
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe combined effect of light irradiation and chloride on the physicochemical properties of silver nanoparticles†
Bojie Yuan,
Minghao Sui *,
Hongtao Lu,
Jingyu Wang and
Jie Qin
*,
Hongtao Lu,
Jingyu Wang and
Jie Qin
Shanghai Institute of Pollution Control and Ecological Security, State Key Laboratory of Pollution Control and Resource Reuse, School of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, People's Republic of China. E-mail: minghaosui@tongji.edu.cn
First published on 24th December 2019
Abstract
Understanding the effect of various environmental factors on the transformation of silver nanoparticles (Ag NPs) is crucial to determine their toxicity and fate in the environment. Ag NPs are inevitably exposed to sunlight and will be in contact with chloride (Cl−), a ubiquitous ligand in natural water, once released into the environment. In this study, the combined effect of Cl− and light on various physicochemical properties (optical property, dissolution, morphology, surface charge) of different sizes Ag NPs was studied. The results showed that light irradiation, in the presence of Cl−, led to a great decrease in the concentration of dissolved Ag and a remarkable increase in the zeta potential of Ag NPs, as well as the generation of some tiny Ag NPs and fusion aggregates. AgCl was suggested to rapidly coat onto Ag NPs after exposure to Cl−. And the AgCl layer was obviously destroyed by photoreduction under light irradiation. Meanwhile, the Ag NP size exhibited a great impact on the destruction of the AgCl layer. It was further observed that the AgCl layer gradually re-formed when the light was removed, which suggested that Ag NPs might present different states during the daytime and at night in aquatic environments.
Introduction
Nanomaterials have been widely employed in medical, commercial and industrial products in the past several decades due to their unique chemical and physical properties.1–5 Among these nanomaterials, silver nanoparticles (Ag NPs) with outstanding optical, catalytic, electrical, and antibacterial properties are currently one of the most widely used engineered nanoparticles. As a result, Ag NPs will be inevitably released from the related products of Ag NPs during production, use and washing and eventually introduced into the environment.6 Ag NPs have been reported to be highly toxic to a variety of model organisms by many studies.7–12 Hence, there is a growing concern about the potential risk of released Ag NPs to human and ecological health. Therefore, it is crucial to understand the transformation and fate of Ag NPs in the environment.After releasing into the environment, Ag NPs are inevitably exposed to sunlight. It was reported that light irradiation had a strong effect on various properties of Ag NPs. Under light irradiation, the dissolution of Ag NPs was obviously accelerated.13 Furthermore, the dissolution rate was found to be affected by irradiation conditions (UV-365 > xenon lamp > UV-254) and surface coating (bare-Ag NPs > PVP-Ag NPs > citrate-Ag NPs).14 In addition, exposing Ag NPs to light irradiation also induced the destabilization of Ag NPs and promoted formation of aggregates.15,16
The behavior and fate of Ag NPs in aquatic environment are also sensitive to environment conditions. In the presence of natural organic materials (NOMs), the stability of Ag NPs was enhanced due to the electrostatic and steric repulsion caused by the adsorption of NOMs on the surface of Ag NPs.17–20 Owing to the photoreduction of NOMs, Ag+ would be reduced to Ag0, which also greatly suppressed the dissolution of Ag NPs.17–19,21 Cl−, a ligand with strong affinity for oxidized silver, ubiquitously presents in natural water and Ag NPs are inevitably in contact with Cl− in aquatic environment. It was reported that Cl− could significantly affect the dissolution of Ag NPs, and the kinetics of dissolution were closely related to the ratio of Cl/Ag.22 At low Cl/Ag ratio, Cl− reacted with dissolved silver and AgCl coating was generated on Ag NPs.22,23 The formation of AgCl coating, a passivating layer, would remarkably suppress the dissolution rate of Ag NPs.24 However, the dissolution rate of Ag NPs was obviously enhanced at high Cl/Ag ratio owing to the generation of soluble AgClx(x−1)− species. The increase in the amount of dissolved silver also led to increased toxicity.22
Additionally, the intrinsic properties of Ag NPs, like particle size, also have significant influence on its behavior. Particles size was found to have an inverse effect on Ag NPs dissolution: small Ag NPs exhibited higher solubility than large ones.25,26 Small Ag NPs with higher surface-to-volume ratio have higher surface energy and reactivity than large particles. Ivanova et al. have demonstrated that the standard redox potential of metal nanoparticles decreased with decreasing size.27
Many studies have focused on the fate and behavior of Ag NPs during light irradiation,17–19,28 however, little is known about the combined effect of light irradiation and Cl−, as well as particle size, on various physicochemical properties of Ag NPs. Here, polyvinyl pyrrolidone (PVP) coated Ag NPs with different size (20, 40 and 57 nm) were synthesized. And then, we investigated the evolution of physicochemical properties (optical property, dissolution, zeta potential and morphology) of three sizes Ag NPs after exposing to Cl− solutions under light irradiation, and this study was expected to provide more information on the behavior and fate of Ag NPs in natural water.
Materials and methods
Materials
Tannic acid (TA), poly(vinylpyrrolidone) (PVP, K29-32, Mw = 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), trisodium citrate (Na3C6H5O7·5H2O), hydrogen peroxide (H2O2), sodium chloride (NaCl) and sodium nitrate (NaNO3) were purchased from Aladdin Chemistry Co. Ltd. Silver nitrate (AgNO3) was purchased from Sigma-Aldrich. Ultrapure water (Aqualore 2S, USA, 18.2 MΩ cm) was used to prepare all solutions.
000), trisodium citrate (Na3C6H5O7·5H2O), hydrogen peroxide (H2O2), sodium chloride (NaCl) and sodium nitrate (NaNO3) were purchased from Aladdin Chemistry Co. Ltd. Silver nitrate (AgNO3) was purchased from Sigma-Aldrich. Ultrapure water (Aqualore 2S, USA, 18.2 MΩ cm) was used to prepare all solutions.
Synthesis and characterization of Ag NPs
According to the method proposed by Bastus et al.,29 PVP coated Ag NPs with different sizes were synthesized. And then, Ag NPs were dispersed in ultrapure water and stored in dark conditions at 4 °C for later use. The total concentration of Ag NPs stock suspension was measured by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7700). Samples were digested with HNO3 and H2O2 before ICP-MS analysis. Transmission electron microscopy (TEM, JEM2011, Jeol) was used to characterize the morphology of synthesized Ag NPs. TEM size distributions were obtained from more than 200 particles of each Ag NPs type. The UV-vis spectra from 300–700 nm were collected by using a UV-3600 spectrophotometer. Hydrodynamic diameter and the polydispersity index (PDI) of Ag NPs, as well as zeta potential, were determined by a zeta potential analyzer (Nano Z, Malvern).The light irradiation experiment
The experiments were carried out in a solar simulator equipped with a 500 W Xe lamp to simulate the sunlight. The reaction vessels were 50 mL quartz tubes and rotated around the light source to ensure each sample obtain similar levels of light exposure. Meanwhile, the incident light would pass through the cooling water layer to remove infrared rays before reaching the reaction vessels. The reaction vessels filled with 30 mL Ag NPs suspensions (6.4 mg L−1, 59.3 μM) and accompanied with magnetic stirring. And then, 0.5 mM of NaCl was introduced to the reaction solutions. The dark control was conducted in the same simulator without light irradiation. At selected time point, samples were taken from the reaction tubes to determine the UV-vis spectra, concentration of dissolved Ag (Ag(dis)) and zeta potential of particles. In addition, the H2O2 mediated oxidation experiments were conducted after the characterization of UV-vis spectra. H2O2 (200 mg L−1) was introduced into the sample, and then the change of absorbance over time at the maximum absorption wavelength was monitored immediately. Ag(dis) was separated from Ag NPs by Amicon centrifugal ultrafilters (Amicon Ultra-15 3K, Millipore, Billerica, MA). Samples were centrifuged at 15000 rpm for 10 min in centrifugal ultrafilters. All Ag in collected solutions was defined as Ag(dis) in this study and the concentration of Ag(dis) was measured by ICP-MS after acidifying with HNO3. It was worth mentioning that some tiny Ag NPs with a size smaller than the pore size of the filter membrane might be generated during light irradiation. Hence, Ag(dis) in this study included mainly free Ag+ and some tiny Ag NPs that could pass the filter membrane. After 9 h of light and dark treatment, samples were also characterized by TEM to monitor the change of morphology of Ag NPs. X-ray diffraction (XRD) of Ag NPs before and after being incubated with NaCl in dark and light conditions was collected by an X-ray diffract meter (D-8 Advance, Bruker).
Results and discussion
Characterization of Ag NPs
TEM images of prepared three sizes Ag NPs were showed in Fig. 1. All of the prepared Ag NPs were in quasi-spherical shape and presented monodispersed. TEM size distributions of synthesized Ag NPs were statistically obtained, which ranged in diameters from 13 to 25 nm, 34 to 45 nm, and 51 to 61 nm, respectively. And the average diameters were 20 ± 6 nm (nAg-20), 40 ± 6 nm (nAg-40) and 57 ± 5 nm (nAg-57). In situ DLS showed hydrodynamic diameters of 43.6 nm, 61.2 nm and 82.2 nm with PDI of 0.184, 0.135 and 0.088, respectively, suggesting that the three sizes of Ag NPs were well monodispersed without significant aggregation. Compared with TEM size, the larger hydrodynamic diameter of Ag NPs is probably due to the presence of some aggregates and PVP coating which decrease the diffusivity of particles.25 The UV-vis measurements revealed that the surface plasmon resonance (SPR) peaks of synthesized Ag NPs were single peak with sharp shape, which also demonstrated that the products were well size-distributed and highly monodispersed.30 In addition, the absorbance peaks of Ag NPs were 401, 416, and 435 nm for nAg-20, nAg-40, and nAg-57, respectively. The position of extinction peaks presented red-shifting as particle size increased, which was in consistence with previous studies.26 The zeta potential of prepared Ag NPs was −7.05 mV, −8.37 mV and −10.4 mV for nAg-20, nAg-40, and nAg-57, respectively. Similar zeta potential values have been also reported in many studies on PVP-coated Ag NPs.22,31–33 The negative charge of PVP-coated Ag NPs might be attributed to the adsorption of some side products on the surface of Ag NPs during the synthesis of Ag NPs.34 The surface area of single Ag NPs increases with the increase of particle size. Thus, more side products might adsorb on the surface of large Ag NPs, resulting in the zeta potential of large Ag NPs become more negative than small one.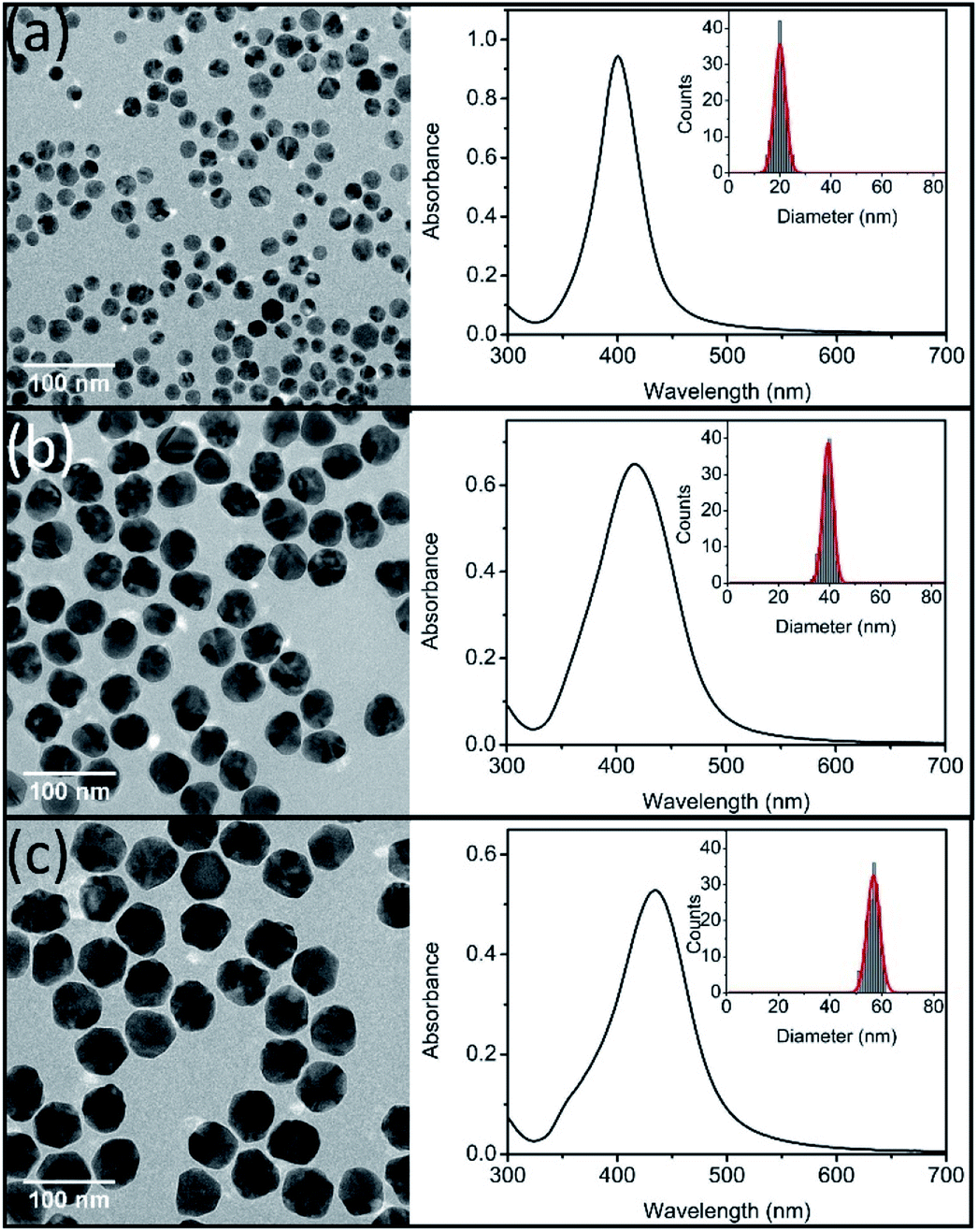 | ||
| Fig. 1 TEM images (left), UV-vis spectra (right) and size distributions (inset) of synthesized Ag NPs (nAg-20 (a), nAg-40 (b), and nAg-57 (c)). | ||
Effect on UV-vis spectra of Ag NPs
The evolution of UV-vis spectra of Ag NPs was monitored during 9 h of treatment in 0.5 mM NaCl. As illustrated in Fig. S1,† in dark conditions, a slight decline was observed in the absorption peak intensity of the three sizes of Ag NPs. It has been reported that the addition of electrolytes (such as NaCl) could promote the dissolution of Ag NPs.23,35 In the presence of electrolytes, Ag+ absorbed on Ag NPs would be redistributed and/or replaced by the electrolyte ions and the dissolution of surface oxide layer was promoted.23 This process also exposed the internal metallic Ag, leading to the enhancement of further dissolution of Ag NPs,35 which should be responsible for the slight decrease of absorption peak (Fig. S1†). However, under light irradiation, it was interesting to observe that the absorption peak of Ag NPs didn't decline in the presence of 0.5 mM NaCl for nAg-40 and nAg-57, and even the absorption peak of nAg-20 presented a slight increase (Fig. 2). To further investigate the reason for the evolution of absorption peak during light treatment, the experiments were conducted in ultrapure water (to check the role of light) and 0.5 mM NaNO3 (to check the role of Na+). The results showed that the absorption peak of Ag NPs presented obvious decline after exposing to light, especially in the presence of NaNO3 (Fig. S2†). Therefore, the abnormal evolution of Ag NPs absorption peak during light irradiation in 0.5 mM NaCl should be attributed to the presence of Cl−.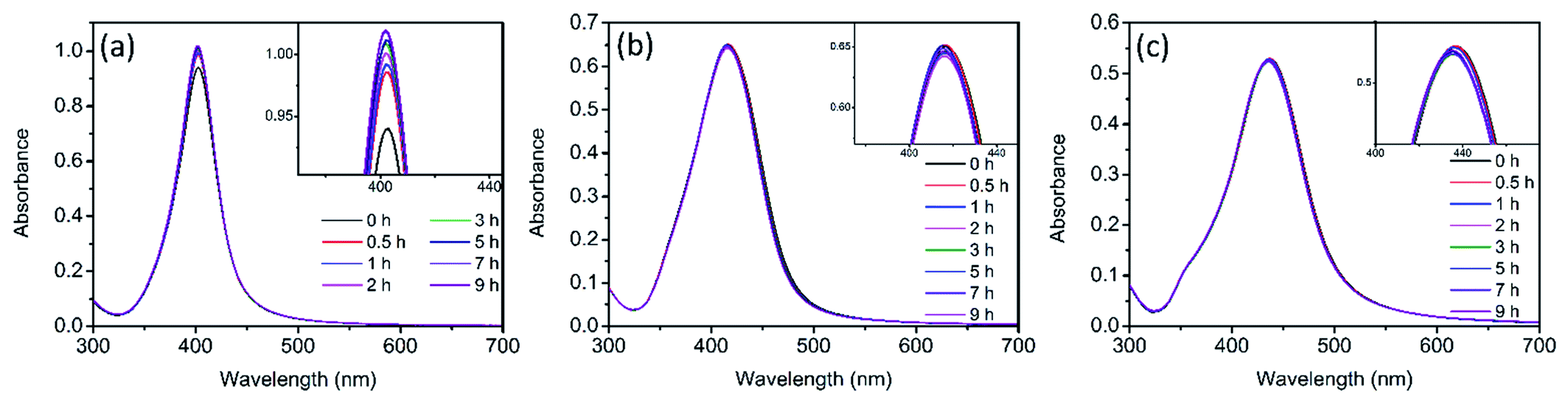 | ||
| Fig. 2 UV-vis spectra of Ag NPs (nAg-20 (a), nAg-40 (b), nAg-57 (c)) over time during light irradiation in 0.5 mM Cl−. | ||
Effect on Ag(dis) concentration
Fig. 3 showed the evolution of Ag(dis) concentration for Ag NPs suspensions in 0.5 mM Cl− solution during 9 h of dark and light treatment. It should first notice that more dissolved Ag presented in pristine Ag NPs suspensions with the decreasing of Ag NPs size. At the beginning, 315.2 μg L−1 of Ag(dis) was detected for nAg-20, while the concentration of Ag(dis) was 191.1 μg L−1 and 132.8 μg L−1 for nAg-40 and nAg-57. From Fig. 3, obviously, the concentration of Ag(dis) exhibited different changing trend in dark and light conditions. During dark treatment, obvious decrease in the concentration of Ag(dis) was observed. Many studies have suggested that the introduction of Cl− to Ag NPs suspensions would lead to the generation of AgCl coating on Ag NPs.22,32 In our study, XRD presented in Fig. S3† confirmed the generation of AgCl as evidenced by the appearing of diagnostic AgCl diffraction peaks after incubating with NaCl. Therefore, the decrease of Ag(dis) concentration should be resulted from the generation of AgCl. The introduction of NaCl was also believed to enhance the dissolution of Ag NPs. However, no increase in Ag(dis) concentration was observed, which might be ascribed to the strong affinity between Cl− and Ag+. The amount of Ag+ consumed by the formation of AgCl was much greater than that released from Ag NPs. Therefore, the concentration of Ag(dis) decreased after the addition of NaCl. In addition, the generation of a passivating layer (AgCl coating) on Ag NPs could remarkably inhibit the dissolution of Ag NPs.24 Hence, the decline in absorption peak of Ag NPs slowed down as the time of contact with NaCl increased (Fig. S1†). | ||
| Fig. 3 Ag(dis) concentration of different size Ag NPs over time during light and dark treatment in 0.5 mM Cl−. | ||
Under light irradiation, the evolution of Ag(dis) concentration was different from that in dark conditions. During light treatment, the concentration of Ag(dis) remarkably decreased and almost no Ag(dis) could be detected after only 3 h light treatment. Yin et al. have reported that the co-occurrence of Cl− could enhance the photo-conversion of Ag+ into Ag NPs owing to the formation of AgCl.36 AgCl, as a common photocatalyst, can be excited under UV irradiation and the photogenerated electrons can reduce the Ag+ of the AgCl lattice to Ag0. This process has been demonstrated in many studies.37–41 Therefore, we speculated that the great decreasing of Ag(dis) concentration after exposing to simulated sunlight irradiation might be attributed to the Cl−-assisted photoreduction of free Ag+. After 9 h of light irradiation, much weaker AgCl XRD patterns than that under dark were observed (Fig. S3†), which was consistent with our speculation. Plenty of studies have reported that dissolved Ag (commonly regarded as the source of Ag NPs toxicity) exhibited highly toxicity to many aquatic organisms.11,22,42 Hence, in aquatic environment, the toxicity of Ag NPs is expected to be diminished to a certain degree.
Effect on the morphology of Ag NPs
TEM images of Ag NPs in 0.5 mM Cl− electrolyte solutions were obtained after 9 h of treatment in dark and light irradiation. In dark control, no obvious change on Ag NPs was observed (Fig. S4†). However, after exposing to light irradiation, many tiny nanoparticles were found in TEM images (Fig. 4). On the basis of discussion above, these tiny nanoparticles were believed to be newly produced Ag NPs from the Cl−-assisted photoreduction of Ag+. Since much Ag+ presented in pristine nAg-20 suspensions (Fig. 3), plenty of tiny Ag NPs were formed in nAg-20 after light treatment, which should also be responsible for the slight increase of absorption peak of nAg-20 (Fig. 2a). Some tiny Ag NPs were also generated in nAg-40 and nAg-57, but no increase in the absorption peak was observed (Fig. 2b and c). We speculated that this might be attributed to the large difference between the size of re-formed tiny Ag NPs and pristine Ag NPs in nAg-40 and nAg-57. Hence, the absorbance of these tiny Ag NPs had little contribution to the absorption peak of nAg-40 and nAg-57. From TEM images, we also found that some Ag NPs adhered to each other and formed irregularly shaped fusion aggregates. The photoreduction of Ag+ to Ag0 under light irradiation might generate nanobridges to connect nearby particles together and led to the formation of fusion aggregates. Much more fusion aggregates were observed in nAg-20, which should be resulted from the presence of much higher Ag(dis) concentration and particles density in nAg-20. | ||
| Fig. 4 TEM images of Ag NPs (nAg-20 (a), nAg-40 (b), and nAg-57 (c)) after 9 h of light irradiation in 0.5 mM Cl−. | ||
Effect on the surface charge of Ag NPs
Zeta potential measurements for Ag NPs in Cl− electrolyte solutions during 9 h of dark and light treatment were measured. As displayed in Fig. 5, zeta potential of Ag NPs presented increasing (more negative) in the dark conditions. Since the concentration of Cl− (0.5 mM) was much higher than the concentration of Ag+ (the concentration of total Ag in this study was 59.3 μM), AgCl was most likely terminated by Cl−, resulting in negative charge of AgCl. With the formation of AgCl coating on the surface of Ag NPs, zeta potential of Ag NPs increased. Similar results have also been reported in many studies.22,32,43 Compare to dark conditions, more increase in the zeta potential of Ag NPs was observed under light irradiation (Fig. 5). Two possible reasons might be responsible for the remarkable increase of zeta potential during light treatment: (1) light irradiation promoted the formation of AgCl on the surface of Ag NPs. The dissolution of Ag NPs has been reported to be obviously accelerated under light.13 Ag+ was captured by Cl− once released from Ag NPs, promoting the formation of AgCl coating on Ag NPs. However, as discussed above, AgCl might also be photoreduced to Ag0 under sunlight and the weak AgCl diffraction peak was observed in XRD patterns after light irradiation (Fig. S3†). It suggested that just a little AgCl existed. Hence, promoting the formation of AgCl layer under light seemed not to be the reason for the remarkable increasing of zeta potential. (2) Light irradiation promoted the adsorption of Cl− on Ag NPs. Cl−, as a nucleophilic reagent, could donate a pair of electrons to the unoccupied orbital of Ag and adsorb on the surface of Ag NPs. The adsorption of Cl− on Ag NPs might be promoted under light irradiation, thereby leading to greatly increasing in zeta potential.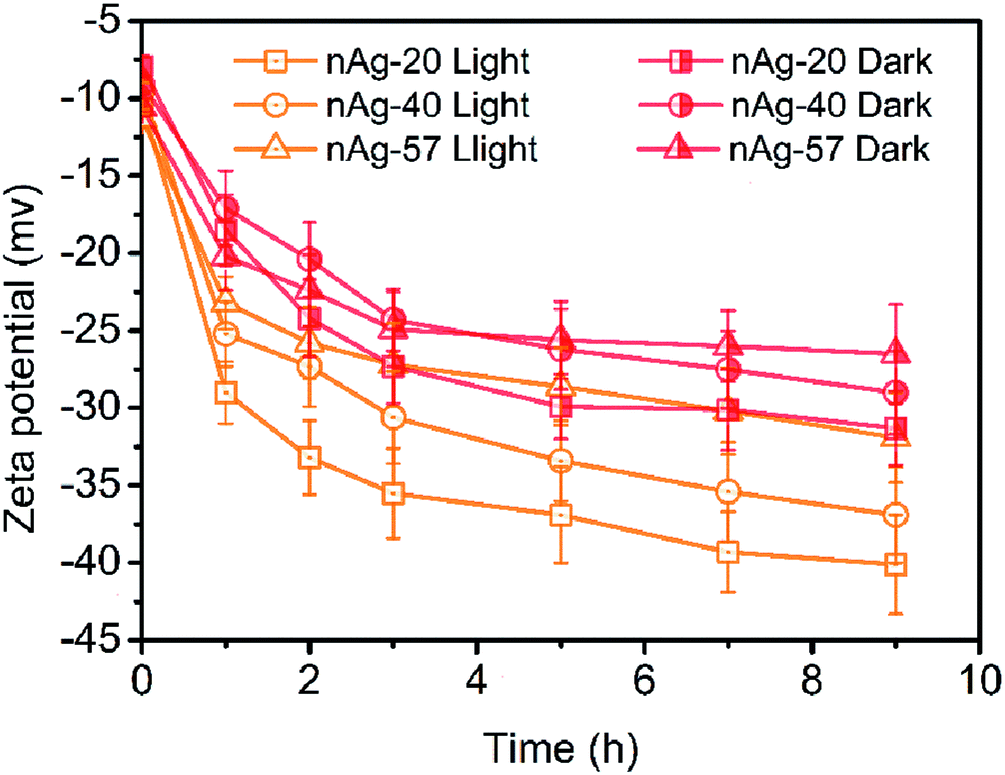 | ||
| Fig. 5 Zeta potential of Ag NPs over tie during light and dark treatment in 0.5 mM Cl−. | ||
The increase in zeta potential of Ag NPs could enhance the stability of Ag NPs due to the extra electrostatic repulsion. Although some fusion aggregates were observed after light irradiation in our study, the possibility of the formation of fusion aggregates in real aquatic environment was significantly reduced due to much lower concentration of Ag NPs presented in real aquatic environment than that used in this study. Thus, it is expected that Ag NPs may be dispersed as a single particle and suspended in natural water for a long time, displaying a persistent potential risk to aquatic vertebrates and invertebrates.
Study of AgCl on the surface of Ag NPs
In the presence of Cl−, light irradiation was found to exhibit a remarkable influence on various physicochemical properties of Ag NPs. In dark conditions, AgCl layer was believed to generate on the surface of Ag NPs after the addition of Cl−. However, the change on the surface of Ag NPs during light treatment was still unclear. The presence of AgCl on the surface of Ag NPs required further investigation. As a passivating layer, the AgCl coating could efficiently block the contact of inner Ag NPs with oxidants, thereby preventing Ag NPs from further oxidation.24 Here, we used H2O2 mediated oxidation experiments to verify the presence of AgCl layer. After introducing H2O2, a strong oxidant, the change of absorbance at the maximum absorption wavelength was recorded, and the result was shown in Fig. 6. The blank control (pristine Ag NPs) was conducted in 0.5 mM NaNO3 to ensure the same ionic strength, and obvious decrease in the absorbance was observed. It should be noted that oxidation of a thin surface layer of Ag NPs by H2O2 might result in the deterioration of the free electron density on the surface, thereby strong dampening of SPR. The major part of Ag NPs remains unoxidized but no absorbance can be observed. Thus, the decrease of absorbance might be attributed to the formation of “white” Ag NPs rather than the dissolution of Ag NPs. To verify whether Ag NPs was dissolved by H2O2 in our system, the size and morphology of nAg-57 after being treated with H2O2 was investigated by TEM (Fig. S5†). The size of residual particles significantly decreased and the morphology of particles was greatly damaged, indicating that the actual oxidation of Ag NPs occurred. And the concentration of dissolved Ag increased from 153.6 μg L−1 to 2694.5 μg L−1 after reacting with H2O2 for 180 s. Hence, the decreasing of absorbance should be resulted from actual dissolution of Ag NPs. Therefore, the dissolution of Ag NPs could be qualitatively reflected by the decreasing of absorbance, and He et al. also used this method to characterize the decay of Ag NPs after the introduction of H2O2.44 In dark control, as shown in Fig. S6,† three sizes of Ag NPs presented remarkable resistance to the oxidation of H2O2 after the addition of Cl− and just a little decreasing in absorbance was observed, which further confirmed the formation of AgCl layer on the surface of Ag NPs. Moreover, during 9 h incubation of Ag NPs with Cl− in dark, samples, taken at various time points, exhibited almost the same decreasing trend in absorbance after introducing H2O2. The sample at 0 h was a sample taken immediately after the introduction of Cl− to Ag NPs suspensions. Therefore, the AgCl layer was supposed to be formed on the surface of Ag NPs as soon as Ag NPs contacted with Cl−. The strong affinity between Cl− and Ag+ should be responsible for the rapid formation of AgCl layer.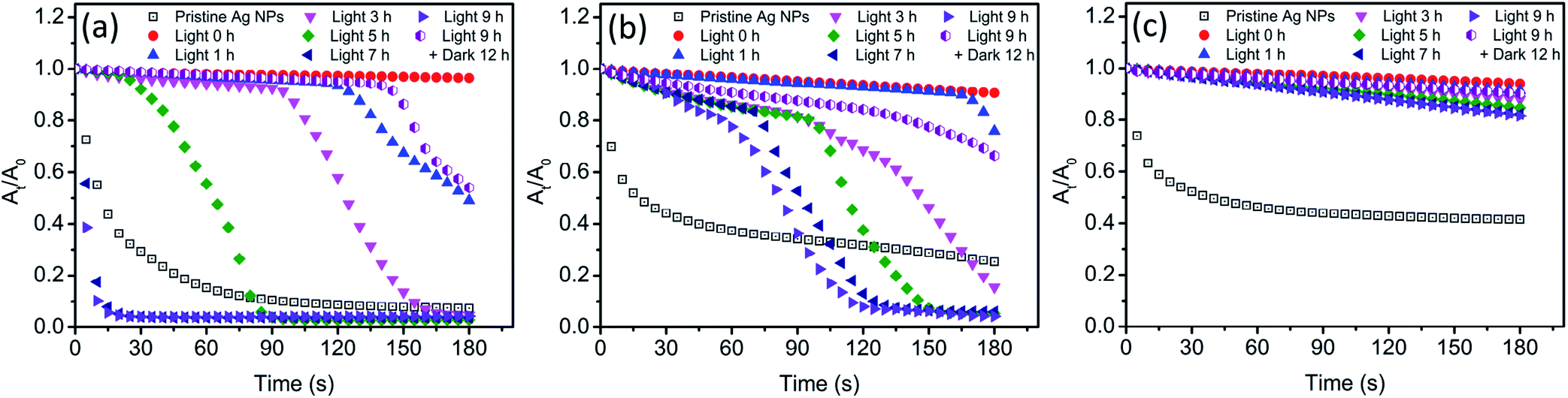 | ||
| Fig. 6 The residual rate of Ag NPs (nAg-20 (a), nAg-40 (b), nAg-57 (c)) absorbance (At/A0) with time after introducing H2O2 (200 mg L−1), where A0 and At were the absorbance of Ag NPs at the maximum absorption wavelength before and after oxidation period of time, respectively. Before the introduction of H2O2, Ag NPs were incubated with 0.5 mM Cl− under light irradiation. | ||
After light irradiation, the antioxidant ability of Ag NPs was remarkably reduced and the absorbance of Ag NPs obviously decreased (Fig. 6). It seemed that the AgCl layer on Ag NPs was destroyed during light treatment. The photoreduction of AgCl as proposed above was believed to be the reason for the destruction of AgCl layer. Hence, the inner Ag NPs was exposed to outside, which allowed H2O2 to erode along the exposed part to the interior Ag NPs. And then the core–shell structure of Ag–AgCl was further destroyed. With the loss of AgCl protection, Ag NPs was significantly oxidized by H2O2, leading to a sharp decline in the absorbance of Ag NPs (Fig. 6a and b). Meanwhile, we also noticed that this sharp decline was much more remarkable when compared with the decreasing of absorbance in blank control. Moreover, after 7 h of irradiation, the decreasing rate of nAg-20 absorbance was found to be obviously higher than blank control. The results suggested that Ag NPs was easier to be oxidized after lighting treatment in the presence of Cl−. Previous study has demonstrated that the oxidation of Ag NPs would be facilitated in the presence of nucleophiles owing to the adsorption of nucleophiles on Ag NPs, and the oxidation rate was dependent on the strength of Ag-nucleophile.45 Hence, in our study, the enhancement of H2O2-mediated dissolution of Ag NPs might be resulted from the adsorption of Cl−. It should also be the reason for the remarkable increase in the value of Ag NPs zeta potential during light irradiation (Fig. 5). Due to the strong interaction between Cl− and Ag NPs, the adsorption of Cl− might also replace the group of PVP absorbed on Ag NPs, leading to the detachment of PVP.46 As the prolonging of irradiation time, the time point when Ag NPs began to be significantly dissolved by H2O2 was constantly advanced (Fig. 6a and b). After 7 h of irradiation, nAg-20 was remarkably dissolved as soon as the introducing of H2O2, which indicated that almost no AgCl presented on Ag NPs. It illustrated that the degree of the damage of the AgCl layer during light irradiation was positively related to the irradiation time. In addition, we also found that the size of Ag NPs had a great effect on the destruction of AgCl layer during light irradiation. As Ag NPs size increased, the effect of light irradiation on H2O2-mediated decrease in Ag NPs absorbance was weakened. After 9 h of irradiation, just a slight enhancement was observed in the decreasing of nAg-57 absorbance, which implied that only slight photoreduction was occurred on AgCl layer. It might also be one of the reasons that less fusion aggregates were observed in the TEM images of large Ag NP than small one (Fig. 4). Under light, Ag NPs absorb visible photons and produce photogenerated electrons and holes, which could be separated by the surface plasmon resonance (SPR)-induced local electromagnetic field.38,41 The photogenerated electrons might transfer from Ag NPs to AgCl layer, promoting the decomposition of AgCl. Small Ag NPs absorb shorter wavelength light, with higher energy, than large Ag NPs, inducing more intense SPR. We speculated that photogenerated electrons were easier to transfer from small Ag NPs to AgCl layer, and then the AgCl layer on small Ag NPs was destroyed more seriously during light treatment. After 9 h of irradiation, the light was removed and samples were kept in dark for 12 h. Then, we found that the antioxidant ability of Ag NPs was enhanced and the H2O2-mediated decreasing of Ag NPs absorbance was obviously inhibited compare with the sample after 9 h of irradiation, which implied that AgCl was re-formed on the surface of Ag NPs in dark conditions. During the incubation in dark, surface Ag atoms were expected to be oxidized by dissolved O2 and then reacted with Cl− to form AgCl, which should be the reason of the re-formation of AgCl layer.
Schematic diagram
Based on the experimental results and discussion above, a possible schematic diagram was proposed to describe the change on Ag NPs after exposing to Cl− containing water under light and dark conditions (Fig. 7). In the presence of Cl−, Ag+ was captured by Cl− and a AgCl layer rapidly coated on the surface of Ag NPs. This process also resulted in a decrease in dissolved Ag concentration and an increase in zeta potential of Ag NPs. The formation of the AgCl layer, a passivating coating, could prevent Ag NPs from further oxidation, even in the presence of a strong oxidant, H2O2. However, under sunlight, the AgCl layer was destroyed due to photoreduction. AgCl could be excited by the UV light in sunlight and produced photoelectrons. The photogenerated electrons reduced the Ag+ of the AgCl lattice to form Ag0. In addition, Ag NPs absorbed the visible light in sunlight and produced photoelectrons by SPR. These photoelectrons might transfer from Ag NPs to AgCl layer, promoting the photoreduction of AgCl. The photoreduction of AgCl led to a great decrease in dissolved Ag concentration and the formation of some tiny Ag NPs, as well as the destruction of AgCl layer on Ag NPs. The destruction of AgCl layer was greatly affected by irradiation time and Ag NPs size. With the prolonging of irradiation time, the AgCl layer was more seriously destroyed. And the AgCl layer on small Ag NPs was more easily photoreduced than large one. With the loss of AgCl protection, inner Ag NPs was exposed to the outside and could be oxidized by external oxidants (like H2O2 and dissolved O2). Under sunlight, the adsorption of Cl− on Ag NPs was also promoted, which led to a great increase in the zeta potential of Ag NPs. Moreover, Cl−, adsorbed on Ag NPs, would donate a pair of electrons to the unoccupied orbital of Ag, shifting the Fermi level of Ag NPs upward.45 Ag NPs with higher Fermi level was more easily to loss e−. Therefore, the oxidation of Ag NPs (nAg-20 and nAg-40) induced by H2O2 was obviously enhanced after light treatment. Unlike the violent reaction between H2O2 and Ag NPs, the reaction of dissolved O2 with Ag NPs was relatively mild, which allowed surface silver atoms to interact with adsorbed Cl− after being oxidized. Therefore, after removing light, the AgCl layer gradually re-formed on Ag NPs owing to the oxidation of dissolved O2.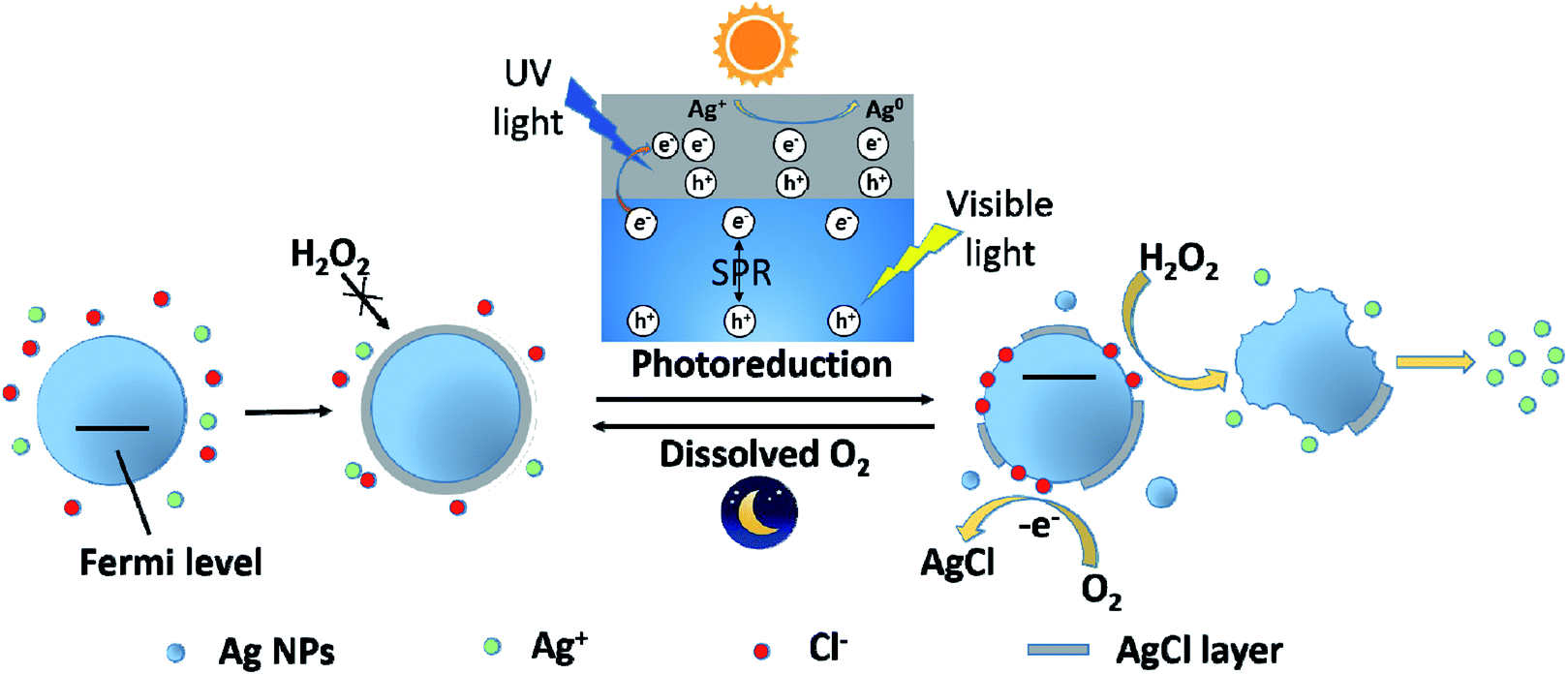 | ||
| Fig. 7 Schematic diagram illustrating the photoreduction and re-formation of AgCl layer on the surface of Ag NPs under light and dark conditions. | ||
Conclusion
In aquatic environment, Ag NPs are inevitably affected by various environmental factors, such as sunlight and Cl−. Our study demonstrated that light irradiation had a great impact on various physicochemical properties of Ag NPs in the presence of Cl−. After exposing to Cl−, AgCl layer was rapidly formed on the surface of Ag NPs, leading to a decrease in the concentration of dissolved Ag and an increase in zeta potential of Ag NPs. Under light irradiation, the decrease in dissolved Ag concentration was obviously enhanced owing to the photoreduction of AgCl. The AgCl layer on Ag NPs was also destroyed, and the adsorption of Cl− on Ag NPs was promoted, resulting in the great increase of zeta potential. In addition, we also found that the destruction of AgCl layer during light treatment was significantly affected by Ag NPs size. AgCl layer on large Ag NPs was much more difficult to be photoreduced than that on small one. Due to the protection of AgCl layer, large Ag NPs are expected to be more stable and exist longer time than small one in aquatic environment. After removing light, surface silver atoms of Ag NPs were gradually oxidized by dissolved O2 and AgCl layer re-formed on the surface of Ag NPs. It was suggested that the destruction of AgCl layer during daytime and re-formation at night was a circle process and Ag NPs might present different states in aquatic environment during daytime and night.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key Research and Development Project (Grant 2019YFC0408801), the Major National Water Pollution Control and Management Project (Grant 2014ZX07405-003, 2017ZX07201001).References
- M. N. Nadagouda, T. F. Speth and R. S. Varma, Acc. Chem. Res., 2011, 44, 469–478 CrossRef CAS PubMed.
- T. Benn, B. Cavanagh, K. Hristovski, J. D. Posner and P. Westerhoff, J. Environ. Qual., 2010, 39, 1875–1882 CrossRef CAS PubMed.
- Q. J. Huang and Y. Zhu, Adv. Mater. Technol., 2019, 4(5), 1800546 CrossRef.
- F. Lu and D. Astruc, Coord. Chem. Rev., 2018, 356, 147–164 CrossRef CAS.
- H. Y. Jin, C. X. Guo, X. Liu, J. L. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- N. C. Mueller and B. Nowack, Environ. Sci. Technol., 2008, 42, 4447–4453 CrossRef CAS PubMed.
- T. D. Huang, M. H. Sui, X. Yan, X. Zhang and Z. Yuan, Colloids Surf., A, 2016, 509, 492–503 CrossRef CAS.
- E. J. Gubbins, L. C. Batty and J. R. Lead, Environ. Pollut., 2011, 159, 1551–1559 CrossRef CAS.
- L. Y. Yin, Y. W. Cheng, B. Espinasse, B. P. Colman, M. Auffan, M. Wiesner, J. Rose, J. Liu and E. S. Bernhardt, Environ. Sci. Technol., 2011, 45, 2360–2367 CrossRef CAS PubMed.
- K. J. Lee, P. D. Nallathamby, L. M. Browning, C. J. Osgood and X. H. N. Xu, ACS Nano, 2007, 1, 133–143 CrossRef CAS.
- X. Y. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-Kim and J. N. Meyer, Environ. Sci. Technol., 2012, 46, 1119–1127 CrossRef CAS.
- P. V. AshaRani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- N. Grillet, D. Manchon, E. Cottancin, F. Bertorelle, C. Bonnet, M. Broyer, J. Lerme and M. Pellarin, J. Phys. Chem. C, 2013, 117, 2274–2282 CrossRef CAS.
- Y. Li, W. Zhang, J. F. Niu and Y. S. Chen, Environ. Sci. Technol., 2013, 47, 10293–10301 CAS.
- Y. W. Cheng, L. Y. Yin, S. H. Lin, M. Wiesner, E. Bernhardt and J. Liu, J. Phys. Chem. C, 2011, 115, 4425–4432 CrossRef CAS.
- J. P. Shi, B. Xu, X. Sun, C. Y. Ma, C. P. Yu and H. W. Zhang, Aquat. Toxicol., 2013, 132, 53–60 CrossRef PubMed.
- W. C. Hou, B. Stuart, R. Howes and R. G. Zepp, Environ. Sci. Technol., 2013, 47, 7713–7721 CrossRef CAS PubMed.
- W. Zhou, Y. L. Liu, A. M. Stallworth, C. Ye and J. J. Lenhart, Environ. Sci. Technol., 2016, 50, 12214–12224 CrossRef CAS PubMed.
- S. J. Yu, Y. G. Yin, J. B. Chao, M. H. Shen and J. F. Liu, Environ. Sci. Technol., 2014, 48, 403–411 CrossRef CAS PubMed.
- X. Y. Yang, S. H. Lin and M. R. Wiesner, J. Hazard. Mater., 2014, 264, 161–168 CrossRef CAS PubMed.
- B. M. Angel, G. E. Batley, C. V. Jarolimek and N. J. Rogers, Chemosphere, 2013, 93, 359–365 CrossRef CAS PubMed.
- C. Levard, S. Mitra, T. Yang, A. D. Jew, A. R. Badireddy, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2013, 47, 5738–5745 CrossRef CAS PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26, 16690–16698 CrossRef CAS PubMed.
- C. M. Ho, S. K. W. Yau, C. N. Lok, M. H. So and C. M. Che, Chem.–Asian J., 2010, 5, 285–293 CrossRef CAS PubMed.
- R. Ma, C. Levard, S. M. Marinakos, Y. W. Cheng, J. Liu, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752–759 CrossRef CAS PubMed.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- O. S. Ivanova and F. P. Zamborini, J. Am. Chem. Soc., 2010, 132, 70–72 CrossRef CAS PubMed.
- A. R. Poda, A. J. Kennedy, M. F. Cuddy and A. J. Bednar, J. Nanopart. Res., 2013, 15(5), 1673 CrossRef.
- N. G. Bastus, F. Merkoci, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef CAS.
- L. Li, J. Sun, X. R. Li, Y. Zhang, Z. X. Wang, C. R. Wang, J. W. Dai and Q. B. Wang, Biomaterials, 2012, 33, 1714–1721 CrossRef CAS PubMed.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS PubMed.
- A. M. E. Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef PubMed.
- J. Farkas, P. Christian, J. A. Gallego-Urrea, N. Roos, M. Hassellov, K. E. Tollefsen and K. V. Thomas, Aquat. Toxicol., 2011, 101, 117–125 CrossRef CAS.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2012, 28, 1095–1104 CrossRef CAS PubMed.
- Y. G. Yin, W. Xu, Z. Q. Tan, Y. B. Li, W. D. Wang, X. R. Guo, S. J. Yu, J. F. Liu and G. B. Jiang, Environ. Pollut., 2017, 220, 955–962 CrossRef CAS PubMed.
- L. Han, P. Wang, C. Z. Zhu, Y. M. Zhai and S. J. Dong, Nanoscale, 2011, 3, 2931–2935 RSC.
- R. F. Dong, B. Z. Tian, C. Y. Zeng, T. Y. Li, T. T. Wang and J. L. Zhang, J. Phys. Chem. C, 2013, 117, 213–220 CrossRef CAS.
- P. Wang, B. B. Huang, Y. Dai and M. H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- L. H. Dong, D. D. Liang and R. C. Gong, Eur. J. Inorg. Chem., 2012, 3200–3208, DOI:10.1002/ejic.201200172.
- B. Z. Tian, R. F. Dong, J. M. Zhang, S. Y. Bao, F. Yang and J. L. Zhang, Appl. Catal., B, 2014, 158, 76–84 CrossRef.
- Z.-m. Xiu, Q.-b. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS.
- D. H. Lin, S. Ma, K. J. Zhou, F. C. Wu and K. Yang, J. Colloid Interface Sci., 2015, 450, 272–278 CrossRef CAS PubMed.
- D. He, S. Garg and T. D. Waite, Langmuir, 2012, 28, 10266–10275 CrossRef CAS PubMed.
- P. Mulvaney, T. Linnert and A. Henglein, J. Phys. Chem., 1991, 95, 7843–7846 CrossRef CAS.
- Y. Tang, W. He, G. Y. Zhou, S. X. Wang, X. J. Yang, Z. H. Tao and J. C. Zhou, Nanotechnology, 2012, 23(35), 355304 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis spectra of Ag NPs during dark treatment in 0.5 mM NaCl, UV-vis spectra of Ag NPs during light treatment in ultrapure water and 0.5 mM NaNO3, XRD patterns of Ag NPs, TEM images of Ag NPs after treating with 0.5 mM NaCl for 9 h in dark conditions and the residual rate of Ag NPs absorbance with time after introducing H2O2 (dark control). See DOI: 10.1039/c9ra09261g |
| This journal is © The Royal Society of Chemistry 2020 |