DOI:
10.1039/C9RA07482A
(Paper)
RSC Adv., 2020,
10, 4579-4588
Simultaneous quantification of five biomarkers in ethanolic extract of Cassia occidentalis Linn. stem using liquid chromatography tandem mass spectrometry: application to its pharmacokinetic studies
Received
17th September 2019
, Accepted 19th January 2020
First published on 29th January 2020
Abstract
Cassia occidentalis L. stem extract is used as a purgative, febrifuge, and diuretic, and in the treatment of flu, fever, fracture and bone diseases. Pharmacological studies prove the osteogenic and antiresorptive effects of Cassia occidentalis L. ethanolic extract (COEE), which may be due to apigenin, apigenin-6-C-glucopyranoside, luteolin, 3′,4′,7-trihydroxyflavone and emodin. The objectives of this study was to develop a selective and sensitive LC-MS/MS method and validate for the simultaneous determination of the above five biomarkers in rat plasma after oral administration of COEE at a dose of 500 mg kg−1. The analytes were separated on a Phenomenex Luna C18 column (4.6 × 150 mm, 3.0 μm) with an isocratic mobile phase consisting of methanol-10 mM ammonium acetate buffer (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :05, v/v). Run time was for 5.5 min with LLOQ of 1 ng mL−1 for all the analytes. The mass spectrometer was operating in negative ionization mode for quantification of the analytes. The calibration curves were linear (r2 > 0.99) for all the analytes. The intra- and inter-day precisions were less than 8.17% and the relative error was between −8.57% and 7.28%. Analytes were rapidly absorbed in the oral pharmacokinetic study. The biomarkers were stable in simulated gastric and intestinal fluids but underwent metabolism in rat liver microsomes. This is the first report on in vivo oral pharmacokinetics and in vitro stability studies of osteogenic compounds present in COEE. These results will be helpful for further understanding of pharmacodynamic behaviour of COEE and the bioanalytical method will be useful for further preclinical/clinical trials.
:05, v/v). Run time was for 5.5 min with LLOQ of 1 ng mL−1 for all the analytes. The mass spectrometer was operating in negative ionization mode for quantification of the analytes. The calibration curves were linear (r2 > 0.99) for all the analytes. The intra- and inter-day precisions were less than 8.17% and the relative error was between −8.57% and 7.28%. Analytes were rapidly absorbed in the oral pharmacokinetic study. The biomarkers were stable in simulated gastric and intestinal fluids but underwent metabolism in rat liver microsomes. This is the first report on in vivo oral pharmacokinetics and in vitro stability studies of osteogenic compounds present in COEE. These results will be helpful for further understanding of pharmacodynamic behaviour of COEE and the bioanalytical method will be useful for further preclinical/clinical trials.
1. Introduction
Medicinal plants play a key role in healthcare. These plants are showing medicinal value due to the presence of phytochemicals and exhibit definite pharmacological action.1 Cassia occidentalis L., also popularly known as Senna occidentalis, belongs to the family Caesalpiniaceae that is widespread in South Asia and South America. Cassia occidentalis L. herbal products are widely marketed across the globe, such as Cassia Virgínica® (Laperli, Recife, Brazil) and Liv.52 DS (The Himalaya Drug Company, India) for the therapy of flu and fever, and as antihepatotoxicity formulation, respectively.2 Stems of Cassia occidentalis L. have been ethno-traditionally used in South India and South America.3–5 Acute, sub-acute and reproductive toxicity of Cassia occidentalis L. was evaluated in Wistar rats and it was found to be non-toxic.3,6 Earlier whole plant of Cassia occidentalis L. extraction was performed by using Soxhlet apparatus and its yield was reported 1.49%.7 Till now, most studies of Cassia occidentalis L. have been based on pharmacology, toxicology or phyto-chemistry but no attention has been paid to the pharmacokinetics of the multiple components in it, neither in vitro nor in vivo.
In the literature, several HPLC-UV and LC-MS/MS methods have been developed to separate and quantify apigenin (APG), apigenin-6-C-glucopyranoside (AP6CG), luteolin (LT), 3′,4′,7-trihydroxyflavone (THF) and emodin (Emd) from rat plasma either individually or in a combination of any two of these. An LC-MS/MS method for measuring four flavonoids and a phenolic acid in rat plasma after gastric administration of Herba Desmodii styracifolii extract was developed.8 The above described LC-MS/MS method had a high lower limit of quantification (LLOQ; 1.70 and 0.84 ng mL−1 for APG and AP6CG, respectively) and a long run time (>10 min). Another LC-MS/MS method for quantification of only vitexin and AP6CG in rat plasma was developed, which had high LLOQ (2 ng mL−1 for both) and long run time (7.5 min).9 For quantification of Emd and 2 others anthraquinones in rat plasma, both LLOQ (0.02 μM) and run time (18 min) had higher in the reported LC-MS/MS method.10 Other analytical methods are mostly for the individual analysis of either any one of them. In other words, LC-MS/MS method for the simultaneous determination of APG, AP6CG, LT, THF and Emd in rat plasma samples after the oral administration of the Cassia occidentalis L. ethanolic extract (COEE) have not been developed (Fig. 1).
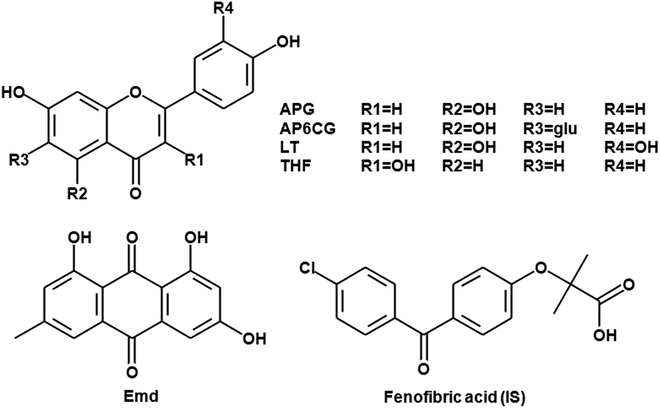 |
| | Fig. 1 Chemical structure of analytes and IS. | |
In the present study, a sensitive and short LC-ESI-MS/MS method was developed and validated for the simultaneous determination of the concentrations of APG, AP6CG, LT, THF and Emd in rat plasma for the first time with shorter run time. The novel method was utilised for the evaluation of the oral pharmacokinetics after the administration of COEE to rats and relevant in vitro studies. The results may be helpful for further pharmacodynamics investigation of COEE and the bioanalytical method may be useful for phase 1 clinical trial according to phyto-pharmaceutical guideline of DCG(I).
2. Experimental
2.1 Materials
APG, AP6CG, LT, THF, Emd, fenofibric acid, dimethyl sulphoxide, NADPH and ammonium acetate were procured from Sigma Aldrich, USA. LC-MS/MS grade methanol and acetonitrile were purchased from Merck (Darmstadt, Germany). Cassia occidentalis was collected from the surrounding areas of Lucknow, India (geographical location 27.08080 N and 80.89590 E) and stems were separated and dried under shade. Cassia occidentalis specimen has been deposited in CSIR-CDRI herbarium (voucher specimen number KRA-CDRI-25009).
2.2 Apparatus and operational conditions
2.2.1 Liquid chromatography. Shimadzu HPLC apparatus consisted of SIL-HTc auto-sampler and LC-20AD binary pumps (Shimadzu, Kyoto, Japan), which were used for injecting 10 μL aliquots of the processed samples on to Phenomenex Luna C18 column (4.6 × 150 mm, 3.0 μm). The mobile phase consisting of methanol and 10 mM ammonium acetate buffer in the ratio of 95:05 (v/v) was run in isocratic mode at a flow rate of 0.6 mL min−1. Aqueous mobile phase was duly filtered through 0.22 μm Millipore filter (Billerica, USA) and degassed ultrasonically for 15 min prior to use. Separations were performed on column maintained at 40 °C. Run time was for 5.5 min. Fenofibric acid (50 ng mL−1) was used as internal standard (IS).
2.2.2 Mass spectrometry. The LC-MS/MS analysis was performed using API 4000 QTRAP mass spectrometer (Applied Biosystems, MDS Sciex, Toronto, Canada). The electrospray ionization source was operated in the negative ion mode. The MRM analysis was done by observing the precursor ion to product ion transition of m/z 269.3/116.9 for APG, 431.2/311 for AP6CG, 269.1/224.9 for Emd, 284.9/133 for LT, 268.9/132.8 for THF, and 316.9/230.9 for IS. The instrument parameters, viz. auxiliary gas, nebulizer gas, curtain gas and collision gas, were set at 50, 50, 30 and high, respectively. Compound parameters, viz. declustering potential, entrance potential, collision energy and collision exit potential were −76, −10, −49 and −5 V for APG, −100, −10, −20 and −15 V for AP6CG, −100, −10, −50 and −20 V for LT, −100, −10, −40 and −10 V for THF, −100, −10, −40 and −10 V for Emd and −55, −10, −12 and −10 V for IS, respectively. Nitrogen was used in case of curtain and collision gas, whereas zero gas served as the nebulizing gas. All the data were controlled and synchronized by Analyst 1.6 software (Applied Biosystems, MDS Sciex, Toronto, Canada).
2.3 Preparation of COEE
C. occidentalis stem was separated, dried under shade and powdered. The powder (12 kg) was percolated for 24 h at room temperature with 95% ethanol (40 L). The percolate was filtered, concentrated under reduced pressure to yield (6%) dried semi-solid ethanolic extract. To calculate the administered dose, the contents of APG, AP6CG, LT, THF, and Emd in C. occidentalis extract were quantitatively measured by an external standard method utilising the same chromatographic conditions as described above. The results show that the concentrations of APG, AP6CG, LT, THF, and Emd in the extract were 992.8 ± 3.5, 1477.6 ± 15.4, 891.8 ± 268.3, 253.7 ± 76.3 and 80 ± 19.3 ng mg−1, respectively.
2.4 Preparation of calibration standards (CS) and quality control (QC) samples
The primary stock solutions of APG, AP6CG, LT, THF, and Emd were prepared in dimethylsulfoxide separately by dissolving appropriate amounts. Then, the stock solutions were mixed and diluted with acetonitrile to prepare a final mixed standard solution containing 10 μg mL−1 of each analyte. Working stocks were prepared by serial dilution of final stock solution with acetonitrile in the calibration range of 10–2000 ng mL−1 for all the analytes. For the validation of the method, three concentrations of the standard solution containing 30, 750, and 1500 ng mL−1 were used to prepare the QC plasma samples. All of the solutions were prepared fresh.
Preparation of CS samples involved spiking 10 μL of working stocks of corresponding concentrations to 90 μL blank rat plasma. The final concentrations of the analytes in the CS ranged from 1–200 ng mL−1 for APG, AP6CG, LT, THF, and Emd. The QC samples containing low, medium and high concentrations were prepared similarly at the concentrations of 3, 75, and 150 ng mL−1.
2.5 Sample preparation
The extraction procedure optimised for plasma samples involved protein precipitation followed by liquid–liquid extraction. In brief, all the CS, QC and plasma samples were cleaned as follows: 100 μL of plasma sample was incubated with 50 of μL β-glucuronidase enzyme (2 mg mL−1 in 100 mM ammonium acetate buffer pH 5), and 50 μL ammonium acetate buffer (100 mM, pH 5) for 2 hours at 37 °C on water shaking bath. Incubated mixture was precipitated with 200 μL of acetonitrile containing IS by vortexing for 15 s and acidified with 100 μL 0.1% formic acid in 10 mM ammonium formate. This was vortexed for 5 min at 1200 rpm. Later liquid–liquid extraction was performed by adding 2 mL of ethyl acetate and vortexing for 5 min. Then the samples were centrifuged at 8000 rpm for 5 min and the partitioned supernatant (1.5 mL) was transferred to clean ria vials and dried with turbo vap at 37 °C. The dried residue was reconstituted with 100 μL of mobile phase and 10 μL of this is injected into LC-MS/MS.
2.6 Method validation
The method was fully validated (specificity, sensitivity, linearity, recovery, accuracy, precision, dilution integrity, carry-over effect and stability) according to the Guidance for Industry: Bioanalytical Method Validation of USFDA.11 All the validation studies were performed as previously reported by Valicherla et al.12 and Riyazuddin et al.13
2.7 Application of the LC-ESI-MS/MS quantitative analysis
2.7.1 Oral pharmacokinetic study. Sprague Dawley (SD) rats were obtained from the National Laboratory Animal Centre of the CSIR-CDRI, India. They were housed in well-ventilated cages placed in environmentally controlled room (temperature 24 ± 2 °C, relative humidity 40–60%, 12 h light/dark cycle). All the animal experiments were carried out as per the approval and guidelines of the Institutional Animal Ethical Committee (IAEC) at CSIR-CDRI (IAEC approval no. IAEC/2017/286). The rats were administered COEE (suspended with 0.5% carboxymethyl cellulose sodium) at a dose of 500 mg kg−1 intragastrically. Blood samples (approximately 0.25 mL) were collected at 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, 24, 48, and 72 h post-dosing from the retro-orbital plexus into heparin containing micro-centrifuge tubes. The whole blood was immediately centrifuged at 5000 g for 10 min at 4 °C and the plasma was separated and stored at −70 ± 10 °C until analysis. As mentioned above, plasma samples were processed and data was approved on the grounds of QCs performance (five QCs each at three concentration levels).
2.7.2 In vitro stability studies. Stability of APG, AP6CG, LT, THF, and Emd present in COEE was evaluated in simulated gastro-intestinal fluids, plasma and rat liver microsomes (RLM). Simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) composition was as per the United States Pharmacopeia (2012). In brief, SGF was prepared by dissolving sodium chloride (0.2 g), purified pepsin (3.2 g) and hydrochloric acid (0.7 mL) and diluting in water to 100 mL. This test solution had a pH of 1.2. Similarly SIF was prepared by dissolving monobasic potassium phosphate (0.68 g) in 25 mL of water and added to 0.77 mL of sodium hydroxide (0.2 N) and 50 mL water. Later pancreatin (1 g) was added, pH was adjusted to 6.8 and volume was makeup to 100 mL with water.14 Plasma stability was performed in fresh plasma collected from non-treated rats. Microsomal stability was performed in RLM prepared in house and the conditions are similar to as reported by Riyazuddin et al.15 Briefly, the contents of reaction mixture are RLM (0.5 mg mL−1), 40 mM MgCl2, 1 mM NADPH in 0.8 mL of 50 mM Tris–HCl buffer (pH 7.4). All the stability studies were performed at 37 °C in a shaking water bath incubator and the reactions were initiated by spiking 100 μg mL−1 of COEE into respective reaction media. Samples were collected at different time points and were processed as described above.
3. Results and discussion
3.1 Method development
3.1.1 Mass spectrometry. A 4000 QTRAP mass spectrometer coupled with an ESI source was utilised for the quantification of APG, AP6CG, LT, THF, Emd and IS. To optimise mass spectrometer conditions, analytes and IS were directly injected by a syringe pump in an ESI full scan mode and the ion intensity was quite high in negative mode. The predominantly yielded [M − H]− ions were m/z 269.3, 431.2, 284.9, 268.9, 269.1 and 316.9 for APG, AP6CG, LT, THF, Emd and IS, respectively. The most abundant and stable product ions in the Q3 MS spectra for APG, AP6CG, LT, THF, Emd and IS were observed at m/z 116.9, 311, 133, 132.8, 224.9 and 230.9, respectively. The product ion spectra of analytes and IS have been shown in Fig. 2. The optimization of MS parameters such as declustering potential, entrance potential, collision energy, collision cell exit potential, ion source temperature, nebulizer gas, auxiliary gas, ion spray voltage, collision gas and curtain gas were performed using the automatic tuning tool in order to obtain the maximum sensitivity. In conclusion, the predominant transitions at m/z 269.3/116.9 for APG, 431.2/311 for AP6CG, 269.1/224.9 for Emd, 284.9/133 for LT, 268.9/132.8 for THF, and 316.9/230.9 for IS were utilised in MRM scan mode.
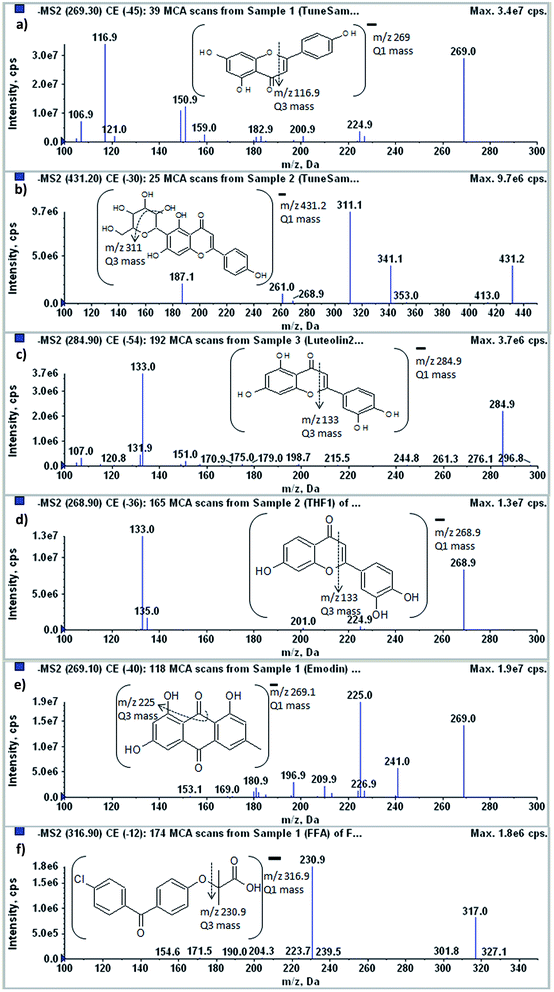 |
| | Fig. 2 Mass fragmentation pattern in negative mode of (a) apigenin, (b) apigenin-6-C-glucopyranoside, (c) luteolin, (d) 3′,4′,7-trihydroxyflavone, (e) emodin and (f) fenofibric acid representing their Q1 and Q3 (m/z) values. | |
3.1.2 Chromatography. Various mobile phase compositions such as organic phase containing acetonitrile, methanol, and a combination of both acetonitrile and methanol with aqueous phase consisting of water, 10 mM ammonium acetate, 0.1% formic acid along with different types of chromatographic columns (Phenomenex Luna C18, 150 × 4.6 mm, 5 μm; Phenomenex Luna C18, 150 × 4.6 mm, 3 μm, and Supelco Ascentis Phenyl 100 × 3 mm, 3 μm) were correlated in terms of their capability to create a sharp peak and maximum signal intensity within a short run time. The results showed that simple isocratic elution of methanol and 10 mM ammonium acetate (95:05, v/v) with Phenomenex Luna C18, 150 × 4.6 mm, 3 μm yielded retention time less than 5.5 min allowing high throughput analysis. The optimised mobile phase was pumped at 0.6 mL min−1 flow rate. The extract ion chromatograms (XIC) formed by the multiple reaction monitoring of the blank, standards and the sample are provided in Fig. 3.
 |
| | Fig. 3 Representative extract ion MRM chromatograms of (a) apigenin, (b) apigenin-6-C-glucopyranoside, (c) luteolin, (d) 3′,4′,7-trihydroxyflavone, (e) emodin and (f) fenofibric acid in blank plasma, blank plasma spiked with analytes at the LLOQ and IS, and plasma sample obtained after the oral administration of ethanolic extract of C. occidentalis. | |
3.2 Processing of the plasma samples
Processing of plasma samples is a crucial step for accurate and reliable LC-MS/MS analysis. The use of protein precipitation (PP) and liquid–liquid extraction (LLE) methods were compared for analyte and IS extraction. Acceptable recovery and lack of matrix effect were obtained by the combination of PP and LLE. Acetonitrile was used for PP and ethyl acetate for LLE. Using of acetonitrile acidified with formic acid further enhanced the extraction efficiency which may be due to suppression of protein adsorption and promotion of release of analyte and IS from proteins.8 The glucuronide conjugates of analytes were hydrolyzed by β-glucuronidase enzyme. The free form of an analytes can be quantified easily instead of complex.16
3.3 Method validation
3.3.1 Specificity. MRM chromatograms of blank rat plasma, blank plasma spiked with analytes and IS, and plasma samples from pharmacokinetic study after oral administration of COEE are shown in Fig. 3. Under the optimized conditions, the retention times of APG, AP6CG, LT, THF, Emd and IS were 3.02, 2.62, 2.5, 2.89, 3.85 and 2.43 min, respectively. There was no significant interference of endogenous elements in blank plasma specifying that the developed method was specific to the analytes.
3.3.2 Linearity and sensitivity. As shown in Table 1, calibration curves of the all the five analytes exhibited good linear regression across the corresponding concentration range. The LLOQ of all the analytes was determined as 1 ng mL−1. Using the linearity equation obtained from the calibrations, the unknown concentrations of the osteogenic compounds in the plasma samples of the oral pharmacokinetic study of COEE were determined.
Table 1 The linear ranges, LLOQs and regression equations for the determination of the analytes in rat plasma
| Analyte |
Linear range (ng mL−1) |
LLOQ (ng mL−1) |
Regression equation |
Correlation coefficient (r2) |
| APG |
1–200 |
1 |
y = 10.2 × 10−3x + 4.59 × 10−2 |
0.9979 |
| AP6CG |
1–200 |
1 |
y = 3.71 × 10−4x + 5.43 × 10−3 |
0.9977 |
| LT |
1–200 |
1 |
y = 4.48 × 10−3x + 2.41 × 10−3 |
0.9918 |
| THF |
1–200 |
1 |
y = 10.2 × 10−3x + 4.59 × 10−2 |
0.9979 |
| Emd |
1–200 |
1 |
y = 9.4 × 10−3x + 2.51 × 10−2 |
0.9973 |
3.3.3 Matrix effects and extraction recovery. As displayed in Table 2, the mean extraction recoveries of APG, AP6CG, LT, THF, and Emd at three different concentration levels were spread across 84.62% to 107.06%, with RSD values less than 8.69%. When the liquid–liquid extraction method was used for sample preparation, ion suppression or enhancement caused by endogenous material may be lesser than protein precipitation method. This is apparent from Table 2 where the matrix effect ranged from 85.93% to 105.66%, with RSD values less than 13.36%.
Table 2 Recoveries and matrix effect of APG and AP6CG in rat plasma at three QC levels (n = 5)
| Analyte |
Nominal concentration (ng mL−1) |
Extraction recovery (%, mean ± SD) |
RSD (%) |
Matrix effect (%, mean ± SD) |
RSD (%) |
| APG |
3 |
86.30 ± 1.45 |
1.68 |
105.66 ± 9.24 |
7.99 |
| 75 |
96.87 ± 8.69 |
8.69 |
99.30 ± 4.28 |
4.31 |
| 150 |
90.43 ± 1.18 |
1.31 |
98.15 ± 2.71 |
2.76 |
| AP6CG |
3 |
103.51 ± 7.97 |
7.7 |
85.93 ± 8.76 |
10.19 |
| 75 |
91.2 ± 3.32 |
3.65 |
98.15 ± 3.2 |
3.26 |
| 150 |
91.28 ± 4.96 |
5.44 |
104.29 ± 9.76 |
9.36 |
| LT |
3 |
94.98 ± 2.22 |
2.33 |
102.34 ± 3.05 |
2.98 |
| 75 |
84.62 ± 5.85 |
6.91 |
88.72 ± 9.82 |
11.07 |
| 150 |
89.17 ± 1.81 |
2.03 |
102.97 ± 0.69 |
0.67 |
| THF |
3 |
104.57 ± 2.99 |
2.86 |
90.8 ± 6.92 |
7.62 |
| 75 |
94.7 ± 2.45 |
2.59 |
98.81 ± 8.45 |
8.55 |
| 150 |
94.79 ± 1.83 |
1.93 |
89.36 ± 7.84 |
8.77 |
| Emd |
3 |
107.06 ± 4.79 |
4.48 |
95.24 ± 8.14 |
8.54 |
| 75 |
104.76 ± 5.61 |
5.36 |
104.53 ± 13.97 |
13.36 |
| 150 |
98.45 ± 1.93 |
1.96 |
96.83 ± 9.45 |
9.76 |
3.3.4 Precision and accuracy. As shown in Table 3, the precisions (% RSD) of intra-day and inter-day of the all the analytes at three QC levels were no more than 8.17%, whereas the relative error (% RE) of the accuracies ranged from −8.57 to 7.28. All of the results of accuracy and precision were within the acceptable variation criteria of ±15%.
Table 3 The intra- and inter-day precision and accuracies of the analytes in rat plasma (n = 6)
| Analyte |
Spiked concentration (ng mL−1) |
Intra-run |
Inter-run |
| Measured concentration (ng mL−1) |
Precision (% RSD) |
Accuracy (% RE) |
Measured concentration (ng mL−1) |
Precision (% RSD) |
Accuracy (% RE) |
| APG |
3 |
3.03 ± 0.13 |
4.40 |
0.89 |
2.98 ± 0.13 |
4.20 |
−0.72 |
| 75 |
79.27 ± 3.75 |
4.73 |
5.69 |
75.47 ± 2.53 |
3.36 |
0.62 |
| 150 |
158.67 ± 4.04 |
2.55 |
5.78 |
159 ± 7.81 |
4.91 |
6 |
| AP6CG |
3 |
3.10 ± 0.15 |
4.77 |
3.33 |
3.22 ± 0.18 |
5.59 |
7.28 |
| 75 |
71.80 ± 2.10 |
2.92 |
−4.27 |
74.83 ± 3.49 |
2.79 |
1.56 |
| 150 |
146.67 ± 3.21 |
2.19 |
−2.22 |
145.37 ± 5.92 |
4.07 |
−3.09 |
| LT |
3 |
3.06 ± 0.14 |
4.42 |
1.87 |
3.03 ± 0.16 |
5.25 |
0.97 |
| 75 |
75.28 ± 7.84 |
5.42 |
0.37 |
69 ± 4.93 |
7.15 |
−8 |
| 150 |
158.5 ± 11.46 |
7.23 |
5.67 |
145.7 ± 10.35 |
7.1 |
−2.87 |
| THF |
3 |
2.99 ± 0.24 |
8.17 |
−0.2 |
2.93 ± 0.23 |
7.68 |
−2.18 |
| 75 |
71.05 ± 3.42 |
4.81 |
−5.27 |
73.2 ± 2.62 |
3.57 |
−2.4 |
| 150 |
157 ± 3.12 |
1.99 |
4.67 |
149.2 ± 4.88 |
3.27 |
−0.53 |
| Emd |
3 |
3.1 ± 0.14 |
4.46 |
3.27 |
3.04 ± 0.14 |
4.58 |
1.4 |
| 75 |
79.45 ± 4.77 |
6 |
5.93 |
79 ± 1.56 |
1.98 |
5.33 |
| 150 |
137.15 ± 3.73 |
2.72 |
−8.57 |
147.85 ± 3.34 |
2.26 |
−1.43 |
3.3.5 Stability. The outcomes of stability studies of APG, AP6CG, LT, THF, and Emd in rat plasma across various conditions to which they may be exposed during the sample storage and processing have been compiled in Table 4. It was observed that all the analytes were stable within the acceptable limits of ±15% variations across different conditions such as at 4 °C for 24 h in auto-sampler, at 24 °C for 8 h on bench-top, at −70 ± 10 °C for 30 days in ultra-low temperature freezer and after three freeze–thaw cycles from −70 ± 10 °C to room temperature.
Table 4 The stability of analytes in rat plasma and plasma extracts (mean ± SD, n = 3)
| Analyte |
Nominal concentration (ng mL−1) |
Auto-sampler stability (4 °C, 24 h) |
Bench-top stability (24 ± 4 °C, 8 h) |
Long-term stability (−70 ± 10 °C, 30 days) |
Freeze–thaw stability (−70 ± 10 °C, 3 cycles) |
| Measured concentration (ng mL−1) |
Accuracy (% RE) |
Measured concentration (ng mL−1) |
Accuracy (% RE) |
Measured concentration (ng mL−1) |
Accuracy (% RE) |
Measured concentration (ng mL−1) |
Accuracy (% RE) |
| APG |
3 |
3.18 ± 0.03 |
−6 |
2.78 ± 016 |
7.2 |
2.64 ± 0.06 |
12.17 |
2.66 ± 0.1 |
11.2 |
| 75 |
78.53 ± 5.59 |
−4.7 |
68.23 ± 3.23 |
9.03 |
68.38 ± 3.11 |
8.83 |
67.35 ± 2.25 |
10.2 |
| 150 |
135 ± 6.48 |
10 |
136.6 ± 6.06 |
8.93 |
139.25 ± 0.92 |
7.17 |
133.45 ± 2.9 |
11.03 |
| AP6CG |
3 |
2.95 ± 0.21 |
1.6 |
3.13 ± 0.28 |
−4.33 |
2.89 ± 0.36 |
3.83 |
2.91 ± 0.37 |
3.13 |
| 75 |
71.93 ± 2.48 |
4.1 |
78.25 ± 1.15 |
−4.33 |
82.25 ± 3.38 |
−9.67 |
78.5 ± 5.63 |
−4.67 |
| 150 |
146.4 ± 1.73 |
2.4 |
155.85 ± 6.8 |
−3.9 |
145.65 ± 4.23 |
2.9 |
142.95 ± 4.22 |
4.7 |
| LT |
3 |
3 ± 0.16 |
−0.11 |
3.01 ± 0.11 |
−0.33 |
2.82 ± 0.14 |
6 |
2.61 ± 0.36 |
12.89 |
| 75 |
76.63 ± 5.74 |
−2.18 |
79.8 ± 4.94 |
−6.4 |
71.73 ± 1.37 |
4.36 |
73.7 ± 2.14 |
1.73 |
| 150 |
164 ± 6.24 |
−9.33 |
143.33 ± 2.08 |
4.44 |
143.33 ± 2.31 |
4.44 |
148.33 ± 4.62 |
1.11 |
| THF |
3 |
3.23 ± 0.2 |
−7.67 |
3.1 ± 0.23 |
−3.33 |
2.9 ± 0.09 |
3.43 |
2.86 ± 0.28 |
4.63 |
| 75 |
76.5 ± 1.3 |
−2 |
81.5 ± 1.15 |
−8.67 |
71.83 ± 3.49 |
4.23 |
73.1 ± 3.21 |
2.53 |
| 150 |
154.5 ± 1.5 |
−3 |
158 ± 4.33 |
−5.33 |
151.2 ± 3.12 |
−0.8 |
156.9 ± 7.69 |
−4.6 |
| Emd |
3 |
3.04 ± 0.42 |
−1.44 |
2.86 ± 0.06 |
4.8 |
3.02 ± 0.06 |
−0.77 |
2.89 ± 0.13 |
3.77 |
| 75 |
77.83 ± 7.36 |
−3.78 |
73.05 ± 2.6 |
2.6 |
72.9 ± 4.06 |
2.8 |
73.23 ± 1.13 |
2.37 |
| 150 |
158 ± 6.93 |
−5.33 |
140.15 ± 4.14 |
6.57 |
146.5 ± 7.4 |
2.33 |
140.1 ± 3.67 |
6.6 |
3.4 Application of bioanalytical method
3.4.1 Oral pharmacokinetic study. After oral administration of 500 mg kg−1 COEE, the validated LC-ESI-MS/MS method was use to simultaneously determine APG, AP6CG, LT, THF, and Emd in rat plasma at different time points. Fig. 4 shows the mean plasma concentration–time profiles of four flavones and one anthraquinone, and the major pharmacokinetic parameters are listed in Table 5. After COEE was orally administered, APG, LT, THF, and Emd appeared to be absorbed rapidly compared to AP6CG. Similar results of rapid absorption were observed in case of APG and LT when plant extracts were orally administered to rodents.17,18 A second peak was observed in the plasma pharmacokinetic profiles of flavones which might be due to entero-hepatic recirculation as reported earlier in case of flavanoids.19–21 Fig. 4 shows that AP6CG and THF were eliminated within 12 h and LT and Emd were eliminated within 24 h, while only APG could be detected in plasma till 8 h. The pharmacokinetic profile of AP6CG is markedly different from other analytes, possibly owing to the presence of glycone moiety in this molecule.
 |
| | Fig. 4 Mean plasma concentration–time curves of apigenin, apigenin-6-C-glucopyranoside, luteolin, 3′,4′,7-trihydroxyflavone, and emodin after a single oral administration of the ethanolic extract of C. occidentalis. | |
Table 5 Pharmacokinetic parameters of analytes after oral administration of 500 mg kg−1 COEE (mean ± SD, n = 6)
| Parameter |
Apigenin |
Apigenin-6-C-glucopyranoside |
Luteolin |
3′,4′,7-Trihydroxyflavone |
Emodin |
| t1/2 (h) |
2.63 ± 1.44 |
3.18 ± 2.58 |
4.73 ± 0.97 |
0.41 ± 0.3 |
3.94 ± 0.8 |
| Tmax (h) |
0.58 ± 0.29 |
2.08 ± 1.88 |
0.29 ± 0.1 |
0.3 ± 0.11 |
0.58 ± 0.7 |
| Cmax (ng mL−1) |
8.34 ± 4.5 |
7.96 ± 0.55 |
204.02 ± 151.12 |
90.28 ± 56.77 |
21.38 ± 7.44 |
| AUC0–last (h ng mL−1) |
9.81 ± 5.66 |
33.75 ± 21.43 |
542.39 ± 218.46 |
168.1 ± 124.52 |
118.61 ± 33.05 |
| AUC0–∞ (h ng mL−1) |
9.89 ± 5.66 |
43.51 ± 29.25 |
573.1 ± 207.06 |
173.51 ± 124.95 |
123.02 ± 31.35 |
| MRT0–t (h) |
12.8 ± 3.88 |
3.7 ± 2.4 |
7.99 ± 2.15 |
3.86 ± 2.01 |
6.84 ± 1.25 |
3.4.2 In vitro stability studies. The developed analytical method was also used for the analysis of the samples from in vitro studies. The stability of compounds in gastro-intestinal fluid is an essential requirement for oral bioavailability.14,22 It was observed that all the analytes were stable in SGF but in SIF and rat plasma, analytes were moderately stable, as shown in Fig. 5. In SIF, Emd was rather unstable with 50% of the compound remaining after 60 min. In the area of drug discovery and development, compound stability in plasma plays a crucial role. Compounds which have unstable in plasma, exhibiting rapid clearance and shorter t1/2.23 In fresh rat plasma, after incubating for 4 h at 37 °C, the order of stability of analytes is APG > Emd > THF > LT > AP6CG. Drug is cleared off from the body mainly by hepatic metabolism. Several drug metabolizing enzymes i.e., cytochrome P450s, monooxygenases, carboxylestrases, epoxide hydrolase, etc. are present in liver microsomes. These enzymes are used for investigating the metabolism of drugs. The most prominent group of these enzymes is cytochrome P450s, which play an important role in drug metabolism.24,25 In RLM, >90% of APG and Emd underwent phase I metabolism within 60 min whereas 60 and 30% in the cases of AP6CG and THF, respectively. LT did not undergo phase I metabolism in RLM. The in vitro stability studies showed that LT was relatively stable in different conditions and this might be the reason for its abundant plasma levels (high Cmax and AUC values) in the pharmacokinetic study.
 |
| | Fig. 5 Time-dependent depletion of apigenin, apigenin-6-C-glucopyranoside, luteolin, 3′,4′,7-trihydroxyflavone, and emodin in (a) simulated gastric fluid, (b) simulated intestinal fluid, (c) plasma, and (d) RLM. | |
4. Conclusions
A novel LC-ESI-MS/MS method has been developed and validated for simultaneous quantification of APG, AP6CG, LT, THF, and Emd in rat plasma from orally administered COEE with a short run time of 5.5 min. With no report on pharmacokinetics of osteogenic constituents of COEE, this is the first in vivo oral pharmacokinetics and in vitro stability report of four flavones and one anthraquinone present in COEE. These results will be helpful for further pharmacodynamics investigation of COEE and the bioanalytical method will be useful for phase 1 clinical trial according to phyto-pharmaceutical guideline of DCG(I).
Author's contributions
MR, AH and JRG designed the research work; MR, AH, SV, RK, RG, SK, SS performed the research experiments; MR, AH, SV, RK, RG, SK, SS, RM, TN, NC, JRG supervised, analysed and compiled the data; MR, AH, JRG prepared the manuscript.
Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.
Acknowledgements
The authors acknowledge funding from Council of Scientific and Industrial Research, GoI funding under The Phytopharmaceutical Mission (HCP0010 to N. C. and J. R. G.). MR and SV are thankful to CSIR for research fellowship. JRG is thankful to DBT and CSIR for financial support. AH is thankful to ICMR for research fellowship. CDRI communication number: 10031.
References
- J. Ranjithkumar, K. Sivasankari and T. Sekar, J. Chem. Pharm. Res., 2010, 2, 371–377 CAS.
- S. Pal, P. Kumar, E. Ramakrishna, S. Kumar, K. Porwal, B. Kumar, K. R. Arya, R. Maurya and N. Chattopadhyay, J. Ethnopharmacol., 2019, 235, 8–18 CrossRef CAS PubMed.
- M. G. Silva, T. P. Aragão, C. F. Vasconcelos, P. A. Ferreira, B. A. Andrade, I. M. Costa, J. H. Costa-Silva, A. G. Wanderley and S. S. Lafayette, J. Ethnopharmacol., 2011, 136, 341–346 CrossRef PubMed.
- V. Singh, Natl. J. Maxillofac. Surg., 2017, 8, 4 CrossRef PubMed.
- J. Yadav, V. Arya, S. Yadav, M. Panghal, S. Kumar and S. Dhankhar, Fitoterapia, 2010, 81, 223–230 CrossRef CAS PubMed.
- T. Aragao, M. Lyra, M. Silva, B. Andrade, P. Ferreira, L. Ortega, S. Da Silva, J. Da Silva, M. Fraga and A. Wanderley, J. Ethnopharmacol., 2009, 123, 163–166 CrossRef CAS PubMed.
- L. Verma, P. Singour, P. Chaurasiya, H. Rajak, R. Pawar and U. Patil, Pharmacogn. Res., 2010, 2, 132 Search PubMed.
- D. Sun, L. Dong, P. Guo, W. Yan, C. Wang and Z. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 932, 66–73 CrossRef CAS PubMed.
- C. Yan, H. Liu and L. Lin, Biomed. Chromatogr., 2013, 27, 228–232 CrossRef CAS PubMed.
- W. Wu, R. Yan, M. Yao, Y. Zhan and Y. Wang, Biomed. Chromatogr., 2014, 28, 564–572 CrossRef CAS PubMed.
- US FDA, Draft guidance for industry: Bioanalytical method validation, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV), Rockville, MD, 2018 Search PubMed.
- G. R. Valicherla, P. Tripathi, S. K. Singh, A. A. Syed, M. Riyazuddin, A. Husain, D. Javia, K. S. Italiya, P. R. Mishra and J. R. Gayen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1031, 123–130 CrossRef CAS PubMed.
- M. Riyazuddin, A. Husain, G. R. Valicherla, S. Verma, A. P. Gupta, K. S. Praveena, A. Singh, S. R. Avula, K. V. Sashidhara and D. Datta, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 121829 CrossRef PubMed.
- J. E. Coughlin, R. K. Pandey, S. Padmanabhan, K. G. O'Loughlin, J. Marquis, C. E. Green, J. C. Mirsalis and R. P. Iyer, Drug Metab. Dispos., 2012, 40, 970–981 CrossRef CAS PubMed.
- M. Riyazuddin, G. R. Valicherla, A. Husain, M. K. Hussain, M. Shukla, R. Katekar, A. P. Gupta, P. Singh, D. Banerjee and K. Hajela, J. Pharm. Biomed. Anal., 2019, 162, 205–214 CrossRef CAS PubMed.
- J. W. Lampe, S. S. Li, J. D. Potter and I. B. King, J. Nutr., 2002, 132, 1341–1344 CrossRef CAS PubMed.
- K. Duan, Z. Yuan, W. Guo, Y. Meng, Y. Cui, D. Kong, L. Zhang and N. Wang, J. Ethnopharmacol., 2011, 135, 201–208 CrossRef CAS PubMed.
- H. S. Cheruvu, N. K. Yadav, G. R. Valicherla, R. K. Arya, Z. Hussain, C. Sharma, K. R. Arya, R. K. Singh, D. Datta and J. R. Gayen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1081, 76–86 CrossRef PubMed.
- L. Wan, C. Guo, Q. Yu, Y. Li, X. Wang, X. Wang and C. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 855, 286–289 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, Z. Zhang and Z. Lu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3595–3600 CrossRef CAS PubMed.
- K. Shimoi, K. Yoshizumi, T. Kido, Y. Usui and T. Yumoto, J. Agric. Food Chem., 2003, 51, 2785–2789 CrossRef CAS PubMed.
- Y. S. Chhonker, H. Chandasana, R. Mukkavilli, Y. D. Prasad, T. S. Laxman, S. Vangala and R. S. Bhatta, Drug Test. Anal., 2016, 8, 966–975 CrossRef CAS PubMed.
- L. Di, E. H. Kerns, Y. Hong and H. Chen, Int. J. Pharm., 2005, 297, 110–119 CrossRef CAS PubMed.
- S. A. Sheweita, Curr. Drug Metab., 2000, 1, 107–132 CrossRef CAS PubMed.
- K. J. Coe and T. Koudriakova, in Optimization in Drug Discovery, Springer, 2014, pp. 87–99 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Athar Husain†ad,
Saurabh Vermaad,
Roshan Katekarad,
Richa Gargad,
Sudhir Kumarb,
Sabbu Satishb,
Rakesh Maurya
a,
Athar Husain†ad,
Saurabh Vermaad,
Roshan Katekarad,
Richa Gargad,
Sudhir Kumarb,
Sabbu Satishb,
Rakesh Maurya



