction time on the reaction at 100 °C. It was understood that the reaction was completed within 1 h and gave the product 3aa in a quantitative yield when phenylacetylene 2a was used as the coupling partner with 1a. In contrast, with the aliphatic alkyne 2i, the reaction afforded only 10% product yield in 1 h and required 24 h for complete conversion, furnishing the product 3ai in 99% yield. From this study, it was noted that aliphatic alkynes reacted at a relatively slower rate than the aromatic ones (Fig. 8).
 |
| | Fig. 8 Reaction completion time with respect to phenylacetylene 2a and 1-hexyne 2i. Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), alkyne 2 (0.5 mmol), Pd/SOS (0.3 mol% of Pd), K2CO3 (0.5 mmol), DMF (0.5 mL). Isolated product yield. | |
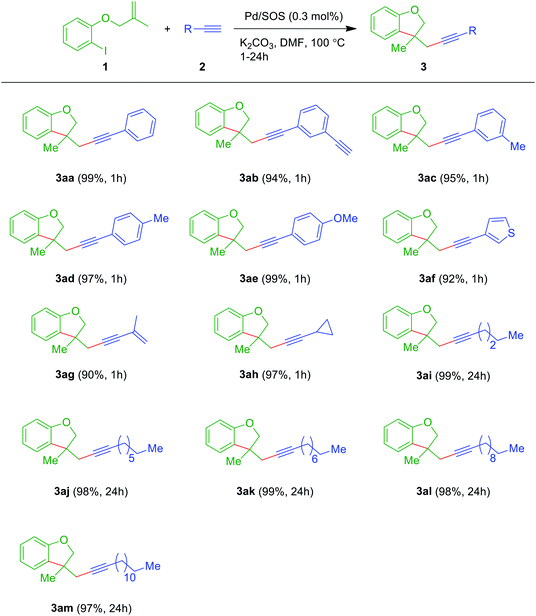
With the optimized reaction conditions in hand (Table 2, entry 1), to check the generality of this domino process, the reaction was explored with other ortho-iodoarylallyl ethers 1 and terminal alkynes 2. To our delight, the reaction was feasible and furnished the products 3aa–3am in excellent to near quantitative yields under the standard reaction conditions (Table 3). As noted above (Fig. 8), the reaction was faster with arylacetylenes 2a–2f than the aliphatic ones 2g–2m. It is worth mentioning that the reaction mixture was exclusively transformed into a single product 3ab containing a free terminal acetylenic C–H, wherein only one acetylene moiety was utilized in the reaction out of two. No other product (i.e., the product in which both alkynes of 2b reacted with two molecules of ether 1a) was formed other than 3ab even by altering the ratios of 1a and 2b. This free terminal acetylenic group would allow further derivatization under traditional coupling reactions. Also, the method was smooth with heteroaryl alkyne 2f. Furthermore, the reaction was found to exhibit excellent chemo-selectivity with enyne 2g, in which an alkyne moiety was involved in Sonogashira coupling over Heck coupling with a terminal ene functional group. Apart from alkyl terminal alkynes, cyclopropyl alkyne 2h was found to be suitable for this domino process.
Table 3 Synthesis of dihydrobenzofurans 3aa–3ama,b
| Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), alkynes 2 (0.5 mmol), Pd/SOS (0.3 mol% of Pd), K2CO3 (0.5 mmol), DMF (0.5 mL). Isolated product yield 3aa–3am. |
 |
After the successful accomplishment of the synthesis of dihydrobenzofurans, we turned our attention to the synthesis of oxindole derivatives. Thus, the reaction was performed with 4 and terminal alkynes 2 under standard reaction conditions. Notably, as anticipated, the reaction was found to be quite successful and furnished the desired oxindoles 5aa–5bo in excellent to near quantitative yields. Also, it was observed that in the case of oxindole synthesis, the required reaction times, particularly for aromatic alkynes, were slightly longer (Table 4).
Table 4 Synthesis of oxindoles 5aa–5boa,b
| Reaction conditions: N-(2-iodophenyl)-N-alkylmethacrylamide (0.25 mmol), alkynes 2 (0.5 mmol), Pd/SOS (0.3 mol% of Pd), K2CO3 (0.5 mmol), DMF (0.5 mL). Isolated product yield 5aa–5bo. |
 |
Recyclability test
To check the sustainability of the catalyst and its retainable activity, a recyclability study was also carried out; the results are shown in Fig. 9. The catalyst was recovered by using centrifugation and washings with ethanol followed by water; it was further dried in a vacuum oven at 60 °C overnight. The recovered Pd/SOS catalyst was then subjected to the next catalytic cycles. The recovered catalyst was found to be active without an appreciable reduction in the product's (3aa) yield under the established conditions. Thus, based on the above-mentioned results, it was confirmed that the Pd/SOS catalyst has enough stability and can be reused.
 |
| | Fig. 9 Recyclability test of the catalyst Pd/SOS. Reaction conditions: 1-iodo-2-((2-methylallyl)oxy)benzene 1a (68 mg, 0.25 mmol), phenylacetylene 2a (51 mg, 0.5 mmol), Pd/SOS (0.3 mol% of Pd), K2CO3 (0.5 mmol), DMF (0.5 mL). Isolated product yield. | |
Mechanism
Based on previous explorations via homogenous catalysis, a conceivable reaction path can be detailed, as depicted in Scheme 3. The Pd(II) intermediate P could be formed upon activating the iodoarene bond of ortho-iodoarylallyl ether 1 by the Pd(0) heterogeneous catalyst. The subsequent intramolecular Heck step would form a bicyclic species Q. The coupling of Q with terminal acetylene 2 would generate R. Lastly, dihydrobenzofuran 3 can be formed via reductive elimination from R, regenerating the Pd(0) nanocatalyst.
 |
| | Scheme 3 Plausible mechanism for the formation of dihydrobenzofurans 3. | |
Conclusion
In this study, we have successfully achieved the synthesis of a novel heterogeneous catalyst. The catalyst was composed of ultra-small Pd nanoparticles on SOS (nano-silica uniformly distributed on the surface of bulk silica). Also, it was characterized by various physicochemical techniques. In addition, the efficiency and applicability of the catalyst have been demonstrated by the domino synthesis of dihydrobenzofurans and oxindoles using a sub-molar quantity of the catalyst (0.3 mol%). Moreover, this reaction showed functional group tolerance. The catalyst was robust and recyclable.
Conflicts of interest
There are no conflicts to declare.
References
- F. Jiang, R. Li, J. Cai, W. Xu, A. Cao, D. Chen, X. Zhang, C. Wang and C. Shu, J. Mater. Chem. A, 2015, 3, 19433 RSC.
- B. Lakshminarayana, J. Chakraborty, G. Satyanarayana and Ch. Subrahmanyam, RSC Adv., 2018, 8, 21030 RSC.
- B. Lakshminarayana, L. Mahendar, J. Chakraborty, G. Satyanarayana and Ch. Subrahmanyam, J. Chem. Sci., 2018, 130, 47 CrossRef.
- B. Lakshminarayana, L. Mahendar, P. Ghosal, G. Satyanarayana and Ch. Subrahmanyam, ChemistrySelect, 2017, 2, 2700 CrossRef CAS.
- S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 2229 CrossRef CAS PubMed.
- M. Dhiman, B. Chalke and V. Polshettiwar, ACS Sustainable Chem. Eng., 2015, 3, 3224 CrossRef CAS.
- N. Bayal, B. Singh, R. Singh and V. Polshettiwar, Sci. Rep., 2016, 6, 24888 CrossRef CAS PubMed.
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652 CrossRef CAS PubMed.
- A. S. L. Thankamony, C. Lion, F. Pourpoint, B. Singh, A. J. P. Linde, D. Carnevale, G. Bodenhausen, H. Vezin, O. Lafon and V. Polshettiwar, Angew. Chem., Int. Ed., 2015, 54, 2190 CrossRef CAS PubMed.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
- Q. B. Li, F. T. Zhou, Z. G. Liu, X. F. Li, W. D. Zhu and J. W. Xie, J. Org. Chem., 2011, 76, 7222 CrossRef CAS PubMed.
- M. Sickert, H. Weinstabl, B. Peters, X. Hou and M. Lautens, Angew. Chem., Int. Ed., 2014, 126, 5247 CrossRef.
- Z. Lu, C. Hu, J. Guo, J. Li, Y. Cui and Y. Jia, Org. Lett., 2009, 12, 480 CrossRef PubMed.
- K. Ramesh, S. Basuli and G. Satyanarayana, Eur. J. Org. Chem., 2018, 2018, 2171 CrossRef CAS.
- K. Ramesh and G. Satyanarayana, Green Chem., 2018, 20, 369 RSC.
- B. Lakshminarayana, L. Mahendar, P. Ghosal, B. Sreedhar, G. Satyanarayana and Ch. Subrahmanyam, New J. Chem., 2018, 42, 1646 RSC.
- D. Eisenberg and W. Kauzman, Science, 1969, 166, 861, DOI:10.1126/science.166.3907.861.
- A. K. Kronenberg and G. H. Wolf, Tectonophysics, 1990, 172, 255 CrossRef.
- J. Fukuda, Water in Rocks and Minerals – Species, Distributions, and Temperature Dependences, 2012, DOI:10.5772/35668.
- Y. Wu, L. Zhang, G. Li, C. Liang, X. Huang, Y. Zhang, G. Song, J. Jia and C. Zhixiang, Mater. Res. Bull., 2001, 36, 253 CrossRef CAS.
- J. Lin, H. Chen, T. Fei and J. Zhang, Colloids Surf., A, 2013, 421, 51 CrossRef CAS.
- D. C. Manatunga, R. M. de Silva and K. N. de Silva, Appl. Surf. Sci., 2016, 360, 777 CrossRef CAS.
- B. Li, Y. Zhang, L. Fu, T. Yu, S. Zhou, L. Zhang and L. Yin, Nat. Commun., 2018, 9, 1076 CrossRef PubMed.
- B. Lakshminarayana, G. Satyanarayana and Ch. Subrahmanyam, ACS Omega, 2018, 3, 13065 CrossRef CAS PubMed.
- E. Meku, C. Du, Y. Sun, L. Du, Y. Wang and G. Yin, J. Electrochem. Soc., 2016, 163, F132 CrossRef CAS.
- J. A. L. López, J. C. López, D. V. Valerdi, G. G. Salgado, T. Díaz-Becerril, A. P. Pedraza and F. F. Gracia, Nanoscale Res. Lett., 2012, 7, 604 CrossRef PubMed.
- F. H. Cincotto, T. C. Canevari, A. M. Campos, R. Landers and S. A. Machado, Analyst, 2014, 139, 4634 RSC.
- X. Zhou and T. Shi, Appl. Surf. Sci., 2012, 259, 566 CrossRef CAS.
- B. Ulgut and S. Suzer, J. Phys. Chem. B, 2003, 107, 2939 CrossRef CAS.
- Y. Zhao, J. Zhao, Z. Su, X. Hao, Y. Li, N. Li and Y. Li, J. Mater. Chem. A, 2013, 1, 8029 RSC.
- H. Y. Wu, K. J. Lin, P. Y. Wang, C. W. Lin, H. W. Yang, C. C. M. Ma and T. R. Jan, Int. J. Nanomed., 2014, 9, 4257 CAS.
- Y. Zhang, X. Xia, X. Cao, B. Zhang, N. H. Tiep, H. He, S. Chen, Y. Huang and H. J. Fan, Adv. Energy Mater., 2017, 7, 1700220 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09429f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
G. Satyanarayana
,
G. Satyanarayana