
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development of isoguanosine: from discovery, synthesis, and modification to supramolecular structures and potential applications
Tingting Ding†
,
Fan Tang†,
Guangcheng Ni,
Jiang Liu*,
Hang Zhao * and
Qianming Chen
* and
Qianming Chen
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Chinese Academy of Medical Sciences Research Unit of Oral Carcinogenesis and Management, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan 610041, P. R. China. E-mail: zhaohangahy@scu.edu.cn; liujiang@scu.edu.cn
First published on 10th February 2020
Abstract
Isoguanosine (isoG), an isomer of guanosine (G), differs from G by the translocation of the C2 carbonyl and C6 amino groups. This minor translocation of functional groups results in significant differences in the properties of G and isoG. Like G, isoG can self-assemble into various supramolecular structures such as tetramers and decamers in the presence of various cations. And it shows a wide range of applications including in ionophores, genetics, gel formation, and cancer treatment. Although there have been many studies and reviews concerning G in recent years, to the best of our knowledge, there have been no reviews that summarized isoG works. Herein, the discovery, synthesis, modification, supramolecular structures and potential applications of isoG are systematically reviewed in this article, in order to provide a reference for future studies.
1 Introduction
Nucleic acids are one of the most basic substances of life, not only comprising genetic material but also playing a crucial role in protein biosynthesis. Nucleic acids are biopolymers composed of nucleotide monomers. Nucleotides in living organisms are composed of a five-carbon sugar, three phosphate molecules and a nitrogenous base (adenine, guanine, cytosine, thymine or uracil, Fig. 1). Compared with the other four bases, guanine has more unique properties because of its unique 2-amino and 6-carbonyl groups. Its 2-amino and 6-carbonyl groups can act as a hydrogen bond donor and receptor, respectively, resulting in its assembly into different structures, such as the cation-templated G-quartet and G-quadruplex.1,2 G-quartets have various applications in ionophore and nanostructure formation. And G-quadruplexes are present in telomeres, suppressing the growth of tumors as telomerase inhibitors. They can also take part in gel formation, and recognize proteins and small molecules. In addition, G-quadruplexes are found in promoter regions of genes and in some untranslated regions of messenger RNA (mRNA).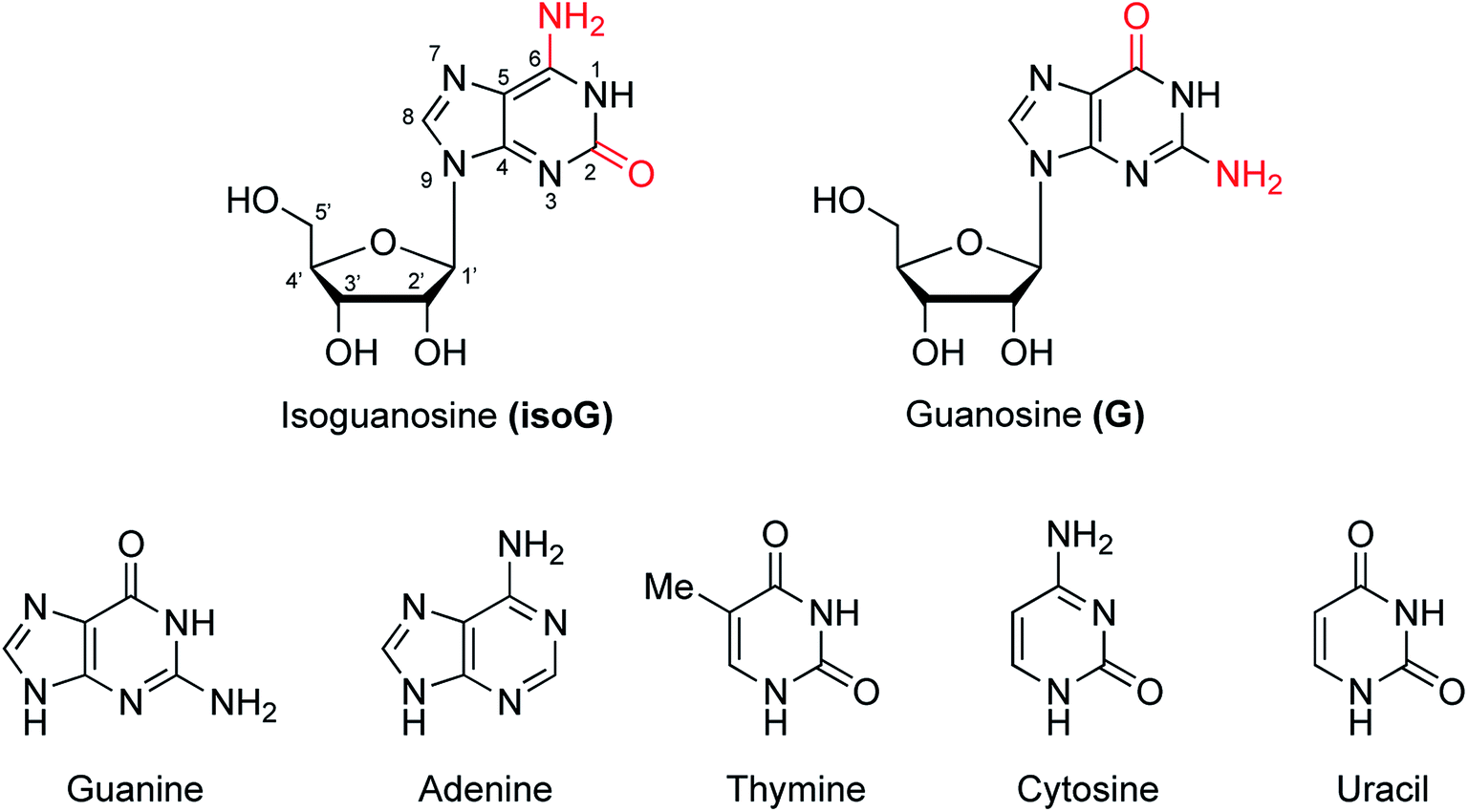 | ||
| Fig. 1 Structures of isoG and G as well as the five natural bases. | ||
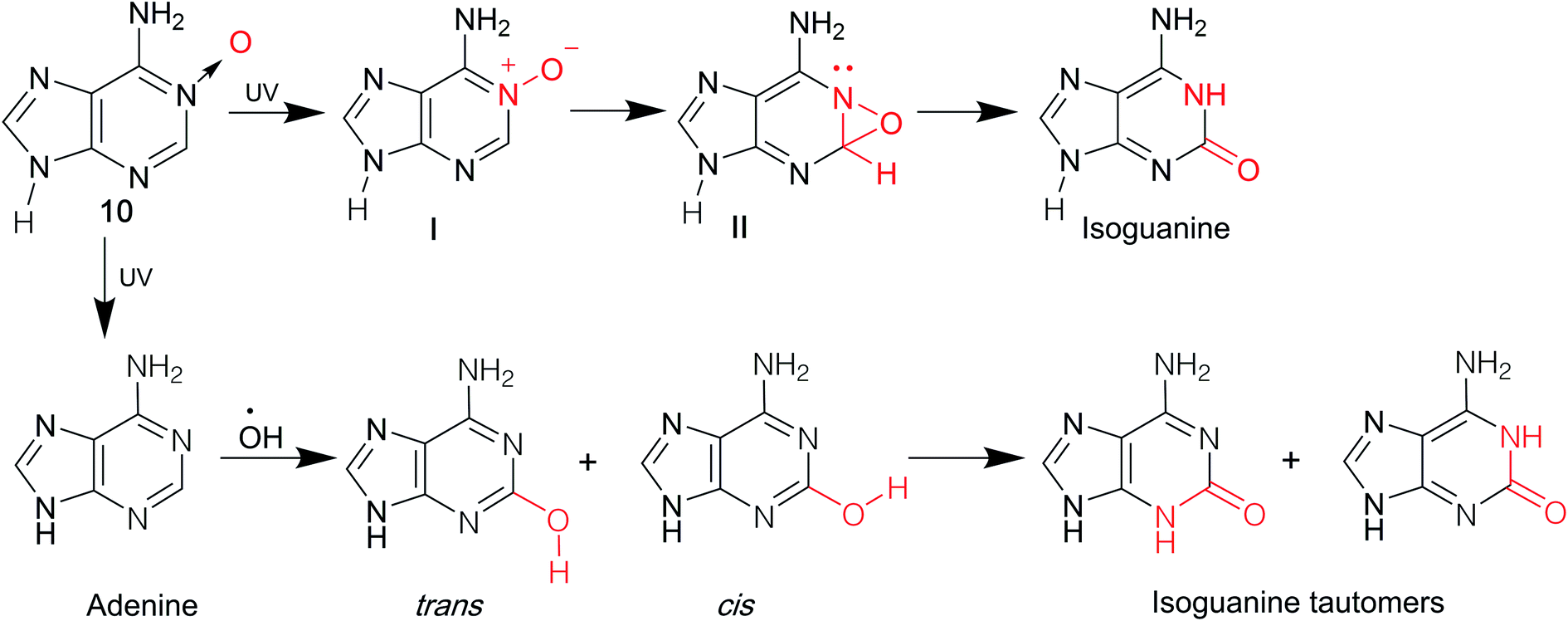
isoG is a natural isomer of G, differing from G because the C2 carbonyl and C6 amino groups are transposed (purine numbering is widely used in this paper) (Fig. 1). This minor change leads to significant differences between isoG and G. isoG, is also known as crotonoside, 2-hydroxyadenosine (2-OH-Ado), 2-oxoadenosine (2-oxo-Ado), 1,2-dihydro-2-oxo-adenosine (1,2-dihydro-2-oxo-Ado), 2-oxy-6-amino-isoguanine-D-riboside and 6-amino-9-β-D-ribofuranosyl-9H-purine-2-oxo-isoguanine riboside. Significant research into isoG has been carried out since Fischer3 first synthesized isoguanine in 1897. isoG/isoguanine were firstly separated from natural substances such as croton beans (Crotonis oleum),4 butterfly wings5 and marine mollusks.6 Later, isoG and its derivatives were synthesized from three categories of methods, one is from the protected 2,6-diamino-9-β-D-ribofuranosylpurine 1,7 adenosine 1-oxygen 2,8 the protected 6-chloroxanthosine 3 (Fig. 3)9 by the introduction of functional groups to the base. The second is from G by exchanging C2 amino and C6 carbonyl groups,10 and the third is from 5-amino-1-(β-D-ribofuranosyl)-imidazole-4-carboxamide (AICA riboside, 4, Fig. 3) to construct the nitrogen heterocycle.11 In addition, researchers have modified the sugar or base groups of isoG/isoguanine to explore differences among them and the potential unique properties. In the meantime, the structures, self-assembly properties of isoG with/without metal cations (M+) have been studied in monomer and multi-molecular layers. isoG can self-assemble into different cation-templated supramolecular structures, such as (isoG)4·M+ (ref. 12) and (isoG)10·M+,13 in a different manner from G. Finally, its potential applications in fields of ionophore, gel-formation, and anticancer treatments etc. are also of great significance and have been systematically reviewed. We hope that this review can not only give a glimpse of the comprehensive development surrounding isoG, but also inspire further research into this fascinating topic.
2 Discovery process of isoG
In 1897, Fischer3 synthesized many purines and identified a strong similarity between guanine and one particular product 2-oxo-6-aminopurine, which had not been discovered in nature (Fig. 2). Believing that 2-oxo-6-aminopurine was oxidized adenine, he suggested that 2-oxo-6-aminopurine would most likely be discovered in animals. And 2-oxo-6-aminopurine may have already been mistaken for guanine. Then in 1927, Buell14 et al. found that oxyadenine, a purine isolated from pig blood, is consistent with 2-oxo-6-aminopurine. However, the presence of 2-oxo-6-aminopurine in pig blood could not be confirmed. In 1932, during a study of croton seeds, Cherbuliez and Bernhard4 accidently discovered a new glucoside, which had not been discovered in nature. They identified it as 2-hydroxy-6-amino-purine-D-riboside. However, the yields obtained from the seeds were variable and very low, and glycosyl crystals could not be obtained by hydrolysis. Subsequently, in 1935, Spies15 et al. successfully enriched the new glucoside and obtained the crystals, and identified the structure as the D-ribose form. In 1939, Spies16 confirmed the observations of Fischer3 and Cherbuliez4 et al. that isoguanine is converted to xanthine by hydrochloric acid but is resistant to nitric acid, whereas guanine is converted to xanthine in nitric acid. This reactivity was evidence of the 2-oxo-6-aminopurine structure that Bendich had observed, i.e., that 2,6-diaminopurine can only be converted to isoguanine rather than xanthine in the reaction with excess nitrous acid. In 1939, Falconer17 et al. confirmed the ribose was linked to the base at N9 position. Afterwards, isoguanine was separated from the butterfly wings of Prioneris thestylis by Purrmann5 in 1940 and by Pettit18 et al. in 1976. In 1951, experiments by Davoll7,19 et al. into optical properties of these compounds revealed that isoG was consistent with 9-β-D-ribofuranosylisoguanine. Later, in a study of Diaulula sandiegensis in 1981, Fuhrman6 et al. confirmed the presence of isoG in extracts of the digestive glands of the sea slug Diaulula sandiegensis. However, the yield was very low, only one milligram of crystals can be obtained from per gram of dry tissue.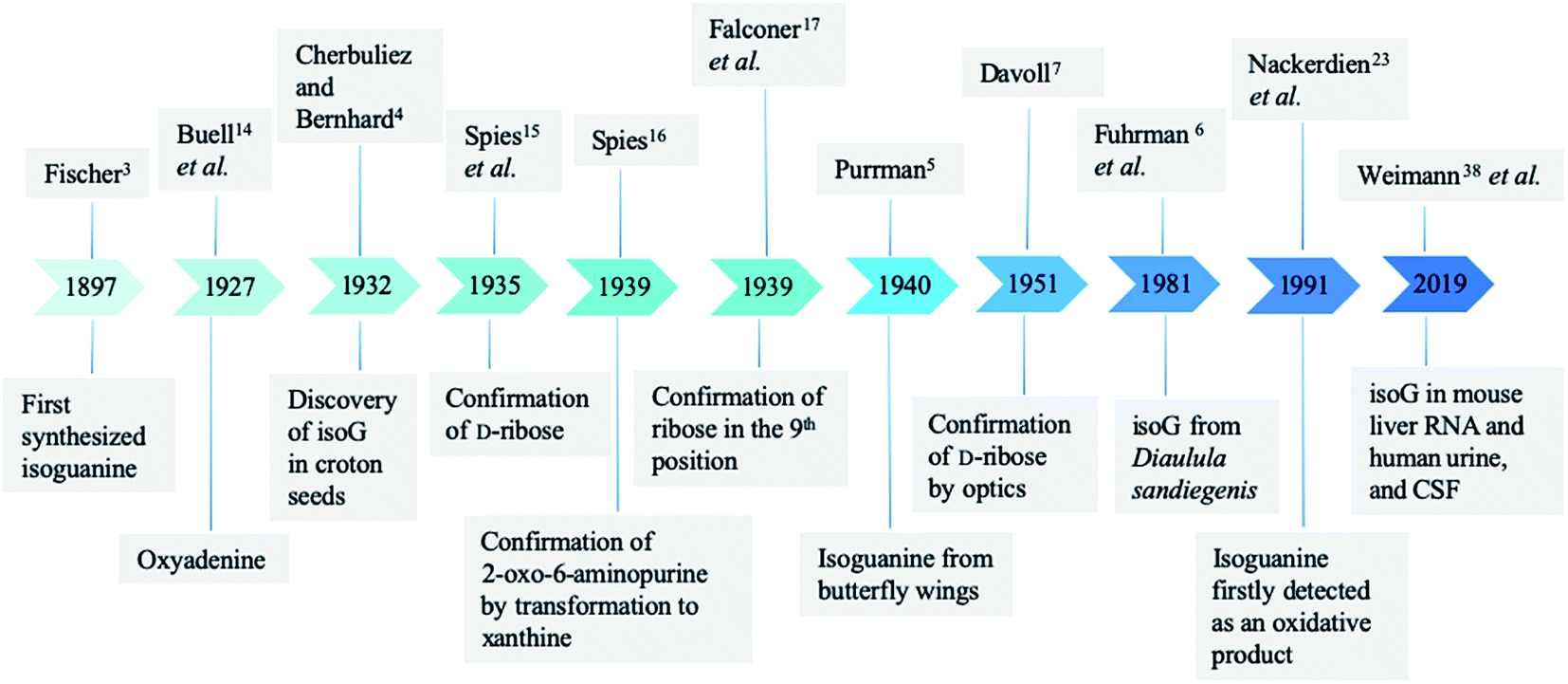 | ||
| Fig. 2 Natural discovery of isoguanine/isoG and the confirmation of structure. | ||
Interestingly, although isoG is a nucleoside that has been isolated from a variety of species, it does not exist as a natural nucleic acid. In 1950, Brown20 et al. demonstrated that isoguanine is not a precursor of nucleoside biosynthesis in mice. In 1952, they reported that isoguanine could not be used as a pure source of purines in Lactobacillus casei, nor could it be integrated into nucleic acid.21 Notably, C14 crotonoside has been used to investigate its role in the metabolism of Escherichia coli (E. coli). The results indicate that isoguanine and its derivatives play an important role in nucleic acid metabolism.21,22
In 1991, using gas chromatography/mass spectrometry (GC-MS) in isolated human chromatin isoguanine, Nackerdien23 et al. firstly detected 2-OH-Ade within Ni(II)/Co(II)–H2O2-treated DNA. And in 1992, Dizdaroglu24 reported that in the presence of Fe(III), Cu(II), Ni(II) and Co(II), isoguanine (2-hydroxyadenine) was a product of H2O2-treated mammalian chromatin both in vivo and in vitro. Oxidation reaction commonly occurs in cells because of the presence of reactive oxygen species (ROS), which plays an important role in DNA damage. In this respect, it is logical that isoguanine is more abundant in human cancerous tissue than in normal tissue, which is reported by Olinski25,26 et al. in 1992 and 1994. In 1993, Dizdaroglu27 et al. identified isoguanine in mice at levels of only a few oxidation sites per 105 DNA bases. They also identified isoguanine as an oxidation product of hepatic chromatin exposed to γ-ray in pregnant rats and fetuses of theirs. In 1996, using GC-MS, Jaruga28 et al. reported that isoguanine is produced as a DNA oxidation product of adenine in E. coli, as well as in cultured human lymphoblast cells. These have drawn attention toward isoguanine as a marker for oxidative damage formation, repair, mutagenicity.29–34
In 2002, Cadet35 et al. demonstrated that after γ-ray radiolysis and Fenton reaction of free nucleosides, the yield of 2′-deoxyisoguanosine (isoGd) was low, compared to the yield of 8-oxo-2′-deoxy-adenosine, expect in the presence of ferrous ion (Fe2+)/ethylenediaminetetraacetic acid (EDTA) with oxygen or cuprous ion (Cu+). However, Fe2+/EDTA with oxygen or Cu+ are typically at very different conditions from those found in cells. They concluded that isoGd cannot be detected under a limit of per 107 bases in human DNA and E. coli. They also reported that the process of conventional GC-MS, which involves acid hydrolysis and derivation at high temperature, may have caused an overestimation of isoguanine/isoG.35–37 Notably, in 2019, using the ultra-performance liquid chromatography (UPLC) and electrospray ionization isotope dilution tandem mass spectrometry, Weimann38 et al. firstly identified and quantified the ribonucleoside isoG in mouse liver RNA, human urine, and cerebrospinal fluid (CSF). However, the amount of detected isoG is more than or equal to that of 8-oxo-G, which is most easily oxidized, and isoGd was not detected. These data are opposed to the common hypothesis that the free nucleoside isoG may originate from oxidation reaction. As a result, the reason why isoG exists in mammals still remains a matter of study.
3 Syntheses of isoG
As a special nucleoside, isoG is expected to present applications in various areas. However, its yield from natural sources is too low. Thus, many synthetic methods have been reported (Table 1), and these synthetic methods can be divided into three categories: (1) the introduction of the functional groups to existing heterocyclic 9-β-D-ribofuranosylpurine nucleosides (Fig. 3), such as 2,6-diamino-9-β-D-ribofuranosylpurine 1,7 adenosine 1-oxide 2,8 the protected 6-chloroxanthosine 3,39 (2) G as an initial reactant to build isoG by exchanging the C2 amino and C6 carbonyl groups, (3) the construction of nitrogen heterocycle with AICA riboside (4, Fig. 3) as a precursor for base synthesis to introduce oxo group at C2 position. Notably, the third method is challenging because it is difficult to obtain AICA. These syntheses will be discussed in detail in the following parts.| Year | Authors | Initial substrates | Productivity | References |
|---|---|---|---|---|
| a The yield is not mentioned. | ||||
| 1897 | Fischer | 6-Oxy-2,8-dichlorpurine | —a | 3 |
| 1951 | Davoll | 2,6-Diamino-9-β-D-glucopyranosylpurine/2,6-diamino-9-β-D-ribofuranosyl purine | 55% 9-β-D-Glucopyranosylisoguanine/57% 9-β-D ribofuranosylisoguanine | 7 |
| 1964 | Brown et al. | Adenosine 1-oxide | Variable | 8 |
| 1965 | Ravindranathan et al. | 2,6-Diaminopurine riboside | —a | 40 |
| 1968 | Montgomery et al. | The protected 2,6-diaminopurine nucleoside | By-product | 42 |
| 1995 | Napoli et al. | The protected 6-chloroxanthosine | 20% isoG | 43 |
| 1995 | Napoli et al. | 2′,3′,5′-tri-O-acetylxanthosine | 76% isoG | 43 |
| 1997 | Napoli et al. | The protected 6-chloroxanthosine | 80% isoG | 48 |
| 2012 | Cheng et al. | Adenine | —a | 44 |
| 1985 | Nair et al. | G | 34% isoG | 39 |
| 1991 | Divakar et al. | G | 64% isoG | 10 |
| 1968 | Yamazaki et al. | 4-Amino-5-imidazole carbonitrile (AICN) | —a | 45 |
| 1976 | Yamazaki et al. | AICA (riboside) | By-product | 46 |
| 1987 | Reese et al. | The protected AICA riboside | —a | 9 |
| 1987 | Chern et al. | AICA (riboside) | 77% isoG/(—a) isoguanine | 11 |
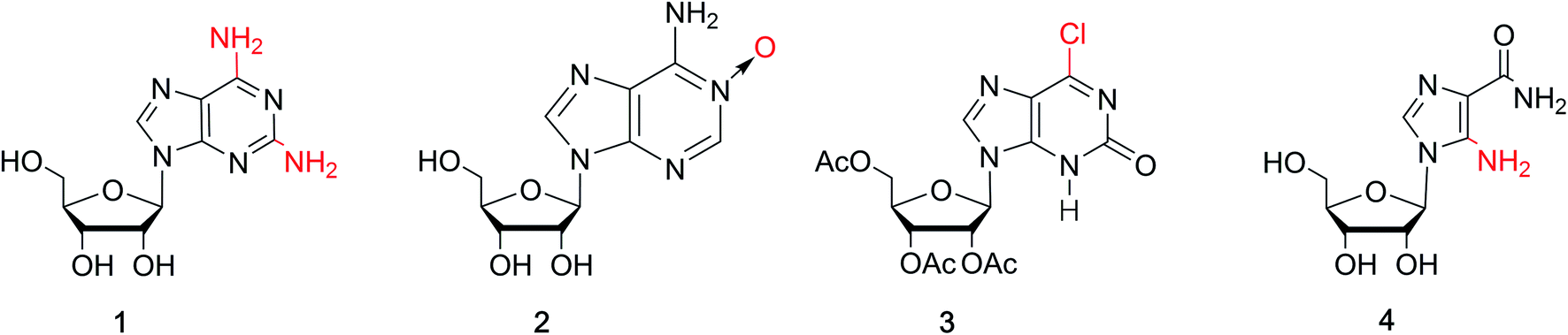 | ||
| Fig. 3 Common substrates for the synthesis of isoG. | ||
3.1 Introducing functional groups on the existing heterocycle
Isoguanine was initially synthesized by Fischer3 in 1897. It can resist the deamination by nitrous acid, and the reaction of 2,6-diaminopurine with excess nitrous acid only yields isoguanine not xanthine. In 1951, Davoll7,19 et al. reported that 9-β-D-glucopyranosyl-isoguanine can be obtained in 55% yield by the reaction of 2,6-diamino-9-β-D-glucopyranosylpurine with nitrous acid, while 9-β-D-ribofuranosyl-isoguanine can be obtained in 57% yield from the reaction of 2,6-diamino-9-β-D-ribofuranosylpurine (1, Fig. 3) and nitrous acid (Fig. 4). However, the overall yields are low and harmful heavy metal ions (Hg and Pb) are used. So in 1965, Ravindranathan40 et al. synthesized isoG using the way of Davoll but using much charcoal instead of a lead salt for purification. On account of the relevance of ultraviolet (UV) light and the mutations in nucleic acids, in 1964, Brown8 et al. reported a photochemical preparation of isoG from adenosine 1-oxide (2, Fig. 4). Adenosine 1-oxide is more sensitive to UV light than adenosine. However, this method gives variable proportions of two major products. One product is adenosine (6, Fig. 4) via the direct loss of oxygen. The other product is isoG via the rearrangement of oxygen atom to the neighboring carbon atom. Subsequently in 1975, Mantsch41 et al. concluded that if nucleotides of the initial substance 2 is irradiated with UV light under alkaline condition (0.01 M NH4OH), isoG nucleotides can be the corresponding primary products. This is also the case for adenine 1-oxide (10, Fig. 5).8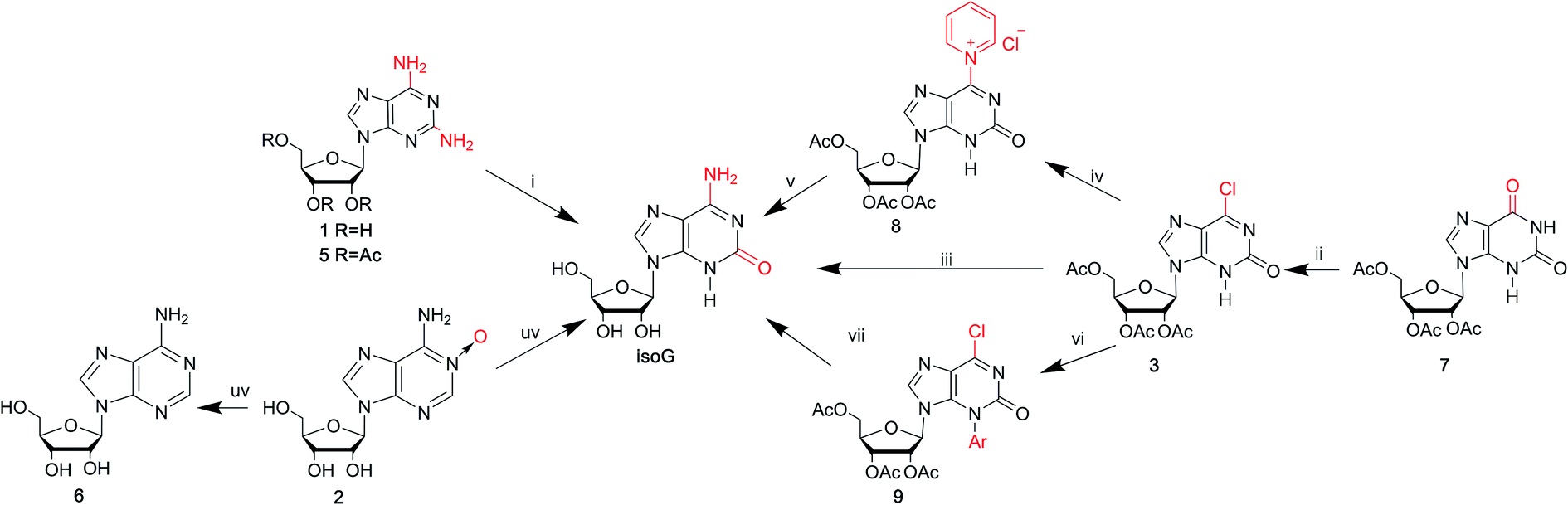 | ||
Fig. 4 Syntheses of isoG via introducing functional groups. (i) Nitrous acid for compound 1; hydrogen and palladium-on-charcoal catalyst, 40% fluoroboric acid, sodium nitrite for compound 5; (ii) PPh3 (2 mol equiv.), CCl4, 3 h, reflux; (iii) aq. NH3 (32%), 3 days, room temperature; (iv) pyridine–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 2 h, 50 °C; (v) aq. NH3 (32%), 5 h, 50 °C; (vi) 2,4-dinitrochlorobenzene in DMF solution, 1.5 h, 80 °C; (vii) NH4OH. :1), 2 h, 50 °C; (v) aq. NH3 (32%), 5 h, 50 °C; (vi) 2,4-dinitrochlorobenzene in DMF solution, 1.5 h, 80 °C; (vii) NH4OH. | ||
 | ||
| Fig. 5 Syntheses of isoguanine via introducing functional groups. | ||
In 1968, in a synthesis of 2-fluoroadenosine by Montgomery42 et al., isoG was reported to be a by-product, relying on a protected 2,6-diaminopurine nucleoside (5, Fig. 4). Generally, the introduction of N to the assigned activated nucleoside bases of guanine, cytosine and adenine is easy but this is not the case for isoG. A significant intermediate 3 can be obtained from 2′3′5′-tri-O-acetylxanthosine 7 (Fig. 4). Nevertheless, in 1995, Napoli43 et al. reported a direct route from the protected 6-chloroxanthosine 3 to isoG, but this gives a very low yield of 20%. So, they synthesized isoG through the reagent to nucleophilic attack on the pyridinium α-carbon, and obtained the intermediate (8, Fig. 4). Then isoG was isolated by HPLC in 76% overall yield by ring opening (Zincke reaction) of the compound 8. Then in 1997, they proposed another way by introducing a strong electron-withdrawing group in the heterocyclic to enhance the reactivity of C6 towards aqueous ammonia, in a yield of 80%, with an important intermediate (9, Fig. 4). In 2012, Cheng44 et al. published a new two-step method based on adenine. A mixture of isoguanine tautomers was obtained (Fig. 5). Reactions had been proved where OH radicals attack adenine on the C2, C4, C5, and C8 positions, and the reactions are spontaneous and exothermic. In the first step, attack by OH radicals at the C2 position of adenine forms a favorable low-energy complex (Fig. 5). In the second step, the enol cis–trans forms are converted into the corresponding N1H/N3H keto forms.
3.2 G to isoG by exchanging C2 amino and C6 carbonyl groups
As mentioned above, the route from 2,6-diaminopurines to isoG requires harmful heavy metal ions (Hg and Pb) and the overall yields are low. isoG was reported as a by-product in a synthesis of nucleosides from 2,6-diaminopurines, as well as from adenosine-1-oxide, both in variable yields. In 1985, Nair39 et al. found a new reproducible and efficient method for the synthesis of isoG starting from G (Fig. 6). This method contains five steps. Firstly, the sugar is protected by selective acetylation. Secondly, reaction with phosphorus oxychloride and N,N-dimethylaniline yields the 6-chloropurine nucleoside (11, Fig. 6). Thirdly, n-pentyl nitrite reacts with diiodomethane to obtain protected 2-iodo-6-chloropurine nucleoside (12, Fig. 6). Then, 2-iodoadenine (13, Fig. 6) is obtained by reacting with ethanolic ammonia. And finally, in the last key step in this process, photo-induced hydration occurs. The product isoG was isolated by HPLC. However, this method gives a low yield about 34%. In addition, in the fifth step, the requirement of UV irradiation limits the spread of this synthetic route. Therefore, in 1991, Divakar10 et al. developed an improved five-step route (Fig. 6) starting from G, which achieved isoG with a 64% overall yield involving the same 6-chloropurine nucleoside (11, Fig. 6), with an important intermediate 14. This means that, for the latter reaction, inavailability of direct synthesis to 6-chloroxanthosine is not required. In contrast, a better yield is obtained. Moreover, conditional complex photochemical steps are avoided.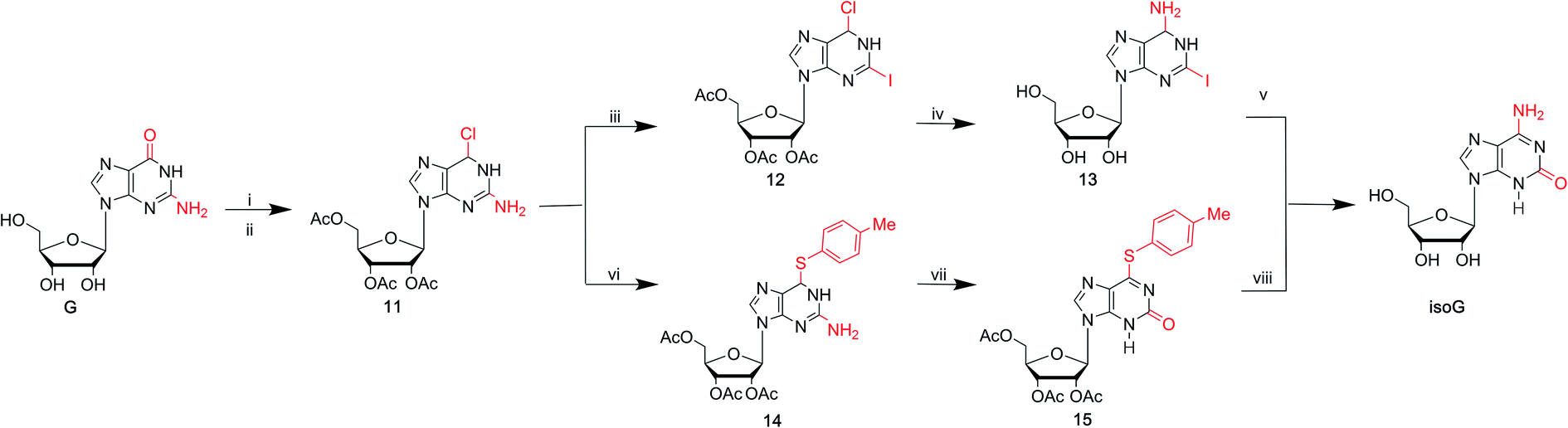 | ||
| Fig. 6 Syntheses of isoG from G. (i) Ac2O, pyridine, DMF, 70 °C; (ii) POCl3, N,N-dimethylaniline, CH3CN, 100 °C; (iii) n-C5H11ONO, CH2I2, heating; (iv) NH3, C2H5OH; (v) H2O, UV; (vi) toluerthliol, TEA, DMF, 100 °C; (vii) 50% AcOH, NaNO2, 50 °C; (viii) ammonia, ethanol, 70 °C. | ||
3.3 isoG from AICA by constructing a nitrogen heterocycle
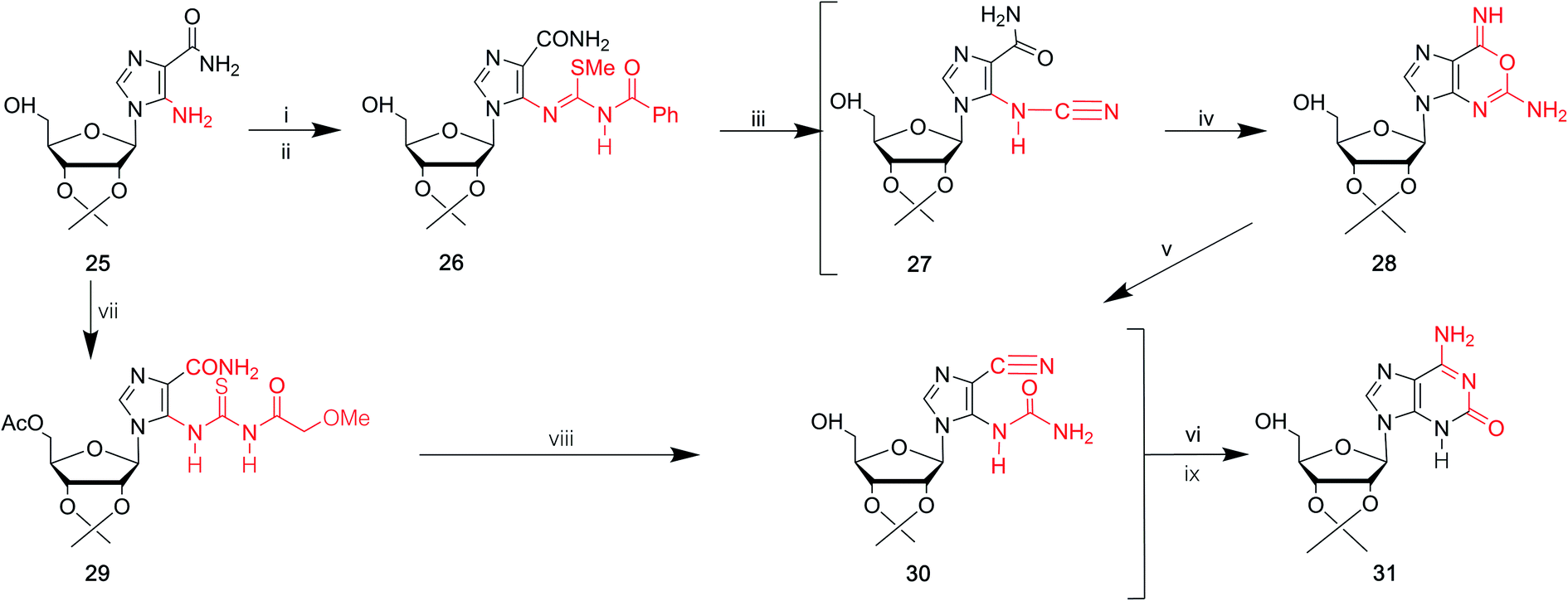
In 1968, Yamazaki45 et al. found that isoG/isoguanine can be obtained from 4-amino-5-imidazole carbonitrile (AICN, 16, Fig. 7) and its protected riboside (21, Fig. 7), but the yield is too low. For a long time, there have been great efforts to synthesize guanine from AICA. In 1976, isoguanine was discovered as an unexpected product in a synthetic process of guanine by Yamazaki46 et al. from AICA. Isoguanine was obtained from an intermediate substance 4-cyanamido-imidazole-carboxamide (20, Fig. 7) in weakly alkaline or neutral conditions. Whereas in alkaline conditions, the compound 20 was converted into guanine. They synthesized isoG depending on the protected AICA riboside (25, Fig. 8), in low yield with three plausible and non-isolated intermediates (27, 28, 30, Fig. 8). Subsequently in 1987, Reese9 et al. reported that the compound 30 can also be obtained from compound 25 via the intermediate putative methoxyacetyl thioureido derivative 29 (Fig. 8). Moreover, in 1987, enlightened by a synthesis of 1-methyl-isoG, Chern11 et al. proposed that an N,N′-dicyclohexylcarbodiimide (DCC)-mediated cyclodesulfurative method would allow milder reaction conditions and produce a higher yield of isoG. They reported an improved, more efficient route in milder reaction conditions using AICA 32 and its riboside 4 to synthesize isoguanine and isoG, respectively (Fig. 9). When AICA/AICA riboside 32/4 reacts with benzoyl isothiocyanate in dimethylformamide (DMF) at room temperature, it can be converted to benzoylthiourea intermediates 33/34, respectively. Next, DCC was added to this system and isoguanine/isoG can be obtained via stirring, washing and separation. And isoG can be obtained in a high overall yield of 77%. In 1991, they also synthesized several 1-substituted isoG through similar DCC-mediated ring-closure reactions.47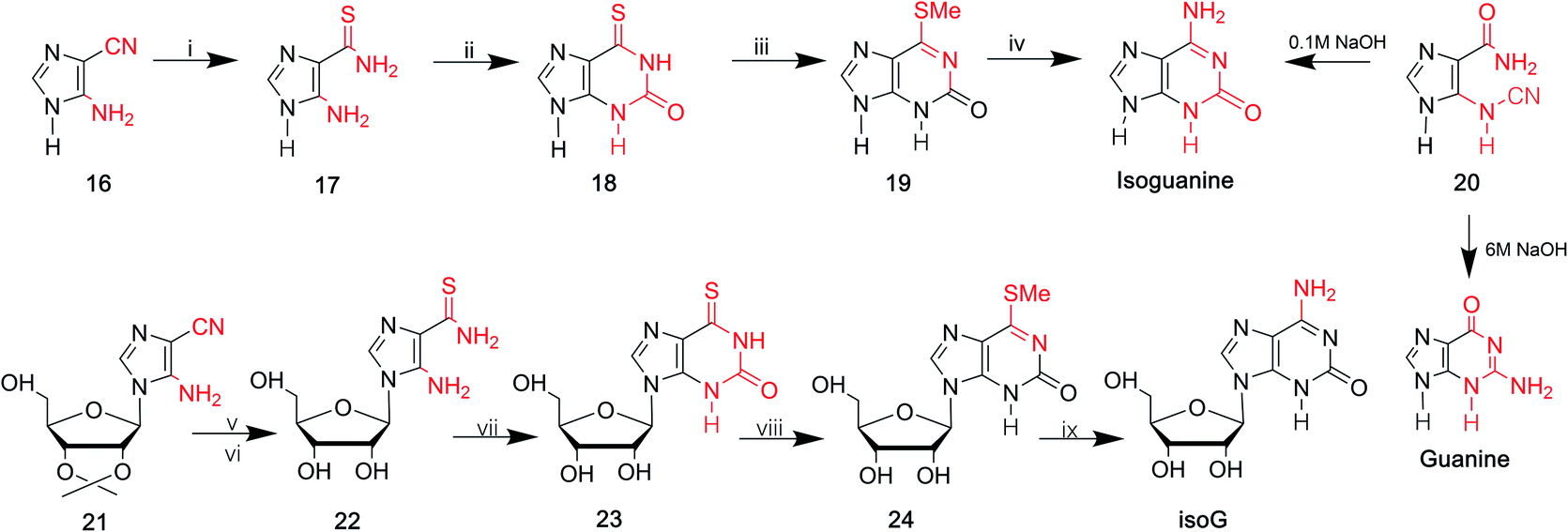 | ||
| Fig. 7 Syntheses of isoG/isoguanine from AICN. (i) H2S, KOH; (ii) fusion with urea; (iii) CH3I, alkaline solution; (iv) ammonia, autoclave at 130 °C, 3 h; (v) H2S; (vi) acid condition, hydrolyzation; (vii) diethyl carbonate; (viii) CH3I; (ix) ammonia. | ||
 | ||
| Fig. 8 The syntheses of isoG from the protected AICA riboside. (i) Benzoyl isothiocyanate; (ii) methyl iodide, alkali; (iii)–(vi) 0.1 M NaOH, heating, reflux; (vii) acetic anhydride, in pyridine solution, then methoxyacetyl isothiocyanate; (viii) mercury(II) perchlorate, pyridine, in tetrahydrofuran solution, room temperature, then methanolic ammonia; (ix) N1,N1,N3,N3-tetramethylguanidine, water, in tetrahydrofuran, room temperature. | ||
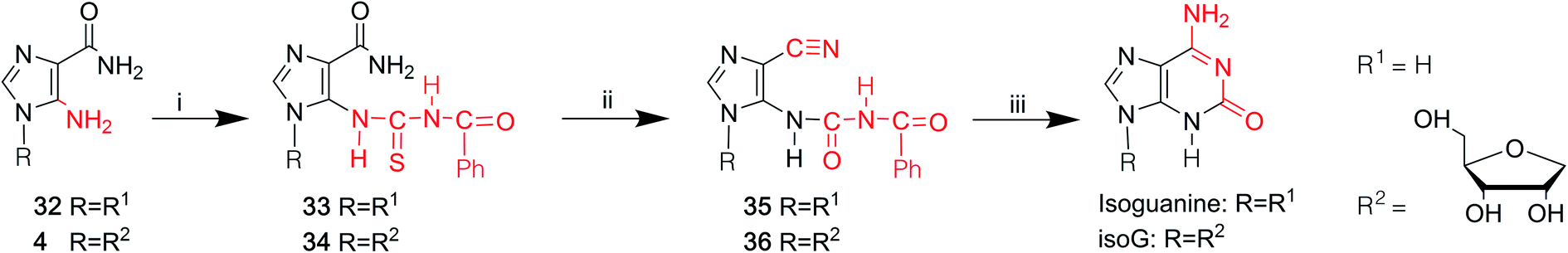 | ||
| Fig. 9 Syntheses of isoguanine/isoG from AICA and its riboside. (i) Benzoyl isothiocyanate, DMF, room temperature; (ii) dicyclohexylcarbodiimide, DMF, room temperature; vacuo, 50 °C, hot toluene; (iii) ethanol, 33% aqueous ammonia. | ||
In summary, as shown in Table 1, over time, the syntheses of isoG/isoguanine have developed toward milder reaction conditions and higher yield. To date, the most outstanding synthetic route to isoG is that involving 6-chloroxanthosine by Napoli48 et al., with a yield of 80%, under mild conditions.
4 The modified isoG
In the above sections, we have discussed the discovery of natural isoG, as well as a variety of synthetic methods. isoG contains a nitrogenous base (a purine ring) and a carbohydrate residue. Minor modification to the base or sugar may lead to great differences in the higher structures and properties compared to the parent compound. Thus, a number of base-modified (Fig. 10) or sugar-modified (Fig. 11) analogs of isoG have been discovered in nature or synthesized. Summaries are shown in Table 2. And some of their properties are discussed in the following part.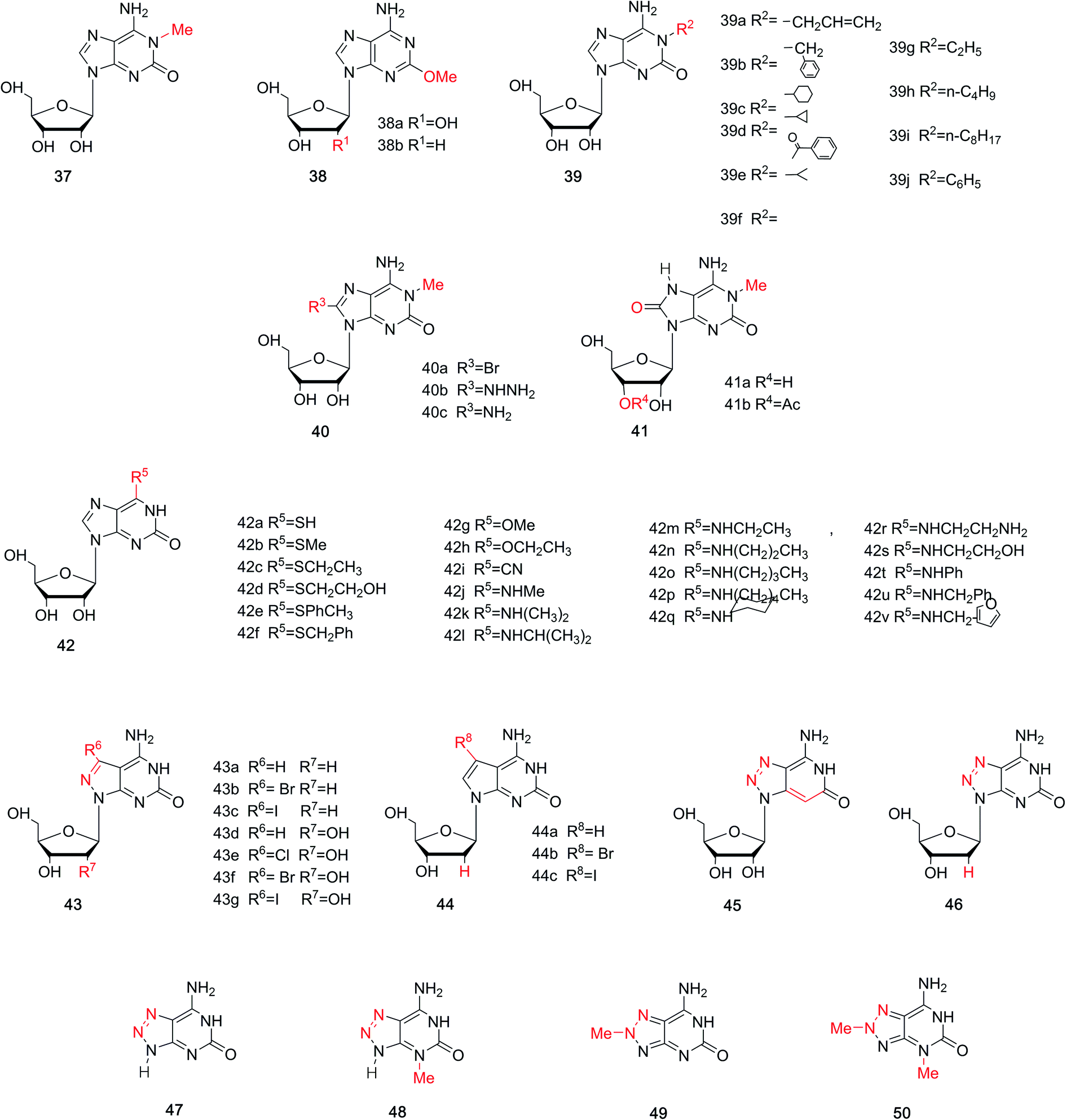 | ||
| Fig. 10 Base-modified derivatives of isoG/isoguanine. | ||
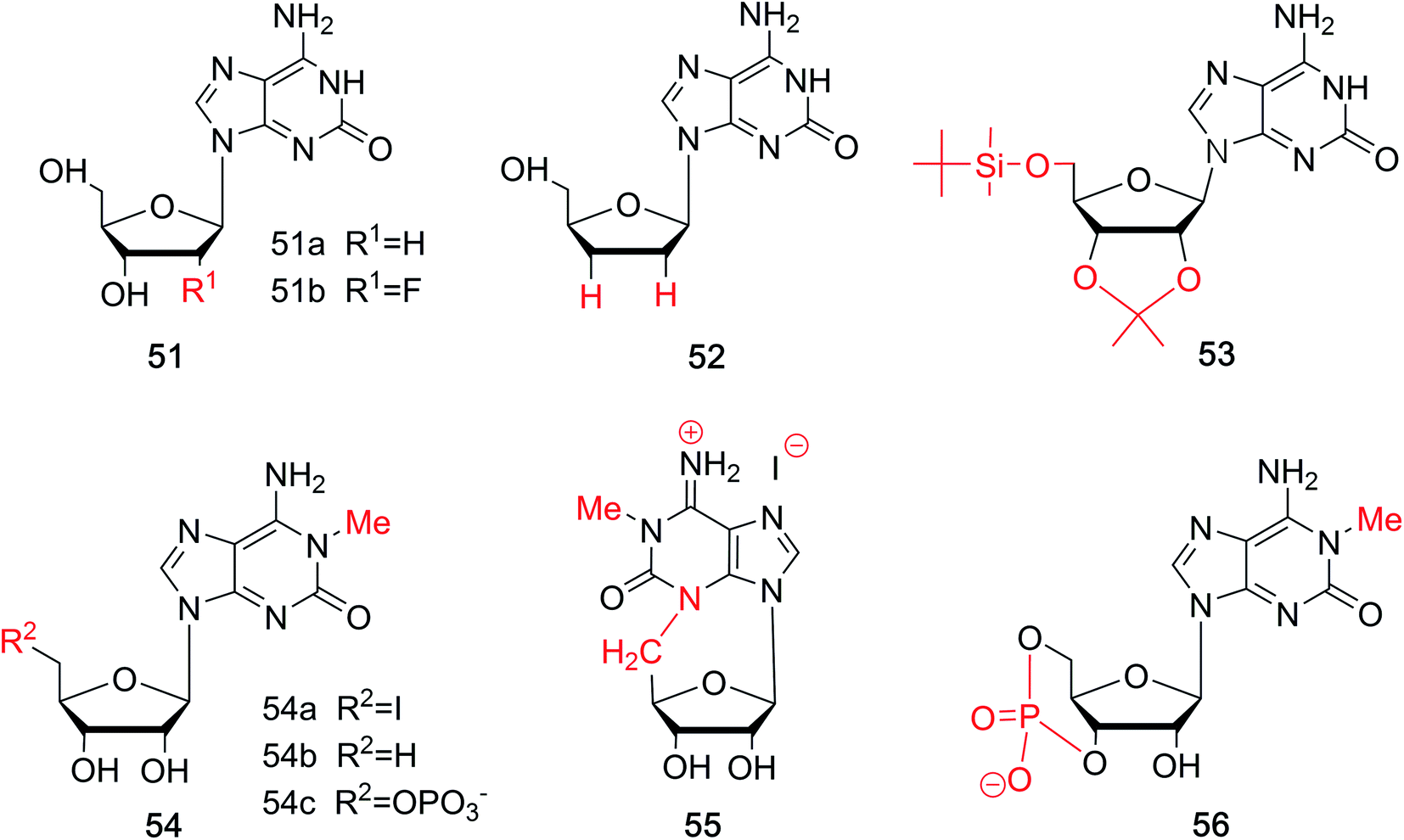 | ||
| Fig. 11 Sugar-modified derivatives of isoG. | ||
| Analogs | Naming | From | First | Authors | References |
|---|---|---|---|---|---|
| a Synthesized. | |||||
| Base-modified | 1-Methyl-isoG 37 | Natural | 1979/1980 | Fuhrman et al./Quinn et al. | 49 and 50 |
| Spongosine 38a | Natural | 1951 | Bergmann et al. | 52 | |
| 2′-Deoxyspongosine 38b | Natural | 1994 | Searle et al. | 55 | |
| N1-Substituted isoG 39 | Syna | 1991/1981 | Chern et al./Bartlett et al. | 48 and 56 | |
| 8-Substituted-1-methyl-isoG 40 | Syna | 1981/2017 | Bartlett et al./Firsova et al. | 56 and 57 | |
| 8-Oxo-1-methyl-isoG 41a | Natural | 2017 | Thomas et al. | 57 | |
| 3′-o-Acetyl-8-oxo-1-methyl-isoG 41b | Natural | 2017 | Thomas et al. | 57 | |
| 6-Substituted isoG 42 | Syna | 1994 | Sung Jun Lee et al. | 58 | |
| 7-Halogenated-8-aza-7-deaza-isoguanosine derivatives 43 | Syna | 2003/2007 | Seela et al. | 67 and 68 | |
| 7-Halogenated-7-deaza-isoguanosine derivatives 44 | Syna | 2007 | Seela et al. | 68 | |
| 8-Aza-3-deaza-isoG 45 | Syna | 2004 | Jeselnik et al. | 69 | |
| z8isoGd 46 | Syna | 2010 | Seela et al. | 73 | |
| 8-Aza-isoguanine 47 | Syna | 1970 | Fox et al. | 74 | |
| N3-Methyl-8-aza-isoguanine 48 | Syna | 1970 | Fox et al. | 74 | |
| N8-Methyl-8-aza-isoguanine 49 | Syna | 1970 | Fox et al. | 74 | |
| N3,N8-Dimethyl-8-aza-isoguanine 50 | Syna | 1970 | Fox et al. | 74 | |
| Sugar-modified | isoGd 51a | Syna | 1973 | Kazimierczuk et al. | 76 |
| FisoGd 51b | Syna | 2015 | Seela et al. | 78 | |
| 2′3′-Dideoxy-isoG 52 | Syna | 1999 | Kim et al. | 81 | |
| 5′-(tert-Butyldimethylsilyl)-2′,3′-O-isopropylidene-substituted isoG 53 | Syna | 1995 | Davis et al. | 82 | |
| 5′-Substituted-1-methyl-isoG 54 | Syna | 1981 | Bartlett et al. | 56 | |
| 3,5-Anhydro-l-methyl-isoG hydriodide 55 | Syna | 1981 | Bartlett et al. | 56 | |
| 1-Methyl-isoG cyclic 3′,5′-phosphate 56 | Syna | 1981 | Bartlett et al. | 56 | |
4.1.1 1-Methyl-isoG (doridosine). Dorid nudibranchs are shell-less sea slugs. Interestingly, they have long lives despite lacking the typical protection given by a shell. Thus, there has been significant conjecture about their defense mechanism. In 1979, a tissue analysis by Fuhrman49 et al. revealed that only the digestive gland extract is toxic, and this acts as the unique defense mechanism of Dorid nudibranch. In 1980, Quinn50 et al. isolated a new pharmacologically active substance from the Australian sponge Tedania digitate, in a yield of 0.71% from the crude extract. They also developed a synthesis of doridosine from AICA riboside (4, Fig. 3) by acetylation in four steps, giving a yield of 39%. Meanwhile, they identified doridosine as identical to 1-methyl-isoG (37, Fig. 10) by spectrum, chemical degradation, synthetic analysis. In 1981, Fuhrman51 et al. concluded that the substances isolated from nudibranch and sponge are indeed the same. Moreover, 1-methyl-isoG 37 has been shown to have several pharmacological effects related to hypotension, hypothermia and anti-inflammatory, and these will be discussed in Section 6.3.
4.1.2 2-Methoxyadenine (spongosine). Nucleosides from marine organism have been found to have various biological activities, and this has aroused great interests among scholars. In a study of Carribean sponge Cryptotethya crypta in Florida,52,53 a mixture of two unnatural nucleosides spongothymidine and spongouridine were obtained. And these were identified as 3-β-arabofuranosides of thymidine and uridine, respectively. The development of materials has promoted elucidation of the composition and structures of these purines and the carbohydrate. Then in 1951, Bergmann53 et al. firstly isolated spongosine (38a, Fig. 10) from Cryptotethya crypta. It was indentified as pentosylmethyl-aminooxypurine. In 1956, they determined the structure of D-riboside of 2-methoxy-6-aminopurine (2-methoxyadenine) using UV absorption spectra and the synthesis of 2-methyladenine.54 They concluded that it is not only the first methoxy purine found in nature, but also the first occurrence of methoxy substance isolated from animals. In addition, in 1994, Searle55 et al. firstly identified 2′-deoxy-spongosine [9β-(2′-deoxy-D-ribofuranosyl)-2-methoxyadenine, 38b, Fig. 10] as a natural product from Western Australia.
4.1.3 More analogs. In terms of the great effects of 1-methyl-isoG 37, many analogs of 1-methyl-isoG have been prepared from the protected imidazole nucleoside to explore the structure–activity correlation. As shown in Fig. 10, some N1-substituted isoG 39 have been developed to explore the importance of N1-methyl-substituted compounds in marine products. Some of these have been synthesized in good yield, by Bartlett56 et al. in 1981 and Chern47 et al. in 1991. In addition, in 1981, using 1-methyl-isoG 37 as a starting material, C8-substituted-1-methyl-isoG 40 was prepared by Bartlett56 et al. Their effects on cardiovascular response and muscle relaxation, etc. will be discussed later in Section 6.3. Moreover, in 2017, Thomas57 et al. identified 8-oxo-1-methyl-isoG 41a and 3′-O-acetyl-8-oxo-1-methyl-isoG 41b as natural derivatives of isoG in a sponge from the Northeastern Atlantic, Clathria (Microciona) strepsitoxa. And these compounds showed no significant cytotoxic or antimicrobial activity. In addition, in 1994, Lee58 et al. synthesized a series of 6-substituted isoG derivatives 42 and isoG by one-step nucleophilic substitution from the compound 15 (Fig. 6). These were synthesized to explore the antitumor activities, and the results will be discussed later in Section 6.3.
8-Aza-7-deazapurines and their 7-halogenated derivatives have been reported to possess excellent biological and physical properties, such as antiparasitic and antiviral effects, because of their structural similarity to purines in nucleic acids. And earlier studies showed that 7-substituents of 8-aza-7-deazapurine nucleosides and 7-deazapurine nucleosides incorporated oligodeoxynucleosides could increase antiparallel strand (aps) duplexes stability.59–66 Thus, in 2003 and 2007, Seela67,68 et al. synthesized 7-halogenated 8-aza-7-deazaisoguanosine derivatives 43 and 7-halogenated 7-deaza-isoguanosine derivatives 44 from 8-aza-7-deazapurine-4,6-diamine riboside derivatives. Their tautomerism will be discussed later in Section 5.1. Moreover, in 2004, 8-aza-3-deaza-isoG 45 was synthesized by Jeselnik69 et al. to study the influence of the lack of N3 on the hydrogen-bonding modes. 8-Azapurine nucleosides, including 8-azaadenine, 8-azaguanine, and their corresponding nucleosides, have been reported to be fluorescent. And they show various applications as fluorescent probes and in biochemical process.70–72 Consequently, in 2010, Seela73 et al. synthesized 8-aza-2′-deoxyisoguanosine (z8isoGd, 46) from the protected 8-aza-2′-deoxyguanosine (z8Gd) in a yield of 27%. z8isoGd shows great fluorescence that becomes stronger in alkaline conditions than at neutral pH.73 Moreover, 8-azaisoguanine 47 and its N-methyl derivatives N3-, or N8-methyl-8-azaisoguanine and N3,N8-dimethyl-8-azaisoguanine 48–50 were synthesized in the method of Fox74 et al. In 2012, Wierzchowski75 et al. reported these compounds 48–50 also showed fluorescence, which is highly sensitive to the microenvironment.
Fluorine-substituted nucleosides have been reported to be efficient antiviral compounds. The fluorine–carbon bond is particularly stable, and its introduction to the sugar moiety can increase the lipophilicity of a drug.77 Fluorine is generally incorporated into the 2′-position of the sugar, and it has the near radius as hydrogen and can exist in duplexes. Apart from these, fluorine also has a significant effect on the sugar conformation because of its strong electronegativity. Based on the characteristic of fluorine mentioned above, through deamination of 2,6-diamino-2′-deoxy-2′-fluoropurine nucleoside, Seela78 et al. first synthesized 2′-fluorine-2′-deoxyisoguanosine (FisoGd, 51b, Fig. 11) in a yield of 79%. Studies concerning the stability of its duplexes will be discussed in Section 5.3.4.
Some 2′3′-dideoxynucleoside derivatives have been reported to show effects such as inhibiting reverse transcription.79,80 Inspired by these findings, in 1999, Kim81 et al. synthesized 2′,3′-dideoxy-isoG (52, Fig. 11) from G in a yield of 10%. And studies on its biological activities are ongoing. Besides, in 1995, the protected isoG derivatives 5′-(tert-butyldimethylsilyl)-2′,3′-O-isopropylidene-substituted isoG (53, Fig. 11) has been prepared by Davis82 et al. to study the self-assembly of isoG. So hydrogen bonds of isoG can be researched at different temperature and in different solvents. 1-Methyl-isoG (37, Fig. 10) has been identified as a natural product. And in 1981, using 1-methyl-isoG as a starting material, Bartlett56 et al. prepared several N9-substituted doridosine compounds (54–56, Fig. 11). The effects of these compounds are shown later in Section 6.3. All the derivatives mentioned above can be found in Table 2 for their original sources.
5 Structure properties of isoG
The discovery of isoG and isoguanine in nature and their structural determination have been discussed above. In addition, we have discussed the low content and low extraction efficiency of these compounds in natural products, which led to the development of a series of synthetic methods. Next, some of the special structural properties of isoG, including its tautomerism at the single molecule level and the resulting unnatural pairing in nucleic acids, its self-assembled structures such as tetramers, the more highly ordered “sandwich-like” decamer structure, the isoG4 quadruplexes formed in helix, the parallel-stranded (ps) and antiparallel-stranded (aps) chains containing isoG will be discussed as follows.5.1 Tautomerism enol–keto of isoG
The tautomeric phenomenon of nitrogen heterocycle has been widely investigated, for example, the N1H, N3H and O6H forms of guanine.83,84 In a study of poly (isoG) by Golas85 et al. in 1976, the study of the tautomeric forms of isoG seems particularly important to identify correct base pairings containing isoG. In 1976, Sepiol86 et al. reported that the tautomerism of isoG is affected by concentration, temperature, and solvent polarity. As the polarity of the solution changes, the enol ratio can vary from 5% in aqueous medium to 80% in polar solvents. They proposed that isoG is often present in solution as a mixture of enol and keto forms. isoG shows a UV absorption 310 nm for the N1H keto form and 270 nm for the O2H enol form. In general, the enol form (O2H) is favored in lower polarity solvents such as dioxane or at higher temperature in chloroform solution.67,86 In contrast, the N1H-2-oxo-6-amino form (I, Fig. 12) is mainly present in polar solvents. However, the N3H form of isoG was not found until Eschenmoser and co-workers confirmed its existence in 1993.87–89 In 1997, Switzer84 et al. reported that isoG may possess nine tautomerism forms (I–IX, Fig. 12). The tautomeric forms I–V are the five most stable forms by gas-phase calculation. They also concluded that cis-enol configuration of isoguanine is more stable than trans-enol configuration. Notably, if isoG is methylated, the enol–keto shift is faster, and the ketone 6-amino-N3H form of 9-methylisoguanine has been confirmed in solid state by Banerjee90 et al. in 1978. However, researchers have not successfully isolated the enol form of 9-methylisoguanine.90 In 2012, Schaefer44 et al. reported that the cis/trans enol isoguanine can be converted to the corresponding N1H and N3H ketone forms, respectively (Fig. 5).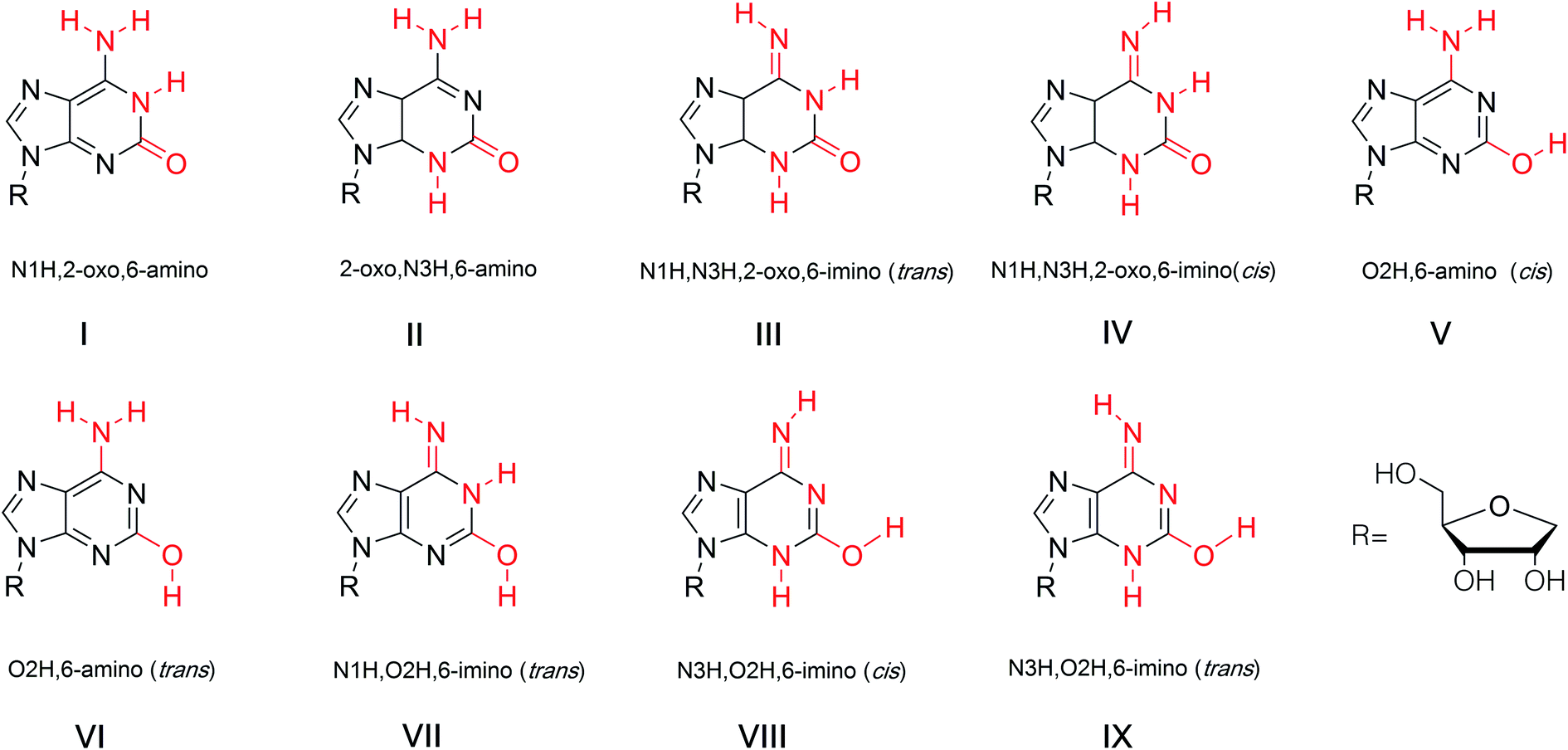 | ||
| Fig. 12 Tautomerism of isoG. | ||
In a study of pyrazolo[3,4-d]pyrimidine by Seela68,91 et al. in 2007, it is concluded that for isoG and it derivatives, a higher ratio of the ketone form results in a stronger mismatch recognition ability in duplexes. Compared to isoGd (51a, Fig. 11) and 7-deaza-isoGd 44a, 7-halogenated-7-deaza-isoGd (44b, 44c, Fig. 10) prefers the ketone form, thus, the compounds 44b and 44c show a stronger mismatch ability. The proportion of the ketone form of 8-aza-7-deaza-isoG and its 7-halogenated derivatives (43, Fig. 10) are higher than the original isoG, so the mismatch recognition ability is higher. However, the effect of 7-halogenation on the tautomerism of 8-aza-7-deaza-isoG 43d is less than that on 7-deaza-isoGd 44a. Thus, 7-halogen substitution should not change the base recognition of 8-aza-7-deaza-isoG derivatives 43a/43d. Pyrazolo[3,4-d]-pyrimidines 43d–43g is more enolized than pyrrolo[2,3-d]pyrimidines 44, while isoG itself shows the highest enol content. For compound 43d–43g the equilibrium constants [KTAUT (keto/enol)] range from 400 to 1200, whereas for compound 43a–43c they range from 1000 to 10000. Therefore, these compounds are expected to exhibit better mismatch recognition than isoG.
5.2 Mispairing
Many researchers have studied the tolerance of base mutation that do not affect the original complementary pairing of oligonucleotides, for the purpose of expanding the genetic alphabet, designing tools for monitoring biological mechanism, and creating new gene systems. A common method is to study the pairing by inserting irregular bases. As mentioned above, isoG is an unnatural nucleoside, so the study of its base pairing is of great significance for subsequent applications. In 1962, Rich92 et al. hypothesized that isocytosine (isoC) and isoG, the isomers of C and G, respectively, may make up a third Watson–Crick base pair. The complementary isoG–isoC pair would form a similar geometry to that of G–C. But there were questions as to why the isoG–isoC is not a constituent of nucleic acids. Subsequently, in 1990, Benner93 et al. confirmed the existence of the isoG–isoC base pair, as well as a second pair, isoG–T, in vitro polymerase experiments. Afterwards, evidence for isoG–isoC pair without enzymes was obtained by Horn94 et al. and Switzer95 et al. in 1995. The base pairing of isoG in the enol form is stronger than that in the ketone form, which is the first experimental evidence that tautomerism can lead to base pairing differences. The keto–enol tautomerism of isoG and the possible cis and trans conformers result in various types of mispairing and different hydrogen-bonding modes. It has been reported that in different tautomeric forms, isoGd can pair with several natural bases, deoxycytidine, deoxythymidine, deoxyuridine, deoxyguanosine, and deoxyadenosine (Cd, Td, Ud, Gd, Ad, respectively), as well as with some special unnatural bases such as isoCd and 5-aza-7-deaza-Gd in different templates (Fig. 13).84,94,96–98 On the basis of the tautomerism of the bases described above, the possible hydrogen-bonding of isoguanine–isocytosine, isoguanine–uracil, isoguanine–cytosine were put forward (Fig. 13).84 And in 2003, the impact of the temperature and the adjacent base pairs on base pairings of isoguanine was investigated by Maciejewska99 et al. They concluded both affect base-pairing by changing the polarity of the solution around the isoguanine to cause tautomerism. The temperature was found to have only a slight influence on the base pairing. The 3′-adjacent base affects the tautomerism, while the 5′-neighbor can not, because the 5′-terminal is not at the polymerase active site for the stacking with template.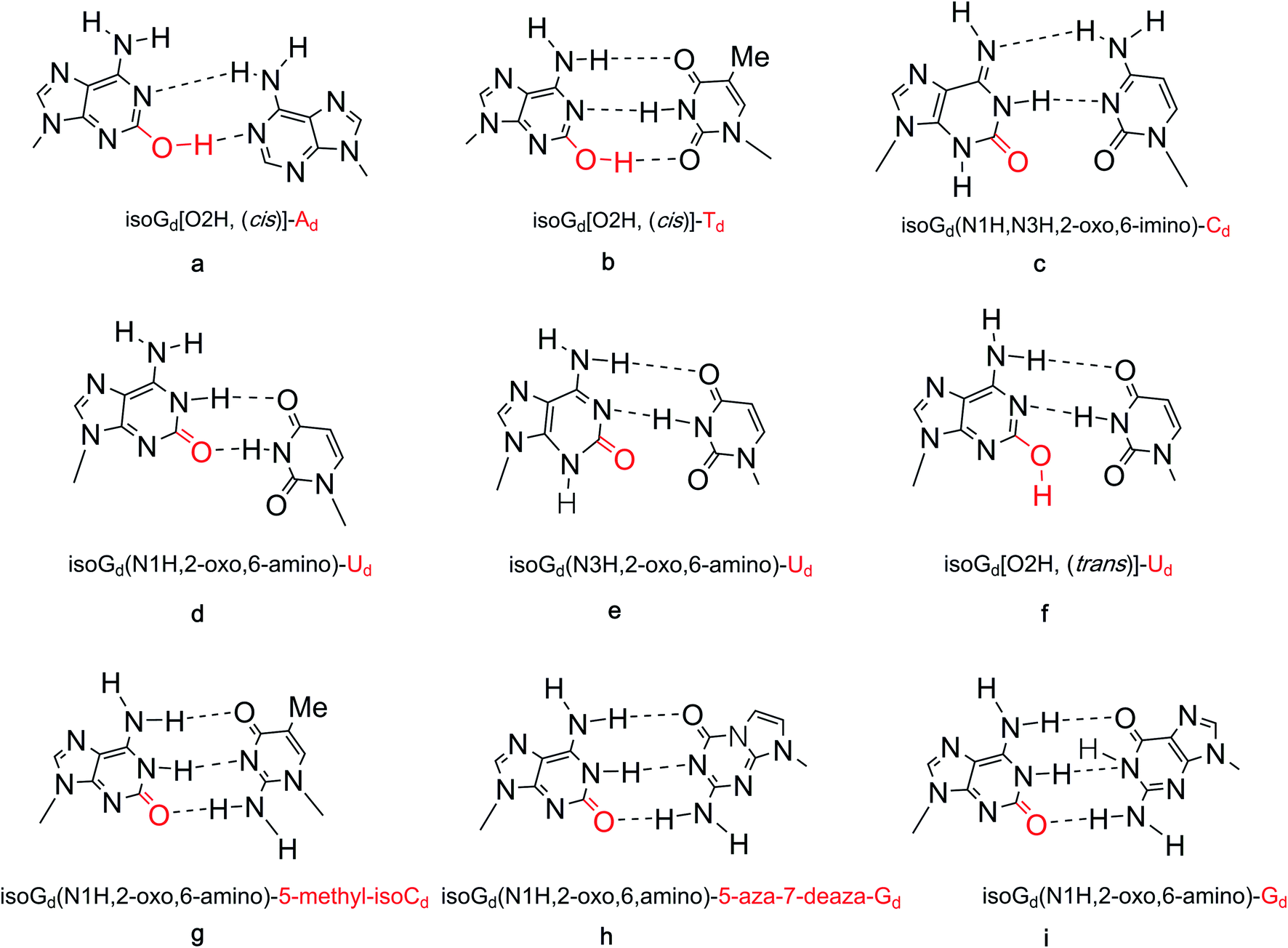 | ||
| Fig. 13 Base pairs of isoG. | ||
In 1986, Jaworski96 et al. carried out experiments to explore the interaction energies of isoguanine–isocytosine and isoguanine–uracil, and compared them with energies of natural base pairs guanine–cytosine, adenine–thymine/adenine–uracil. It was found that isoguanine–uracil interaction energy is smaller than that of guanine–cytosine and isoguanine–isocytosine but stronger than that of adenine–thymine/adenine–uracil pairs. Additionally, isoguanine–isocytosine pairing is slightly stronger than guanine–cytosine pairing. And isoGd-5-methyl-isoCd pair was reported to be isoenergetic with the G–C pair, and each base can effectively identify mismatches in synthetic oligonucleotides, by Horn94 et al. in 1995 and Switzer84 et al. in 1997. The isoguanine–cytosine and uracil–adenine base pairs have the same energy. In 1997, using ab initio data from calculations combined with thermodynamic data, Switzer84 et al. concluded the reason why isoguanine–isocytosine shows unnatural stability may be that isoguanine adopts a carbonyl-imino form.
Isoguanine is a modified base, so its enzymatic recognition and removal have aroused wide interest among scholars. Various DNA and RNA enzymes can catalyze the recognition of different bases by hydrogen bonding interactions. Thus, the enzymological properties of these varied base pairs have been investigated to explore whether they are incorporated into duplexes by DNA and RNA polymerases, and, if so, under what conditions. It has been demonstrated that various enzymes catalyze the entry of isoG into the nucleotide chains in vitro (Table 3). For the first time, Switzer100 et al. reported that d-isoGTP can be incorporated opposite T/isoC by the Klenow fragment (KF) of E. coli DNA polymerase (pol) I and T7 RNA polymerase in 1989. Then in 1993, they reported that in different templates, avian myeloblastosis virus (AMV) reverse transcriptase can enhanced the formation of isoG–isoC, and isoG–U base pairs via T7 RNA pol.101 And T7 RNA pol has a preference in catalyzing the incorporation of U over isoC. However, T4 DNA pol rejected isoguanine's incorporation under reported experimental conditions. Moreover, in 1993, Dervan102 et al. reported that 5-methyl-isoCd can direct the incorporation of N6-(6-aminohexyl)-isoG into the transcribed RNA by T7 RNA pol. And this amino group affords site-specifically modified RNA sequences beneficial for future studies after transcription. Until then, the possibility of isoG opposite A/G and the incorporation by mammalian polymerase were not investigated. Subsequently, in 1995, in vitro DNA synthesis, Kamiya31 et al. reported that deoxythymidine monophosphate (dTMP) and deoxyadenosine monophosphate (dAMP) can be incorporated to pair with isoG by KF of E.coli DNA pol I, recombinant rat DNA pol β and calf thymus DNA pol α. And deoxyguanosine monophosphate (dGMP) can pair with isoG by KF of E.coli DNA pol I. They also concluded that d-isoGTP can mismatch with T/C (showing a preference for T) using mammalian (calf) DNA pol α.29 Whereas d-isoGTP can only pair with T by KF of E.coli DNA pol I. In 1996, using calf thymus DNA pol α and recombinant rat DNA pol β, they confirmed the dTMP and deoxycytidine monophosphate (dCMP) pairs with isoG.103 Whereas dTMP and dGMP were incorporated to form base pairs with isoG by KF of DNA pol I. In addition, human immunodeficiency virus 1 reverse transcriptase (HIV-1RT) has a high tolerance for nonstandard bases, indicating that it promotes nonstandard base pairing containing isoGd, and thus preventing HIV-infected cells from being recognized and dying in the cell's S phase.104,105 Then in 1998, Lutz106 et al. reported that compared to eukaryotic DNA polymerases, the incorporation of T or U by HIV-1RT to mismatch with d-isoGTP is more efficient, whether dATP exists or not. In 2000, Kamiya107,108 et al. reported the 2-OH-dATP can pair with T and G using the α (catalytic) subunit of E. coli DNA pol III and KF of DNA pol I. Above all, the incorporation of isoG depends on the types of polymerases, as well as the sequence contexts in various templates.
| a T4 pol do not catalyze any nucleosides opposite isoG under experimental conditions. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Enzymes | E. coli DNA pol I | E. coli DNA pol III | DNA pol β | DNA pol α | T7 RNA pol | AMV reverse transcriptase | T4 pol | HIV-1RT |
| Mispairing with isoG | T/isoC/A/G | T/G | T/A/C | T/A/C | U/T/isoC | isoC | —a | T/U |
| References | 29, 31, 100, 103, 107 and 108 | 107 and 108 | 31 and 103 | 29, 31 and 103 | 100 and 101 | 101 | 101 | 106 |
In 1975, Mantsch41 et al. reported that isoguanine nucleotides are not recognized by the mitochondrial endometrial translocation enzyme system, but they can be well recognized by mitochondrial phosphatases in the inter-membrane space. They are also involved in phosphoryl group transfer reactions. These reactions are catalyzed by pyruvate kinase, phosphofructokinase and hexokinase. Additionally, in 1998, Kamiya109 et al. reported that isoguanine opposite thymine/cytosine cannot be moved by a glycosylase-type mechanism in rat organs. In bacteria such as in E. coli, there are some specific enzymes for the removal of unnatural base from damaged nucleic acid. In 2011, Raushel110 et al. reported that cytosine deaminase (CDA) from E. coli can catalyze the deamination of isoguanine to xanthine. However, in 2000, Kamiya111,112 et al. demonstrated that the removal of isoguanine by the MutY and MutM proteins of E. coli is slow because of poor affinity. Surprisingly, they reported that among the known akin nucleotide substrates, the nucleotide forms of isoG (2-OH-ATP and 2-OH-dATP, where ATP is adenosine triphosphate and dATP is deoxyadenosine triphosphate) are substrates with the highest affinity for the human MTH1 (hMTH1) protein, which is a hydrolytic enzyme.113,114 Moreover, 2-OH-dADP and 8-OH-dGDP (dADP and dGDP are deoxyadenosine diphosphate and deoxyguanosine diphosphate, respectively) can competitively inhibit the effect of hMTH1 on 2-OH-dATP and 8-OH-dGTP (dGTP is deoxyguanosine triphosphate), which indicates that hMTH1 takes effect on the damaged substances via the same receptor.114
5.3 Self-assembling of isoG
We have discussed the tautomeric forms of isoG in different solvents and the various base pairs catalyzed by different DNA and RNA enzymes above. Now, we will describe several types of advanced structures such as tetramers, decamers, oligonucleotides, and ps/aps strands that form based on the self-assembly properties of isoG. Self-assembly is a process where substances combine into a supramolecular system through intermolecular forces, such as electrostatic and hydrophobic interactions. The discoveries of poly(isoguanylic acid), the oligonucleotide d(T4isoG4T4), and G-quartets indicate that isoG may also self-assemble into supramolecular structures. In fact, both G and isoG can self-assemble into supramolecular systems, such as supramolecular gel by hydrogen-bonding, π–π stacking, and hydrophobic interactions. And these structures show a wide range of applications in material science, medical chemistry, and nanotechnology etc. As mentioned before, isoG differs from G in that the C2 carbonyl and C6 amino groups are transposed. This minor change causes significant differences between isoG and G. In particular, the variation in hydrogen bond donors and acceptors affects the hydrogen-bonded systems, resulting in great changes in self-assembled supramolecular structures. For G, the angle between the hydrogen bond donor (N1 amide and N2 amino) on its Watson–Crick face and the receptor (N7 and O6) in the Hoogsteen face is 90°, and this is favorable for the formation of planar tetrads (Fig. 14).115,116 On the contrary, the isoG donor–receptor angle of 67° prefers cyclic pentad (Fig. 14). In 1994, Davis82 et al. raised that for isoG, there are two prerequisites for self-association. One is that the hydrogen-bonding receptors of isoG (O2–C2–N3) are close to 180°, whereas the N7–C5–C6–O6 bond of G is nonplanar. And the ribose of isoG, as well as the hydrogen-bond acceptors, are more likely to form hydrogen bonds in the lower position of purine.117,118 The other prerequisite is the formation of hydrogen bond between the N6H proton and adjacent isoG monomer O2′. In contrast, G is unlikely to form such a hydrogen bond because the hydrogen bond acceptor O6/N7 and the glycosyl group are in opposite positions in the heterocycle.82 This is also an important reason why isoG self-assembled oligonucleotides are more stable than the corresponding G self-assembled oligonucleotides.119 It has also been proposed that isoG has three variable N protons that are important for supporting the self-assembly.82 C6NHA and C6NHB (Fig. 14) rotates slowly, allowing the exocyclic amino group to form a strong hydrogen bond, and the third proton N1H is confirmed based on nuclear Overhauser effect (NOE) and UV studies in CDCl3.82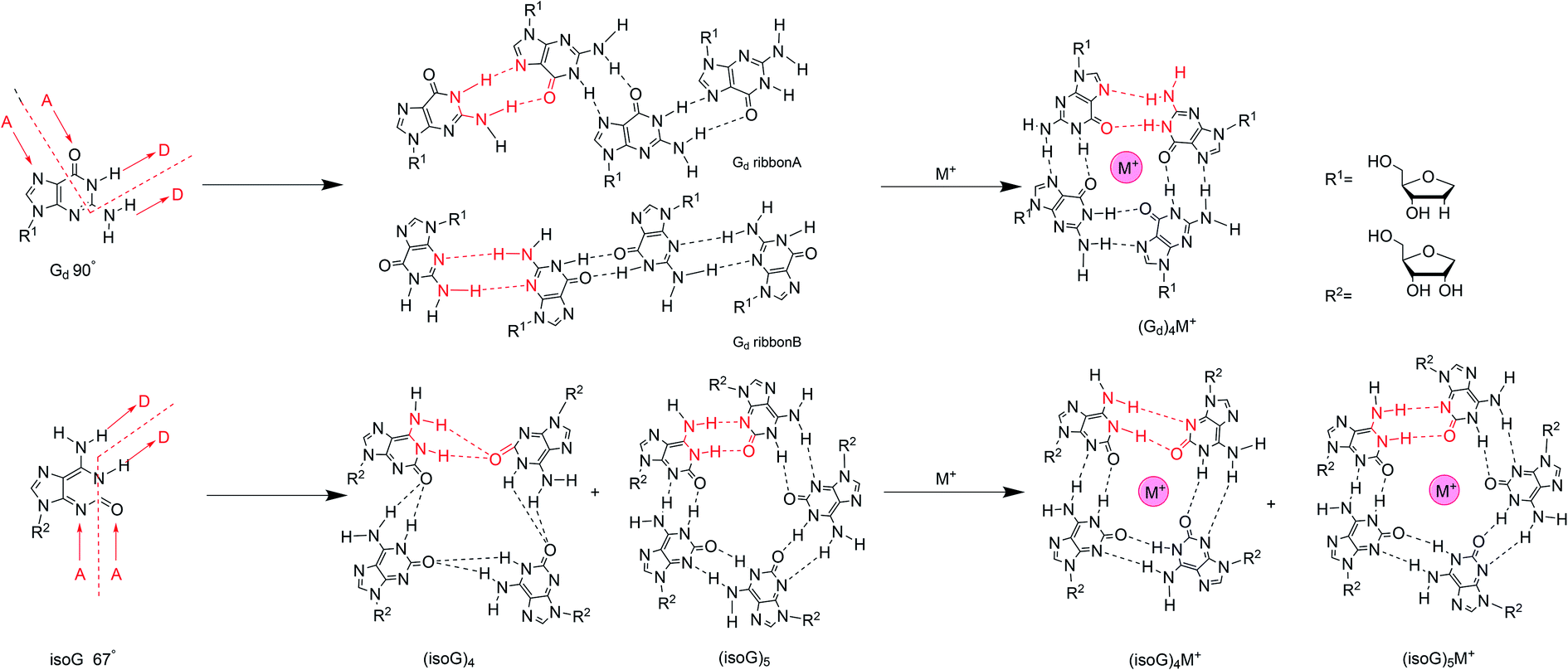 | ||
| Fig. 14 Self-assembly of isoG and G with/without cations. | ||
Similar to the interaction of the cations with G, alkali ions have a great influence on the self-assembled structures of isoG. Different ions have been titrated into pure isoG mononucleoside in d6-acetone to identify the different structures.82 Tetramerization with lithium ion (Li+) is unstable because Li+ is too small for the central cavity. Li+ may be combined with the O2 or N3 hydrogen-bond donors to hinder hydrogen-bond formation. A low concentration of Na+ can stabilize the tetramer, probably via ionic coupling. But no new NMR peaks have been observed with Na+, which indicates that no higher-ordered self-assemblies, such as stacking structures, are formed in the presence of Na+. Nevertheless, high concentrations of Na+ destroy isoguanine tetramers, probably for the same reason as that for Li+. Notably, the addition of K+/barium ion (Ba2+) form octamers, presenting a new peak with a sandwich structure.12,82 And K+/Ba2+ enhanced the thermal stability compared to the original tetramer. Moreover, the Ba2+ octamer is more stable than K+ octamer.
The hydrogen-bonding modes are different in the isoG tetramer in the presence and absence of cations. Next, the hydrogen bonds in alkali-metal-free isoG tetrad and pentad structures will be introduced. In 2003, Meyer124 et al. proposed that the hydrogen-bond modes in tetrad and pentad are different (Fig. 14), one difference is that the N1–H1⋯O2 hydrogen bond (H bond) in the tetrad is longer than that in the pentad. The other is the tetrad N6–H6⋯O2 H bond forms a crossing hydrogen bond with the N1–H1⋯O2 H bond. Whereas two hydrogen bond, N1–H1⋯O2 and N6–H6⋯N3 are formed without crossing in the pentad. Besides, the second H bond N6–H6⋯N3 in pentad is shorter than the second bond N6–H6⋯O2 in tetrad. When the ions are added to the isoG solution, there are no crossing hydrogen bonds occurring. In other words, alkali ions can affect the crossing hydrogen-bond mode of the protected isoG tetramer (53)4 to form independent N6–H6⋯N3 and N1–H1⋯O2 hydrogen bonds (Fig. 14). This is not surprising because base-cation linkages are usually stronger than base–base linkages for isoG. That is, the cation interactions are the dominant contribution to the interaction energy. As a result of metal cations present in the cavity, the base–base hydrogen bond in the tetrad is shorter than in the pentad, and the distance between cation and O2 is also shorter. It indicates the hydrogen bond distance is shortened in the presence of alkali ions, and this effect is more pronounced for Li+ and decreases with increasing ion size. From Li+ to cesium ion (Cs+), the distance between O2 and the cation increases. Further, larger ions result in smaller connection energies between the ion and C2 carbonyl group.
The protected isoG tetramer (53)4·M+ is stabilized by the hydrogen-bond system of the outer ring and the inner ring (Fig. 14). The inner ring is composed of four hydrogen bonds formed between the imino N1H proton and the C2 carbonyl oxygen of an adjacent isoG. The outer ring is comprised of hydrogen bonds between N6H and N3H of the adjacent monomer. Four nonbonded C2 oxygen atoms point to the cavity, interacting with metal alkali ions, and selectively bind to monovalent or divalent cations to form a stable complex. The presence of multiple metal ions causes different changes to hydrogen-bond model, and metal cations are often required to stabilize hydrogen-bonded macrocycles. The self-assembled structures of isoguanine with different metal ions varies. For example, the tetramer of isoG is strongly nonplanar except with Li+, whereas the pentamer is often planar. In fact, most nonplanar structures converge into planar structures, so most of the known tetrad/pentad structures are planar, which is more stable. However, there is no selectivity over monovalent and divalent ions. That is, there is no difference in affinity for different charge numbers. Moreover, the tetrad has strong selectivity and preference for small ions. However, for large ions, there is no preference between the tetrad and pentad. The energy of (isoG)4·Li+ tetrad is higher than that of its pentad, whereas the energy of the pentad with a larger radius is higher than the corresponding tetrad energy. The energies of the planar tetrad structures are relatively low; i.e., they are more stable. And the metal-free and (isoG)5·K+/rubidium ion (Rb+) pentad energies are also relatively low. Li+ and Na+ ions are too small for the pentameric cavity, whereas Cs+ is too large to be in the cavity so it may sit over the isoG5 pentad plane. The protected isoG (53, Fig. 11) can also self-associate in the gas phase and studies into this fact are ongoing.
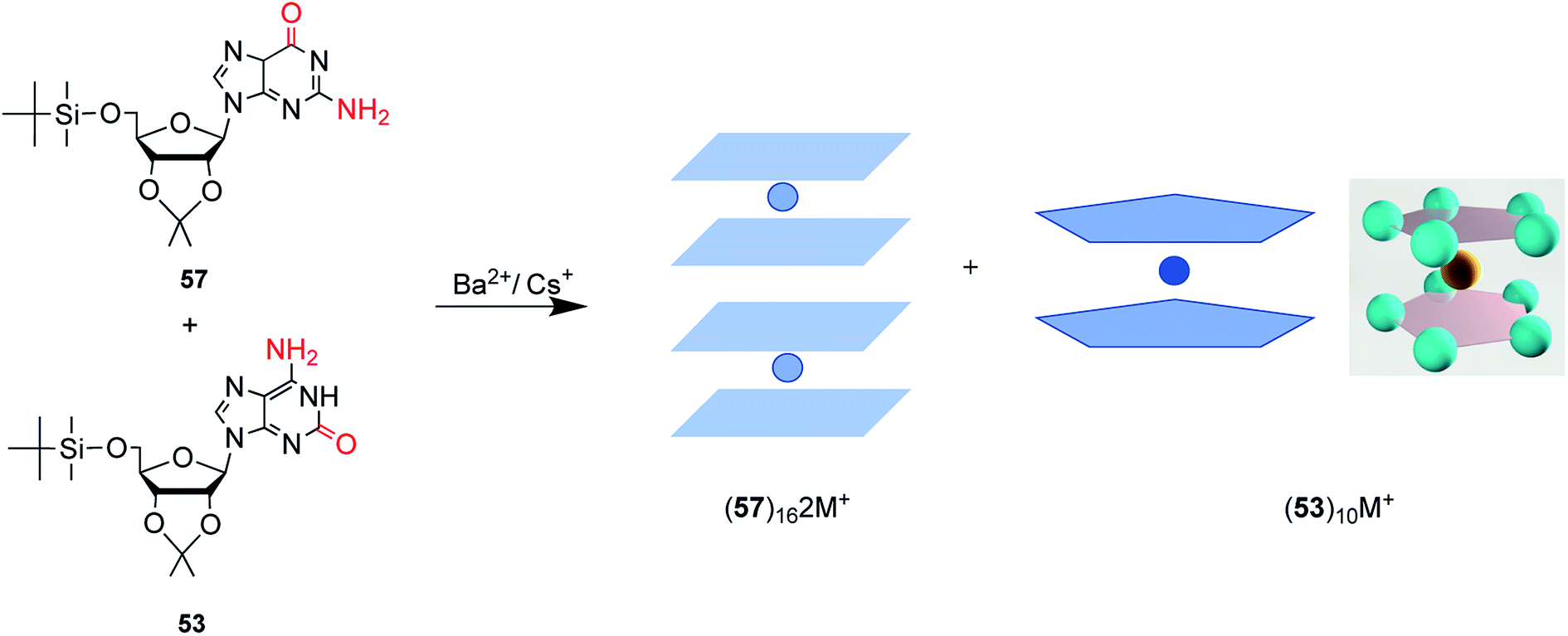 | ||
| Fig. 15 The protected G and isoG self-assemble and stack into hexadecamer and decamer respectively in the presence of Ba2+. | ||
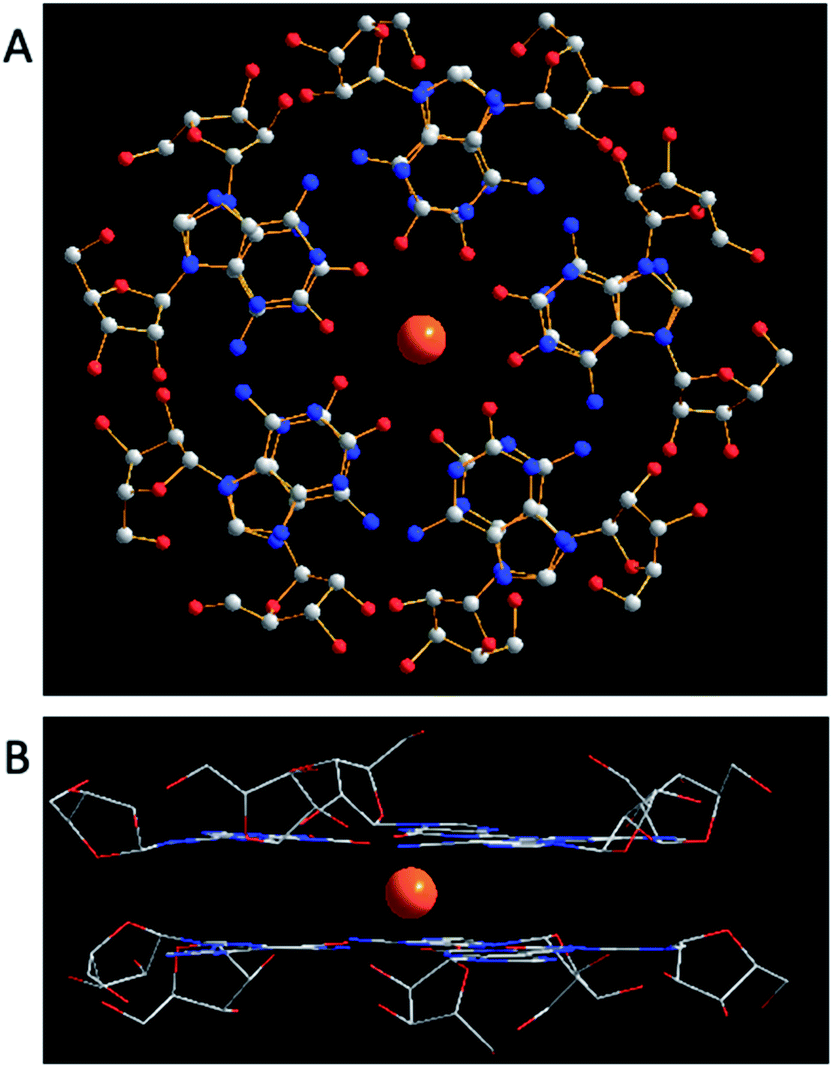 | ||
| Fig. 16 (A) A top view of X-ray crystal structure of (53)10·Cs+. Two pentamers directly overlap with ten chemically equivalent isoG units, having Cs+ (yellow) and ten carbonyl groups in the center. C atoms are grey, N atoms are blue, O atoms are red. (B) A sandwich model with Cs+ as the bisector of the plane. Anions are omitted for clearness. | ||
isoG is selective for the largest alkali cation, Cs+ (r = 1.67 Å), whereas G-quartets have a preference for K+ (r = 1.33 Å).132 Importantly, in the presence of Cs+, isoG only formed the (53)10·Cs+ structure. However, for other cations such as Li+, (53)10·M+ and (53)10·2M+ structures are formed, and stay nonstop transition between each other.132 Notably, (53)10·2K+ directly comes from two pentamers rather than by the addition of more K+. Furthermore, in 2014, Rodriguez133 et al. reported that small dendritic molecules containing isoG can also self-assemble into a decameric nucleodendrimers, even without cations. And they demonstrated the existence of G analogs can promote the formation of decameric nucleodendrimers, but are not involved in decameric structures. Experiments to explore the abilities of different ions to induce nucleodendrimers identified that the isoG-based nucleodendrimers show a preference for Cs+ over K+, which reflects the fact that the cavity size is more suitable for the large radius of Cs+. In addition to the above, in 2002, from the NMR spectrum, Davis134 et al. concluded that G and isoG self-assemble independently in the presence of Cs+/Ba2+, rather than form crosslinked structures (Fig. 15). However, in CD2Cl2 where there is no cation, G and isoG are crosslinked.13,129,134
Moreover, in 2000, Davis13 et al. found that (D,L)-isoG (53, 59, Fig. 17) undergoes enantiomeric self-recognition in solution containing Cs+. Then in 2001, they found that the protected (D,L)-G (57, 58, Fig. 17) can self-associate with diastereoselectivity in Ba2+-containing solutions, yielding two homochiral G-quadruplexes (57)16·2Ba2+·pic− and (58)16·2Ba2+·4pic−.126 But in the presence of K+, (D,L)-G forms a diastereomeric mixture. As mentioned above, hydrogen bonds can take shape between neighboring isoG ribose residue to help conquer the entropic demands in relation to enantiomeric self-sorting, whereas this does not occur for G. Such hydrogen bonds can transfer conformational information between pentameric isoG to ensure homochiral conformation. And the achiral Cs+ promotes the enantioselectivity of isoG, thus promoting formation of the Cs+ sandwich meso decamer, (53)5·Cs+·(59)5·Ph4B−, as determined by X-ray crystal structure analysis. However, enantioselectivity is not possible with other ions.
 | ||
| Fig. 17 The D and L structures of the protected G and isoG. | ||
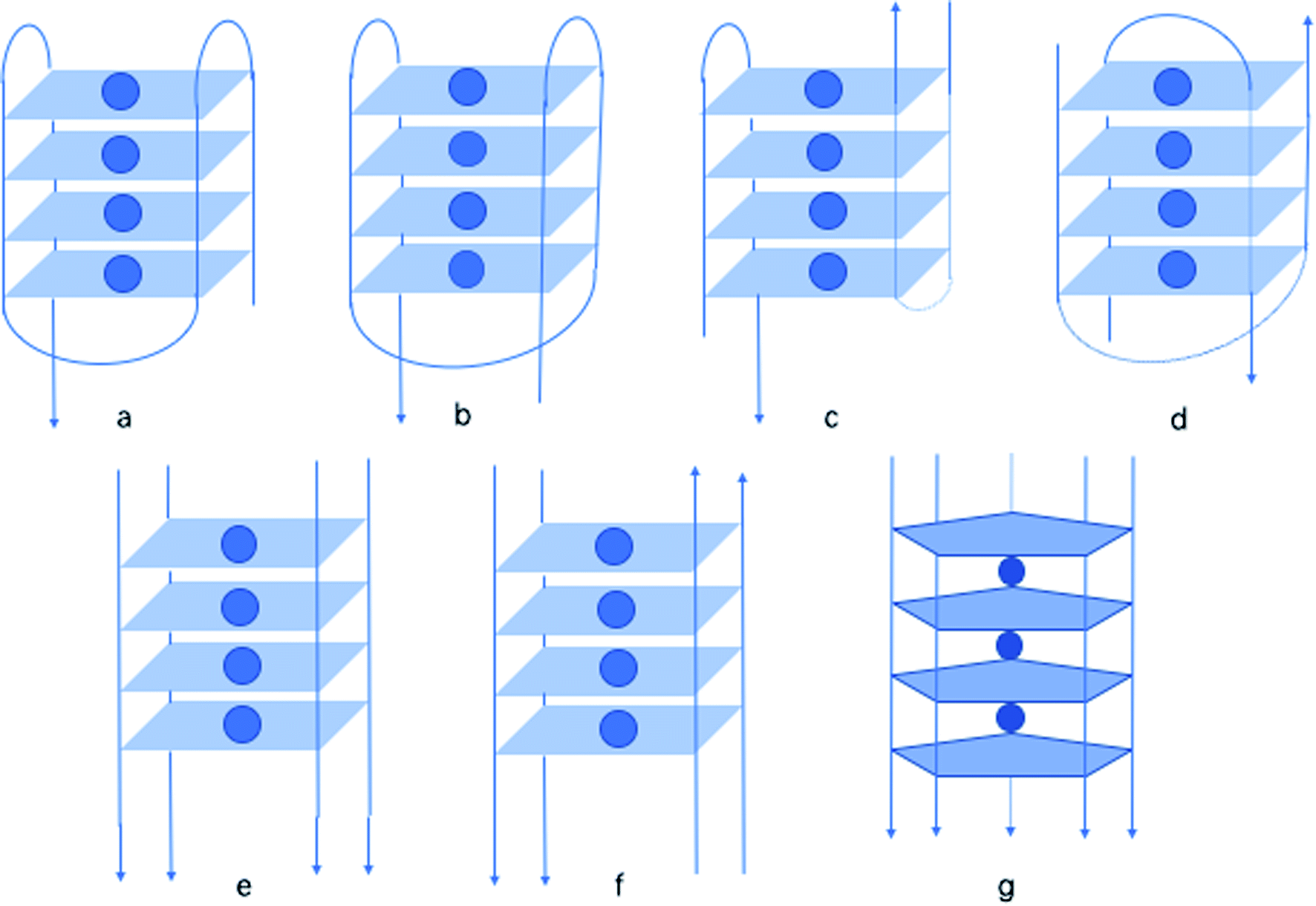 | ||
| Fig. 18 A variety of oligonucleotide structures: (a) monomer chair, (b) monomer basket, (c) dimer chair, (d) dimer basket, (e) tetramer parallel, (f) tetramer antiparallel, (g) pentaplex. | ||
In 1997, by native polyacrylamide gel electrophoresis (PAGE), Roberts141 et al. reported that similar to G, isoG also shows a tendency to form tetraplexes, showing the similar monomer, dimer, tetramer structure (Fig. 18a–f). Notably, they found that isoG can even form mixed quartets with G, which may induce the formation of mixed tetraplexes. Moreover, isoG-tetraplex may show higher stability than G-tetraplex in the presence of K+. And isoG-tetraplex shows a preference for K+ over Na+. Afterwards, in 1998, Seela98 et al. concluded that oligonucleotide tetraplex d(T4isoG4T4)4 has an ion preference in the following order: K+ > Rb+ > Cs+ > Na+ > Li+. In contrast, the order for d(T4G4T4)4 is K+ > Rb+ > Na+ > Cs+ > Li+. In 1999, Switzer115 et al. reported that ionic identity can determine the self-assembly of DNA strands into quadruplexes or pentaplexes. In the presence of Cs+, isoG self-assembles into pentaplexes. Whereas, in the presence of Na+ and K+, it forms quadruplexes. However, it was found that G self-assembles into almost the same quadruplexes in Cs+ and K+, which is consistent with the results of Sen142 et al. in 1993. Interestingly, in 2001, Seela143 et al. found that 8-aza-7-deaza-isoGd (43a, Fig. 10) can self-assemble into d(T4(43a)4T2). And the compound 43a can form a pentaplex (Fig. 18g) in the presence of Cs+, and a tetraplex in the presence of Na+, K+ and Rb+. This is different from d(T4isoG4T2) because the d(T4isoG4T2) can self-assemble into both four-stranded and five-stranded structure in Rb+ solution. In addition, in 1997, they reported that 7-deaza-isoGd (44a, Fig. 10) can also form a tetraplex, identical in hydrogen-bonding structure to d(T4isoG4T4)4, but the former is less stable because of the heterocycle change.144 This observation is also a confirmation that N7 does not participate in the isoG hydrogen-bonding system. Moreover, in 1997, they proved that isoGd is sensitive to acid and can be easily hydrolyzed under acidic conditions.144–146 But 7-deaza and 8-aza-7-deaza purines can add to the stability of the glycosylic bond. Moreover, 7-deaza-isoGd can form stronger tetraplexes in the presence of Na+/K+ comparing to isoGd, respectively. The 7-halogen substitution is capable of enhancing stabilities of the base pairs (even in mismatches), glycosylic bond, and duplexes of 8-aza-7-deaza-isoG derivatives. However, 7-deaza-isoGd show the topmost glycosylic bond stability but the lowest duplex stability.145
 | ||
| Fig. 19 Ps duplexes with isoC–G/isoG–C and aps duplexes with isoC–isoG. | ||
6 Applications
In the above parts, the discovery of isoG, its synthesis, modifications, tautomerism, and different mismatches, as well as the self-assembly properties of isoG, have been introduced. Based on the above characteristics, the applications of isoG have been investigated, such as the formation of supramolecular gels, the role of base-pairing in duplexes, its use as an ionophore, and its use as an antitumor agent. In the following sections, these applications will be discussed in detail.6.1 isoG-based supramolecular hydrogels
Hydrogels have been an important topic for centuries because of their diverse applications in many fields, including in nanoscience, medicine, and the cosmetics industry. Hydrogels are viscoelastic solid-like materials composed of an elastic crosslinked network, and the major component solvent.153 Hydrogels can be divided into two categories. One is polymer hydrogels, whose shape is maintained by strong chemical bonds crosslinked long chains, which cannot be broken easily and are not thermally reversible. The other one is low molecular weight hydrogels (LMWH). LMWH formed by the self-assembly of materials through noncovalent interactions, such as hydrogen-bonding, π–π stacking, hydrophobic forces, and van der Waals interactions. LMWH, also known as supramolecular hydrogels or physical hydrogels, will be discussed in this paper. Nucleosides are desirable candidates for building supramolecular hydrogels because of their ability to self-assemble via noncovalent interactions, as well as their biocompatibility in cells.The gelation of guanylic acid was firstly identified in 1910 by Bang.154 G and some of its derivatives can self-assemble into various supramolecular gels based on its unique hydrogen bond donors and receptors.154 There have been many reports concerning G-based hydrogels and their applications in diagnostics and nanoscience. But these G-based hydrogels all show short lifespans. So finding new nucleosides that can form long-life supramolecular hydrogels is crucial. Like G, isoG can form gels as well. In a synthesis of isoG in 1951, Davoll7 noted the formation of a “gelatinous material” during the deamination of 2,6-diaminopurine, but he did not characterize this material further by biochemistry. It was preliminarily described that the aggregation state of nucleosides turns viscous at higher concentrations and when cooled. Then in 1965, Ravindranathan40 et al. synthesized isoG and gave proof for the phenomenon observed by Davoll.7 They reported the formation of asymmetry, ruled, ordered, spiral and temperature-depending structures where the nucleosides were uncharged. And these structures are similar to the gel formed by 5′-monophosphate-guanosine (5′-GMP), as identified by infrared spectroscopy. However, the preference of isoG for alkali ions is different from that of G. They also concluded that the purine ring may be hydrogen-bonded with the amino group of another molecule, and the carbonyl group does not participate in the formation of the ordered structure.
As shown in Table 4 and Fig. 20, remarkably, in 2017, Seela155 et al. found that isoG-based hydrogels formed in various solutions containing Li+, Na+, K+, Rb+ and Cs+, but not in water. And all hydrogels present significant longer lifespans (two or three months) compared to those of G (several days), as well as higher thermodynamic stability and wider pH range. The rheological results suggested that the stabilities of isoG-based gels are strikingly higher than those of G. And scanning electronic microscope (SEM) images revealed that isoG-based hydrogels form helix-like cylinders, whereas G-based gels form individual flat ribbons. As reported, hydrogels formed by isoG at physiological Na+ concentration showed a good small molecule loading and releasing capacity in phosphate buffered saline (PBS) buffer. This suggested their potential applications in drug delivery and as ionophores carrying different ions. In addition, supramolecular hydrogel systems of isoGd (51a, Fig. 11) solutions containing Li+, K+ and Rb+ have been established. However, in Na+ and Cs+ solutions, these hydrogels can not take shape. Moreover, the derivative FisoGd (51b, Fig. 11) can also form supramolecular hydrogels in the presence of Li+, Na+, K+ and Rb+. While in solutions containing Cs+, precipitates form, indicating the ion dependence of the three nucleosides-based gels is different.
| H2O | Li+ | Na+ | K+ | Rb+ | Cs+ | |
|---|---|---|---|---|---|---|
| a Precipitation.b Crystallization.c Solution.d Viscous solution.e Not experimented. | ||||||
| isoG | Sc | Gel | Gel | Gel | Gel | Gel |
| G | —e | Cryb | Cryb | Gel | Cryb | Cryb |
| isoGd | Gel | Gel | Sc | Gel | Gel | Sc |
| Gd | Cryb | Cryb | Cryb | Gel | VSd | Cryb |
| FisoGd | Gel | Gel | Gel | Gel | Gel | Sc |
| z8isoGd | Prea | Prea | Prea | Gel | Prea | Prea |
| z8Gd | Prea | Sc | Sc | Sc | Sc | Sc |
| G:isoG (1:1) |
—e | Prea | Cryb | Gel | Gel | Cryb |
| G:isoG (1:3) |
—e | Cryb | Cryb | Gel | Gel | Cryb |
| G:isoG (3:1) |
—e | Cryb | Cryb | Gel | Cryb | Cryb |
 | ||
| Fig. 20 The development of hydrogels formed by isoG. | ||
Hydrogels with fluorescence have aroused much attention due to its latent applications in drug delivery, molecule marking, and tissue engineering. As discussed before, z8isoGd, z8Gd are virtually fluorescent in alkaline environment and at neutral pH, similar to those reported for 8-aza-G and relevant 8-azapurine nucleosides.70–73 Notably, isoGd and Gd are not fluorescent. In addition, z8isoGd and z8Gd both show self-assembly properties. So experiments have been carried out by Seela156 et al. in 2017 to prepare smart (stimulus-responsive) gels with fluorescence, which can be regulated by external changes, such as pH, heat and nucleoside mimics. Remarkably, z8isoGd formed the first G-similar hydrogel with fluorescence properties even in solid state. And the hydrogel also shows a longer lifetime and higher thermal stability than isoG and G hydrogels. However, z8Gd does not form a gel under experimental conditions. z8isoGd hydrogel has a great selectivity for K+, and its pentad and tetrad structures are the basis of its self-assembly into gels. SEM images revealed that z8isoGd gel forms dense fibers and bundles, whereas z8Gd forms nanotubes. It makes great significance that the fluorescence hydrogel formed by z8isoGd can take response to external changes such as pH, UV, and heat. Additionally, in terms of the ability to form gels even at a very low KCl concentrations, z8isoGd hydrogel is expected to be applied in various fields of nanobiotechnology, chemistry, biology and medicine. Interestingly, although N7 does not participate in the isoG hydrogen-bonding system, the absence of N7 in 7-deaza-isoGd results in this system being unable to form a hydrogel. Moreover, the addition of 1.0 equivalent of 7-deaza-isoGd can break the hydrogels formed by isoG and the fluorescence hydrogels formed by z8isoGd.
Self-healing is an ability to self-repair when the external stimuli, such as pH, external force and temperature, are moved. It is one of the most attractive properties of hydrogels and empowers them with diverse applications in many fields like tissue engineering. For example, so-called injectable hydrogels undergo shear-thinning on injection, but the hydrogels recover soon after injection.157 There have been many reports on self-healing polymer hydrogels, but only a few studies concerning the self-healing supramolecular nucleoside hydrogels have been published. In 2018, Das158 et al. developed a self-healing arylboronate esters mediated G-quartet hydrogel as promising 3D-bioink. However, the applications of G-based hydrogels are limited for their short lifetime within hours and low stabilities. This has drawn attention to isoG, an isomer of G. However, the injection of isoG hydrogels is challenging.159 Recently, inspired by the self-assembled tetramers formed by mixing d(T4G4T4) and d(T4isoG4T4) verified by Seela140 et al. in 1996, the tetraplexes formed by d(T8isoG4T) and d(T4G4T) in the presence of K+ proved by Roberts141 et al. in 1997, and the self-healing co-gel developed by mixing G and Gd by Adhikari160 et al. in 2014, Zhao159 et al. successfully constructed a self-healing supramolecular co-hydrogel in 2018 by simply mixing G and isoG with 1:1 and 1:3 molar ratios in the presence of K+. In terms of ion dependence, both mixtures can form co-gels in the presence of K+ and Rb+. But the 3:1 could only produce a co-gel in the presence of K+. Furthermore, the lifespan of the gels varied from hours to months. SEM images show that these co-gels form flower-like structures. Rheological and injection experiments concluded the excellent self-healing properties, short recovery time, and syringeability of the co-gel. Moreover, G2isoG2-quartets are possibly formed inside the self-healing supramolecular nucleoside co-gel.
Furthermore, it is widely known that isoG possesses antitumor property toward several cancer lines (details are shown in Section 6.5);58,161 and the injectable property could greatly promote the application of these supramolecular gels in antitumor treatment. In addition, borate ester is well-known as a dynamic covalent bond to construct self-healing hydrogels. Based on these, in 2017, Sadler162 et al. developed an anticancer hydrogel (dopamine-conjugated platinum IV-G4K+-borate ester, Pt-G4K+B hydrogel) by using borate ester linkage. Inspired by above-mentioned findings, recently, Zhao163 et al. developed another type of dual-functional co-gel called isoguanosine-borate-guanosine (isoGBG) hydrogel (Fig. 21), which possesses delivery and antitumor activities. isoGBG hydrogel displays excellent stability, self-healing property and biocompatibility, as well as highly antitumor activity and excellent inhibition of tumor recurrence in vivo. Moreover, the mechanism was preliminarily explored, and it was concluded that the mechanism of inhibiting oral cancer cells might be through the caspase-dependent signaling pathway to induce apoptosis via regulating dephosphorylation of epidermal growth factor receptor (EGFR) (Fig. 21).
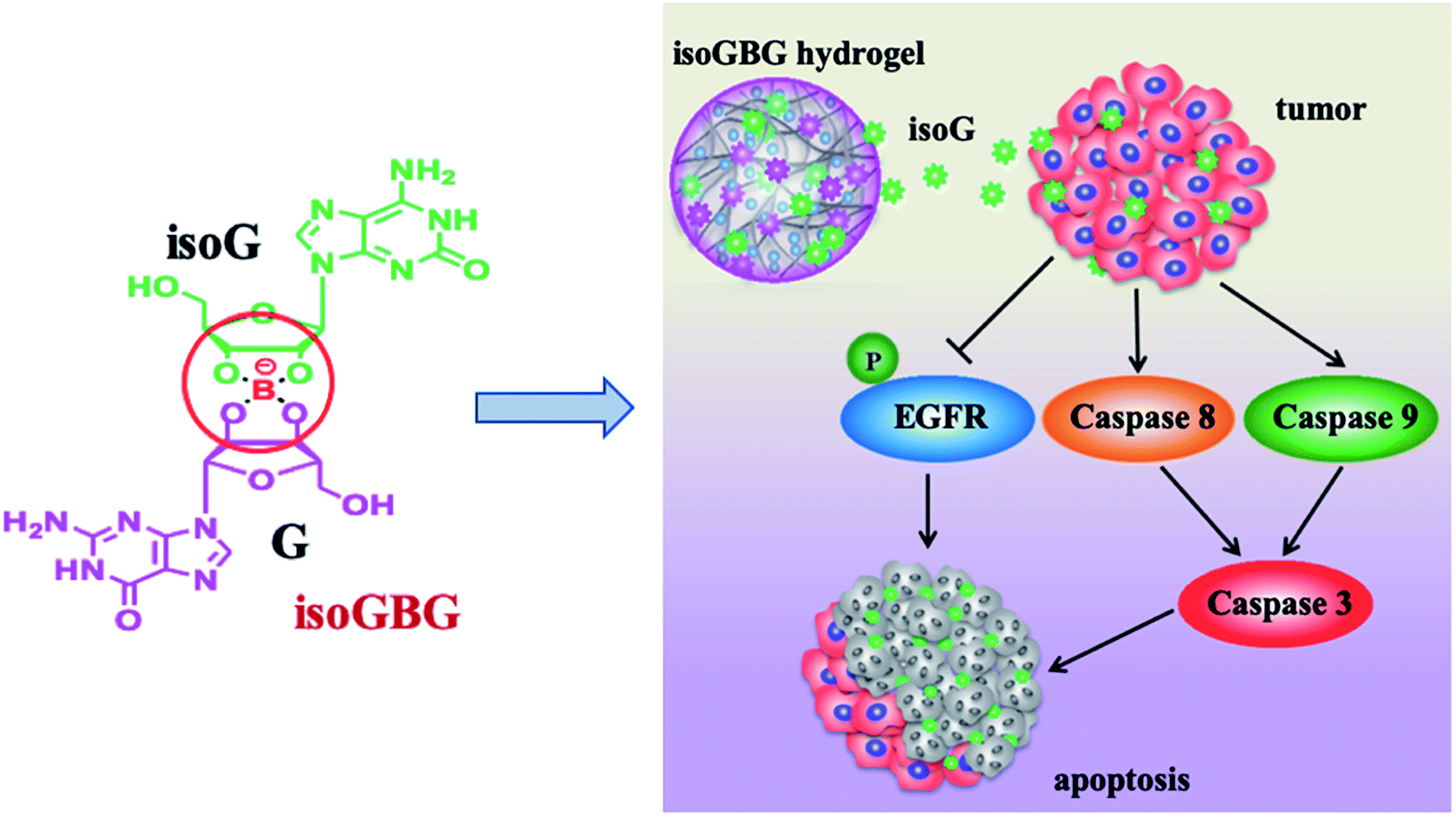 | ||
| Fig. 21 The dual-functional isoGBG displays antitumor activity, may via the caspase-dependent signaling pathway to induce apoptosis via regulating dephosphorylation of EGFR. | ||
6.2 Applications of isoG mispairing
In Section 5.2, different mismatches and the incorporation of isoguanine have been discussed. Although isoG can be integrated into DNA/RNA by multiple enzymes in various templates, isoG–isoC is not accepted as a natural base pair. The three reasons why isoG–isoC cannot be a natural base pair are: (1) tautomerism causes unstable base pairing;96,101 (2) deoxygenated isoC is easily degraded to deoxygenated U;101 (3) isoG–isoC cannot form an R-type triple-chain structure.164 Although the tautomerism of isoG limits its stable transmission as a gene letter, it enables the existence of gene mismatch, suggesting isoG's various alternative applications. In 1996, for the first time, Kamiya103 et al. suggested that isoG may be mutagenic and lead to A to C, A to G and A to T transversions in cells. Then in 1997, isoG is proved to induce mutagenicity in E. coli,32 as well as in living COS-7 cells,165 but it does not hinder the replication. These indicate the mutation caused by isoG can occur in both eukaryotic and prokaryotic cells. Interestingly, the mutagenicity is affected by the sequence and strands that the base is located in. 8-OH-dGTP is a significant marker of DNA oxidation. In 1998, Kamiya166 et al. demonstrated that the mutagenicity induced by 2-OH-dATP in E. coli was more significant than that caused by 8-OH-dGTP, which also gave a confirmation of 2-OH-dATP-G base pair.Interestingly, the nonstandard isoG–isoC base pair contributes to the stability of duplexes. In 2001, Turner97 et al. reported this effect has a sequence dependency in duplexes because of the large difference in electron density. In 2004, Prudent167,168 et al. proved that isoG–isoC stabilizes the G–C double helix and can be applied to polymerase chain reaction (PCR) to detect the mutant genes. Beyond that, in 1992, Benner169 et al. raised that it can help regulate ribosome translation. 91% of isoG-containing RNA could be read, which is higher than the control, because the frameshift with isoG skips the isoC. However, the transfer RNA (tRNA) containing the meaningless CUA codon is meaningless and results in translation termination. This property can also aid the development of nonstandard base pairs to extend gene dictionaries and provide a deeper understanding of translation termination. Moreover, in 1993, Switzer101 et al. reported that the tautomerism of isoG leads to various mispairings, which may enhance the antiviral activity. The stability of base-pairs of z8isoGd (46, Fig. 10) with 5-methyl-isoC, Ad, Td, Cd and Gd can be monitored by fluorescence,73 in other words, the fluorescence signal is correlated with DNA base paring stability. Thus, z8isoGd can be applied as a sensor with fluorescence for detecting mismatch DNA duplexes, even more complicated DNA structures,73 such as in triplexes reported by Seela170 et al. in 2010.
6.3 Applications of 1-methyl-isoG
In 1981, Fuhrman171 et al. reported that isoG presents negative inotropic and chronotropic effects in the myocardium. And those effects can be antagonized by theophylline. The effects of negative inotropic and chronotropic activity ranked in the order isoG > 1-methyl-isoG > adenosine > 2-methoxyadenosine. And these substances may act on the same receptor. In addition, as reported by Emerson172 et al. in 1949 and Brown173 et al. in 1952, isoG can produce smooth muscle relaxation, hypotension and bradycardia effects that are more potent and longer-lasting than those of adenosine. Remarkably, 1-methyl-isoG (37, Fig. 10) was also found to have muscle relaxation, hypothermia, hypotension, anti-inflammatory, activating adenosine cyclase (AC) and anti-allergic effects (Fig. 22).30,50,51,56,174–177 In 1981, as shown in Table 5, a comparison of effects of the derivatives of 1-methyl-isoG have been explored by Cook56 et al. Moreover, the effects of halogenated analog 2-chloroadenosine are greater.30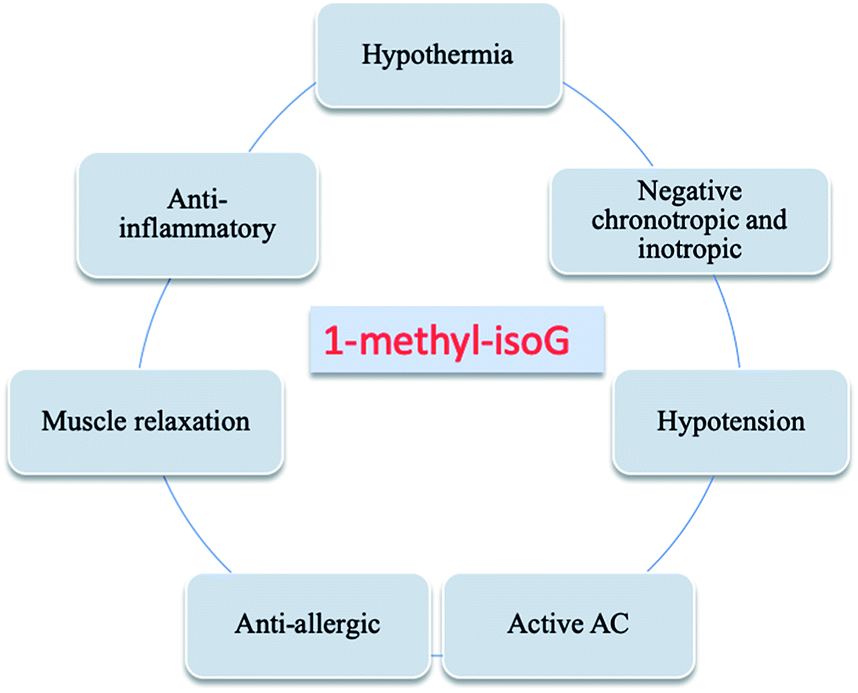 | ||
| Fig. 22 Effects of 1-methyl-isoG. | ||
| Number | Muscle relaxation | Hypothermia | Hypotension | Anti-inflammatory | Anti-allergic |
|---|---|---|---|---|---|
| a Effective.b Limited effect.c Invalid.d Not experimented. | |||||
| 37 | Ya | Ya | Ya | Ya | Ya |
| 38a | Ya | Ya | Ya | Weakb | Ya |
| 39g | Nc | Nc | Ya | —d | Ya |
| 39h | Nc | Nc | Weakb | —d | Ya |
| 39i | Nc | Nc | —d | —d | Weakb |
| 39j | Nc | Nc | —d | —d | Ya |
| 40a | Nc | Nc | —d | —d | —d |
| 40b | Nc | Nc | Weakb | —d | —d |
| 40c | Nc | Nc | Weakb | —d | —d |
| 54a | Nc | Nc | —d | —d | —d |
| 54b | Nc | Nc | —d | —d | —d |
| 54c | Ya | Ya | Ya | Ya | Ya |
| 55 | Nc | Nc | —d | Weakb | Ya |
| 56 | Ya | Ya | Ya | —d | Weakb |
These effects induced by 1-methyl-isoG are more potent than adenosine because 1-methyl-isoG can resist the action of adenosine deaminase. So 1-methyl-isoG can exist in the extracellular fluid for a long time, whereas adenosine has been degraded into inactive inosine.51 And there is a positive correlation between the potency of resistance to adenosine deaminase and the muscle relaxation and cardiovascular effects.30,51,178 However, in 1985, Radulovacki179 et al. reported 1-methyl-isoG does not have a strong hypnosis effect similar to adenosine. And, in 1981, Spence180 et al. showed that the effects of 1-methyl-isoG and adenosine on neuromuscular transmission in the vas deferens are similar. This is probably because the enzyme activity of adenosine deaminase in the vas deferens is inherently low, and thus the degradation is low. The ability of 1-methyl-isoG to cause the accumulation of cyclic adenosine monophosphate (cAMP) in brain is not effective as that of adenosine,181 but neither of these two nucleosides show inhibition of rat brain cAMP phosphodiesterase (PDE). Besides, in 1980, Taylor30 et al. proved that accumulated adenylate cyclase (AC) caused by 1-methyl-isoG may be associated with cardiovascular/relaxation effects.
In 1981, Marwood177 et al. showed that 1-methyl-isoG has equal negative inotropic and chronotropic effects in guinea-pig isolated atria. Nevertheless, adenosine has a greater negative inotropic effect. The active site of 1-methyl-isoG is only on the atria, whereas adenosine can also act on ventricle.51 Furthermore, 1-methyl-isoG showed a biphasic response on cardiac output, which means a slow increase followed by a rapid initial decrease.177 The impact of 1-methyl-isoG in lowering response electrical stimulation in isolated perfused rat tail arteries is similar to that of adenosine. In 1980, Jamieson178 et al. demonstrated that 1-methyl-isoG shows an inhibition of nerve-mediated stimulation, which is induced by electricity or nicotine in isolated guinea-pig ileum. But it is not useful for acetylcholine (ACh) and histamine-mediated contraction. Moreover, the site of action is at the post ganglionic nerve terminals because the effect disappears in isolated mammalian motor axons. Further, in 1981, Spence176 et al. demonstrated that the inhibitory dose for central excitation and monosynaptic or polysynaptic spinal reflex in conscious animals (central pathways) is much lower than that of neuromuscular junction. This means that muscle relaxation may be related to the central pathway (probably the spinal pathway).
In 1980, Taylor182 et al. reported 1-methyl-isoG does not compete with adenosine for uptake sites to activate adenosine cyclase. This indicates that it does not attenuate the effect of adenosine by competition, but because 1-methyl-isoG is not degraded and remains for a long time in the extracellular fluid. 1-Methyl-isoG was thought to act on a benzodiazepine receptor, but that the hypnotic effect was not the same as benzodiazepine ruled that out by Hall183 et al. in 1987. Those effects above are dose-dependent.30,51,176 And theophylline, a widely-used blocker of adenosine receptors, also acts as a dose-dependent antagonist of the effects of 1-methyl-isoG.51,178,179 So 1-methyl-isoG may act on the same receptor as adenosine in the intestinal smooth muscle and pig brain.
6.4 Ionophores
Nuclear waste remnants from the Second World War, nuclear testing, and the nuclear industry have left a large amount of radioactive contamination, mostly Cs+ and Ra2+. They are challenging to remove, leaving behind huge potential risk for human being and pollution of water and soil. Compounds that are highly selective for Cs+ are required because of the large quantity of Na+ and K+ in nuclear waste. In 1990s, crown ethers have been developed to bind Cs+ by McDowell184 et al. and Sachleben185–188 et al. But the Cs+ selectivity are modest resulting from their flexibility. So rigid macrocycles, especially calixarene crowns and their derivatives with different binding groups attached, have been used as recycling ligands. Rigid macrocycles show better complexation and selectivity for Cs+ over other ions such as Li+, Na+, K+ and Rb+.189 In addition, in 1995, calixarene carriers were reported to have rattling cell membrane penetration in polymer inclusion membrane (PIMs) by Reinhoudt190 et al. However, these macrocycles are expensive to synthesize and the ionophore recovery, as well as cations release, are difficult. Nowadays, noncovalent ionophores containing isoG are widely used. They show greater affinity and selectivity for Cs+ compared to those of calixarene and its derivatives. In 1997, Davis189 et al. reported the self-assembled (isoG)10·M+ ionophore shows significant selectivity for Cs+ in competition experiments. And (isoG)10·M+ can even extract bound Cs+ from a calixarene crowns ether. Thus, these compounds can be used for purification and metal nuclear waste disposal.Further, in 2000, Davis80 et al. then reported that in the (isoG)10·Cs+ system (Fig. 15 and 16), Cs+ and the protected isoG 53 are in constant exchange with the outside world. However, the exchange rate of Cs+ itself is 40000 times faster than the compound 53, as shown by multinuclear NMR spectroscopy. So the ion carrier does not disintegrate during ion exchange. The ions can be easily dissociated from the noncovalent ionophore when the solvent polarity changes. The same exchange process is true for other metal ions. Specially, Cs+ possesses an advantage that it can even slow down the isoG exchange rate. In 2001, they proved that the protected isoG 53 assembly can not only release Cs+ more easily without dissociation, but also show a stronger affinity for larger cations such as Cs+ and Ba2+ than K+, Sr2+ and Na+.191 They also demonstrated that (53)10·Cs+ can also promote selective transport through organic polymer inclusion membrane (PIMs) and bulk liquid membranes (BIMs), with high transport rates and excellent selectivity for Cs+. This cation-binding selectivity is attributed to the geometric hydrogen-bonding mode composed of donors and receptors. As discussed earlier, G forms G-quartets, whereas isoG prefers the formation of pentads. The sizes of these macrocyclic cavities of self-assembled ionophores determine the cation-binding selectivity. Moreover, in 2004, they reported that in the case of G, the selectivity for Ra2+ prior to magnesium ion (Mg2+) and calcium ion (Ca2+) requires an existing lipophilic anion.192 Nevertheless, isoG shows preferential selectivity for Cs+ over Mg2+, Ca2+, Sr2+ and Ba2+ without any additional environmental conditions. In addition, a significant radium ion (226Ra2+) preference over other monovalent alkali cations was observed for both 57 and 53, with a “precipitation prevention/dissolving” property over a wide pH range. And, in 2005, Reinhoudt193 et al. reported that Ra2+ can be selectively extracted from gas-field produced water by isoG-based self-assemblies.
6.5 Antitumor activity
In 1994, Kim161 et al. found that isoG has strong anti-cancer effects against P338, L5178Y, Sp2/O, HL60, Lymphoma Raji, both in vitro and in vivo. And isoG shows especial effectiveness against ascites tumor and malignant solid tumor. It is generally believed that isoG enters the cell by simple diffusion or diffusion combined with active transporters. The phosphorylation of isoG in the cytoplasm produces active nucleotides that inhibit tumor growth. Moreover, in 1994, they also reported that most of 6-substituted isoG derivatives (42, Fig. 10) show low cytotoxicity against cancerous Pogras 1, HeLa and Molt-4 cells, except those with amino, sulfur and furfurylamine substituents.58 This suggests that high cytotoxicity depends on the N2H or SH perssad at C6 position on the heterocyclic ring. In particular, when these substituents are displaced by nonpolar alkyl groups, the cytotoxicity decreases. Then in 1995, they demonstrated that the combination of berberine with isoG monophosphate greatly increases the antitumor effect.194 However, in 2004, Nowak195 et al. reported that similar to other telomerase inhibitors, isoGd showed low potency and specificity in inhibiting tumor cell growth.6.6 More applications
Apart from applications in gel formation, ionophore, and as antitumor agent, in 1973, isoG was reported to be an inhibitor of inosine monophosphate (IMP) pyrophosphorylase by Hagen.196 And in 1974, Holy197 et al. reported that isoG can also inhibit inducible E. coli binding site. In addition, in 1975, Mantsch41 et al. reported that its 5′-di- and -tri-phosphates can strongly bind and inhibit glutamic acid dehydrogenase. In 1988, Wissler198 et al. reported that isoG also acts as an essential constituent of Cu(II)-containing ribonucleic acid produced by cultured pig macrophages. This unusual Cu(II)-containing extracellular RNA was a formidable angiogenesis factor, and acted as an adenosine A1-receptor agonist. Besides, in 1999, Davis131 et al. raised that the protected isoG (53, Fig. 11) self-assembly can act as phase transfer catalysts. And in 2018, Anna174 et al. proposed that by strongly affecting the highly ordered structure of the G-quartet in TBA, isoG greatly affects the anticoagulant activity of the thrombin binding aptamer (TBA).7 Summary and outlook
In summary, since Fischer first synthesized isoguanine in 1897, there have been many reports focusing on the comparison of isoG with G. In this review, we have detailed the discovery, and synthesis of base- or sugar-modified derivatives of isoG, its monomer structure and aggregates, and the diverse applications of isoG/isoguanine. Since the yields of isoG/isoguanine in natural sources such as croton beans, butterfly wings and marine mollusk are too low, a series of synthetic methods of isoG/isoguanine by introducing functional groups to the heterocycle of G and AICA have been proposed. And various base- or sugar-modified derivatives have been synthesized or found in nature. Additionally, some unique properties of isoG and its derivatives including enol–keto tautomerism, mispairing, self assembly into tetramer (isoG4), decamer (isoG10) and isoG4 quadruplex, and fluorescence suggest its potential diverse applications in the formation of supramolecular hydrogels, in ps and aps duplexes and even triplexes, also in waste-collection as an ionophore. In addition, further applications such as antitumor activity, inhibition of IMP-pyrophosphorylase, and mutation-inducing have been talked about. As the structure and properties of isoG become better understood, there will be further studies and more applications will be identified. For example, because of its unique hydrogen bond acceptors and donors, isoG is expected to have a wider range of applications in the field of supramolecular self-assembly, the formation of hydrogel and ionophore. Additionally, isoG is most likely to play a significant role in genetic disease monitoring probes because of the variable nonstandard pairs caused by its tautomerism and its incorporation by different enzymes. Moreover, benefited from its antitumor ability and self-assembly properties, the preparation of supramolecular gels integrating antitumor activity and drug delivery appears extremely significative. Thus, isoG may play a significant role in the clinical treatment of various cancers and other therapies involving smooth muscle relaxation, hypotension, and bradycardia. In conclusion, the unique properties of isoG, such as its use in supramolecular assemblies, as a nonstandard base and a medicine with antitumor activity, will broaden its applications in diverse fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the National Natural Science Foundations of China (81922020, 81970950, 81621062).References
- M. Chen, W. Lin, L. Hong, N. Ji and H. Zhao, BioMed Res. Int., 2019, 2019 DOI:10.1155/2019/6258248.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1897, 30, 2226–2254 CrossRef CAS.
- E. Cherbuliez and K. Bernhard, Helv. Chim. Acta., 1932, 15, 464–471 CrossRef CAS.
- R. Purrmann, Justus Liebigs Ann. Chem., 1940, 544, 182–190 CrossRef CAS.
- F. A. Fuhrman, G. J. Fuhrman, R. J. Nachman and H. S. Mosher, Science, 1981, 212, 557–558 CrossRef CAS PubMed.
- J. Davoll, J. Am. Chem. Soc., 1951, 73, 3174–3176 CrossRef CAS.
- G. B. Brown, G. Levin and S. Murphy, Biochemistry, 1964, 3, 880–883 CrossRef CAS PubMed.
- C. B. Reese, Y. S. Sanghvi and R. Kuroda, J. Chem. Soc., Perkin Trans. 1, 1987, 1527–1531 RSC.
- K. J. Divakar, M. Mottahedeh, C. B. Reese, Y. S. Sanghvi and K. A. D. Swift, J. Chem. Soc., Perkin Trans. 1, 1991, 4, 771–774 RSC.
- C. Ji-Wang, L. Horng-Yuh, H. Min and S. Fang-Jy, Tetrahedron Lett, 1987, 28, 2151–2154 CrossRef.
- S. Tirumala and J. T. Davis, J. Am. Chem. Soc., 1997, 119, 2769–2776 CrossRef CAS.
- X. Shi, J. C. Fettinger, M. Cai and J. T. Davis, Angew. Chem., Int. Ed., 2000, 39, 3124–3127 CrossRef CAS.
- M. V. Buell and M. E. Perkins, J. Biol. Chem., 1927, 72, 745–749 Search PubMed.
- J. R. Spies and N. L. Drake, J. Am. Chem. Soc., 1935, 57, 774 CrossRef CAS.
- J. R. Spies, J. Am. Chem. Soc., 1939, 61, 350–351 CrossRef CAS.
- R. Falconer, J. M. Gulland and L. F. Story, J. Am. Chem. Soc., 1939, 1784, 10.1039/jr9390001784.
- G. R. Pettit, R. H. Ode, R. M. Coomes and S. L. Ode, Lloydia, 1976, 39, 363–367 CAS.
- J. Davoll and B. A. Lowy, J. Am. Chem. Soc., 1951, 73, 1650–1655 CrossRef CAS.
- A. Bendich, G. B. Brown, F. S. Philips and J. B. Thiersch, J. Biol. Chem., 1950, 183, 267–277 CAS.
- B. A. Lowy, J. Davoll and G. B. Brown, J. Biol. Chem., 1952, 197, 591–600 CAS.
- S. Friedman and J. S. Gots, Arch. Biochem. Biophys., 1951, 32, 227–229 CrossRef CAS PubMed.
- Z. Nackerdien, K. S. Kasprzak, G. Rao, B. Halliwell and M. Dizdaroglu, Cancer Res., 1991, 5837–5842 CAS.
- M. Dizdaroglu, Mutat. Res., 1992, 275, 331–342 CAS.
- R. Olinski, T. Zastawny, J. Budzbon, J. Skokowski, W. Zegarski and M. Dizdaroglu, FEBS Lett., 1992, 309, 193–198 CrossRef CAS.
- P. Jaruga, T. H. Zastawny, J. Skokowski, M. Dizdaroglu and R. Olinski, FEBS Lett., 1994, 341, 59–64 CrossRef CAS.
- T. Mori, Y. Hori and M. Dizdaroglu, Int. J. Radiat. Biol., 1993, 64, 645–650 CrossRef CAS.
- P. Jaruga and M. Dizdaroglu, Nucleic Acids Res., 1996, 24, 1389–1394 CrossRef CAS PubMed.
- H. Kamiya and H. Kasai, J. Biol. Chem., 1995, 270, 19446–19450 CrossRef CAS PubMed.
- J. Baird-Lambert, J. F. Marwood, L. P. Davies and K. M. Taylor, Life Sci., 1980, 26, 1069–1077 CrossRef CAS PubMed.
- H. Kamiya, T. Ueda, T. Ohgi, A. Matsukage and H. Kasai, Nucleic Acids Res., 1995, 23, 761–766 CrossRef CAS PubMed.
- H. Kamiya and H. Kasai, Nucleic Acids Res., 1997, 25, 304–311 CrossRef CAS PubMed.
- H. Kamiya, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2010, 703, 32–36 CrossRef CAS PubMed.
- H. Kasai, Free Radicals Biol. Med., 2002, 33, 450–456 CrossRef CAS.
- S. Frelon, T. Douki and J. Cadet, Free Radical Res., 2002, 36, 499–508 CrossRef CAS PubMed.
- J. Cadet, T. Douki and J. L. Ravanat, Environ. Health Perspect., 1997, 105, 1034–1039 CAS.
- J. Cadet, T. Douki, J.-L. Ravanat and J. Wagner, Bioanal. Rev., 2012, 4, 55–74 CrossRef.
- A. Weimann, G. McLeod, T. Henriksen, V. Cejvanovic and H. E. Poulsen, Scand. J. Clin. Lab. Invest., 2019, 79, 1–8 CrossRef PubMed.
- V. Nair and D. A. Young, J. Org. Chem., 1985, 50, 406–408 CrossRef CAS.
- R. V. Ravindranathan and H. T. Miles, Biochim. Biophys. Acta, 1965, 94, 603–606 CrossRef CAS.
- H. H. Mantsch, I. Goia, M. Kezdi, O. Barzu, M. Dansoreanu, G. Jebeleanu and N. G. Ty, Biochemistry, 1975, 14, 5593–5601 CrossRef CAS PubMed.
- J. A. Montgomery and K. Hewson, J. Org. Chem., 1968, 33, 432 CrossRef CAS PubMed.
- L. De Napoli, D. Montesarchio, G. Piccialli, C. Santacroce and M. Varra, J. Chem. Soc., Perkin Trans. 1, 1995, 15, 45–47 Search PubMed.
- Q. Cheng, J. Gu, K. R. Compaan and H. F. Schaefer, Chem.–Eur. J., 2012, 18, 4877–4886 CrossRef CAS PubMed.
- A. Yamazaki, I. Kumashiro, T. Takenishi and M. Ikehara, Chem. Pharm. Bull., 1968, 16, 2172–2181 CrossRef CAS.
- A. Yamazaki, M. Okutsu and Y. Yamada, Nucleic Acids Res., 1976, 3, 251–260 CrossRef CAS PubMed.
- J. W. Chern, G. S. Lin, C. S. Chen and L. B. Townsend, J. Org. Chem., 1991, 56, 4213–4218 CrossRef CAS.
- L. De Napoli, A. Messere, D. Montesarchio, G. Piccialli and M. Varra, Nucleosides Nucleotides, 1997, 16, 183–191 CrossRef CAS.
- F. A. Fuhrman, G. J. Fuhrman and K. Deriemer, Biol. Bull., 1979, 156, 289–299 CrossRef CAS PubMed.
- R. J. Quinn, R. P. Gregson, A. F. Cook and R. T. Bartlett, Tetrahedron Lett., 1980, 21, 567–568 CrossRef CAS.
- Y. H. Kim, R. J. Nachman, L. Pavelka, H. S. Mosher, F. A. Fuhrman and G. J. Fuhrman, J. Nat. Prod., 1981, 44, 206–214 CrossRef CAS.
- W. Bergmann and R. J. Feeney, J. Org. Chem., 1951, 16, 981–987 CrossRef CAS.
- W. Bergmann and D. C. Burke, J. Org. Chem., 1955, 20, 1501–1506 CrossRef CAS.
- W. Bergmann and D. C. Burke, J. Org. Chem., 1956, 21, 226–228 CrossRef CAS.
- P. Searle and T. Molinski, J. Nat. Prod., 1994, 57, 1452–1454 CrossRef CAS.
- R. T. Bartlett, A. F. Cook, M. J. Holman, W. W. McComas, E. F. Nowoswait, M. S. Poonian, J. A. Baird-Lambert, B. A. Baldo and J. F. Marwood, J. Med. Chem., 1981, 24, 947–954 CrossRef CAS PubMed.
- D. Firsova, K. Calabro, P. Lasserre, F. Reyes and O. P. Thomas, Tetrahedron Lett., 2017, 58, 4652–4654 CrossRef CAS.
- S. Lee, J. Kim, Y. Cho and J. Kim, Arch. Pharmacal Res., 1994, 17, 170–174 CrossRef CAS.
- C. Buhr, R. Wagner, D. Grant and B. Froehler, Nucleic Acids Res., 1996, 24, 2974–2980 CrossRef CAS.
- G. Balow, V. Mohan, E. A. Lesnik, J. F. Johnston, B. P. Monia and O. L. Acevedo, Nucleic Acids Res., 1998, 26, 3350–3357 CrossRef CAS PubMed.
- F. Seela and H. Thomas, Helv. Chim. Acta, 1995, 78, 94–108 CrossRef CAS.
- N. Ramzaeva and F. Seela, Helv. Chim. Acta, 1996, 79, 1549–1558 CrossRef CAS.
- F. Seela and Y. Chen, Chem. Commun., 1996, 2263–2264, 10.1039/CC9960002263.
- G. Becher, J. He and F. Seela, Helv. Chim. Acta, 2001, 84, 1048–1065 CrossRef CAS.
- F. Seela and G. Becher, Helv. Chim. Acta, 1999, 82, 1640–1655 CrossRef CAS.
- F. Seela and G. Becher, Chem. Commun., 1998, 2017–2018 RSC.
- F. Seela and R. Kröschel, Nucleic Acids Res., 2003, 31, 7150–7158 CrossRef CAS PubMed.
- F. Seela and K. Xu, Org. Biomol. Chem., 2007, 5, 3034–3045 RSC.
- M. Jeselnik, S. Jaksa and J. Kobe, ChemInform, 2004, 35, 153–160 CrossRef.
- J. Wierzchowski, J. Sepioł, D. Sulikowski, B. Kierdaszuk and D. Shugar, J. Photochem. Photobiol., A, 2006, 179, 276–282 CrossRef CAS.
- F. Seela, A. M. Javelakar and I. Munster, Helv. Chim. Acta, 2005, 88, 751–765 CrossRef CAS.
- F. Seela, I. Munster, U. Loechner and H. Rosemeyer, Helv. Chim. Acta, 1998, 81, 1139–1155 CrossRef CAS.
- D. Jiang and F. Seela, J. Am. Chem. Soc., 2010, 132, 4016–4024 CrossRef CAS.
- H. U. Blank, I. Wempen and J. J. Fox, J. Org. Chem., 1970, 35, 1131–1138 CrossRef CAS.
- J. Wierzchowski, G. Mędza, J. Sepioł, M. Szabelski and D. Shugar, J. Photochem. Photobiol., A, 2012, 237, 64–70 CrossRef CAS.
- Z. Kazimierczuk and D. Shugar, Acta Biochim. Pol., 1973, 20, 395–402 CAS.
- Z. Kazimierczuk, R. Mertens, W. Kawczynski and F. Seela, Helv. Chim. Acta, 1991, 74, 1742–1748 CrossRef CAS.
- S. A. Ingale, P. Leonard, Q. N. Tran and F. Seela, J. Org. Chem., 2015, 80, 3124–3138 CrossRef CAS PubMed.
- K. Ono, M. Ogasawara, Y. Iwata, H. Nakane, T. Fujii, K. Sawai and M. Saneyoshi, Biochem. Biophys. Res. Commun., 1986, 140, 498–507 CrossRef CAS PubMed.
- M. Cai, V. Sidorov, Y. F. Lam, R. A. Flowers and J. T. Davis, Org. Lett., 2000, 2, 1665–1668 CrossRef CAS PubMed.
- S. Kim, S. Lee, W. Sun, S. Oh and J. Kim, Arch. Pharmacal Res., 1999, 22, 619–623 CrossRef CAS PubMed.
- T. Davis, S. Tirumala, J. R. Jenssen, E. Radler and D. Fabris, J. Org. Chem., 1995, 60, 4167–4176 CrossRef.
- W. Pfleiderer, Justus Liebigs Ann. Chem., 1961, 647, 167–173 CrossRef.
- C. Roberts, R. Bandaru and C. Switzer, J. Am. Chem. Soc., 1997, 119, 4640–4649 CrossRef CAS.
- T. Golas, M. Fikus, Z. Kazimierczuk and D. Shugar, Eur. J. Biochem., 1976, 65, 183–192 CrossRef CAS PubMed.
- J. Sepiol, Z. Kazimierczuk and D. Shugar, Z. Naturforsch., C: Biosci., 1976, 31, 361–370 CAS.
- R. Krishnamurthy, S. Pitsch, M. Minton, C. Miculka, N. Windhab and A. Eschenmoser, Angew. Chem., Int. Ed., 1996, 35, 1537–1541 CrossRef CAS.
- J. Hunziker, H. J. Roth, M. Böhringer, A. Giger, U. Diederichsen, M. Göbel, R. Krishnan, B. Jaun, C. Leumann and A. Eschenmoser, Helv. Chim. Acta, 1993, 76, 259–352 CrossRef CAS.
- A. Eschenmoser, Pure Appl. Chem., 1993, 65, 1179–1188 Search PubMed.
- A. Banerjee, W. Saenger, B. Lesyng, Z. Kazimierczuk and D. Shugar, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2472–2477 CrossRef.
- F. Seela, X. Peng and K. Xu, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1569–1572 CrossRef CAS.
- Horiz. Biochem, ed. A. Rich, M. Kasha and B. Pullman, 1962, pp. 103–126 Search PubMed.
- A. P. Joseph, A. B. Steven, K. Tilman and E. M. Simon, Nature, 1990, 343, 33–37 CrossRef.
- T. Horn, C.-A. Chang and M. L. Collins, Tetrahedron Lett., 1995, 36, 2033–2036 CrossRef CAS.
- C. Roberts, R. Bandaru and C. Switzer, Tetrahedron Lett., 1995, 36, 3601–3604 CrossRef CAS.
- A. Jaworski, J. S. Kwiatkowski and B. Lesyng, Int. J. Quantum Chem., 1985, 28, 209–216 CrossRef.
- X. Chen, R. Kierzek and D. H. Turner, J. Am. Chem. Soc., 2001, 123, 1267–1274 CrossRef CAS.
- F. Seela, C. Wei, A. Melenewski and E. Feiling, Nucleosides Nucleotides, 1998, 17, 2045–2052 CrossRef CAS.
- A. Maciejewska, K. Lichota and J. Kusmierek, Biochem. J., 2003, 369, 611–618 CrossRef CAS.
- C. Switzer, S. Moroney and S. A. Benner, J. Am. Chem. Soc., 1989, 111, 8322–8323 CrossRef CAS.
- C. Y. Switzer, S. E. Moroney and S. A. Benner, Biochemistry, 1993, 32, 10489–10496 CrossRef CAS PubMed.
- Y. Tor and P. Dervan, J. Am. Chem. Soc., 1993, 115, 4461–4467 CrossRef CAS.
- H. Kamiya and H. Kasai, FEBS Lett., 1996, 391, 113–116 CrossRef CAS.
- J. Horlacher, M. Hottiger, V. N. Podust, U. Hübscher and S. A. Benner, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6329–6333 CrossRef CAS PubMed.
- M. J. Lutz, H. A. Held, M. Hottiger, U. Hübscher and S. A. Benner, Nucleic Acids Res., 1996, 24, 1308–1313 CrossRef CAS PubMed.
- M. J. Lutz, J. Horlacher and S. A. Benner, Bioorg. Med. Chem. Lett., 1998, 8, 499–504 CrossRef CAS PubMed.
- H. Kamiya and H. Kasai, Nucleic Acids Res., 2000, 28, 1640–1646 CrossRef CAS PubMed.
- H. Kamiya, H. Maki and H. Kasai, Biochemistry, 2000, 39, 9508–9513 CrossRef CAS PubMed.
- Y. Tsurudome, T. Hirano, H. Kamiya, R. Yamaguchi, S. Asami, H. Itoh and H. Kasai, Mutat. Res., 1998, 408, 121–127 CAS.
- D. S. Hitchcock, A. A. Fedorov, E. V. Fedorov, L. J. Dangott, S. C. Almo and F. M. Raushel, Biochemistry, 2011, 50, 5555–5557 CrossRef CAS PubMed.
- T. Ohtsubo, K. Nishioka, Y. Imaiso, S. Iwai, H. Shimokawa, H. Oda, T. Fujiwara and Y. Nakabeppu, Nucleic Acids Res., 2015, 43, 3870–3871 CrossRef CAS PubMed.
- H. Kamiya and H. Kasai, J. Radiat. Res., 2000, 41, 349–354 CrossRef CAS PubMed.
- K. Fujikawa, H. Kamiya, H. Yakushiji, Y. Nakabeppu and H. Kasai, Nucleic Acids Res., 2001, 29, 449–454 CrossRef CAS PubMed.
- K. Fujikawa, H. Kamiya, H. Yakushiji, Y. Fujii, Y. Nakabeppu and H. Kasai, J. Biol. Chem., 1999, 274, 18201–18205 CrossRef CAS PubMed.
- J. C. Chaput and C. Switzer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10614–10619 CrossRef CAS.
- C. Switzer and J. C. Chaput, Methods, 2001, 23, 141–148 CrossRef CAS PubMed.
- P. Schuster, G. Zundel and C. Sandorfy, The Hydrogen Bond, North Holland Co., New York, 1976, p. 403 Search PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Systems, Springer-Verlag, New York, 1991, pp. 103–110 Search PubMed.
- F. Seela, C. Wei and A. Melenewski, Origins Life Evol. Biospheres, 1997, 27, 597–608 CrossRef CAS PubMed.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- T. Giorgi, F. Grepioni, I. Manet, P. Mariani, S. Masiero, E. Mezzina, S. Pieraccini, L. Saturni, G. P. Spada and G. Gottarelli, Chem.–Eur. J., 2002, 8, 2143–2152 CrossRef CAS.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- T. J. Pinnavaia, C. L. Marshall, C. M. Mettler, C. L. Fisk, H. T. Miles and E. D. Becker, J. Am. Chem. Soc., 1978, 100, 3625–3627 CrossRef CAS.
- M. L. Meyer and J. Suhnel, J. Phys. Chem. A, 2003, 107, 1025–1031 CrossRef CAS.
- S. L. Forman, J. C. Fettinger, S. Pieraccini, G. Gottarelli and J. T. Davis, J. Am. Chem. Soc., 2000, 122, 4060–4067 CrossRef CAS.
- X. Shi, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2001, 123, 6738–6739 CrossRef CAS PubMed.
- X. Shi, J. C. Fettinger and J. T. Davis, Angew. Chem., Int. Ed., 2001, 40, 2827–2831 CrossRef CAS PubMed.
- X. Shi, J. C. Fettinger and J. T. Davis, Angew. Chem., 2001, 2909–2913 CrossRef.
- X. Shi, K. M. Mullaugh, J. C. Fettinger, Y. Jiang, S. A. Hofstadler and J. T. Davis, J. Am. Chem. Soc., 2003, 125, 10830–10841 CrossRef CAS PubMed.
- M. Cai, A. L. Marlow, J. C. Fettinger, D. Fabris, T. J. Haverlock, B. A. Moyer and J. T. Davis, Angew. Chem., Int. Ed., 2000, 39, 1283–1285 CrossRef CAS.
- A. L. Marlow and J. T. Davis, Tetrahedron Lett., 1999, 40, 3539–3542 CrossRef CAS.
- T. Evan-Salem, L. Frish, F. W. B. Van Leeuwen, D. N. Reinhoudt, W. Verboom, M. S. Kaucher, J. T. Davis and Y. Cohen, Chem.–Eur. J., 2007, 13, 1969–1977 CrossRef CAS PubMed.
- V. Abet, R. Evans, F. Guibbal, S. Caldarelli and R. Rodriguez, Angew. Chem., 2014, 126, 4962–4966 CrossRef.
- M. Cai, X. Shi, V. Sidorov, D. Fabris, Y.-F. Lam and J. T. Davis, Tetrahedron, 2002, 58, 661–671 CrossRef CAS.
- E. H. Blackburn and J. W. Szostak, Annu. Rev. Biochem., 1984, 53, 163–194 CrossRef CAS PubMed.
- J. R. Williamson, M. K. Raghuraman and T. R. Cech, Cell, 1989, 59, 871–880 CrossRef CAS PubMed.
- F. Aboul-Ela, A. I. H. Murchie, D. G. Norman and D. M. J. Lilley, J. Mol. Biol., 1994, 243, 458–471 CrossRef CAS PubMed.
- A.-E. Fareed, I. H. M. Alastair and M. J. L. David, Nature, 1992, 360, 280–282 CrossRef.
- W. Yong and D. J. Patel, Biochemistry, 1992, 31, 8112–8119 CrossRef PubMed.
- F. Seela, C. Wei and A. Melenewski, Nucleic Acids Res., 1996, 24, 4940–4945 CrossRef CAS.
- C. Roberts, J. C. Chaput and C. Switzer, Chem. Biol., 1997, 4, 899–908 CrossRef CAS.
- E. A. Venczel and D. Sen, Biochemistry, 1993, 32, 6220–6228 CrossRef CAS.
- F. Seela and R. Kröschel, Bioconjugate Chem., 2001, 12, 1043–1050 CrossRef CAS PubMed.
- F. Seela and C. Wei, Chem. Commun., 1997, 1869–1870 RSC.
- F. Seela, C. Wei, A. Melenewski, Y. He, R. Kröschel and E. Feiling, Nucleosides Nucleotides, 1999, 18, 1543–1548 CrossRef CAS.
- F. Seela and C. Wei, Helv. Chim. Acta, 1999, 82, 726–745 CrossRef CAS.
- N. B. Ramsing and T. M. Jovin, Nucleic Acids Res., 1988, 16, 6659–6676 CrossRef CAS PubMed.
- F. Seela, B. Gabler and Z. Kazimierczuk, Collect. Czech. Chem. Commun., 1993, 58, 170–173 CrossRef.
- F. Seela, C. Wei and A. Melenewski, Nucleosides Nucleotides, 1997, 16, 1523–1527 CrossRef CAS.
- X.-L. Yang, H. Sugiyama, S. Ikeda, I. Saito and A. H. J. Wang, Biophys. J., 1998, 75, 1163–1171 CrossRef CAS PubMed.
- F. Seela, Y. He and C. Wei, Tetrahedron, 1999, 55, 9481–9500 CrossRef CAS.
- S. S. Pujari and F. Seela, J. Org. Chem., 2013, 78, 8545–8561 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- I. Bang, Biochem. Z., 1910, 26, 293–311 CAS.
- H. Zhao, A. H. Schäfer and F. Seela, ChemPlusChem, 2017, 82, 826–833 CrossRef CAS.
- H. Zhao, D. Jiang, A. H. Schäfer and F. Seela, ChemPlusChem, 2017, 82, 778–784 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2011, 8, 260–272 RSC.
- A. Biswas, S. Malferrari, D. M. Kalaskar and A. K. Das, Chem. Commun., 2018, 54, 1778–1781 RSC.
- F. Tang, H. Feng, Y. Du, Y. Xiao, H. Dan, H. Zhao and Q. Chen, Chem.–Asian J., 2018, 13, 1962–1971 CrossRef CAS PubMed.
- B. Adhikari, A. Shah and H.-B. Kraatz, J. Mater. Chem. B, 2014, 2, 4802–4810 RSC.
- J. Kim, S. Lee, Y. Han, J. Moon and J. Kim, Arch. Pharmacal Res., 1994, 17, 115–118 CrossRef CAS PubMed.
- V. Venkatesh, N. K. Mishra, I. Romero-Canelon, R. R. Vernooij, H. Shi, J. P. C. Coverdale, A. Habtemariam, S. Verma and P. J. Sadler, J. Am. Chem. Soc., 2017, 139, 5656–5659 CrossRef CAS.
- H. Zhao, H. Feng, J. Liu, F. Tang, Y. Du, N. Ji, L. Xie, X. Zhao, Z. Wang and Q. Chen, Biomaterials, 2019, 230 DOI:10.1016/j.biomaterials.2019.119598.
- M. M. Cox, Mutat. Res., 1997, 384, 15–22 CAS.
- H. Kamiya and H. Kasai, Biochemistry, 1997, 36, 11125–11130 CrossRef CAS PubMed.
- M. Inoue, H. Kamiya, K. Fujikawa, Y. Ootsuyama, N. Murata-Kamiya, T. Osaki, K. Yasumoto and H. Kasai, J. Biol. Chem., 1998, 273, 11069–11074 CrossRef CAS.
- S. C. Johnson, C. B. Sherrill, D. J. Marshall, M. J. Moser and J. R. Prudent, Nucleic Acids Res., 2004, 32, 1937–1941 CrossRef CAS.
- S. C. Johnson, D. J. Marshall, G. Harms, C. M. Miller, C. B. Sherrill, E. L. Beaty, S. A. Lederer, E. B. Roesch, G. Madsen, G. L. Hoffman, R. H. Laessig, G. J. Kopish, M. W. Baker, S. A. Benner, P. M. Farrell and J. R. Prudent, Clin. Chem., 2004, 50, 2019–2027 CrossRef CAS PubMed.
- J. D. Bain, S. Christopher, C. Richard and A. B. Steven, Nature, 1992, 356, 537–539 CrossRef CAS PubMed.
- F. Seela, D. Jiang and S. Budow, ChemBioChem, 2010, 11, 1443–1450 CrossRef CAS PubMed.
- F. A. Fuhrman and G. J. Fuhrman, Comp. Biochem. Physiol., C: Comp. Pharmacol., 1982, 72, 203–210 CrossRef CAS.
- P. L. Ewing, F. Schlenk and G. A. Emerson, J. Pharmacol. Exp. Ther., 1949, 97, 379–383 CAS.
- D. A. Clarke, J. Davoll, F. S. Philips and G. B. Brown, J. Pharmacol. Exp. Ther., 1952, 106, 291–302 CAS.
- K. Weronika, C. Tomasz and P. Anna, PLoS One, 2018, 13, e0197835 CrossRef.
- F. A. Fuhrman, G. J. Fuhrman, Y. H. Kim, L. A. Pavelka and H. S. Mosher, Science, 1980, 207, 193–195 CrossRef CAS PubMed.
- P. Buckle and I. Spence, Naunyn-Schmiedeberg's Arch. Pharmacol., 1981, 316, 64–68 CrossRef CAS PubMed.
- J. F. Marwood, Clin. Exp. Pharmacol. Physiol., 1981, 8, 575–584 CrossRef CAS.
- D. Jamieson and P. Davis, Eur. J. Pharmacol., 1980, 67, 295–300 CrossRef CAS.
- M. Radulovacki, R. M. Virus, D. Rapoza and R. C. Crane, Neuropharmacology, 1985, 24, 547–549 CrossRef CAS.
- P. Buckle and I. Spence, Naunyn-Schmiedeberg's Arch. Pharmacol., 1982, 319, 130–135 CrossRef CAS.
- M. Huang, H. Shimizu and J. W. Daly, J. Med. Chem., 1972, 15, 462–466 CrossRef CAS.
- L. P. Davies, K. M. Taylor, R. P. Gregson and R. J. Quinn, Life Sci., 1980, 26, 1079–1088 CrossRef CAS.
- L. P. Davies, J. Baird-Lambert and J. G. Hall, Neuropharmacology, 1987, 26, 493–497 CrossRef CAS.
- W. J. McDowell and G. N. Case, Anal. Chem., 1992, 64, 3013–3017 CrossRef CAS.
- J. C. Bryan, K. Kavallieratos and R. A. Sachleben, Inorg. Chem., 2000, 39, 1568–1572 CrossRef CAS PubMed.
- J. C. Bryan, R. A. Sachleben and B. P. Hay, Inorg. Chim. Acta, 1999, 290, 86–94 CrossRef CAS.
- Y. Deng, R. A. Sachleben and B. A. Moyer, J. Chem. Soc., Faraday Trans., 1995, 91, 4215–4222 RSC.
- R. A. Sachleben, Y. Deng, D. R. Bailey and B. A. Moyer, Solvent Extr. Ion Exch., 1996, 14, 995–1015 CrossRef CAS.
- J. T. Davis, S. K. Tirumala and A. L. Marlow, J. Am. Chem. Soc., 1997, 119, 5271–5272 CrossRef CAS.
- A. Casnati, A. Pochini, R. Ungaro, F. Ugozzoli, F. Arnaud, S. Fanni, M.-J. Schwing, R. J. M. Egberink, F. d. Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 2767 CrossRef CAS.
- S. Lee, J. Lamb, M. Cai and J. Davis, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 51–57 CrossRef CAS.
- F. W. B. van Leeuwen, W. Verboom, X. Shi, J. T. Davis and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 16575–16581 CrossRef CAS.
- F. W. B. van Leeuwen, C. J. H. Miermans, H. Beijleveld, T. Tomasberger, J. T. Davis, J. T. Davis, W. Verboom and D. Reinhoudt, Environ. Sci. Technol., 2005, 39, 5455–5459 CrossRef CAS.
- S. Lee, J. Kim, S. Lee and J. Kim, Arch. Pharmacal Res., 1995, 18, 138–139 CrossRef CAS.
- R. Nowak, M. Sawardo-Rochowska, Z. Kazimierczuk and R. J. Nowak, Eksperimental'naia Onkologiia, 2004, 26, 20–23 CAS.
- C. Hagen, Biochim. Biophys. Acta, 1973, 293, 105–110 CrossRef CAS.
- J. Doskocil and A. Holý, Nucleic Acids Res., 1974, 1, 645–652 CrossRef CAS PubMed.
- J. H. Wissler, S. Kiesewetter, E. Logemann, M. Sprinzl and L. M. G. Hellmeyer Jr, Biol. Chem., 1988, 369, 948–949 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |